
Abstract
Following trauma, local hemostasis and thrombosis act to induce physiological wound healing and innate immune responses, respectively, to impede the dissemination of damage-associated molecular patterns (DAMPs) into the systemic circulation. However, if overwhelmed by systemic inflammation caused by extensive tissue damage and tissue hypoperfusion, both of these processes cause pathologic changes, which manifest as systemic disseminated intravascular coagulation (DIC). High levels of DAMPs and inflammatory cytokines activate both extrinsic and intrinsic coagulation pathways. Impaired anticoagulation pathways induce insufficient control of coagulation, leading to systemic thrombin generation and ultimately consumption coagulopathy. Fibrin(ogen)olysis due to tissue-type plasminogen activator is highly active in the early phase of trauma due to endothelial hypoperfusion and hypoxia in DIC with the fibrinolytic phenotype, contributing to the oozing-type coagulopathic bleeding. Persistently high levels of plasminogen activator inhibitor-1 expressed in the endothelium change DIC with the fibrinolytic phenotype into the thrombotic phenotype, which is followed by microvascular thrombosis and then multiple organ dysfunction syndrome. Microvascular thrombosis has been observed in both types of DIC, but especially in the fibrinolytic phenotype, where it can be exacerbated by antifibrinolytic therapy. DIC should be diagnosed by DIC scoring systems, and key to managing DIC is treating the trauma itself and hemorrhagic shock. The mechanisms of hemostatic changes in trauma are multifactorial; the coexistence of hypothermia, acidosis, and dilution aggravate DIC and lead to so-called trauma induced coagulopathy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Trauma
- Disseminated intravascular coagulation (DIC)
- Coagulopathy
- Coagulation
- Fibrinolysis
- Anticoagulation
- Thrombin
- Plasmin
Historical Perspective
Disseminated intravascular coagulation (DIC) was first described clinically in a case of sepsis following trauma and was termed as “ DIC syndromes” in 1961 [1]. On the basis of autopsy evidence, MacKay lists trauma as one of the most common causes of DIC and he discusses the pathogenesis of DIC based on a thorough understanding of the clinical courses of coagulation changes seen after trauma [2]. As early as the 1970s, Flute [3] correctly pointed out that trauma stimulates blood coagulation and fibrinolysis concurrently, and that fibrinolysis is the compensatory mechanism for fibrin formation in the blood. When this compensating mechanism fails, DIC may cause microvascular thrombosis with bleeding and tissue necrosis resulting from a defibrination syndrome. The pathogenesis of DIC in trauma is considered to be due to the entry of thromboplastic materials, such as the products of tissue damage, into the circulation, which in turn lead to platelet aggregation, coagulation activation, and fibrin deposition. If sufficiently severe to induce the consumption of hemostatic components, systemic intravascular coagulation results [2–4]. So, even by the early 1970s, the definition, pathogenesis, clinical aspects, and treatment of DIC as well as the clinical conditions associated with it, such as trauma, had already been described.
Until the early 1980s, whole blood was transfused, which provided adequate amounts of most coagulation factors. During massive transfusion of packed red blood cells (PRBC) the dilution of coagulation factors occurs long before thrombocytopenia develops [5]. Trauma specialists are well aware of the fact that injury and hemorrhagic shock, not hemodilution, induce the early coagulopathy of trauma [6–10]. In 1985, Ordog et al. clearly demonstrated that coagulation abnormalities during hemorrhagic shock are attributable to the trauma itself, independent of fluid and blood replacement; this was reported to most likely be DIC [2, 6, 11]. In the late 1980s, the use of whole blood was almost completely replaced by PRBC, which have no coagulation factors. Around a decade later, Hiippala [12] revealed that hypofibrinogenemia develops first, is followed by other coagulation factor deficits and later by thrombocytopenia. Therefore, the use of plasma transfusion became the primary intervention for abnormal bleeding [12]. Despite these changes in transfusion strategies and the importance of Hiippala’s report, the incorrect notion that plasma should be transfused late in the resuscitation process continued to be advocated until recently [13]. This partly explains the prevailing notion of dilutional coagulopathy and the lack of acceptance of an endogenously induced DIC according to trauma or shock itself [14]. In view of this historical perspective, although multiple factors contribute to trauma induced coagulopathy, it is DIC induced consumption coagulopathy, and not dilution coagulopathy, that is important pathogenetically, with hypothermia and acidosis subsequently modifying the processes of DIC.
Definition and Diagnosis of DIC
The Scientific and Standardization Committee (SSC) on DIC of the International Society on Thrombosis and Haemostasis (ISTH) defines DIC as an acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction [15]. The most important aspects of this definition are the “activation of coagulation with loss of localization” and “damage to the microvasculature,” referring in turn to thrombin generation in the circulation and to extensive damage to endothelial cells, which results in insufficient coagulation control. Figure 13.1 shows the basic concept of DIC in which circulating blood is hypercoagulable due to systemic thrombin generation due to insufficient anticoagulation mechanisms, but is hypocoagulable outside the vessels and difficult to clot following injury due to consumption coagulopathy [16].

The properties of blood inside and outside the vessels under normal conditions and DIC. Reprinted with modifications, with permission from Clinics in DIC by Matsuda T; Shinkoh-Igaku Shuppan Co., Ltd., Tokyo, 1983
The DIC diagnostic criteria of ISTH and the Japanese Association for Acute Medicine (JAAM) have been prospectively validated in critically ill patients, including those with trauma [16–18]. The JAAM DIC scoring system has proven diagnostic validity for DIC in the early phase of trauma and has better diagnostic sensitivity than the ISTH scoring system. In addition, the JAAM DIC score on admission to the emergency department (ED) can independently predict death and the need for massive transfusion in trauma patients [19–21]. These features of the JAAM diagnostic criteria may be dependent on the deletion of fibrinogen as a scoring criteria, inclusion of sensitive systemic inflammation criteria, and the addition of a dynamic component such as decreasing rate of platelet count. The ISTH scoring system includes a table of the “clinical conditions that may be associated with DIC” as a mandatory clause, and restricts the use of the scoring algorithm in patients without underlying diseases. JAAM presents the same table while also adding another table with the title of “clinical conditions that should be carefully ruled out” in order to increase the specificity of the scoring system. Tables 13.1 and 13.2 show the ISTH and JAAM DIC scoring systems, respectively [15, 18].
