
Abstract
Disseminated intravascular coagulation (DIC) is an acquired syndrome characterized by systemic inflammation, activation of coagulation, insufficient anticoagulation control, increased followed by inhibition of fibrinolysis, and endothelial injury. Damage-associated molecular patterns (DAMPs), such as histones, and neutrophil extracellular traps, comprising neutrophil DAMPs and neutrophil elastase, induce all changes observed in DIC and thus, play pivotal roles in the pathogenesis of DIC. In DIC, platelet dysfunction and consumption coagulopathy induce oozing-type bleeding, enhancing surgical bleeding. A decreased oxygen delivery to cells and tissues due to microvascular thrombosis associated with endothelial injury and direct cellular injury by histones induces multiple organ dysfunction syndrome (MODS). Trauma has been a leading cause of DIC for over 50 years. DIC influences the outcome of trauma patients in two ways: a fibrinolytic phenotype immediately after injury exacerbates bleeding via increased fibrin(ogen)olysis, which progresses to a thrombotic phenotype giving rise to MODS in the late stage. Many studies have shown an increased transfusion requirement and higher rates of MODS and mortality in DIC than in non-DIC. To improve outcomes, DIC and trauma should be simultaneously treated, targeting patients with definitively diagnosed DIC and a high disease severity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Coagulopathy
- Disseminated intravascular coagulation (DIC)
- Intensive care unit (ICU)
- Organ dysfunction
- Sepsis
- Trauma
- Treatment
In this chapter, the readers will learn the following points concerning DIC in trauma:
-
The definition, diagnosis, phenotypes, and time courses of DIC.
-
The pathophysiology of cytokines, PARs, DAMPs, and NETs involved in DIC.
-
The multiple effects of thrombin on platelets, coagulation, fibrinolysis, and inflammation.
-
How DIC-induced MODS and critical bleeding affect a patient’s outcome.
-
How to manage DIC in order to improve a patient’s outcome.
1 Introduction
Disseminated intravascular coagulation (DIC) is an acquired syndrome characterized by systemic activation of coagulation not restricted to the site of insults and can be caused by non-infectious and infectious insults [1]. The two major insults that evoke DIC are trauma and sepsis, which induce systemic inflammatory response syndrome (SIRS) [1, 2]. In the early 1990s, SIRS was considered the main cause of organ dysfunction affecting a patient’s outcome [2]. Based on this concept, many randomized controlled trials targeting the control of SIRS were performed; however, none of these trials managed to achieve their aims, thereby suggesting that a change in the treatment strategies for controlling only SIRS [3]. Concurrently with this paradigm shift, tight molecular links between inflammation and coagulation were detected, in which thrombin plays a central role, resulting in multiple organ dysfunction syndrome (MODS) and eventually death [4]. DIC represents dysregulated inflammatory and coagulofibrinolytic responses to the insults such as trauma and sepsis; therefore, DIC can induce the development of MODS via the bidirectional interplay between inflammation and coagulation [1].
Penner summarized the trauma studies, including head trauma, published during the 1990s [5]. In those studies, DIC patients showed high inflammatory cytokines levels and increased systemic thrombin generation as measured by thrombin antithrombin complex (TAT) or prothrombin fragment 1 + 2 (PF1 + 2) immediately after trauma. These changes were associated with MODS and higher mortality rates than in non-DIC patients. In 2001, the International Society on Thrombosis and Haemostasis (ISTH) published a definition and diagnostic criteria for DIC [6]. This official communication positioned trauma (polytrauma, neurotrauma, fat embolism, etc.) as the main cause of DIC and stated that generalized inflammatory responses to insults with the release of cytokines from multiple inflammatory cells lead to extensive damage of the microvascular endothelium, which can result in organ dysfunction. Following this announcement, close relationships among trauma, inflammatory responses, microvascular disturbances, DIC, and MODS have been identified by the mid 2000s [7,8,9].
The ISTH again confirmed that tissue damage, including major trauma, is a cause of DIC with a high level of evidence about two decades after the first announcement [10]. DIC is an old disease with a history of over half a century; however, it is still a key disease to be recognized and diagnosed in the critical care setting [11]. In this chapter, the management of DIC in the intensive care unit (ICU) will be reviewed and discussed with the goal of improving the prognosis of critically ill trauma patients.
2 Trauma-Induced Coagulopathy and DIC
Trauma-induced coagulopathy is defined as the pre-stage of full-blown DIC, such as sepsis-induced coagulopathy. Three subcommittees of the ISTH published an official announcement, noting that dysregulated inflammatory and coagulofibrinolytic responses to trauma converge the trauma-induced coagulopathy in the final pathway of DIC [12] (Fig. 33.1). If trauma is sufficiently severe, however, DIC develops immediately after trauma without first proceeding through the stage of trauma-induced coagulopathy [13]. Expanding our understanding of these relationships between trauma-induced coagulopathy and DIC will likely provide therapeutic benefit to severely injured trauma patients.

Relationship between trauma-induced coagulopathy (TIC) and disseminated intravascular coagulation (DIC). TIC is defined as the pre-stage of full-blown DIC. If trauma is sufficiently severe, dysregulated inflammatory and coagulofibrinolytic responses to trauma converge in the final pathway of DIC. Exogenously induced secondary coagulopathies such as anemia-, hypothermia-, acidosis-, and dilution-induced coagulopathies, modify DIC
Another point of note is that trauma-induced coagulopathy comprises primary and secondary coagulopathies [12, 14] (Table 33.1). Trauma-itself-induced primary coagulopathy, namely DIC, is modified by the anemia-, dilution-, hypothermia-, and acidosis-induced secondary coagulopathies.
3 The Definition and Diagnosis
3.1 The Definition
The ISTH defined DIC as “DIC is an acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction” [6]. The key points of this definition are systemic thrombin generation not restricted to the insult’s site and endothelial cell activation followed by subsequent injury.
The ISTH further states that DIC is accompanied by the loss of tight junctions between endothelial cells which gives rise to capillary leak syndrome [6]. Excessive thrombin generation and endothelial injury with microvascular thrombus formation leads to decreased oxygen delivery to the cells and tissues and subsequent exhaustion of platelets and consumption of coagulation factors, causing MODS and oozing-type bleeding [6]. Therefore, DIC has long been recognized as “thrombohemorrhagic disorder” [15,16,17]. The definition of DIC established by the ISTH has provided a logical base concerning the pathophysiology of DIC.
3.2 The Diagnosis
The ISTH proposed overt DIC diagnostic criteria involving two steps; assessing underlying clinical conditions that may be associated with DIC and applying an algorithm for the diagnosis of DIC [6]. Before using the algorithm, a risk assessment to check whether or not a patient has an underlying disorder is mandatory. If a patient has such a disorder, algorithm may be applied. The ISTH overt DIC scoring system was prospectively validated in diverse patients including trauma admitted to the ICU [18]. The results confirmed a sufficient diagnostic accuracy of the ISTH scoring system for diagnosing DIC in ICU patient clinically suspected of having this syndrome.
To diagnose DIC during its latent period before progression to full-blown overt DIC, the ISTH proposed non-overt DIC diagnostic criteria as well [6]. However, the prospective validation of non-overt DIC diagnostic criteria failed to prove the progression of non-overt DIC to overt DIC [19, 20]. To overcome this drawback associated with non-overt DIC diagnostic criteria and the low sensitivity for diagnosing DIC using the overt DIC diagnostic algorithm, the Japanese Association for Acute Medicine (JAAM) established the JAAM DIC diagnostic criteria. The JAAM diagnostic criteria have been prospectively validated in a critical care setting several times [20,21,22]. These studies showed that the JAAM DIC diagnostic criteria were able to identify DIC patients with high sensitivity and moderate specificity, and that the DIC diagnosed according to the JAAM criteria progresses to the ISTH overt DIC. Of note, the JAAM DIC diagnostic criteria can be applied at an early stage of trauma as well as a late stage with acceptable validity for the DIC diagnosis [23,24,25]. The ISTH and JAAM DIC diagnostic criteria can be found elsewhere [6, 21].
4 Phenotypes and Time Courses
4.1 Phenotypes
DIC comprises fibrinolytic and thrombotic phenotypes [1, 26,27,28]. DIC essentially is a thrombotic phenotype, and that DIC with a fibrinolytic phenotype is defined as the coexistence of DIC and pathological systemic fibrin(ogen)olysis [26, 28]. A condition wherein one insult simultaneously evokes DIC (with a thrombotic phenotype) and pathological systemic fibrin(ogen)olysis is called DIC with a fibrinolytic phenotype (Fig. 33.2).

Two phenotypes of disseminated intravascular coagulation (DIC). DIC essentially is a thrombotic phenotype; DIC with a fibrinolytic phenotype is defined as the coexistence of DIC and pathological systemic fibrin(ogen)olysis due to tissue hypoperfusion, systemic hypoxia/ischemia, etc. A condition wherein one insult simultaneously evokes DIC (with thrombotic phenotype) and pathological systemic fibrin(ogen)olysis is called DIC with a fibrinolytic phenotype. Importantly, massive thrombin generation due to the activation of the tissue factor-dependent coagulation pathway and insufficient anticoagulation controls always underlie both types of DIC. PAI-1 plasminogen activator ingibitor-1, TFPI tissue factor pathway inhibitor, t-PA tissue-type plasminogen activator. (Modified with permission (Creative Commons Attribution International License) [28])
Typical conditions that induce DIC with a fibrinolytic phenotype are acute promyelocytic leukemia [29], a long hypoxic state (e.g., asphyxia and drowning) [30], cardiac arrest and resuscitation [31], postpartum hemorrhagic shock [32], isolated traumatic brain injury (iTBI) [33, 34], and severe trauma [35, 36]. The assembly of plasminogen and tissue-type plasminogen activator (t-PA) on promyelocytic leukemia cell surface-expressed annexin II promotes the conversion of plasminogen to plasmin [1, 29]. Common pathomechanisms increasing fibrin(ogen)olysis in asphyxia, drowning, cardiac arrest and resuscitation, postpartum hemorrhagic shock, and severe trauma are prolonged hypoxia- and ischemia-induced massive thrombin generation as well as marked t-PA release from endothelial Waibel-Palade bodies [1, 36,37,38]. Neurons and other cell types within the central nervous system synthetize and store t-PA in the granules and are rich in tissue factor, both of which are immediately released into the circulation in iTBI, causing DIC and systemic fibrin(ogen)olysis [39, 40]. All conditions, except for acute promyelocytic leukemia, aggravate fibrin(ogen)olysis due to the time delay between the immediate release of t-PA and the delayed expression of plasminogen activator inhibitor-1 (PAI-1) mRNA [1, 7, 13, 35, 36].
4.2 Time Courses
The time courses in coagulofibrinolytic changes after trauma are shown in Fig. 33.3 [7, 35, 36]. The left side shows the physiologic state of hemostasis and wound healing, while the right side shows the pathological changes observed in DIC. These physiological and pathological states should be and can be distinguished using DIC diagnostic criteria. It is important to recognize, as the ISTH warned, that many published studies have discussed these two conditions without clear separation, confusing our understanding of trauma-induced coagulopathy and DIC [36, 41]. The main differences between these two time courses concerning the dynamics of thrombin generation and inhibition of fibrinolysis by PAI-1. Thrombin generation and inhibition of fibrinolysis transiently continues for several days under conditions of the normal hemostasis and wound healing but persist until DIC is improved under DIC conditions.