Phenotypes and Time Courses of DIC
DIC can be subdivided into the fibrinolytic (hemorrhagic) and antifibrinolytic (thrombotic) phenotypes [14, 22, 23]. DIC in the early phase of trauma manifests a fibrinolytic phenotype, which contributes to coagulopathic bleeding and is associated with a poor prognosis [14, 18]. DIC in the late phase of trauma has a thrombotic phenotype that also affects prognosis as it leads to organ dysfunction [14, 16, 24]. The synergistic activation of primary and secondary fibrin(ogen)olysis by tissue-type plasminogen activator (tPA) is considered to be the cause of DIC with the fibrinolytic phenotype [22, 23], while plasminogen activator inhibitor-1 (PAI-1) mediated inhibition of fibrinolysis is considered to be the cause of DIC with the thrombotic phenotype [16, 23, 24]. Activation of coagulation and an ineffective anticoagulation system are common to both phenotypes (Fig. 13.2) [25]. For further description of fibrinolysis mechanisms seen in trauma induced coagulopathy refer to Chap. 9 ( Fibrinolysis-Moore).

The two phenotypes of DIC. Although the activation of the tissue-factor dependent pathway as the initial step of the coagulation cascade and the presence of insufficient anticoagulation systems are the same, DIC can be subdivided into the fibrinolytic (broken line) and thrombotic (straight line) phenotypes. In DIC with the fibrinolytic phenotype, DIC and systemic fibrin(ogen)olysis coexist. Annexin II expression on the promyelocytes increases the t-PA activity in patients with acute promyelocytic leukemia. Microvascular thrombosis- and shock-induced hypoxia/ischemia in endothelial cells accelerate t-PA release from endothelial Weibel–Palade bodies. Neutrophil elastase-derived fibrinolysis and consumption-induced α2-plasmin inhibitor α2 plasmin inhibitor depression enhance fibrin(ogen)olysis. Reprinted from reference [25]
Use of the DIC diagnostic criteria distinguishes the pathological reaction of DIC from physiological hemostasis and wound healing [14, 16, 26]. Figure 13.3 (left) shows normal changes in hemostasis and wound healing, while Fig. 13.3 (right) shows the abnormal hemostatic responses associated with DIC from immediately after trauma to the late phase of trauma [14, 16, 26].

Schematic diagrams of the variations in thrombin activity (A, measured by fibrinopeptide A-FPA), plasmin activity (B, fibrinopeptide B β15-42-FPBβ15-42), fibrin formation, and secondary fibrinolysis (C, D-dimer) from day 0 (in the emergency department) to day 4. Left, normal changes in hemostasis and wound healing. There are three phases of fibrinolysis: early activation, impairment (D, PAI-1: fibrinolysis shutdown), and reactivation. Normally, both thrombin activity and PAI-1 are completely shut off by days 3–5 after trauma, followed by the reactivation of fibrinolysis. Right, pathological changes in DIC. Massive thrombin activation persists until day 4 after trauma; increased activation of plasmin as well as excessive fibrinolysis are present on day 0 (* DIC with the fibrinolytic phenotype), followed by impairment of fibrinolysis due to persistent elevation of PAI-1 released from endothelial cells with transcription (** DIC with the thrombotic phenotype). Reprinted with permission [26]
Pathogenesis of DIC
Innate Immunity, Inflammation, and Coagulation at the Injury Site
Trauma can produce a systemic inflammatory response syndrome (SIRS) characterized by proinflammatory cytokine release and activation of leukocytes and endothelial cells, processes which are understood as innate immunity [27]. Close interactions between innate immunity, inflammation, and coagulation have been recognized [28, 29]. Innate immune cells have evolved cell-specific prothrombotic pathways that are activated after insults and operate in intact blood vessels to protect the host from nonself (pathogen-associated molecular patterns, PAMPs ) and altered-self (damage-associated molecular patterns, DAMPs); this concept is referred to as immunothrombosis [30]. During the responses to PAMPs and DAMPs, monocytes and their microparticles express tissue factor, which activates the extrinsic coagulation pathway [31, 32]. Neutrophils are recruited to the sites of inflammation and are activated [33], and then release neutrophil extracellular traps (NETs) , which are comprised of a matrix of DNA, histones, nucleosomes, and neutrophil elastase, thereby promoting thrombosis [34]. Histones induce platelet activation and also promote thrombin generation both by the recruitment of platelet and the impairment of thrombomodulin-dependent protein C activation [35–37]. NETs can also activate the intrinsic coagulation pathway by activating FXII to form FXIIa [38], which then promotes the activation of complement pathways. The generated C3a and C5a further promote thrombosis and platelet activation [39]. In addition, extracellular RNA derived from damaged cells constitutes a procoagulant cofactor for the activation of the FXII/FXI coagulation pathway [40]. The neutrophil elastase that is present on NETs induces the degradation and inactivation of tissue factor pathway inhibitor (TFPI) [41, 42] and the thrombomodulin expressed on the endothelium [43, 44]. These processes further promote thrombin generation. For further description of DAMPs as well as of neutrophils and the innate immune system during injury, please refer to Chap. 11 (DAMPs-Esmon) and Chap. 10 (Neutrophils-Yaffe), respectively.
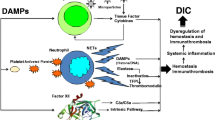
Activation of coagulation and insufficient control of it lead to immunothrombosis at injury sites with inflammation, impeding the dissemination and tissue invasion of PAMP and DAMPs as well as pathogens and damaged cells themselves [30, 42]. In trauma, thrombin escaping into the circulation from the injury sites is controlled by antithrombin, TFPI, and thrombomodulin present in intact endothelial cells, as depicted in the cell-based model of hemostasis [45]. However, when systemic inflammation caused by both extensive injury and shock overwhelms these control mechanisms that restrict hemostasis and inflammation locally, DIC ensues [45]; Figs. 13.4 and 13.5 outline t hese processes.