Schematic diagrams of the variations in thrombin activity (A, measured by fibrinopeptide A, FPA), plasmin activity (B, fibrinopeptide B β15–42, FPBβ15–42), fibrin formation and secondary fibrinolysis (C, D-dimer) from day 0 (in the emergency department) to day 4. Left, normal changes in hemostasis and wound healing. There are three phases of fibrinolysis: early activation, impairment (D, PAI-1: fibrinolytic shutdown), and reactivation. Normally, both the thrombin activity and PAI-1 are completely shut off by days 3–5 after trauma, followed by the reactivation of fibrinolysis. Right, pathological changes in DIC. There is a time delay between immediate t-PA-induced massive plasmin generation and the induction of PAI-1mRNA, which causes systemic hyperfibrin(ogen)olysis (asterisk, DIC with a fibrinolytic phenotype), followed by the impairment of fibrinolysis due to persistent elevation of PAI-1 released from endothelial cells via transcription (double asterisk, DIC with a thrombotic phenotype). Persistent and systemic thrombin generation always underlies these changes in fibrinolysis. DIC disseminated intravascular coagulation, PAI-1 plasminogen activator inhibitor-1, t-PA tissue-type plasminogen activator. (Modified with permission [7])
Figure 33.3 (right) further depicts the two phenotypes of DIC. The time delay between the immediate increases in plasmin generation due to t-PA release and delayed elevation of PAI-1 (single asterisk) indicates DIC with a fibrinolytic phenotype, and persistent increases in both thrombin generation and PAI-1 (double asterisk) indicate DIC with a thrombotic phenotype. Furthermore, DIC with a fibrinolytic phenotype exists only for a couple of hours after presentation to the emergency department [13, 31]; however, in cases of severe trauma, the fibrinolytic phenotype progresses to the thrombotic phenotype without complication of sepsis [42].
5 Pathophysiology
Although DIC develops in diverse underlying conditions, once initiated, the pathomechanisms that give rise to DIC are similar, regardless of the setting [1]. The modern pathophysiology of DIC and its characteristics in trauma were established around the 1990s [1, 5, 6, 35]. Recent advances have been described in studies which highlighted bidirectional interplay among innate immunity, inflammation, and coagulofibrinolytic responses, in which histones and neutrophil extracellular traps (NETs) play central roles, constituting the main pathophysiology of DIC [43, 44].
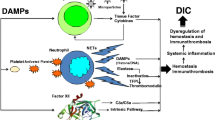
Trauma induces innate immune responses via the altered-self, danger-associated molecular patterns (DAMPs), which are recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), NOD-like receptors (NLRs), and RIG-I-like receptors (RLRs). Signal transductions of PRRs leads to the expressions of pro-inflammatory cytokines (TNF-α, IL-1, IL-6) and chemokines (IL-8) as well as anti-inflammatory cytokines (IL-10) [28, 45, 46]. Innate immune responses activate inflammation, coagulation, endothelium, and complement pathways to maintain body homeostasis with the balance of inflammation and anti-inflammation called SIRS and compensatory anti-inflammatory syndrome (CARS), respectively [2, 47]. At the site of insults such as trauma and sepsis, hemostatic thrombosis and immunothrombosis formed by the activation of coagulation cascades and activated neutrophil-released NETs control the insults (hemorrhage and infection), delimits and fixes the insult-induced injuries, and protects the dissemination of DAMPs, pathogen-associated molecular patterns (PAMPs), and microorganisms into the circulation [28, 45, 46, 48]. However, if the insult is sufficiently severe, local physiological thrombosis spreads throughout the whole body and then pathological DIC associated with SIRS ensues [28, 48] (Fig. 33.4).

Trauma elicits nonspecific innate immune inflammatory responses that limit and repair tissue damage. The figure depicts a simplified schematic representation of the activation of pattern recognition receptors by DAMPs and their signaling through the adaptor proteins. This cascade promotes the transcription of several pro-inflammatory cytokines and chemokines, leading to local and systemic inflammatory responses. Local inflammation begins as an adaptive host response, serving to promote the host defense and physiologic hemostasis and fibrin-mediated host defense called immunothrombosis. Spillover of the DAMPs and inflammatory cytokines into the circulation induces SIRS, which activates systemic coagulation, suppresses fibrinolysis, and overwhelms the anticoagulant control mechanisms that restrict hemostasis locally, giving rise to DIC. Importantly, DAMPs themselves can activate coagulation and impair anticoagulation pathways through endothelial damage. ACS apoptosis-associated speck-like protein containing caspase recruit domain, DAMPs damage-associated molecular patterns, DIC disseminated intravascular coagulation, MAVS mitochondrial antiviral signaling, MODS multiple organ dysfunction syndrome, MyD88 myeloid differentiation factor 88, NLRs nucleotide-binding oligomerization domain-containing receptors, RLRs retinoic acid inducible gene-I-like receptors, SIRS systemic inflammatory response syndrome, STING stimulator of interferon gene, TRIF toll/IL-1 receptor homology domain-containing adaptor inducing interferon β, TLRs toll-like receptors. (Modified with permission [45])
5.1 Cytokines
Important pro-inflammatory cytokines that induce SIRS following trauma are TNF-α, IL-1, and IL-6. Immediately after trauma, significant increases in these cytokines have been confirmed in DIC and severely injured patients [49, 50]. TNF-α and IL-1 induce tissue factor expression on monocytes and endothelial cells and downregulate endothelial protein C receptor (EPCR) leading to the suppression of the protein C anticoagulant pathway [51, 52]. IL-6 is also able to induce the activation of coagulation [53]. These inflammatory cytokines further activate neutrophils and endothelial cells [51], which upregulate or express adhesion molecules such as selectins, integrins, and immunoglobulin superfamily which elicit platelets, neutrophils, and endothelial cells interactions, leading to the release of neutrophil-mediated reactive oxygen species and neutrophil elastase [54,55,56]. Activated platelets- and endothelial cells-upregulated P-selectin induces the expression of tissue factor on monocytes [1, 57]. Neutrophil-released reactive oxygen species and elastase injure endothelial cells, bringing about the dysfunction of anticoagulation systems comprising tissue factor pathway inhibitor (TFPI), antithrombin/glycosaminoglycan, and protein C/thrombomodulin on the endothelial cells and the glycocalyx [58, 59]. Furthermore, TNF-α induces immediate t-PA release followed by persistent expression of PAI-1, which impairs fibrinolysis [60,61,62].
Taken together, these findings indicate that inflammatory cytokines are capable of causing all pathomechanisms of DIC. The following pathomechanisms have been confirmed in DIC after trauma: SIRS [49, 63, 64], activation of platelets and systemic thrombin generation [64, 65], insufficient anticoagulant systems such as TFPI [66], antithrombin [36, 63, 65], protein C [36], activation followed by the impairment of fibrinolysis [31, 49, 67, 68] and endothelial cells activation and injury [35, 69, 70].
5.2 Protease-Activated Receptors (PARs)
Signals of coagulation proteases are transmitted to inflammatory cells via protease-activated receptors (PARs) [71, 72]. The PARs are G-protein coupled receptors with seven transmembrane domains that can sense signals from coagulation proteases with auto-activation mechanisms by the proteases-cleaved and exposed active sequence functioning as tethered ligand. The four known PARs are PAR1, PAR2, PAR3, and PAR4. Tissue factor/FVIIa complex activates PAR2, tissue factor/FVIIa/FXa ternary complex activates PAR1 and PAR2, and FIIa (thrombin) activates PAR1, PAR3, and PAR4, resulting in the inductions of varied effects on platelets, leucocytes, and endothelial cells [73, 74] (Table 33.2). Therefore, the PARs play pivotal roles in bidirectional interplays between inflammation and coagulation [43], which forms vicious cycles in DIC, leading to the development of MODS [1, 74].
5.3 DAMPs and NETs
Two epoch-making studies were published in the 2000s [75, 76]. One showed that NETs kill bacteria and another reported that extracellular histones are major mediators of death through endothelial injury and microvascular thrombosis in sepsis. Following these studies, DAMPs including histones and NETs containing neutrophil DAMPs such as DNA and histones became major mediators of many pathologic disorders including DIC [1, 48, 77, 78].
5.3.1 Cytokines and SIRS
Circulating mitochondrial DAMPs, mitochondrial DNA and formyl peptides, showed immediate and thousands-fold higher increases in severely injured trauma patients than control subjects, which were followed by the TNF-α and IL-6 expression associated with SIRS and organ dysfunction [79, 80]. Circulating histones rising to toxic levels immediately after trauma led to IL-6 release, systemic thrombin generation, endothelial injury, and NETs formation, which induced microvascular thrombosis and organ dysfunction [50]. These studies demonstrate close relationships among innate immunity, SIRS, DIC, and MODS in trauma.
The bidirectional interplays between cell injury-induced histones triggering NETs formation and NETs as a source for localized and systemic histones are important in the pathomechanisms of DIC [48, 78]. The above-mentioned DAMPs-induced expression of cytokines and chemokines via PRRs is well recognized [28, 45, 48]. Histones and NETs also induce expression of TNF-α, IL-1, and L-6 via the signal transductions of PRRs, leading to development of SIRS [50, 81,82,83]. Rapid increases in these cytokines may be due in part to their release from presynthesized storage in addition to the induction of their expressions [50, 81].
5.3.2 Platelets and Coagulation
Histones directly or via interactions with TLR2 and TLR4 induce platelet activation and subsequent aggregation associated with P-selectin release and phosphatidylserine exposure, leading to thrombocytopenia in vivo and in critical illness [84,85,86,87,88]. Activated platelets and NETs collaborate to promote thrombin generation and intravascular coagulation [89].
Tissue factor expression on monocytes and its exposure on the endothelium due to endothelial injury is the most important trigger of DIC [1, 90]. Indeed, DIC patients after trauma shows high tissue factor levels associated with persistent thrombin generation [64, 91]. Extracellular histones induce tissue factor antigen, activity, and mRNA in endothelial cells via TLR2 and TLR4 [92, 93]. Histones are able to promote prothrombin auto-activation, which is an important finding indicating that thrombin is independently generated without the activation of coagulation cascades [94]. Furthermore, histone-induced phosphatidylserine exposures on red blood cells (RBCs) and endothelial cells enhance coagulation activation [93, 95]. Other DAMPs, including DNA contained in NETs and RNA released from injured cells, promote thrombosis dependent on the FXII/FXI-induced activation of contact pathways of coagulation [96,97,98], which is related to a poor prognosis of DIC [99].
5.3.3 Anticoagulant Systems and Endothelial Cells
The impairment of thrombomodulin-dependent protein C activation by histones increases thrombin generation, which is a main pathomechanisms of DIC [100]. DIC induces capillary leak syndrome [1, 6]. Histone-induced endothelial injury contributes to increases in endothelial permeability [50, 75] and mitochondrial DAMPs also increase endothelial permeability [101], which gives rise to loss of antithrombin into the extravascular spaces [102]. Neutrophil elastase, a constituent of NETs, cleaves or degrades TFPI, protein C, and antithrombin [1]. High levels of neutrophil elastase associated with endothelial injury in DIC after trauma have been repeatedly confirmed [67, 69, 70]. Furthermore, in vitro, in vivo, and clinical studies of trauma and sepsis confirmed histone-induced endothelial injury by elevated levels of soluble thrombomodulin and its association with thrombin generation and microvascular thrombosis [50, 84]. These findings indicate that histones and NETs are deeply involved in the three major pathomechanisms of DIC; thrombin generation due to insufficient anticoagulant systems, endothelial injury, and capillary leak syndrome.
5.3.4 Activation and Impairment of Fibrinolysis
Extracellular DNA acts as a template for the activation of fibrinolysis under the conditions of neutralization by endogenous serpins such as PAI-1; however, under conditions of the over expressions of PAI-1, extracellular DNA has antifibrinolytic effects, which suggests the role of DNA in DIC [103]. Cell-free DNA binds to both plasmin and fibrin, forming ternary complex and consequently inhibiting fibrinolysis [104]. DNA, histones, and NETs were shown to delay t-PA-mediated fibrin clot lysis, and the former two further increased the fibrin clot fiber diameter, resulting in the thrombus stability [105, 106]. Although these results seem to be physiological effects of histones and DNA supporting the strength of immunothrombosis at the insult site, under the pathologic condition of DIC with increased levels of PAI-1, these DAMPs may play pathological roles through the inhibition of fibrinolysis.
5.3.5 Brief Summary
In vivo experiments clearly showed that histones were able to decrease platelet counts and fibrinogen levels, and prolong the prothrombin time and activated partial thromboplastin time (APTT), which resulted in platelet and fibrin thrombosis associated with organ dysfunction and low survival probability [84]. Similar results were obtained in an experimental trauma model and in trauma patients [50]. As such, DAMPs, especially histones and NETs constitute the main pathophysiology of DIC [78].
5.4 Multiple Actions of Thrombin
The most important key factor in DIC is thrombin, which controls and influences all other factors associated with DIC [1] (Fig. 33.5). In addition to the fundamental role of thrombin in the formation of fibrin clots, thrombin enhances inflammation and activates platelets through PARs [71, 74]. On the surface of activated platelets, FIXa/FVIIIa (tenase) and FXa/FVa (prothrombinase) complexes induce thrombin burst, which accelerate thrombin generation about hundreds of thousands-folds [107]. The conversion of prothrombotic and inflammatory properties of thrombin into anti-thrombotic and anti-inflammatory actions can be done via complex formation with endothelial thrombomodulin [1, 43]. The thrombin thrombomodulin complex-formed activated protein C suppresses thrombin generation by the proteolytic degradation of FVa and FVIIIa. This complex also enhances the activation of TAFI by thrombin about 1250-fold [108]. The anti-inflammatory properties of thrombin complexed with thrombomodulin are exerted through several pathways, such as the binding of activated protein C and EPCR-induced changes in PAR1 inflammatory properties [1, 43, 109] and cleaving of carboxy-terminal arginine residues of bradykinin, C3a and C5a as well as osteopontin by TAFIa [110,111,112]. Thrombin-mediated fibrinolysis control also should be acknowledged. Thrombin stimulates fibrinolysis via immediate t-PA release from endothelial Waibel-Palade bodies [37, 113]. In addition to the induction of PAI-1 expression in endothelial cells [114], thrombin inhibits fibrinolysis through TAFIa [108, 115, 116]. The thrombin thrombomodulin complex activate TAFI [108], and then TAFIa removes the carboxyl-terminal lysine from fibrin, leading to downregulation of fibrinolysis [115, 116].

Multiple actions of thrombin. Multiple actions of thrombin protect the fine balances in the physiological status to maintain homeostasis. Excessive thrombin generation in DIC upsets this balance and induces multifaceted consequences, leading to inflammation, thrombosis, and bleeding. For this reason, DIC has been described as thrombohemorrhagic disorder. Refer to the text for details. APC activated protein C, DIC disseminated intravascular coagulation, EPCR endothelial protein C receptor, PAI-1 plasminogen activator inhibitor-1, PARs protease-activated receptors, TAFIa activated thrombin activatable fibrinolysis inhibitor, t-PA tissue-type plasminogen activator
Massive thrombin generation from immediately to several days after trauma has been repeatedly confirmed in DIC patients since the early 1990s [7, 9, 64, 65] and recent publications have successfully reproduced these results [13, 117]. Dumber and Chandler elegantly showed that immediate and excessive thrombin generation not restricted to injury sites is due to tissue factor activity in systemic circulation and that reduced antithrombin levels allow systemic thrombin generation to continue once started [118, 119]. Tissue factors in systemic circulation likely cause significant thrombin generation in DIC patients [120]. Substantial evidence of massive thrombin generation in DIC after trauma suggest that the manifestation of bleeding and thrombosis in DIC may be dependent on the fine balance of multiple actions of thrombin [1].
6 MODS and the Prognosis
Esmon et al. [4] announced that the molecular links between inflammation and coagulation are unquestioned and stated that the inflammation-coagulation autoamplification loop progresses to vascular injury and MODS, leading to death. The ISTH fully accept this concept and published statement that inflammation-induced systemic thrombin generation and endothelial injury give rise to MODS [6]. DIC is a frequent complication of SIRS [63] and its prevalence increases in parallel with the severity of inflammation associated with stepwise increases in organ dysfunction [121].
DIC patients after trauma consistently showed significantly higher prevalence of MODS and rates of mortality than in non-DIC patients [49, 65, 66, 68,69,70, 117]. DIC and sustained SIRS for more than 3 days after trauma have shown significant increases in tissue factor levels and thrombin generation for 5 days after trauma associated with high rates of MODS and death [64]. A clinical decision analysis confirmed these results while showing that likelihood ratios of SIRS for more than 3 days and DIC on day 1 for predicting MODS after trauma were 6.25 and 11.6, respectively, and that DIC and sustained SIRS were also significantly associated with high mortality [122]. Stepwise logistic regression analyses and multiple regression analyses with the stepwise method showed that the DIC scores immediately after trauma were independent predictor of massive transfusion and death of trauma patients [24, 25]. A multicenter prospective study confirmed that the area under the receiver operating characteristic curve showed significant performance of DIC score immediately after trauma for predicting MODS, massive transfusion, and patient death [123]. Therefore, irrespective of phenotypes, DIC is a leading cause of MODS and death, and the DIC score has a good performance for predicting poor prognosis in trauma patients. Furthermore, DIC has been recognized as an independent predictor of MODS and death regardless of underlying disorders [1].
6.1 Microvascular Thrombosis
Microthrombosis after trauma, including iTBI, has been well established [28, 124]. In 1969, McKay reviewed DIC in trauma and reported that a tissue examination of the disease revealed either or both platelet or fibrin thrombi in the arterioles, venules, and capillaries of a variety of organs such as the brain, pituitary gland, lungs, liver, kidneys, adrenal glands, and gastrointestinal tracts [16]. Sevitt et al. [125] showed thrombi formation in the lungs of one patient who died within a few hours after trauma, a stage they called acceleration of fibrinolysis, which equals a period of DIC with a fibrinolytic phenotype [13]. They further confirmed frequent capillary microthrombosis within 3 h (24.4%) and during the next 9 h (37.9%) after injury in trauma patients. Within 48 h of injury, a total of 66.7% autopsied patients revealed thrombi formation [126]. In addition, many studies have produced the evidence supporting microvascular thrombosis in severe trauma [127, 128], in iTBI [129] and in DIC associated with such trauma [40, 130]. A study of iTBI comprising 88% of patients diagnosed as DIC demonstrated that large microthrombi were more commonly observed in autopsy in patients who died immediately after iTBI and that in addition to the brain/spinal cord, remote organ microthrombi formation such as in the liver, lungs, kidneys, pancreas, pituitary gland, thymus, and intestine was frequently observed [130]. The results clearly support the notion that coagulofibrinolytic changes in iTBI are not markedly different from those in trauma patients without head injury [131].
Microvascular thrombosis reduces oxygen delivery to cells and tissues, leading to MODS. Many clinical, experimental, and autopsy studies showing close correlations between microvascular thrombosis and tissue injury in many vital organs, including the brain, lungs, liver, and kidneys, support this theory [74]. However, microvascular thrombosis alone does not explain the pathomechanisms of MODS in DIC [132]; the bidirectional interplay between coagulation and inflammation should also be acknowledged. Pro-inflammatory cytokine-induced neutrophils and endothelial interactions and PAR-mediated amplification of coagulation and inflammation are important for MODS in DIC. TNF-α- and IL-1-induced thrombin formation upregulates P-selectin and induces the expression of E-selectin on the endothelium and L-selectin on the neutrophils, initiating neutrophils and endothelial interactions, and further promoting the expressions of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on the endothelial cells [54,55,56, 133]. As a result, activated neutrophils adhering to endothelial cells secrete elastase, myeloperoxidase, and reactive oxygen species, leading to endothelial injury, which promotes thrombin generation and fibrin thrombosis, thus causing MODS [54, 58, 74, 134]. The activation of neutrophils and endothelial injury associated with MODS and the higher mortality rate in post-trauma DIC than in non-DIC patients has been confirmed [69, 70]. Although trauma-related studies are lacking, PARs, especially PAR1, PAR2, and PAR4, contribute to the development of MODS in sepsis through a vicious cycle of inflammation and fibrin thrombosis [73, 74]. Taken together, these findings suggest that microvascular thrombosis associated with endothelial injury is a primary cause of MODS in DIC.
6.2 Histones and NETs
In addition to microvascular thrombosis associated with neutrophil activation-induced endothelial injury, DAMPs, especially histones, and NETs are now considered a major pathogenesis of MODS both in trauma and sepsis, especially in patients complicated with DIC [76,77,78,79, 84, 135]. In clinical settings, histones and NETs are involved in the development of DIC [136,137,138], MODS [139,140,141], and high mortality [138, 142] both in trauma and sepsis. Furthermore, experimental studies have shown that histones can cause brain, cardiac, lung, liver, and renal injuries [50, 78, 84].
The pathomechanisms of MODS in DIC caused by histones and NETs can be attributed to their pro-inflammatory, procoagulant, antifibrinolytic effects as well as endothelial injury associated with insufficient anticoagulant systems [78]. In addition to these indirect mechanisms, histone-mediated direct cellular injuries are deeply involved in the development of MODS. Sera from patients with high levels of histones showed a reduced viability of cells derived from the heart, lung, liver, and kidney as well as cultured endothelial cells [141]. A previous study showed the histone-induced promotion of Ca2+ influx without intracellular Ca2+ mobilization [143]. However, histones bind to endothelial cells and cardiomyocytes, and then induce Ca2+ influx and overload with consequent pore formation, leading to endothelial injury and cardiac dysfunction [50, 140]. In cecal ligation and puncture models of sepsis, the accumulations of histones and neutrophils was observed in lungs, liver, kidneys, and spleen suggesting NETs formation [144]. Taken together, these findings indicate that histones and NETs as a source of histones synergistically cause MODS and DIC both in indirect and direct ways and affect the outcomes of the trauma and sepsis patients. These pathomechanisms in DIC are shown in Fig. 33.6 [78].

Bidirectional interplays between histones and NETs in DIC. Histones and NETs as a source of histones synergistically induce inflammation, platelet and coagulation activation, insufficient anticoagulation control, inhibition of fibrinolysis, and cytotoxic effects on cells. Thrombin generation as a result of these processes plays a central role in the cross talk between inflammation and coagulofibrinolytic changes. DIC disseminated intravascular coagulation, NETs neutrophil extracellular traps. (Modified with permission (Creative Commons Attribution International License) [78])
7 Management
In the ICU, trauma patients suffer from DIC due to two conditions; trauma itself-induced DIC and complicated sepsis-induced DIC. Because DIC with a fibrinolytic phenotype continues only for a few hours after injury and then progresses to a thrombotic phenotype [13, 42], a majority of DIC diagnosed in the ICU are considered to be the thrombotic phenotype. Whether the etiology involves trauma or sepsis, the same managements approach is applied to thrombotic phenotype DIC [1]. The following descriptions are based on the findings of studies on sepsis; however, findings can also be applied to the management of DIC in severely injured trauma patients in the ICU.
7.1 Rationale
7.1.1 Why
Innate immune, inflammatory, and coagulofibrinolytic responses maintain the body’s homeostasis against the insults, such as trauma and sepsis. However, if an insult is sufficiently severe, these physiological responses transform into pathological dysregulated inflammatory and coagulofibrinolytic responses (namely DIC), which affects the patient’s outcome due to the disturbance of homeostasis by the development of MODS (Fig. 33.7). The failures of all randomized controlled trials targeting SIRS [3] led to the understanding that bidirectional interplay between innate immunity, inflammation, and coagulation is key to the improvement of the outcome of critical illnesses [4, 43, 44]. In addition, the need to target not only pathological responses but also all components (including the insult itself and MODS) affecting the outcome as a whole is now recognized worldwide. For these reasons, DIC as pathological responses, as well as the insult itself, such as trauma, and MODS need to be treated simultaneously in order to improve a patient’s outcome [1].

Innate immune inflammation, and coagulation and fibrinolysis maintain body homeostasis via immunothrombosis and hemostatic thrombosis against the insults. However, if the insult is sufficiently severe, physiological body responses become dysregulated and pathological SIRS and DIC develop, giving rise to MODS, which affects the patient’s outcome. To improve the outcome, both the insult and dysregulated inflammatory and coagulofibrinolytic responses, namely SIRS and DIC need to be treated simultaneously. In addition, artificial organ supports, such as a ventilator, renal replacement therapy, etc., are also needed. DIC disseminated intravascular coagulation, MODS multiple organ dysfunction syndrome, SIRS systemic inflammatory response syndrome
7.1.2 To Whom
Megatrials of anticoagulants, activated protein C and antithrombin, for all populations of sepsis have failed [145, 146]. Following these failures, substantial evidences has been accumulated regarding the actual target patient population. Subgroup analyses of these megatrials have shown that anticoagulants are effective only against sepsis-induced DIC [147,148,149]. Following these publications, knowledge concerning immunothrombosis progressing to DIC has spread worldwide, promoting the understanding that ambiguous treatment with anticoagulants at the stage of immunothrombosis may deteriorate the body’s physiologic responses [48]. Furthermore, a meta-analysis and systematic reviews of randomized controlled trials for anticoagulant therapy in sepsis clearly have shown that the specific target populations are neither whole sepsis patients nor patients diagnosed as “coagulopathy” but patients with a definite diagnosis of DIC [150, 151]. Therefore, the treatment target of anticoagulants therapy is definitively diagnosed DIC [1].