The pathophysiological processes of local hemostasis, immunothrombosis, and systemic DIC. Tissue injury promotes local hemostasis and wound healing. Tissue injury also induces microvascular fibrin thrombosis, or immunothrombosis, to protect the host from DAMPs and to restrict DAMPs only to the injured vascular compartment. DIC results when local hemostasis and immunothrombosis are no longer able to anchor thrombin or to restrict the spread DAMPs at the injured site. Reprinted from reference [25]

Trauma elicits nonspecific innate immune inflammatory responses that limit and repair tissue damage after insults. The figure depicts a simplified schematic representation of the engagement of pattern recognition receptors by DAMPs and their signaling through the adaptor proteins. This cascade promotes the transcription of several pro-inflammatory cytokines and chemokines, leading to local and systemic inflammatory responses. Local inflammation begins as an adaptive host response, serving to promote host defense and physiologic hemostasis and wound healing. Spillover of the inflammatory cytokines into the circulation elicits SIRS, which activates systemic coagulation, suppresses fibrinolysis, and overwhelms the anticoagulant control mechanisms that restrict hemostasis locally, giving rise to DIC. Importantly, DAMPs themselves activate coagulation and impair anticoagulation pathways through endothelial damage (reprinted, with modifications, with permission from Gando S, et al. Infectious and non-infectious insults and body responses. Jp J Acute Med 2011;35:747–52). ACS apoptosis-associated speck-like protein containing caspase recruit domain, DAMPs damage-associated molecular patterns, DIC disseminated intravascular coagulation, MAVS mitochondrial antiviral signaling, MODS multiple organ dysfunction syndrome, MyD88 myeloid differentiation factor 88, NLRs nucleotide-binding oligomerization domain containing receptors, RLRs retinoic acid inducible gene-I-like receptors, SIRS systemic inflammatory response syndrome, STING stimulator of interferon gene, TRIF toll/IL-1 receptor homology domain-containing adaptor inducing interferon beta, TLRs toll-like receptors
Systemic Activation of Coagulation
The major DAMPs released into the extracellular environment following tissue injury are histones, mitochondrial DNA, nucleosomes, and high mobility group box 1 (HMGB1) [46]. Mitochondrial DNA at levels thousands times higher than those measured in healthy volunteers have been observed at a median 93 min after trauma [47]. Elevated levels of mitochondrial DNA were associated with SIRS and correlated with Injury Severity Score (ISS) in trauma patients [48, 49]. Elevated levels of histones and HMGB1 have been seen in severely injured trauma patients within 30 min of trauma or immediately after arrival to the ED [50–52]. Toxic levels of histones insult cells, leading to endothelial injury (elevation of soluble thrombomodulin), platelet aggregation, coagulation activation [increase in thrombin–antithrombin complex (TAT)], interleukin-6 (IL-6) release, and NETs formation [52]. These processes result in edema, microvascular thrombosis, and neutrophil accumulation in the lungs. The HMGB1 released by damaged and inflammatory cells at the injury site promotes the development of microvascular thrombosis [53]. An important point is that HMGB1 inhibits the anticoagulant protein C pathway mediated by the thrombin–thrombomodulin complex, and stimulates tissue factor expression on monocytes. Histones also reduce the cofactor activity of both soluble and endothelial thrombomodulin, impairing protein C activation, and thereby stimulating plasma thrombin generation [37]. For further description of the protein C pathway please refer to Chap. 6 (Protein C-Cohen).
Tumor necrosis factor α (TNF-α) and IL-6 are elevated immediately after histone infusion [52, 54]. In fact, TNF-α and IL-1β are increased on arrival to the ED in trauma patients with DIC [55], with an IL-6 surge occurring within 2 h from injury [56]. This early release of IL-6 suggests it is most likely released from pre-synthesized stores [52]. IL-6 is the most important driver of tissue factor expression on monocytes and endothelial cells [57]. TNF-α and IL-1 also have been shown to elicit tissue factor formation and expression on the surface of monocytes and endothelial cells [58]. These inflammatory cytokines subsequently block the protein C anticoagulant pathway by downregulating thrombomodulin and the endothelial protein C receptor (EPCR) on the endothelium [58]. Furthermore, these inflammatory cytokines activate neutrophils and endothelial cells, and the activated neutrophils release neutrophil elastase, which can cleave thrombomodulin, leading to the release of soluble thrombomodulin from the endothelium in a less active form [43, 44, 58, 59].
Although, the mechanisms involved in microparticle formation in-vivo remain essentially unknown, blood contains microparticles derived from a variety of cell types, including platelets, monocytes, and endothelial cells [60]. All microparticles are pro-coagulant because they provide a membrane surface for assembly of coagulation proteases [61]. In severe trauma, activated platelets enhance the microparticle formation associated with platelet and leucocyte interaction [62]. Increased microparticle formation and thrombin generation are observed immediately after trauma and correlate with ISS [63].
These lines of evidence clearly indicate that the release of DAMPs from injured cells and tissues, the DAMP induced release of inflammatory cytokines, and microparticle formation synergistically hamper the mechanisms controlling coagulation by protein C pathways and activate coagulation, leading to SIRS and systemic thrombin generation, namely DIC, immediately after trauma [30, 64].
Impairment of Anticoagulation Pathways and Endothelial Injury
TFPI
The highly activated tissue factor-dependent pathway is not sufficiently prevented by normal TFPI levels in DIC patients after trauma because neutrophil elastase cleaves TFPI within the polypeptide that links the first and second Kunitz domains [65]. This impairs the ability of TFPI to neutralize both FXa and the tissue factor/FVIIa complex. This finding suggests that tissue factor and tissue factor/FVIIa complex are continuously formed at a rate that normal TFPI inhibition cannot match in DIC patients after trauma [65, 66].
Thrombomodulin and Endothelium
Higher levels of neutrophil elastase and soluble thrombomodulin have been confirmed in patients with DIC and in patients with severe traumatic injuries [52, 67, 68]. Soluble thrombomodulin can be formed by the limited proteolysis of endothelial cell membrane thrombomodulin by neutrophil elastase without any evidence of active secretion [43, 44]. The amount of soluble thrombomodulin correlates with the degree of endothelial injury [44]. Moreover, early elevation of TNF-α and IL-1β in DIC patients after trauma causes thrombomodulin downregulation in the endothelium [43, 44, 55]. Traumatic shock-induced hypoxia leads to a reduction in thrombomodulin and the suppression of thrombomodulin mRNA in the endothelium [69, 70]. Therefore, the high soluble thrombomodulin levels in DIC patients suggest a loss of functional thrombomodulin in the endothelium. In addition, soluble thrombomodulin has only 20 % of the activity of thrombomodulin bound to the endothelium [71]. Taken together, these results suggest that endothelial injury and functional loss of both soluble and endothelial thrombomodulin occurs in DIC after trauma.
Protein S and Protein C
Low levels of protein C activity have been repeatedly confirmed from the early to late phases of DIC [16, 18, 20, 67]. The thrombin–thrombomodulin complex activates protein C to generate activated protein C. For activated protein C to function more effectively, it must form a complex with both protein S and EPCR. The anticoagulant activity of protein S is neutralized by the formation of a complex with complement C4b binding protein (C4bBP) . Increased levels of C4bBP as a consequence of the acute phase reaction following inflammatory insults cause a relative protein S deficiency, which contributes to a procoagulant state and lethal DIC [72]. Lower levels of protein S activity associated with thrombin generation (prothrombin fragment 1 + 2, PF1 + 2) have been demonstrated in trauma patients immediately after arrival at the ED [9]. For further description of the protein C pathway please refer to Chap. 6 (Protein C-Cohen).
Activated protein C is immediately inactivated by protease inhibitors, such as the protein C inhibitors, α1-antitrypsin, α2-antiplasmin, and α2-macroglobulin. In cases of DIC, lower protein C and protein S levels, relative protein S deficiency, and impaired functions of both soluble and endothelial thrombomodulin are all implicated in the insufficient conversion of protein C to activated protein C and the inability of activated protein C to function normally. Increases in activated protein C levels do not indicate a shutoff of thrombin generation. In fact, the elevated activated protein C levels (~10 ng/mL) did not reach a concentration sufficient to inhibit thrombin generation (70–80 ng/mL) in s everely injured trauma patients with tissue hypoperfusion [73, 74].