Post hoc analyses of the megatrials showed that anticoagulants have high degree of effectiveness in patients with high risk of death as evaluated by the Acute Physiology and Chronic Health Evaluation II (APACHE II) score, Sequential Organ Failure Assessment (SOFA) score, and Simplified Acute Physiology Score II (SAPS II) [152, 153]. In systematic reviews and meta-analyses for anticoagulants, meta-regression analyses confirmed significant negative correlations between the effect size of anticoagulant therapies and baseline mortality rates in individual studies, suggesting that the beneficial effects of anticoagulants increase with increasing baseline risk [154, 155]. A multicenter cohort study further proved that anticoagulant therapy was associated with a better outcome according to the deterioration of both DIC scores and APACHE II scores [156] (Fig. 33.8). The second key point concerning the target population in addition to a diagnosis of DIC, therefore, is disease severity, and a SOFA score of 13–17 or APACHE II score of 24–29 may be the therapeutic ranges [157].

A three-dimensional chart showing the log-transformed relative hazard ratios with 95% confidence intervals (gray plate) for hospital mortality of the ISTH DIC scores and APACHE II scores with (blue plate) and without (pink plate) anticoagulant therapies. The red arrow indicates the reduction in hazard ratios at the most severe subset in both scores. The result suggests that anticoagulant therapy may be beneficial in patients with DIC and a high disease severity. APACHE II Acute Physiology and Chronic Health Evaluation II, DIC disseminated intravascular coagulation, ISTH International Society on Thrombosis and Haemostasis. (Reprinted with permission [156])
The third key point concerns heparin administration. Concomitant heparin use with anticoagulants consistently induced the deterioration of the drug effects and was associated with bleeding complications [158, 159]. The concomitant use of anticoagulants for DIC and a prophylactic dose of heparin for venous thromboembolism should therefore probably be avoided.
Taken together, these findings indicate that the treatment targets are critically ill patients with DIC and a high trauma severity without concomitant heparin use.
7.1.3 When
Both the ISTH and JAAM recommend conducting repeated evaluation of DIC scores for the diagnosis and subsequent treatments [6, 22]. Nobody object to the treatment of patients with definitively diagnosed DIC with high DIC scores and high prognostic scores, such as SOFA and APACHE II. However, for patients only suspected of having DIC or with low DIC scores, the repeated calculation of the DIC score is necessary. Continuous or worsening of the coagulation score on the first day of sepsis was associated with an increased risk of MODS and mortality rate [160]. A significantly lower survival probability in patients with newly developed DIC and persistent DIC than in those without DIC or those whose DIC improved from days 0 to 3 after the diagnosis of sepsis was repeatedly confirmed [161, 162]. Odds ratios after adjusting for potential confounders of DIC for the association of DIC with the development of MODS and death were consistently higher on day 3 after the diagnosis of DIC than on day 0 [162]. The results of these studies support the importance of the repeated evaluation of DIC scores for making treatment decisions and predicting the outcome of the patients. Taken together, these findings suggest persistent or worsening DIC score is confirmed, then it is time to start DIC treatment.
8 Underlying Disorders
The ISTH proposed the concept of controlled and uncontrolled DIC [6]. The endothelial regulatory network is temporarily activated and overridden under condition of controlled DIC, and this event is quickly reversed once the underlying conditions are removed or treated, i.e., transfusion reaction or abruptio placenta. Uncontrolled DIC occurs when the regulatory network becomes insufficient (TFPI, antithrombin, protein C, thrombomodulin) or injured (endothelial cells), i.e., trauma and sepsis. As defined, controlled DIC can be resolved by the resolution of the underlying disorder; however, in uncontrolled DIC, simultaneous treatments of both DIC and the underlying disorders, such as trauma, is always required [1, 6, 163].
9 Substitution Therapy
Consumption coagulopathy recognized as thrombocytopenia, low fibrinogen levels, and a prolonged prothrombin time and APTT, is more prominent in trauma-induced DIC than in septic DIC due to the synergistic effects of consumption, loss by critical bleeding, and effects of dilution. Hayakawa et al. [164] showed that the platelet counts, prothrombin time, APTT, and fibrinogen reached the critical thresholds for massive bleeding faster in DIC patients than in non-DIC patients during the first 24 h after trauma. Many studies focusing on trauma found that in addition to RBC, DIC patients more frequently needed the transfusion of platelet concentrates, fresh-frozen plasma (FFP), and fibrinogen concentrate than in non-DIC patients [13, 25, 117, 123, 164]. To stop both trauma-induced critical bleeding and DIC-evoked oozing-type bleeding, substitution therapy using platelet concentrate, FFP, or fibrinogen concentrate is required in DIC after trauma. The critical thresholds for initiating each type of substitution therapy are mentioned in the ISTH guidance [163].
Caution should be practiced when the prothrombin complex concentrate (PCC) is applied as substitution therapy [1, 163]. In addition to a lack of essential coagulation factors, such as FV, VIII, and FXIII, PCC does not contain any or contains a very small amount of anticoagulation factors, such as antithrombin, protein C, and protein S [1, 163]. As a result, PCC increases thrombin generation accompanied by thrombocytopenia and a prolonged prothrombin time, potentially inducing or aggravating thromboembolic complications and DIC [165,166,167,168]. A careful check of the constituents of PCC at each ICU is required.
9.1 Anticoagulants
9.1.1 Heparin
A recent trend in anticoagulant use for DIC is that anticoagulant factor concentrates is preferred to unfractionated heparin (UFH) and low-molecular-weight heparin (LMWH). Thus far, no robust clinical study on heparin use for DIC showing an improvement of patient’s outcome has been published. The ISTH guidance recommends a therapeutic dose of heparin be administered to DIC patients, preferring LMWH to UFH based on the results of a small randomized controlled trial [163, 169].
9.1.2 Anticoagulant Factor Concentrates
The ideal anticoagulant factor concentrates to use for the treatment of DIC has been controversial. After the withdrawal of activated protein C from the global market, we now have two anticoagulation factor concentrates available for use; antithrombin and recombinant human soluble thrombomodulin (rhsThrombomodulin) [170]. After the publication of the ISTH guidance recommending the administration of antithrombin or rhsThrombomodulin for DIC (potentially recommended, needs further evidence) [163], many valuable studies on these drugs have been published.
Antithrombin
A systematic review and meta-analysis concluding that there was no evidence antithrombin improves the mortality of patients with sepsis or DIC included serious flaws in their analysis of the largest population of the KyberSept trial, due to the fact that in that analysis, they did not select DIC patients [146, 171, 172]. Post hoc analyses of the KyberSept trial showed that antithrombin improved the outcome without increasing bleeding side effects in patients with DIC at high risk of death and without concomitant use of heparin [149, 153, 159]. Although the KyberSept trial used an extremely high dose of antithrombin, evidences supporting a supplementary dose of antithrombin administration for septic DIC has been published. A supplementary dose of antithrombin improved the DIC score and doubled the DIC recovery rate without any risk of bleeding [173]. Using a nationwide database, Tagami et al. showed that antithrombin was associated with a significant reduction in mortality rates among DIC patients [174, 175]. In addition, a meta-analysis concerning antithrombin for sepsis-induced DIC repeatedly confirmed a significant reduction in the mortality rate [176]. Using antithrombin for DIC is promising, so a multinational prospective randomized trial concerning the efficacy of a supplementary dose of antithrombin for DIC needs to be conducted [176, 177].
rhsThrombomodulin
A phase III randomized double-blind controlled trial showed the superiority of rhsThrombomodulin to heparin for improving DIC and alleviating bleeding symptoms in patients with infection or hematological malignancies [178]. Following this trial, a phase IIb study restricting participants to those with sepsis and suspected DIC was conducted, which encourage us to conduct further trials on the rhsThrombomodulin in sepsis-associated coagulopathy including DIC [179]. This phase IIb study identified three factors that were associated with a reduced mortality among septic patient; the prothrombin time international normalized ratio (PTINR) >1.4, thrombocytopenia, and dysfunction in at least one organ. However, a phase III trial of rhsThrombomodulin failed to reduce the mortality in patients with sepsis-associated coagulopathy diagnosed with using above-mentioned three factors [180]. Post hoc analyses showed three issues likely associated with the negative results; concomitant heparin use, patients no longer meeting the inclusion criteria for platelet counts and PTINR at the starting point of the drug, and no stratification of the patients by thrombin generation levels [180, 181]. Subgroup analyses of the phase III trial after adjusting for these three factors showed a significant reduction in the mortality rates in rhsThrombomodulin group compared with the control group [180, 182,183,184]. The new terms “sepsis-associated coagulopathy” and “sepsis-induced coagulopathy” cooperating three items identified in phase IIb trial were established and rhsThrombomodulin therapy for patients who met these criteria showed a reduction in mortality [185]. Further study will need to be performed in order to confirm the positive effect of rhsThrombomodulin on these two coagulopathies and DIC.
9.2 Antifibrinolytics
Antifibrinolytic agents are contraindicated in DIC with the thrombotic phenotype [1, 163]. However, antifibrinolytics are considered for use in DIC with the fibrinolytic phenotype diagnosed at a very early stage of trauma and in cases with acute promyelocytic leukemia [1, 163]. The CRASH-2 trial showed a reduction in the risk of death in bleeding trauma patients who used tranexamic acid within 3 h after injury [186, 187]. The coagulofibrinolytic changes in iTBI patients are the same as those in patients without brain injury, so, DIC immediately after iTBI also belongs to fibrinolytic phenotype [33, 131]. The CRASH-3 trial demonstrated significant reductions in head-injury-related death due to the administration of tranexamic acid, especially in patients with mild-to-moderate head injury and those who received early tranexamic acid treatment [188].
The CRASH-2 and CRASH-3 were not intended to include DIC patients; however, the mechanisms underlying the effects of tranexamic acid on the fibrinolytic systems and restricted effective timeframe within a few hours after injury support the notion that tranexamic acid may improve excessive bleeding in DIC patients with a fibrinolytic phenotype at very early stage of trauma and in those with iTBI.
9.3 Histones and NETs
Histones and NETs are promising targets for improving DIC, organ dysfunction, and the outcome. Some experimental studies have shown that anti-histone antibody ameliorated histone-induced IL-6 release, thrombin generation, endothelial injury, organ dysfunction, and survival probability [50, 76, 140]. rhsThrombomodulin binds to histones and was shown to improve histone-induced platelet aggregation and thrombocytopenia, microvascular thrombosis, organ dysfunction, and the survival probability [84]. Nonanticoagulant heparin prevented the cytotoxic effects of histones and improved the mortality rates in mouse model of sepsis and sterile infection [189]. It further attenuated the histone-induced pro-inflammatory cytokines (IL-6, IL-8) production, tissue factor generation, and C3a formation in a whole blood model [190]. Peptidylarginine deiminase 4 (PAD-4) is a key protein for NETs formation (NETosis) as a citrullinating enzyme of arginine residues of histones that results in chromatin decondensation and the release of neutrophil DNA (NETs) [191]. In addition, PAD4 is now known to regulate pathological thrombosis [191]. Therefore, PAD4 inhibitors prevent the formation of NETs and thrombosis. Already formed NETs DNA could be degraded using DNase. Aside from rhsThrombomodulin, these drugs are still in the experimental phase; however, their potential efficacy is promising for DIC treatment [78, 191].
10 Conclusions
DIC is defined as dysregulated inflammatory and coagulofibrinolytic responses to the insults and is deeply involved in the outcome of critically ill patients due to the development of bleeding and MODS. DIC involves systemic thrombin generation, insufficient anticoagulation pathways, and increased fibrinolysis (followed by its impairment) in association with endothelial injury. DAMPs, especially histones released from injured cells and activated neutrophil-formed NETs containing neutrophil DNA, histones, and elastase are considered the main pathomechanisms involved in DIC. Histones and NETs synergistically induce systemic inflammation, platelet and coagulation activation, dysfunction of anticoagulant systems, and inhibition of fibrinolysis, leading to microvascular thrombosis. Histone-induced direct cellular injury, including that of endothelial cells, as well as the reduction in oxygen delivery due to microvascular thrombosis give rise to MODS. Platelet dysfunction, consumption coagulopathy, and hyperfibrin(ogen)olysis induce oozing-type of bleeding. DIC can be diagnosed using a diagnostic scoring system. Definitively diagnosed DIC with a high disease severity and persistent or worsening DIC is the true target of the treatment with anticoagulant factor concentrates in trauma patients admitted to the ICU.
Even today, it is important to keep in mind the fact that DIC equals a sign that “Death Is Coming” [192].
Key Concept
-
DIC, defined as dysregulated inflammatory and coagulofibrinolytic responses to an insult and known as thrombohemorrhagic disorder, markedly affects a patient’s outcome due to microvascular thrombosis-induced organ dysfunction and critical bleeding.
Take Home Messages
-
Trauma is a leading cause of DIC.
-
Bidirectional interplays among innate immunity, inflammation, and coagulation play important roles in the pathophysiology of DIC.
-
Histones, as well as NETs involved in inflammation, activation of coagulation, impairment of anticoagulation controls, and the inhibition of fibrinolysis, lead to microvascular thrombosis and endothelial cell injury.
-
Microvascular thrombosis- and endothelial cell injury-caused MODS and consumption coagulopathy-induced critical bleeding affects a patient’s outcome.
-
To improve a patient’s outcome, the simultaneous treatments of trauma-itself, DIC, and MODS is necessary.
References
Gando S, Levi M, Toh CH. Disseminated intravascular coagulation. Nat Rev Dis Primers. 2016;2:16037. https://doi.org/10.1038/nrdp.2016.37.
American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit Care Med. 1992;20:864–74.
Nasraway SA Jr. Sepsis research: we must change course. Crit Care Med. 1999;27:427–30. https://doi.org/10.1097/00003246-199902000-00054.
Esmon CT, Fukudome K, Mather T, Bode W, Regan LM, Stearns-Kurosawa DJ, Kurosawa S. Inflammation, sepsis, and coagulation. Haematologica. 1999;84:254–9.
Penner JA. Disseminated intravascular coagulation in patients with multiple organ failure of non-septic origin. Semin Thromb Hemost. 1998;24:45–52. https://doi.org/10.1055/s-2007-995822.
Taylor FB Jr, Toh CH, Hoots WK, Wada H, Levi M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86:1327–30.
Gando S. Disseminated intravascular coagulation in trauma patients. Semin Thromb Hemost. 2001;27:585–92. https://doi.org/10.1055/s-2001-18864.
Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36:691–709. https://doi.org/10.1016/j.injury.2004.12.037.
Gando S. Tissue factor in trauma and organ dysfunction. Semin Thromb Hemost. 2006;32:48–53. https://doi.org/10.1055/s-2006-933340.
Squizzato A, Gallo A, Levi M, Iba T, Levy JH, Erez O, Ten Cate H, Solh Z, Gando S, Vicente V, Di Nisio M. Underlying disorders of disseminated intravascular coagulation: Communication from the ISTH SSC Subcommittees on Disseminated Intravascular Coagulation and Perioperative and Critical Care Thrombosis and Hemostasis. J Thromb Haemost. 2020;18:2400–7. https://doi.org/10.1111/jth.14946.
Toh CH, Dennis M. Disseminated intravascular coagulation: old disease, new hope. BMJ. 2003;327:974–7. https://doi.org/10.1136/bmj.327.7421.974.
Moore HB, Gando S, Iba T, Kim PY, Yeh CH, Brohi K, Hunt BJ, Levy JH, Draxler DF, Stanworth S, Görlinger K, Neal MD, Schreiber MA, Barrett CD, Medcalf RL, Moore EE, Mutch NJ, Thachil J, Urano T, Thomas S, Scărlătescu E, Walsh M. Defining trauma-induced coagulopathy with respect to future implications for patient management: communication from the SSC of the ISTH. J Thromb Haemost. 2020;18:740–7. https://doi.org/10.1111/jth.14690.
Gando S, Shiraishi A, Wada T, Yamakawa K, Fujishima S, Saitoh D, Kushimoto S, Ogura H, Abe T, Otomo Y. A multicenter prospective validation study on disseminated intravascular coagulation in trauma-induced coagulopathy. J Thromb Haemost. 2020;18:2232–24. https://doi.org/10.1111/jth.14931.
Gando S, Hayakawa M. Pathophysiology of trauma-induced coagulopathy and management of critical bleeding requiring massive transfusion. Semin Thromb Hemost. 2016;42:155–65. https://doi.org/10.1055/s-0035-1564831.
Hardaway RM. Disseminated intravascular coagulation syndromes. Arch Surg. 1961;83:842–50. https://doi.org/10.1001/archsurg.1961.01300180042009.
McKay DG. Trauma and disseminated intravascular coagulation. J Trauma. 1969;9:646–60. https://doi.org/10.1097/00005373-196908000-00002.
Flute PT. Coagulation and fibrinolysis after injury. J Clin Pathol Suppl (R Coll Pathol). 1970;4:102–9. https://doi.org/10.1136/jcp.s3-4.1.102.
Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004;32:2416–21. https://doi.org/10.1097/01.ccm.0000147769.07699.e3.
Toh CH, Downey C. Performance and prognostic importance of a new clinical and laboratory scoring system for identifying non-overt disseminated intravascular coagulation. Blood Coagul Fibrinolysis. 2005;16:69–74. https://doi.org/10.1097/00001721-200501000-00011.
Gando S, Iba T, Eguchi Y, Ohtomo Y, Okamoto K, Koseki K, Mayumi T, Murata A, Ikeda T, Ishikura H, Ueyama M, Ogura H, Kushimoto S, Saitoh D, Endo S, Shimazaki S. A multicenter, prospective validation of disseminated intravascular coagulation diagnostic criteria for critically ill patients: comparing current criteria. Crit Care Med. 2006;34:625–31. https://doi.org/10.1097/01.ccm.0000202209.42491.38.
Gando S, Saitoh D, Ogura H, Mayumi T, Koseki K, Ikeda T, Ishikura H, Iba T, Ueyama M, Eguchi Y, Ohtomo Y, Okamoto K, Kushimoto S, Endo S, Shimazaki S. Natural history of disseminated intravascular coagulation diagnosed based on the newly established diagnostic criteria for critically ill patients: results of a multicenter, prospective survey. Crit Care Med. 2008;36:145–50. https://doi.org/10.1097/01.Ccm.0000295317.97245.2d.
Gando S, Saitoh D, Ogura H, Fujishima S, Mayumi T, Araki T, Ikeda H, Kotani J, Kushimoto S, Miki Y, Shiraishi S, Suzuki K, Suzuki Y, Takeyama N, Takuma K, Tsuruta R, Yamaguchi Y, Yamashita N, Aikawa N. A multicenter, prospective validation study of the Japanese Association for Acute Medicine disseminated intravascular coagulation scoring system in patients with severe sepsis. Crit Care. 2013;17:R111. https://doi.org/10.1186/cc12783.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, Katabami K. Application of the Japanese Association for Acute Medicine disseminated intravascular coagulation diagnostic criteria for patients at an early phase of trauma. Thromb Res. 2009;124:706–10. https://doi.org/10.1016/j.thromres.2009.06.036.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, Katabami K. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res. 2009;124:608–13. https://doi.org/10.1016/j.thromres.2009.06.034.
Oshiro A, Yanagida Y, Gando S, Henzan N, Takahashi I, Makise H. Hemostasis during the early stages of trauma: comparison with disseminated intravascular coagulation. Crit Care. 2014;18:R61. https://doi.org/10.1186/cc13816.
Marder VJ FD, Colman RW, Levi M. Consumptive thrombohemorrhagic disorders. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, ed. Hemostasis and thrombosis: basic principles and clinical practice, 5th edn. Philadelphia: Lippincott Williams & Wilkins, 2006, 1571–1600.
Asakura H. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care. 2014;2:20. https://doi.org/10.1186/2052-0492-2-20.
Gando S, Otomo Y. Local hemostasis, immunothrombosis, and systemic disseminated intravascular coagulation in trauma and traumatic shock. Crit Care. 2015;19:72. https://doi.org/10.1186/s13054-015-0735-x.
Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, Hajjar KA. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340:994–1004. https://doi.org/10.1056/nejm199904013401303.
Schwameis M, Schober A, Schörgenhofer C, Sperr WR, Schöchl H, Janata-Schwatczek K, Kürkciyan EI, Sterz F, Jilma B. Asphyxia by drowning induces massive bleeding due to hyperfibrinolytic disseminated intravascular coagulation. Crit Care Med. 2015;43:2394–402. https://doi.org/10.1097/ccm.0000000000001273.
Gando S, Wada T. Disseminated intravascular coagulation in cardiac arrest and resuscitation. J Thromb Haemost. 2019;17:1205–16. https://doi.org/10.1111/jth.14480.
Kruithof EK, Tran-Thang C, Gudinchet A, Hauert J, Nicoloso G, Genton C, Welti H, Bachmann F. Fibrinolysis in pregnancy: a study of plasminogen activator inhibitors. Blood. 1987;69:460–6.
Wada T, Gando S, Maekaw K, Katabami K, Sageshima H, Hayakawa M, Sawamura A. Disseminated intravascular coagulation with increased fibrinolysis during the early phase of isolated traumatic brain injury. Crit Care. 2017;21:219. https://doi.org/10.1186/s13054-017-1808-9.
Hayakawa M, Maekawa K, Kushimoto S, Kato H, Sasaki J, Ogura H, Matsuoka T, Uejima T, Morimura N, Ishikura H, Hagiwara A, Takeda M, Kaneko N, Saitoh D, Kudo D, Kanemura T, Shibusawa T, Furugori S, Nakamura Y, Shiraishi A, Murata K, Mayama G, Yaguchi A, Kim S, Takasu O, Nishiyama K. Hyperfibrinolysis in severe isolated traumatic brain injury may occur without tissue hypoperfusion: a retrospective observational multicentre study. Crit Care. 2017;21:222. https://doi.org/10.1186/s13054-017-1811-1.
Gando S, Sawamura A, Hayakawa M. Trauma, shock, and disseminated intravascular coagulation: lessons from the classical literature. Ann Surg. 2011;254:10–9. https://doi.org/10.1097/SLA.0b013e31821221b1.
Gando S, Wada H, Thachil J. Differentiating disseminated intravascular coagulation (DIC) with the fibrinolytic phenotype from coagulopathy of trauma and acute coagulopathy of trauma-shock (COT/ACOTS). J Thromb Haemost. 2013;11:826–35. https://doi.org/10.1111/jth.12190.
Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. 2005;15:302–8. https://doi.org/10.1016/j.tcm.2005.09.005.
Gupta N, Zhao YY, Evans CE. The stimulation of thrombosis by hypoxia. Thromb Res. 2019;181:77–83. https://doi.org/10.1016/j.thromres.2019.07.013.
Hijazi N, Abu Fanne R, Abramovitch R, Yarovoi S, Higazi M, Abdeen S, Basheer M, Maraga E, Cines DB, Higazi AA. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood. 2015;125:2558–67. https://doi.org/10.1182/blood-2014-08-588442.
Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–88. https://doi.org/10.1385/ncc:1:4:479.
Gando S, Wada H, Kim HK, Kurosawa S, Nielsen JD, Thachil J, Toh CH. Comparison of disseminated intravascular coagulation in trauma with coagulopathy of trauma/acute coagulopathy of trauma-shock. J Thromb Haemost. 2012;10:2593–5. https://doi.org/10.1111/jth.12011.