Antithrombin
Antithrombin inactivates thrombin and inhibits several proteases in both the extrinsic and intrinsic coagulation pathways, including FIXa, FXa, FXIa, and FXIIa. For further description of antithrombin please refer to Chap. 2 (TAT-Bock). Thus, a reduction in antithrombin can markedly influence the coagulation processes and is a potential risk factor for thrombosis [75]. Insufficient levels of antithrombin, compared with the potential for thrombin generation in the prothrombin complex concentrate, induced DIC characteristics in a pig model of coagulopathy with blunt liver injury [76]. The severity of injury and tissue hypoperfusion are major contributors to the reduction in antithrombin in trauma [77, 78]. Low antithrombin levels are associated with thromboembolic complications, which can develop from DIC [79]. Extremely low levels have been observed in cases of trauma patients immediately after arrival at the ED [8, 21] and for several days thereafter [8, 21, 80].
Two studies showed that a decreased ability to localize hemostasis at the wound site and subsequently generate thrombin systemically results from decreased antithrombin levels in patients with DIC and coagulopathy immediately after trauma [81, 82]. Similarly, a multiple regression analysis demonstrated that low antithrombin levels are an independent determinant of high soluble fibrin levels and a marker of thrombin generation in trauma patients with DIC [80].
These findings also indicate there is much lower availability of the TFPI, antithrombin/glycosaminoglycan, and thrombomodulin/protein C systems for the regulation of thrombin generation and activation in DIC patients. Moreover, higher soluble thrombomodulin levels suggest the presence of extensive damage to microvasculature endot helium. Figure 13.6 summarizes these changes.

The balance between thrombin generation and inhibition. DIC occurs when there is an imbalance between thrombin generation and inhibition. Insufficient coagulation control mechanisms contribute to massive thrombin generation in the circulation, which overwhelms activated protein C mediated inhibition of thrombin generation. Reprinted from reference [25]
Thrombin Generation in the Systemic Circulation
Soluble fibrin and fibrinopeptide A are regarded as accurate markers of thrombin generation and activity because both are formed as a result of the direct action of thrombin on fibrinogen, which leads to fibrin formation. Extremely elevated levels of fibrinopeptide A have been noted in trauma patients with DIC immediately after arrival at the ED [8]. In addition, higher levels of these molecular markers of thrombin generation in the early phase of trauma have been repeatedly confirmed [9, 14, 67, 68]. In two studies, Dunbar and Chandler observed excessive non-wound-related thrombin generation in trauma patients with both DIC and trauma induced coagulopathy immediately after arrival at the hospital (Fig. 13.7) [81, 82]. Their first study showed marked systemic thrombin generation due to circulating procoagulants that initiate thrombin generation systemically, as well as reduced ability to localize hemostasis at the wound site due to the loss of antithrombin. Their second study found that tissue factor activity accounted for approximately 80 % of all procoagulant activity. Importantly, the term “acute coagulopathy of trauma” in the first study was changed to “DIC” in this second one. Reports showing a significant correlation between tissue factor and the markers of thrombin generation, and microparticle formation by activated platelets support these results [62, 83].

Native and tissue factor (TF) stimulated thrombin generation (TG) curves in a normal subject and in a patient with acute coagulopathy of trauma. Native TG: no added tissue factor or phospholipid, contact activation blocked. TF stimulated: sample activated with TF and phospholipid. In normal subjects, little or no thrombin was generated during native TG. In contrast, in patients with acute coagulopathy of trauma, native and TF stimulated TG curves were often similar. This indicates that plasma from those with trauma induced coagulopathy has circulating pro-coagulant activity that can spontaneously initiate coagulation throughout the vascular system, not just at the injured site. Note: The term “acute coagulopathy trauma” used in this study [81] was changed to “DIC” in a subsequent study [82]. Modified with permission [81]
The overall function of the thrombomodulin/protein C anticoagulant pathway can be precisely evaluated by measuring prothrombinase activity [84]. Prothrombinase is a complex comprising FXa, FVa, phospholipids, and Ca2+ and it is the major determinant of thrombin generation from prothrombin. Prothrombinase activity, measured as the thrombin generation rate, decreases in proportion to the amount of thrombin-thrombomodulin complex formation of activated protein C and the subsequent inactivation of FVa [84, 85]. DIC patients after trauma have shown normal prothrombinase activity associated with higher levels of soluble fibrin [80]. These findings suggest that the inhibition of the prothrombinase activity caused by activated protein C-mediated anticoagulation does not overwhelm the activation of the tissue factor systemic thrombin generation or its activation in trauma patients with DIC. This imbalance between thrombin generation (soluble fibrin) and its inhibition (prothrombinase activity) is due to insufficiency in the other anticoagulant mechanisms, such as TFPI and antithrombin, as well as impaired thrombomodulin function due to endothelial injury [80].
Consumption Coagulopathy
The consumptive processes in DIC reflect the multiple actions of thrombin. Increased thrombin generation accounts for decreases in platelets, fibrinogen, FII, FV, FVIII, and FXIII in acute consumption, and the rapid clearance of activated clotting factors in vivo accounts for decreases in other clotting factors such as FIX and FX [22]. Thrombin also induces the release of tPA from endothelial cells, leading to plasmin generation. If plasmin is formed sufficiently in the circulation, it degrades fibrinogen, FV, and FVIII. These lines of evidence support the rapid consumption of thrombin-sensitive hemostatic factors, including platelets, fibrinogen, and Factors V, VIII, and XIII. In pre-DIC and DIC, sensitive and rapid decreases in the levels of FV and FVIII have been observed as a result of thrombin-mediated protein C activation [86, 87]. In DIC due to trauma, platelets are sometimes consumed slowly due to marginalization in blood vessels and release from storage in organs such as the spleen, liver, and lungs [12, 14, 16]. FVIII is known to paradoxically increase in response to clinical insults, including trauma, due to release of von Willebrand factor (VWF) from the endothelial Weibel–Palade bodies [88] and the acute phase behavior of FVIII. VWF immediately interacts with FVIII, prolonging the plasma half-life of FVIII [89]. The consumption of coagulation factors prolongs both the prothrombin time (PT) and activated partial thromboplastin time (PTT); however, the PTT is sometimes normal or even shortened because of the interactions between FVIII with VWF in spite of a prolonged PT in DIC patients.