Murakami H, Gando S, Hayakawa M, Sawamura A, Sugano M, Kubota N, Uegaki S, Jesmin S. Disseminated intravascular coagulation (DIC) at an early phase of trauma continuously proceeds to DIC at a late phase of trauma. Clin Appl Thromb Hemost. 2012;18:364–9. https://doi.org/10.1177/1076029611426138.
Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–8. https://doi.org/10.1046/j.1538-7836.2003.00261.x.
Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost. 2011;9(Suppl 1):182–8. https://doi.org/10.1111/j.1538-7836.2011.04323.x.
Gando S. Role of fibrinolysis in sepsis. Semin Thromb Hemost. 2013;39:392–9. https://doi.org/10.1055/s-0033-1334140.
Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol. 2018;19:327–41. https://doi.org/10.1038/s41590-018-0064-8.
Bone RC. Sir Isaac Newton, sepsis, SIRS, and CARS. Crit Care Med. 1996;24:1125–8. https://doi.org/10.1097/00003246-199607000-00010.
Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013;13:34–45. https://doi.org/10.1038/nri3345.
Gando S, Nakanishi Y, Tedo I. Cytokines and plasminogen activator inhibitor-1 in posttrauma disseminated intravascular coagulation: relationship to multiple organ dysfunction syndrome. Crit Care Med. 1995;23:1835–42. https://doi.org/10.1097/00003246-199511000-00009.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, Wang SS, Brohi K, Kipar A, Yu W, Wang G, Toh CH. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–9. https://doi.org/10.1164/rccm.201206-1037OC.
Esmon CT. Possible involvement of cytokines in diffuse intravascular coagulation and thrombosis. Baillieres Best Pract Res Clin Haematol. 1999;12:343–59. https://doi.org/10.1053/beha.1999.0029.
Boermeester MA, van Leeuwen PA, Coyle SM, Wolbink GJ, Hack CE, Lowry SF. Interleukin-1 blockade attenuates mediator release and dysregulation of the hemostatic mechanism during human sepsis. Arch Surg. 1995;130:739–48. https://doi.org/10.1001/archsurg.1995.01430070061012.
van der Poll T, Levi M, Hack CE, ten Cate H, van Deventer SJ, Eerenberg AJ, de Groot ER, Jansen J, Gallati H, Büller HR, et al. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J Exp Med. 1994;179:1253–9. https://doi.org/10.1084/jem.179.4.1253.
McGill SN, Ahmed NA, Christou NV. Endothelial cells: role in infection and inflammation. World J Surg. 1998;22:171–8. https://doi.org/10.1007/s002689900366.
Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61.
Gearing AJ, Newman W. Circulating adhesion molecules in disease. Immunol Today. 1993;14:506–12. https://doi.org/10.1016/0167-5699(93)90267-o.
Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, Furie B. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A. 1994;91:8767–71. https://doi.org/10.1073/pnas.91.19.8767.
Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–76. https://doi.org/10.1056/nejm198902093200606.
Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454:345–59. https://doi.org/10.1007/s00424-007-0212-8.
Suffredini AF, Harpel PC, Parrillo JE. Promotion and subsequent inhibition of plasminogen activation after administration of intravenous endotoxin to normal subjects. N Engl J Med. 1989;320:1165–72. https://doi.org/10.1056/nejm198905043201802.
Levi M, ten Cate H, van der Poll T, van Deventer SJ. Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA. 1993;270:975–9.
Biemond BJ, Levi M, Ten Cate H, Van der Poll T, Büller HR, Hack CE, Ten Cate JW. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci (Lond). 1995;88:587–94. https://doi.org/10.1042/cs0880587.
Gando S, Kameue T, Nanzaki S, Nakanishi Y. Disseminated intravascular coagulation is a frequent complication of systemic inflammatory response syndrome. Thromb Haemost. 1996;75:224–8.
Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med. 1997;25:1820–6. https://doi.org/10.1097/00003246-199711000-00019.
Gando S, Tedo I, Kubota M. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20:594–600. https://doi.org/10.1097/00003246-199205000-00009.
Gando S, Nanzaki S, Morimoto Y, Ishitani T, Kemmotsu O. Tissue factor pathway inhibitor response does not correlate with tissue factor-induced disseminated intravascular coagulation and multiple organ dysfunction syndrome in trauma patients. Crit Care Med. 2001;29:262–6. https://doi.org/10.1097/00003246-200102000-00006.
Hayakawa M, Sawamura A, Gando S, Kubota N, Uegaki S, Shimojima H, Sugano M, Ieko M. Disseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastase. Surgery. 2011;149:221–30. https://doi.org/10.1016/j.surg.2010.06.010.
Hayakawa M, Sawamura A, Gando S, Jesmin S, Naito S, Ieko M. A low TAFI activity and insufficient activation of fibrinolysis by both plasmin and neutrophil elastase promote organ dysfunction in disseminated intravascular coagulation associated with sepsis. Thromb Res. 2012;130:906–13. https://doi.org/10.1016/j.thromres.2012.01.015.
Gando S, Nakanishi Y, Kameue T, Nanzaki S. Soluble thrombomodulin increases in patients with disseminated intravascular coagulation and in those with multiple organ dysfunction syndrome after trauma: role of neutrophil elastase. J Trauma. 1995;39:660–4. https://doi.org/10.1097/00005373-199510000-00007.
Gando S, Kameue T, Matsuda N, Hayakawa M, Ishitani T, Morimoto Y, Kemmotsu O. Combined activation of coagulation and inflammation has an important role in multiple organ dysfunction and poor outcome after severe trauma. Thromb Haemost. 2002;88:943–9.
Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. https://doi.org/10.1038/35025229.
Riewald M, Ruf W. Orchestration of coagulation protease signaling by tissue factor. Trends Cardiovasc Med. 2002;12:149–54. https://doi.org/10.1016/s1050-1738(02)00153-6.
Pawlinski R, Mackman N. Tissue factor, coagulation proteases, and protease-activated receptors in endotoxemia and sepsis. Crit Care Med. 2004;32:S293–7. https://doi.org/10.1097/01.ccm.0000128445.95144.b8.
Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med. 2010;38:S35–42. https://doi.org/10.1097/CCM.0b013e3181c9e31d.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. https://doi.org/10.1126/science.1092385.
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–21. https://doi.org/10.1038/nm.2053.
Ito T. PAMPs and DAMPs as triggers for DIC. J Intensive Care. 2014;2:67. https://doi.org/10.1186/s40560-014-0065-0.
Alhamdi Y, Toh CH. Recent advances in pathophysiology of disseminated intravascular coagulation: the role of circulating histones and neutrophil extracellular traps. F1000Res. 2017;6:2143. https://doi.org/10.12688/f1000research.12498.1.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. https://doi.org/10.1038/nature08780.
Gu X, Yao Y, Wu G, Lv T, Luo L, Song Y. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PLoS One. 2013;8:e72834. https://doi.org/10.1371/journal.pone.0072834.
Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31. https://doi.org/10.4049/jimmunol.1003930.
Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013;43:3336–42. https://doi.org/10.1002/eji.201243224.
Hu Z, Murakami T, Tamura H, Reich J, Kuwahara-Arai K, Iba T, Tabe Y, Nagaoka I. Neutrophil extracellular traps induce IL-1β production by macrophages in combination with lipopolysaccharide. Int J Mol Med. 2017;39:549–58. https://doi.org/10.3892/ijmm.2017.2870.
Nakahara M, Ito T, Kawahara K, Yamamoto M, Nagasato T, Shrestha B, Yamada S, Miyauchi T, Higuchi K, Takenaka T, Yasuda T, Matsunaga A, Kakihana Y, Hashiguchi T, Kanmura Y, Maruyama I. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS One. 2013;8:e75961. https://doi.org/10.1371/journal.pone.0075961.
Carestia A, Rivadeneyra L, Romaniuk MA, Fondevila C, Negrotto S, Schattner M. Functional responses and molecular mechanisms involved in histone-mediated platelet activation. Thromb Haemost. 2013;110:1035–45. https://doi.org/10.1160/th13-02-0174.
Fuchs TA, Bhandari AA, Wagner DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;118:3708–14. https://doi.org/10.1182/blood-2011-01-332676.
Alhamdi Y, Abrams ST, Lane S, Wang G, Toh CH. Histone-associated thrombocytopenia in patients who are critically ill. JAMA. 2016;315:817–9. https://doi.org/10.1001/jama.2016.0136.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118:1952–61. https://doi.org/10.1182/blood-2011-03-343061.
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, Jenne CN. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129:1357–67. https://doi.org/10.1182/blood-2016-09-741298.
Østerud B, Bjørklid E. The tissue factor pathway in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27:605–17. https://doi.org/10.1055/s-2001-18866.
Shimura M, Wada H, Wakita Y, Nakase T, Hiyoyama K, Nagaya S, Mori Y, Shiku H. Plasma tissue factor and tissue factor pathway inhibitor levels in patients with disseminated intravascular coagulation. Am J Hematol. 1997;55:169–74. https://doi.org/10.1002/(sici)1096-8652(199707)55:4<169::aid-ajh1>3.0.co;2-q.
Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res. 2016;137:211–8. https://doi.org/10.1016/j.thromres.2015.10.012.
Kim JE, Yoo HJ, Gu JY, Kim HK. Histones induce the procoagulant phenotype of endothelial cells through tissue factor up-regulation and thrombomodulin down-regulation. PLoS One. 2016;11:e0156763. https://doi.org/10.1371/journal.pone.0156763.
Barranco-Medina S, Pozzi N, Vogt AD, Di Cera E. Histone H4 promotes prothrombin autoactivation. J Biol Chem. 2013;288:35749–57. https://doi.org/10.1074/jbc.M113.509786.
Semeraro F, Ammollo CT, Esmon NL, Esmon CT. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J Thromb Haemost. 2014;12:1697–702. https://doi.org/10.1111/jth.12677.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. https://doi.org/10.1073/pnas.1005743107.
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Günther A, Engelmann B, Preissner KT. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–93. https://doi.org/10.1073/pnas.0608647104.
Oehmcke S, Mörgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun. 2009;1:225–30. https://doi.org/10.1159/000203700.
Park HS, Gu J, You HJ, Kim JE, Kim HK. Factor XII-mediated contact activation related to poor prognosis in disseminated intravascular coagulation. Thromb Res. 2016;138:103–7. https://doi.org/10.1016/j.thromres.2015.12.011.
Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. 2011;9:1795–803. https://doi.org/10.1111/j.1538-7836.2011.04422.x.
Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 2013;8:e59989. https://doi.org/10.1371/journal.pone.0059989.
Asakura H, Ontachi Y, Mizutani T, Kato M, Ito T, Saito M, Morishita E, Yamazaki M, Aoshima K, Takami A, Yoshida T, Suga Y, Miyamoto K, Nakao S. Decreased plasma activity of antithrombin or protein C is not due to consumption coagulopathy in septic patients with disseminated intravascular coagulation. Eur J Haematol. 2001;67:170–5. https://doi.org/10.1034/j.1600-0609.2001.5790508.x.
Komissarov AA, Florova G, Idell S. Effects of extracellular DNA on plasminogen activation and fibrinolysis. J Biol Chem. 2011;286:41949–62. https://doi.org/10.1074/jbc.M111.301218.
Gould TJ, Vu TT, Stafford AR, Dwivedi DJ, Kim PY, Fox-Robichaud AE, Weitz JI, Liaw PC. Cell-free DNA modulates clot structure and impairs fibrinolysis in sepsis. Arterioscler Thromb Vasc Biol. 2015;35:2544–53. https://doi.org/10.1161/atvbaha.115.306035.
Longstaff C, Varjú I, Sótonyi P, Szabó L, Krumrey M, Hoell A, Bóta A, Varga Z, Komorowicz E, Kolev K. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem. 2013;288:6946–56. https://doi.org/10.1074/jbc.M112.404301.
Varjú I, Longstaff C, Szabó L, Farkas ÁZ, Varga-Szabó VJ, Tanka-Salamon A, Machovich R, Kolev K. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost. 2015;113:1289–98. https://doi.org/10.1160/th14-08-0669.
Hoffman M, Monroe DM III. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–65.
Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–8. https://doi.org/10.1074/jbc.271.28.16603.
Rezaie AR. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17:2059–69. https://doi.org/10.2174/092986710791233706.
Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2002;46:131–4. https://doi.org/10.1111/j.1348-0421.2002.tb02669.x.
Leung LL, Myles T, Nishimura T, Song JJ, Robinson WH. Regulation of tissue inflammation by thrombin-activatable carboxypeptidase B (or TAFI). Mol Immunol. 2008;45:4080–3. https://doi.org/10.1016/j.molimm.2008.07.010.
Bajzar L, Jain N, Wang P, Walker JB. Thrombin activatable fibrinolysis inhibitor: not just an inhibitor of fibrinolysis. Crit Care Med. 2004;32:S320–4. https://doi.org/10.1097/01.ccm.0000126361.00450.b1.
Levin EG, Marzec U, Anderson J, Harker LA. Thrombin stimulates tissue plasminogen activator release from cultured human endothelial cells. J Clin Invest. 1984;74:1988–95. https://doi.org/10.1172/jci111620.
Gelehrter TD, Sznycer-Laszuk R. Thrombin induction of plasminogen activator-inhibitor in cultured human endothelial cells. J Clin Invest. 1986;77:165–9. https://doi.org/10.1172/jci112271.
Wang W, Boffa MB, Bajzar L, Walker JB, Nesheim ME. A study of the mechanism of inhibition of fibrinolysis by activated thrombin-activable fibrinolysis inhibitor. J Biol Chem. 1998;273:27176–81. https://doi.org/10.1074/jbc.273.42.27176.
Bouma BN, Meijers JC. Thrombin-activatable fibrinolysis inhibitor (TAFI, plasma procarboxypeptidase B, procarboxypeptidase R, procarboxypeptidase U). J Thromb Haemost. 2003;1:1566–74. https://doi.org/10.1046/j.1538-7836.2003.00329.x.
Yanagida Y, Gando S, Sawamura A, Hayakawa M, Uegaki S, Kubota N, Homma T, Ono Y, Honma Y, Wada T, Jesmin S. Normal prothrombinase activity, increased systemic thrombin activity, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma-shock. Surgery. 2013;154:48–57. https://doi.org/10.1016/j.surg.2013.02.004.
Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009;49:2652–60. https://doi.org/10.1111/j.1537-2995.2009.02335.x.
Chandler WL. Procoagulant activity in trauma patients. Am J Clin Pathol. 2010;134:90–6. https://doi.org/10.1309/ajcp3wpoyskk6bfe.
Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79:1111–5.
Rangel-Frausto MS, Pittet D, Costigan M, Hwang T, Davis CS, Wenzel RP. The natural history of the systemic inflammatory response syndrome (SIRS). A prospective study. JAMA. 1995;273:117–23.
Gando S, Nanzaki S, Kemmotsu O. Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma: application of clinical decision analysis. Ann Surg. 1999;229:121–7. https://doi.org/10.1097/00000658-199901000-00016.
Wada T, Shiraishi A, Gando S. Disseminated intravascular coagulation (DIC) immediately after trauma predicts a poor prognosis of severely injured patients: a sub-analysis of a multicenter prospective study on disseminated intravascular coagulation in trauma. Intensive Care Med. 2021;11(1):11031.
Gando S. Hemostasis and thrombosis in trauma patients. Semin Thromb Hemost. 2015;41:26–34. https://doi.org/10.1055/s-0034-1398378.
Innes D, Sevitt S. Coagulation and fibrinolysis in injured patients. J Clin Pathol. 1964;17:1–13. https://doi.org/10.1136/jcp.17.1.1.
Eeles GH, Sevitt S. Microthrombosis in injured and burned patients. J Pathol Bacteriol. 1967;93:275–93. https://doi.org/10.1002/path.1700930126.
Hardaway RM. The significance of coagulative and thrombotic changes after haemorrhage and injury. J Clin Pathol Suppl (R Coll Pathol). 1970;4:110–20. https://doi.org/10.1136/jcp.s3-4.1.110.
Nuytinck HK, Offermans XJ, Kubat K, Goris JA. Whole-body inflammation in trauma patients. An autopsy study. Arch Surg. 1988;123:1519–24. https://doi.org/10.1001/archsurg.1988.01400360089016.
Stein SC, Graham DI, Chen XH, Smith DH. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery. 2004;54:687–91; discussion 91. https://doi.org/10.1227/01.neu.0000108641.98845.88.
Kaufman HH, Hui KS, Mattson JC, Borit A, Childs TL, Hoots WK, Bernstein DP, Makela ME, Wagner KA, Kahan BD, et al. Clinicopathological correlations of disseminated intravascular coagulation in patients with head injury. Neurosurgery. 1984;15:34–42. https://doi.org/10.1227/00006123-198407000-00008.
Gando S, Nanzaki S, Kemmotsu O. Coagulofibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070–6; discussion 6–7. https://doi.org/10.1097/00005373-199906000-00018.
Taylor FB Jr, Chang AC, Peer GT, Mather T, Blick K, Catlett R, Lockhart MS, Esmon CT. DEGR-factor Xa blocks disseminated intravascular coagulation initiated by Escherichia coli without preventing shock or organ damage. Blood. 1991;78:364–8.
Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol. 1993;11:767–804. https://doi.org/10.1146/annurev.iy.11.040193.004003.
Aird WC. Endothelial cell dynamics and complexity theory. Crit Care Med. 2002;30:S180–5. https://doi.org/10.1097/00003246-200205001-00002.
Liaw PC, Ito T, Iba T, Thachil J, Zeerleder S. DAMP and DIC: the role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016;30:257–61. https://doi.org/10.1016/j.blre.2015.12.004.
Kim JE, Lee N, Gu JY, Yoo HJ, Kim HK. Circulating levels of DNA-histone complex and dsDNA are independent prognostic factors of disseminated intravascular coagulation. Thromb Res. 2015;135:1064–9. https://doi.org/10.1016/j.thromres.2015.03.014.
Delabranche X, Stiel L, Severac F, Galoisy AC, Mauvieux L, Zobairi F, Lavigne T, Toti F, Anglès-Cano E, Meziani F, Boisramé-Helms J. Evidence of netosis in septic shock-induced disseminated intravascular coagulation. Shock. 2017;47:313–7. https://doi.org/10.1097/shk.0000000000000719.
Abrams ST, Morton B, Alhamdi Y, Alsabani M, Lane S, Welters ID, Wang G, Toh CH. A novel assay for neutrophil extracellular trap formation independently predicts disseminated intravascular coagulation and mortality in critically ill patients. Am J Respir Crit Care Med. 2019;200:869–80. https://doi.org/10.1164/rccm.201811-2111OC.
Alhamdi Y, Abrams ST, Cheng Z, Jing S, Su D, Liu Z, Lane S, Welters I, Wang G, Toh CH. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. 2015;43:2094–103. https://doi.org/10.1097/ccm.0000000000001162.
Alhamdi Y, Zi M, Abrams ST, Liu T, Su D, Welters I, Dutt T, Cartwright EJ, Wang G, Toh CH. Circulating histone concentrations differentially affect the predominance of left or right ventricular dysfunction in critical illness. Crit Care Med. 2016;44:e278–88. https://doi.org/10.1097/ccm.0000000000001413.
Cheng Z, Abrams ST, Alhamdi Y, Toh J, Yu W, Wang G, Toh CH. Circulating histones are major mediators of multiple organ dysfunction syndrome in acute critical illnesses. Crit Care Med. 2019;47:e677–e84. https://doi.org/10.1097/ccm.0000000000003839.
Russell RT, Christiaans SC, Nice TR, Banks M, Mortellaro VE, Morgan C, Duhachek-Stapelman A, Lisco SJ, Kerby JD, Wagener BM, Chen MK, Pittet JF. Histone-complexed DNA fragments levels are associated with coagulopathy, endothelial cell damage, and increased mortality after severe pediatric trauma. Shock. 2018;49:44–52. https://doi.org/10.1097/shk.0000000000000902.
Gamberucci A, Fulceri R, Marcolongo P, Pralong WF, Benedetti A. Histones and basic polypeptides activate Ca2+/cation influx in various cell types. Biochem J. 1998;331(Pt 2):623–30. https://doi.org/10.1042/bj3310623.
Fattahi F, Grailer JJ, Jajou L, Zetoune FS, Andjelkovic AV, Ward PA. Organ distribution of histones after intravenous infusion of FITC histones or after sepsis. Immunol Res. 2015;61:177–86. https://doi.org/10.1007/s12026-015-8628-2.
Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. https://doi.org/10.1056/nejm200103083441001.
Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Pénzes I, Kübler A, Knaub S, Keinecke HO, Heinrichs H, Schindel F, Juers M, Bone RC, Opal SM. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. JAMA. 2001;286:1869–78. https://doi.org/10.1001/jama.286.15.1869.
Fourrier F. Severe sepsis, coagulation, and fibrinolysis: dead end or one way? Crit Care Med. 2012;40:2704–8. https://doi.org/10.1097/CCM.0b013e318258ff30.
Dhainaut JF, Yan SB, Joyce DE, Pettilä V, Basson B, Brandt JT, Sundin DP, Levi M. Treatment effects of drotrecogin alfa (activated) in patients with severe sepsis with or without overt disseminated intravascular coagulation. J Thromb Haemost. 2004;2:1924–33. https://doi.org/10.1111/j.1538-7836.2004.00955.x.
Kienast J, Juers M, Wiedermann CJ, Hoffmann JN, Ostermann H, Strauss R, Keinecke HO, Warren BL, Opal SM. Treatment effects of high-dose antithrombin without concomitant heparin in patients with severe sepsis with or without disseminated intravascular coagulation. J Thromb Haemost. 2006;4:90–7. https://doi.org/10.1111/j.1538-7836.2005.01697.x.
Umemura Y, Yamakawa K, Ogura H, Yuhara H, Fujimi S. Efficacy and safety of anticoagulant therapy in three specific populations with sepsis: a meta-analysis of randomized controlled trials. J Thromb Haemost. 2016;14:518–30. https://doi.org/10.1111/jth.13230.
Murao S, Yamakawa K. A systematic summary of systematic reviews on anticoagulant therapy in sepsis. J Clin Med. 2019;8:1869. https://doi.org/10.3390/jcm8111869.
Ely EW, Laterre PF, Angus DC, Helterbrand JD, Levy H, Dhainaut JF, Vincent JL, Macias WL, Bernard GR. Drotrecogin alfa (activated) administration across clinically important subgroups of patients with severe sepsis. Crit Care Med. 2003;31:12–9. https://doi.org/10.1097/00003246-200301000-00002.
Wiedermann CJ, Hoffmann JN, Juers M, Ostermann H, Kienast J, Briegel J, Strauss R, Keinecke HO, Warren BL, Opal SM. High-dose antithrombin III in the treatment of severe sepsis in patients with a high risk of death: efficacy and safety. Crit Care Med. 2006;34:285–92. https://doi.org/10.1097/01.ccm.0000194731.08896.99.
Kalil AC, LaRosa SP. Effectiveness and safety of drotrecogin alfa (activated) for severe sepsis: a meta-analysis and metaregression. Lancet Infect Dis. 2012;12:678–86. https://doi.org/10.1016/s1473-3099(12)70157-3.
Yamakawa K, Aihara M, Ogura H, Yuhara H, Hamasaki T, Shimazu T. Recombinant human soluble thrombomodulin in severe sepsis: a systematic review and meta-analysis. J Thromb Haemost. 2015;13:508–19. https://doi.org/10.1111/jth.12841.
Yamakawa K, Gando S, Ogura H, Umemura Y, Kabata D, Shintani A, Shiraishi A, Saitoh D, Fujishima S, Mayumi T, Kushimoto S, Abe T, Shiino Y, Nakada TA, Tarui T, Hifumi T, Otomo Y, Okamoto K, Kotani J, Sakamoto Y, Sasaki J, Shiraishi SI, Takuma K, Tsuruta R, Hagiwara A, Masuno T, Takeyama N, Yamashita N, Ikeda H, Ueyama M, Fujimi S. Identifying sepsis populations benefitting from anticoagulant therapy: a prospective cohort study incorporating a restricted cubic spline regression model. Thromb Haemost. 2019;119:1740–51. https://doi.org/10.1055/s-0039-1693740.