In cases of trauma with DIC, prolonged PT reflects a decrease in FV, to a lesser extent decreases in Factors II, VII, and X, and decreases in fibrinogen levels immediately to several days after trauma [8, 14, 16, 18, 19, 55, 67, 80, 83]. Prolonged PTT, which reflects a decrease in Factors V, VIII, and fibrinogen, has also been confirmed immediately after trauma in patients who develop DIC [8]. The FVII antigen has been demonstrated to be consumed at a relatively slow speed for about 8 h in a rabbit model of DIC [90]. Importantly, this translated into a FVIIa level increase to 120 % within 2 h after DIC induction, before declining thereafter. Furthermore, in patients studied upon arrival to the ED [91] FXIII and α2-plasmin inhibitor levels where shown to be markedly decreased in DIC.
Activation and Suppression of Fibrinolysis
DIC and pathological systemic fibrin(ogen)olysis sometimes coexist following an insult such as trauma and is referred to as DIC with the fibrinolytic phenotype [22, 23]. Tissue hypoperfusion causes tPA release from the endothelial Weibel–Palade bodies, which leads to systemic fibrin(ogen)olysis in addition to DIC with secondary fibrinolysis [22, 23, 88]. Increased fibrinolysis, as well as the activation of coagulation in trauma, has long been recognized [3, 92], as it was confirmed in a study of severely injured patients, 40 % of whom had a PT international normalized ratio >1.2 [93]. This study demonstrated increased thrombin generation with concomitant fibrinogen and antithrombin consumption, as well as increased tPA levels, plasmin generation, and fibrinolysis along with α2-plasmin inhibitor consumption, all of which coincided with DIC with the fibrinolytic phenotype. The most important point in the pathogenesis of fibrin(ogen)olysis in the early phase of trauma is that there is a several hour time difference between the immediate release of tPA from the endothelium and the later expression of PAI-1 mRNA, leading to an extreme imbalance in these molecules [94–96]. In support of this imbalance, the levels of PAI-1 antigen and activity were found to be almost identical in patients with and without DIC immediately after trauma, while the levels of tPA and plasmin and α2-plasmin inhibitor complex, a marker of plasmin generation, were significantly increased only in DIC patients [8, 16, 55, 80].
In addition to plasmin, neutrophil elastase mediated fibrinolysis is also involved in the pathogenesis of fibrin(ogen)olysis in DIC with the fibrinolytic phenotype [67]. The lower levels of α2-plasmin inhibitor, FXIII, and fibronectin in DIC patients suggest that there is insufficient inhibition of plasmin, impaired cross linking of fibrin, and delayed wound healing, leading to fragile fibrin formation associated with coagulopathic bleeding [8, 91]. A study showing tissue factor driven fibrin(ogen)olysis without tissue hypoperfusion suggests that secondary fibrinolysis caused by tPA release driven by a massive amount of fibrin formation may also have a role in DIC with the fibrinolytic phenotype [97]. Importantly, thrombomodulin mediated thrombin-activatable fibrinolysis inhibitor (TAFI) activation does not appear to have an important role in the pathogenesis of fibrin(ogen)olysis immediately after trauma [67], which indirectly implicates thrombomodulin/protein C pathway impairment.
Fibrinolysis driven by immediate tPA release is usually followed by PAI-1 suppression of fibrinolysis. After achieving hemostasis, the activation of coagulation and PAI-1 disappears completely and fibrinolytic reactivation occurs to degrade excess fibrin clots during physiological wound healing [8, 14, 16, 26]. However, persistent PAI-1 elevation continues until day 5 after injury in DIC patients, which is referred to as DIC with the thrombotic phenotype [8, 14, 16, 26, 55, 95]. When uncontrolled, DIC with the fibrinolytic phenotype in the early phase of trauma progresses to DIC with the thrombotic phenotype in the late phase of trauma [98]. DIC severity and the presence of organ dysfunction briefly after injury, but with complicating sepsis, are involved in the pathogenesis of this continuous progression. During the thrombotic stage, increased fibrinolysis, reflected by elevated D-dimer levels, cannot match the massive fibrin formation, leading to microvascular thrombosis, hypoperfusion and impaired oxygen delivery, which in turn give rise to multiple organ dysfunction syndrome [24, 95, 99]. Figure 13.3 illustrates these processes [26].
DIC and Microvascular Thrombosis
Trauma and Hemorrhagic Shock
Histological evidence of microvascular thrombosis in DIC, especially in DIC with the thrombotic phenotype, has been reported by clinical, experimental, and autopsy studies [99]. Evidence of DIC with the fibrinolytic phenotype is rarely available in humans and was extensively debated during the 1960s and 1970s [100]. These debates on the inconsistency of thrombus formation had come about because of the existence of hyperfibrin(ogen)olysis in the early phase of trauma and hemorrhagic shock. However, fibrin thrombosis [101], vein thrombi formation [102], platelet aggregation, and emboli formation [103, 104] were repeatedly confirmed in hemorrhagic shock and trauma. Subsequently, platelet and fibrin thrombosis became more evident during antifibrinolytic therapy using tranexamic acid in a dog model of hemorrhagic shock [105] (Fig. 13.8). It should be emphasized that the authors of a report expressing negative opinions about DIC, did in fact conclude that some fibrin thrombi were observed in their histological study [106]. Importantly, signs of inflammation, microthrombus and embolus formation have also been observed within 24 h of injury in human studie s [107, 108].

The inhibition of fibrinolysis by tranexamic acid revealed microvascular thrombosis and thromboemboli formation in large vessels in a dog model of hemorrhagic shock. (a) Section of a branch of the portal vein almost completely filled by a mixed thrombus consisting of platelets and fibrin threads in bundles. (b) Section from lung vessels with thrombotic masses consisting of both platelets and fibrin thread. PTAH-staining. Modified with permission [105]
Isolated Traumatic Brain Injury
Publications have demonstrated that the coagulopathy of isolated traumatic brain injury (iTBI) coincides with the definition of DIC proposed by the ISTH; namely intravascular activation of coagulation, with loss of localization of coagulation, and damage to the microvasculature [15, 109, 110]. Therefore, coagulofibrinolytic changes after iTBI may have some overlap with those observed in trauma patients without brain injury; if sufficiently severe, they give rise to DIC [111].
Microthrombi are frequently present in the brains of iTBI patients who died within 24 h of injury, and were found to be associated with marked changes in platelet count, coagulation, and fibrinolysis markers (Fig. 13.9) [109]. More importantly, systemic microthrombi were seen in the spinal cord, liver, lungs, kidneys, colon, and pituitary gland, indicating the presence of DIC in 88 % of patients after autopsy [109]. Stein et al. examined brain tissue from several sources, including animal models and patients with contused brain tissue removed during surgical decompression, and found a high correlation between the severity of coagulopathy and the presence of intravascular thrombosis, confirming the association between intravascular thrombosis and DIC [110, 112]. These investigators also demonstrated a strong link between intravascular thrombosis and the area of ischemic changes and neuronal death [110, 113]. An animal experiment confirmed that the immediate posttraumatic decrease in peri-contusional blood flow is caused by platelet activation and subsequent microthrombosis in the cerebral circulation [114]. These lines of evidence clearly indicate that a DIC processes occurring immediately after iTBI can contribute to secondary brain injury.