Umemura Y, Yamakawa K. Optimal patient selection for anticoagulant therapy in sepsis: an evidence-based proposal from Japan. J Thromb Haemost. 2018;16:462–4. https://doi.org/10.1111/jth.13946.
Wiedermann CJ, Kaneider NC. Comparison of mechanisms after post-hoc analyses of the drotrecogin alfa (activated) and antithrombin III trials in severe sepsis. Ann Med. 2004;36:194–203. https://doi.org/10.1080/07853890410027943.
Wiedermann CJ. Anticoagulant therapy for septic coagulopathy and disseminated intravascular coagulation: where do KyberSept and SCARLET leave us? Acute Med Surg. 2020;7:e477. https://doi.org/10.1002/ams2.477.
Dhainaut JF, Shorr AF, Macias WL, Kollef MJ, Levi M, Reinhart K, Nelson DR. Dynamic evolution of coagulopathy in the first day of severe sepsis: relationship with mortality and organ failure. Crit Care Med. 2005;33:341–8. https://doi.org/10.1097/01.ccm.0000153520.31562.48.
Ogura H, Gando S, Saitoh D, Takeyama N, Kushimoto S, Fujishima S, Mayumi T, Araki T, Ikeda H, Kotani J, Miki Y, Shiraishi S, Suzuki K, Suzuki Y, Takuma K, Tsuruta R, Yamaguchi Y, Yamashita N, Aikawa N. Epidemiology of severe sepsis in Japanese intensive care units: a prospective multicenter study. J Infect Chemother. 2014;20:157–62. https://doi.org/10.1016/j.jiac.2013.07.006.
Gando S, Shiraishi A, Yamakawa K, Ogura H, Saitoh D, Fujishima S, Mayumi T, Kushimoto S, Abe T, Shiino Y, Nakada TA, Tarui T, Hifumi T, Otomo Y, Okamoto K, Umemura Y, Kotani J, Sakamoto Y, Sasaki J, Shiraishi SI, Takuma K, Tsuruta R, Hagiwara A, Masuno T, Takeyama N, Yamashita N, Ikeda H, Ueyama M, Fujimi S. Role of disseminated intravascular coagulation in severe sepsis. Thromb Res. 2019;178:182–8. https://doi.org/10.1016/j.thromres.2019.04.025.
Wada H, Thachil J, Di Nisio M, Mathew P, Kurosawa S, Gando S, Kim HK, Nielsen JD, Dempfle CE, Levi M, Toh CH. Guidance for diagnosis and treatment of DIC from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013; https://doi.org/10.1111/jth.12155.
Hayakawa M, Gando S, Ono Y, Wada T, Yanagida Y, Sawamura A. Fibrinogen level deteriorates before other routine coagulation parameters and massive transfusion in the early phase of severe trauma: a retrospective observational study. Semin Thromb Hemost. 2015;41:35–42. https://doi.org/10.1055/s-0034-1398379.
Grundman C, Plesker R, Kusch M, Hanschmann KM, Eich S, Seitz R, König H. Prothrombin overload causes thromboembolic complications in prothrombin complex concentrates: in vitro and in vivo evidence. Thromb Haemost. 2005;94:1338–9.
Grottke O, Rossaint R, Henskens Y, van Oerle R, Ten Cate H, Spronk HM. Thrombin generation capacity of prothrombin complex concentrate in an in vitro dilutional model. PLoS One. 2013;8:e64100. https://doi.org/10.1371/journal.pone.0064100.
Grottke O, Braunschweig T, Spronk HM, Esch S, Rieg AD, van Oerle R, ten Cate H, Fitzner C, Tolba R, Rossaint R. Increasing concentrations of prothrombin complex concentrate induce disseminated intravascular coagulation in a pig model of coagulopathy with blunt liver injury. Blood. 2011;118:1943–51. https://doi.org/10.1182/blood-2011-03-343046.
Schöchl H, Voelckel W, Maegele M, Kirchmair L, Schlimp CJ. Endogenous thrombin potential following hemostatic therapy with 4-factor prothrombin complex concentrate: a 7-day observational study of trauma patients. Crit Care. 2014;18:R147. https://doi.org/10.1186/cc13982.
Sakuragawa N, Hasegawa H, Maki M, Nakagawa M, Nakashima M. Clinical evaluation of low-molecular-weight heparin (FR-860) on disseminated intravascular coagulation (DIC)—a multicenter co-operative double-blind trial in comparison with heparin. Thromb Res. 1993;72:475–500. https://doi.org/10.1016/0049-3848(93)90109-2.
Angus DC. Drotrecogin alfa (activated)…a sad final fizzle to a roller-coaster party. Crit Care. 2012;16:107. https://doi.org/10.1186/cc11152.
Allingstrup M, Wetterslev J, Ravn FB, Møller AM, Afshari A. Antithrombin III for critically ill patients: a systematic review with meta-analysis and trial sequential analysis. Intensive Care Med. 2016;42:505–20. https://doi.org/10.1007/s00134-016-4225-7.
Iba T, Thachil J. Is antithrombin III for sepsis-associated disseminated intravascular coagulation really ineffective? Intensive Care Med. 2016;42:1193–4. https://doi.org/10.1007/s00134-016-4288-5.
Gando S, Saitoh D, Ishikura H, Ueyama M, Otomo Y, Oda S, Kushimoto S, Tanjoh K, Mayumi T, Ikeda T, Iba T, Eguchi Y, Okamoto K, Ogura H, Koseki K, Sakamoto Y, Takayama Y, Shirai K, Takasu O, Inoue Y, Mashiko K, Tsubota T, Endo S. A randomized, controlled, multicenter trial of the effects of antithrombin on disseminated intravascular coagulation in patients with sepsis. Crit Care. 2013;17:R297. https://doi.org/10.1186/cc13163.
Tagami T, Matsui H, Horiguchi H, Fushimi K, Yasunaga H. Antithrombin and mortality in severe pneumonia patients with sepsis-associated disseminated intravascular coagulation: an observational nationwide study. J Thromb Haemost. 2014;12:1470–9. https://doi.org/10.1111/jth.12643.
Tagami T, Matsui H, Fushimi K, Yasunaga H. Supplemental dose of antithrombin use in disseminated intravascular coagulation patients after abdominal sepsis. Thromb Haemost. 2015;114:537–45. https://doi.org/10.1160/th15-01-0053.
Wiedermann CJ. Antithrombin concentrate use in disseminated intravascular coagulation of sepsis: meta-analyses revisited. J Thromb Haemost. 2018;16:455–7. https://doi.org/10.1111/jth.13950.
Tagami T. Antithrombin concentrate use in sepsis-associated disseminated intravascular coagulation: re-evaluation of a ‘pendulum effect’ drug using a nationwide database. J Thromb Haemost. 2018;16:458–61. https://doi.org/10.1111/jth.13948.
Saito H, Maruyama I, Shimazaki S, Yamamoto Y, Aikawa N, Ohno R, Hirayama A, Matsuda T, Asakura H, Nakashima M, Aoki N. Efficacy and safety of recombinant human soluble thrombomodulin (ART-123) in disseminated intravascular coagulation: results of a phase III, randomized, double-blind clinical trial. J Thromb Haemost. 2007;5:31–41. https://doi.org/10.1111/j.1538-7836.2006.02267.x.
Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, Hoste E, Levy H, Hirman J, Levi M, Daga M, Kutsogiannis DJ, Crowther M, Bernard GR, Devriendt J, Puigserver JV, Blanzaco DU, Esmon CT, Parrillo JE, Guzzi L, Henderson SJ, Pothirat C, Mehta P, Fareed J, Talwar D, Tsuruta K, Gorelick KJ, Osawa Y, Kaul I. A randomized, double-blind, placebo-controlled, phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013;41:2069–79. https://doi.org/10.1097/CCM.0b013e31828e9b03.
Vincent JL, Francois B, Zabolotskikh I, Daga MK, Lascarrou JB, Kirov MY, Pettilä V, Wittebole X, Meziani F, Mercier E, Lobo SM, Barie PS, Crowther M, Esmon CT, Fareed J, Gando S, Gorelick KJ, Levi M, Mira JP, Opal SM, Parrillo J, Russell JA, Saito H, Tsuruta K, Sakai T, Fineberg D. Effect of a recombinant human soluble thrombomodulin on mortality in patients with sepsis-associated coagulopathy: the SCARLET randomized clinical trial. JAMA. 2019;321:1993–2002. https://doi.org/10.1001/jama.2019.5358.
van der Poll T. Recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy: another negative sepsis trial? JAMA. 2019;321:1978–80. https://doi.org/10.1001/jama.2019.5792.
Levi M, Vincent JL, Tanaka K, Radford AH, Kayanoki T, Fineberg DA, Hoppensteadt D, Fareed J. Effect of a recombinant human soluble thrombomodulin on baseline coagulation biomarker levels and mortality outcome in patients with sepsis-associated coagulopathy. Crit Care Med. 2020;48:1140–7. https://doi.org/10.1097/ccm.0000000000004426.
Yamakawa K, Murao S, Aihara M. Recombinant human soluble thrombomodulin in sepsis-induced coagulopathy: an updated systematic review and meta-analysis. Thromb Haemost. 2019;119:56–65. https://doi.org/10.1055/s-0038-1676345.
Yamakawa K, Levy JH, Iba T. Recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy (SCARLET): an updated meta-analysis. Crit Care. 2019;23:302. https://doi.org/10.1186/s13054-019-2587-2.
Valeriani E, Squizzato A, Gallo A, Porreca E, Vincent JL, Iba T, Hagiwara A, Di Nisio M. Efficacy and safety of recombinant human soluble thrombomodulin in patients with sepsis-associated coagulopathy: a systematic review and meta-analysis. J Thromb Haemost. 2020;18:1618–25. https://doi.org/10.1111/jth.14812.
Shakur H, Roberts I, Bautista R, Caballero J, Coats T, Dewan Y, El-Sayed H, Gogichaishvili T, Gupta S, Herrera J, Hunt B, Iribhogbe P, Izurieta M, Khamis H, Komolafe E, Marrero MA, Mejía-Mantilla J, Miranda J, Morales C, Olaomi O, Olldashi F, Perel P, Peto R, Ramana PV, Ravi RR, Yutthakasemsunt S. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376:23–32. https://doi.org/10.1016/s0140-6736(10)60835-5.
Roberts I, Shakur H, Afolabi A, Brohi K, Coats T, Dewan Y, Gando S, Guyatt G, Hunt BJ, Morales C, Perel P, Prieto-Merino D, Woolley T. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377:1096–101, 101.e1–2. https://doi.org/10.1016/s0140-6736(11)60278-x.
CRASH-3 Trial Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet. 2019;394:1713–23. https://doi.org/10.1016/s0140-6736(19)32233-0.
Wildhagen KC, García de Frutos P, Reutelingsperger CP, Schrijver R, Aresté C, Ortega-Gómez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GA. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood. 2014;123:1098–101. https://doi.org/10.1182/blood-2013-07-514984.
Hogwood J, Pitchford S, Mulloy B, Page C, Gray E. Heparin and non-anticoagulant heparin attenuate histone-induced inflammatory responses in whole blood. PLoS One. 2020;15:e0233644. https://doi.org/10.1371/journal.pone.0233644.
Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123:2768–76. https://doi.org/10.1182/blood-2013-10-463646.
Spero JA, Lewis JH, Hasiba U. Disseminated intravascular coagulation. Findings in 346 patients. Thromb Haemost. 1980;43:28–33.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gando, S., Wada, T. (2022). ICU Management: Disseminated Intravascular Coagulation (DIC). In: Pape, HC., Borrelli Jr., J., Moore, E.E., Pfeifer, R., Stahel, P.F. (eds) Textbook of Polytrauma Management . Springer, Cham. https://doi.org/10.1007/978-3-030-95906-7_33
Download citation
DOI: https://doi.org/10.1007/978-3-030-95906-7_33
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-95905-0
Online ISBN: 978-3-030-95906-7
eBook Packages: MedicineMedicine (R0)