Microthrombi in the meningeal vessels were observed in patients with iTBI. Fibrin stains brown (white arrow), containing with red blood cells, which are colorless to faint blue (black arrow) (Immunoperoxidase stain with antifibrinogen specific antiserum and hematoxylin counterstain, ×400). Modified with permission [109]
Fat Embolism Syndrome
Fat embolism syndrome (FES) typically occurs 12–36 h after long bone and pelvic fractures, although fulminant cases immediately after injury have also been reported. Several lines of evidence indicate the presence of DIC in patients with FES [115–117].
Saldeen et al. observed morphological changes in pulmonary, cerebral, and systemic fat emboli that are associated with pathophysiological characteristics of DIC [118]. Furthermore, fibrin thrombosis in pulmonary vessels has been confirmed more often in patients with FES than in other post-traumatic cases, especially in those in whom FES was considered to be the only explanatory cause of death [119]. The presence of fibrin thrombi in lung vessels was also confirmed in patients with FES and acute respiratory distress syndrome [120]. Hyaline microvascular thrombosis and aggregates of platelets, indicating fibrin thrombi and intravascular coagulation, have been reported in both the cerebral arteries and veins in post-traumatic FES patients [121]. Histopathological analysis of FES in a living body revealed intravascular thrombus formation, with the thrombus consisting of fibrin as well as erythrocytes and leukocytes with lipid granules [122].
Animal Models of DIC
Noble–Collip drum-induced polytrauma without significant hemorrhage has been used to mimic lethal traumatic injury [123]. The model reproduces typical DIC with the fibrinolytic phenotype, with animals exhibiting a decreased platelet count, prolonged PT and PTT, decreased fibrinogen and antithrombin levels, and elevated fibrin/fibrinogen degradation products (FDP) levels [123–125]. Furthermore, elevated tPA levels, shortened euglobulin lysis time, and decreased α2-plasmin inhibitor levels indicate immediate activation of the fibrinolytic system [124, 125]. Decreases in the levels of FXII, prekallikrein, and CH50 suggest the activation of both the intrinsic coagulation pathway and the complement system [125]. Immediately after Noble–Collip drum trauma, tissue factor increases in the circulation, and its mRNA expression has been observed in various organs, indicating the activation of the extrinsic coagulation pathway [126]. The Noble–Collip drum model also exhibits a spontaneous thrombin burst measured by a thrombin generation assay. Systemic thrombin generation accelerates as a result of insufficient control by antithrombin. These data support those of a previous study [81]. Moreover, tPA release driven by hypoperfusion of the endothelium leads to hyperfibrin(ogen)olysis. Meanwhile, there was no evidence of activated protein C-mediated shutdown of thrombin generation in the systemic circulation [127].
A systematic review addressed the question: “what are relevant experimental models with which to study early traumatic coagulopathy?” [128]. In this review, a tissue factor induced DIC model was reported to provide “grade A” evidence, representing a key model for traumatic coagulopathy. This model demonstrated that a massive amount of tissue factor also induces DIC associated with fibrin(ogen)olysis without tissue hypoperfusion [97]. This suggests that trauma itself could give rise to DIC without tissue hypoperfusion [67]. Tissue factor activation of coagulation leads to the generalized consumption of not only platelet and coagulation factors including fibrinogen, but also the inhibitory feedback factors involved in controlling coagulation and fibrinolysis, namely, antithrombin and α2-plasmin inhibitor, respectively [81, 91, 97].
Management of DIC
T he cornerstone of DIC management is specific and vigorous treatment of the underlying disorder, that is, injury itself and hemorrhagic shock [14, 129]. There are clear differences in the treatment of DIC with the fibrinolytic phenotype in the early phase of trauma and DIC with the thrombotic phenotype in the late phase of trauma. Management of the latter DIC type is the same as for typical DIC; anticoagulants, platelet and plasma substitution, and coagulation inhibitor concentrates have been proposed [129]. Our discussion will follow with, treatment of fibrinolytic phenotype DIC through a novel approach.
After careful deliberation of “one concept and six considerations” for hemostatic changes during the early phase of trauma proposed by the SSC on DIC of the ISTH [130], trauma and hemorrhagic shock should be managed through damage control resuscitation, which integrates hemostatic resuscitation, damage control surgery, and in certain cases permissive hypotension [131]. Guidlienes by Spahn et al. review the physiological targets of resuscitation and suggest dosing of fluids, blood products, and pharmacological agents in bleeding trauma patients [132]. Anticoagulants are contraindicated for DIC with the fibrinolytic phenotype. Substitution therapies with plasma transfusion, platelet transfusion, and fibrinogen concentrate or cryoprecipitate transfusion maintain normal platelet counts and function, adequate levels of coagulation factors and endogenous anticoagulants including antithrombin and protein C [14, 129, 130]. Guidance for treatment of DIC published by the ISTH recommends the transfusion of plasma, which includes anticoagulant factors such as protein C, protein S, and antithrombin [129]. For further description of plasma transfusion please refer to Chap. 20. The use of agents that are capable of restoring dysfunctional anticoagulant pathways in DIC patients with sepsis has been extensively studied. However, there has been no study on the use of anticoagulant factor concentrates such as activated protein C, recombinant human thrombomodulin, and antithrombin in trauma induced DIC. Supra-normal levels of anticoagulant factors foster bleeding. At present, plasma transfusion to maintain normal levels of protein C and antithrombin may be a reasonable strategy for treating DIC after trauma.
Tranexamic acid can reduce the risk of death in bleeding trauma patients [129, 133] and should be given as early as possible because any delay in administration after trauma reduces its efficacy and may actually be harmful [134]. These studies provide the theoretical basis for antifibrinolytic therapy in DIC with the fibrinolytic phenotype in the early phase of trauma [129]. Since the publication of the CRASH-2 results, there has been considerable discussion about how tranexamic acid should be used in practice. Some authors suggest limiting tranexamic acid use to specific patient subgroups, such as those with low blood pressure or laboratory evidence of hyperfibrinolysis [135, 136]. A theoretical argument has been published, stating that biological insight into how the treatment works is more relevant when applying research results to patient care rather than the application of statistical reasoning [137]. When administering tranexamic acid in the trauma setting, it is important to realize that the challenge for clinicians is not to identify a specific subgroup of patients but to detect those patients that will die from coagulopathic bleeding [137].
Trauma Induced Coagulopathy and DIC
The introduction of the concept of acute coagulopathy of trauma-shock (ACOTS) was proposed following a reacknowledgement that trauma itself and hemorrhagic shock can induce coagulopathy [138, 139]. Following the proposal of ACOTS, which is now a part of the more global term “trauma induced coagulopathy,” there has been some confusion with the use of this terminology. Integration of this nomenclature is described in Table 13.3. Trauma induced coagulopathy consists of varying disease conditions that induce coagulopathy, each with a condition specific pathogenesis. Differences and similarities between DIC and the ACOTS have been extensively debated [14, 16, 25, 130, 140]. A detailed discussion on this point is beyond the scope of this chapter, but it is important to acknowledge that DIC has been the main pathophysiology of trauma induced coagulopathy [25, 140].
Conclusion
The main pathophysiological mechanism of trauma-induced coagulopathy is considered to be DIC. Disseminated intravascular coagulation in the early phase of trauma presents itself as a fibrinolytic phenotype, is associated with hyperfibrin(ogen)olysis and consumption coagulopathy, and contributes to massive hemorrhage (Fig. 13.10) [16]. This type of DIC progresses to DIC with the thrombotic phenotype during the late phase of trauma and drives multiple organ dysfunction. To understand the pathogenesis and appropriate management of DIC, deep insights are needed into the interplay between innate immunity, inflammation, and coagulation and fibrinolysis.

Mechanisms of DIC with the fibrinolytic phenotype. Left, there is a balance between activation of coagulation, anticoagulation, and fibrinolysis during physiological hemostasis; right, DIC with the fibrinolytic phenotype. Consumption induced decreases in protein C, antithrombin, and TFPI and functional loss of both soluble and endothelial thrombomodulin severely impairs anticoagulation, enhancing systemic thrombin generation. TM thrombomodulin, sTM soluble TM, TF tissue factor, PC protein C, TFPI tissue factor pathway inhibitor. Modified with permission [16]
References
Hardaway RM. Disseminated intravascular coagulation syndrome. Arch Surg. 1961;83:842–50.
Mackay DG. Trauma and disseminated intravascular coagulation. J Trauma. 1969;9:646–60.
Flute PT. Coagulation and fibrinolysis after injury. J Clin Path (R Coll Pathol). 1970;23 Suppl 4:102–9.
Damus PS, Salzman EW. Disseminated intravascular coagulation. Arch Surg. 1972;104:262–5.
Murray DJ, Olson J, Strauss R, Tinker JH. Coagulation changes during packed red blood cell replacement of major blood loss. Anesthesiology. 1988;69:839–45.
Bergentz SE, Leandoer L. Disseminated intravascular coagulation in shock. Ann Chir Gynecol Fenn. 1971;60:175–9.
Blombäck M, Eklund J, Hellgren M, Lagerkranser M, Swedenborg J. Blood coagulation and fibrinolytic factors as well as their inhibitors in trauma. Scand J Clin Lab Invest. 1985;45 Suppl 178:15–23.
Gando S, Tedo I, Kubota M. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20:594–600.
Engelman DT, Gabram SGA, Allen L, Ens GE, Jacobs LM. Hypercoagulability following multiple trauma. World J Surg. 1996;20:5–10.
Guay J, Ozier Y, de Moerloose P, Samana CM, Bélisle S, Hardy JF. Polytrauma and hemostatic abnormalities. Can J Anesth. 1998;45:683–91.
Ordog GJ, Wasserberger J, Balasubramanium S. Coagulation abnormalities in traumatic shock. Ann Emerg Med. 1985;14:650–5.
Hiippala S. Replacement of massive blood loss. Vox Sang. 1998;74 Suppl 2:399–407.
Ledgerwood AM, Lucas CE. A review of studies on the effects of hemorrhagic shock and resuscitation on the coagulation profile. J Trauma. 2003;54:S68–74.
Gando S, Sawamura A, Hayakawa M. Trauma, shock, and disseminated intravascular coagulation. Ann Surg. 2011;254:10–9.
Taylor Jr FB, Toh CH, Hoots WK, Wada H, Levi M. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86:1327–30.
Gando S, Wada H, Thachil J. Differentiating disseminated intravascular coagulation (DIC) with the fibrinolytic phenotype from coagulopathy of trauma and acute coagulopathy of trauma-shock (COT/ACOTS). J Thromb Haemost. 2013;11:826–35.
Bakhtiari K, Meijers JCM, de Jonge E, Levi M. Prospective validation of the International Society of Thrombosis and Haemostasis scoring system for disseminated intravascular coagulation. Crit Care Med. 2004;32:2416–21.
Gando S, Saitoh D, Ogura H, Mayumi T, Koseki K, Ikeda T, Ishikura H, Iba T, Ueyama M, Eguchi Y, Otomo Y, Okamoto K, Kushimoto S, Endo S, Shimazaki S. Japanese Association for Acute Medicine disseminated intravascular coagulation (JAAM DIC) study group: natural history of disseminated intravascular coagulation diagnosed based on the newly established diagnostic criteria for critically ill patients: results of a multicenter, prospective survey. Crit Care Med. 2008;6:145–50.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, Katabami K. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res. 2009;124:608–13.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, Katabami K. Application of the Japanese Association for Acute Medicine disseminated intravascular coagulation diagnostic criteria for patients at an early phase of trauma. Thromb Res. 2009;124:706–10.
Oshiro A, Yanagida Y, Gando S, Henzan N, Takahashi I, Makise H. Hemostasis during the early stage of trauma: comparison with disseminated intravascular coagulation. Crit Care. 2014;18:R61.
Marder VJ, Feinstein DI, Colman RW, Levi M. Consumptive thrombohemorrhagic disorders. In: Colman RW, Marder VJ, Clowes AW, George JN, Goldhaber SZ, editors. Hemostasis and thrombosis. Basic principles and clinical practice. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006. p. 1571–600.
Asakura H. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care. 2014;2:20.
Levi M. Disseminated intravascular coagulation. Crit Care Med. 2007;35:2191–5.
Gando S, Otomo Y. Local hemostasis, immunothrombosis and systemic disseminated intravascular coagulation in trauma and traumatic shock. Crit Care. 2015;19:72.
Gando S. Disseminated intravascular coagulation in trauma patients. Semin Thromb Haemost. 2001;27:585–91.
Castellheim A, Brekke OL, Espevik T, Harboe M, Mollnes TE. Innate immune responses to danger signals in systemic inflammatory response syndrome and sepsis. Scand J Immunol. 2009;69:479–91.
Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–8.
Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost. 2011;9 Suppl 1:182–8.
Engelmann B, Massberg S. Thrombosis and intravascular effector of innate immunity. Nat Rev Immunol. 2013;3:34–45.
Rivers RP, Hathaway WE, Weston W. The endotoxin-induced coagulant activity of human monocytes. Br J Haematol. 1975;30:311–6.
Müller I, Klocke A, Alex M, Kotzsch M, Luther T, Morgenstern E, Zieseniss S, Zahler S, Preissner K, Engelmann B. Intravascular tissue factor initiates coagulation via circulating microvesicles and platelets. FASEB J. 2003;17:476–8.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Wrobleski SK, Wakerfield TW, Hartwig JH, Wanger DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5.
Fuchs TA, Bhandari AA, Wanger DD. Histones induce rapid and profound thrombocytopenia in mice. Blood. 2011;18:3708–14.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;18:1952–61.
Ammollo CT, Semeraro F, Xu J, Esmon NL, Esmon CT. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost. 2011;9:1795–803.
von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Latinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wanger DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelet cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–35.
Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–92.
Kannemeier C, Shibamiya A, Nakazawa F, Trusheim H, Ruppert C, Markart P, Song Y, Tzima E, Kennerknecht E, Niepmann M, von Bruehl ML, Sedding D, Massberg S, Günther A, Engelmann B, Preissner KT. Extracellular RNA constitutes a natural procoagulant cofactor in blood coagulation. Proc Natl Acad Sci U S A. 2007;104:6388–93.
Rapaport SI, Rao VM. Initiation and regulation of tissue factor-dependent blood coagulation. Arterioscler Thromb. 1992;12:1111–21.
Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner K, Engelmann B. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. 2010;16:887–96.
Ishii H, Majerus PW. Thrombomodulin is present in human plasma and urine. J Clin Invest. 1985;76:2178–81.
Ishii H, Uchiyama H, Kazama M. Soluble thrombomodulin antigen in conditioned medium is increased by damage of endothelial cells. Thromb Haemost. 1991;65:618–23.
Hoffman M, Monroe III DM. A cell-based model of hemostasis. Thromb Haemost. 2001;85:958–65.
Manson J, Thiemermann C, Brohi K. Trauma alarmins as activators of damage-induced inflammation. Br J Surg. 2012;99 Suppl 1:12–20.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–8.
Yamanouchi S, Kudo D, Yamada M, Miyagawa N, Furukawa H, Kushimoto S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: time course and the association with clinical status. J Crit Care. 2013;28:1027–31.
Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, Gillespie MN, Richards WO. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. 2013;258:591–8.
Cohen MJ, Brohi K, Calfee CS, Rhan P, Chesebro BB, Christiaans SC, Carles M, Howard M, Pittet JF. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174.
Kutcher ME, Xu J, Vilardi RF, Ho C, Esmon CT, Cohen MJ. Extracellular histone release in response to traumatic injury: implications for compensatory role of activated protein C. J Trauma Acute Care Surg. 2012;73:1389–94.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, Wang SS, Brohi K, Kipar A, Yu W, Wang G, Toh CH. Circulating histones are mediators of trauma-associated lung injury. Am J Crit Care Med. 2013;187:160–9.
Ito T, Kawahara K, Nakamura T, Yamada S, Nakamura T, Abeyama K, Hashiguchi T, Maruyama I. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost. 2007;5:109–16.
Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol. 2011;187:2626–31.
Gando S, Nakanishi Y, Tedo I. Cytokines and plasminogen activator inhibitor-1 in posttrauma disseminated intravascular coagulation: relationship to multiple organ dysfunction. Crit Care Med. 1995;23:1835–42.
Jawa RS, Anillo S, Huntoon K, Baumann H, Kulayat M. Interleukin-6 in surgery, trauma, and critical care. Part II. Clinical implications. J Intensive Care Med. 2011;26:73–87.
van der Poll T, de Jonge E, Levi M. Regulatory role of cytokines in disseminated intravascular coagulation. Semin Thromb Hemost. 2001;27:639–51.
Esmon CT. Possible involvement of cytokines in diffuse intravascular coagulation and thrombosis. Clin Haematol. 1999;12:343–59.
Boehme MWJ, Deng Y, Raeth U, Bierhaus A, Ziegler R, Stremmel W, Nawroth PP. Release of thrombomodulin from endothelial cells by concerted action of TNF-alpha and neutrophils: in vivo and in vitro studies. Immunology. 1996;87:134–40.
Burnier L, Fontana P, Kwak BR, Angelillo-Scherrer A. Cell-derived microparticles in haemostasis and vascular medicine. Thromb Haemost. 2009;101:439–51.
Owens III AP, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–97.
Ogura H, Kawasaki T, Tanaka H, Koh T, Tanaka R, Ozeki Y, Hosotsubo H, Kuwagata Y, Shimazu T, Sugimoto H. Activated platelets enhance microparticle formation and platelet-leucocyte interaction in severe trauma and sepsis. J Trauma. 2001;50:801–9.
Park MS, Owen BAL, Ballinger BA, Sarr MG, Schiller HJ, Zietlow SP, Jenkins DH, Ereth MH, Owen WG, Heit JA. Quantification of hypercoagulable state after blunt trauma: microparticle and thrombin generation are increased relative to injury severity, while standard markers are no. Surgery. 2012;151:831–6.
Allam R, Kumar SVR, Darisipudi MN, Anders HJ. Extracellular histones in tissue injury and inflammation. J Mol Med. 2014;92:465–72.
Gando S, Nanzaki S, Morimoto Y, Ishitani T, Kemmotsu O. Tissue factor pathway inhibitor does not correlate with tissue-factor induced disseminated intravascular coagulation and multiple organ dysfunction syndrome in trauma patients. Crit Care Med. 2001;29:262–6.
Petersen LC, Valentin S, Hedner U. Regulation of the extrinsic pathway system in health and disease: the role of factor VIIa and tissue factor pathway inhibitor. Thromb Res. 1995;79:1–47.
Hayakawa M, Sawamura A, Gando S, Kubota N, Uegaki S, Shimojima H, Sugano M, Ieko M. Disseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastase. Surgery. 2011;149:221–30.
Gando S, Nakanishi Y, Kameue T, Nanzaki S. Soluble thrombomodulin increases in patients with disseminated intravascular coagulation and in those with multiple organ dysfunction syndrome after trauma: role of neutrophil elastase. J Trauma. 1995;39:660–4.
Ogawa S, Shreeniwas R, Butura C, Brett J, Stern DM. Modulation of endothelial function by hypoxia: perturbation of barrier and anticoagulant function, and induction of a novel factor X activator. Adv Exp Med Biol. 1990;281:303–12.
Ogawa S, Gerlach H, Esposito C, Pasagian-Macaulay A, Brett J, Stern D. Hypoxia modulates the barrier and coagulant function of cultured bovine endothelium. Increased monolayer permeability and induction of procoagulant properties. J Clin Invest. 1990;85:1090–8.
Öhlin AK, Larsson K, Hansson M. Soluble thrombomodulin activity and soluble thrombomodulin antigen in plasma. J Thromb Haemost. 2005;3:976–82.
Taylor FB, Chang A, Ferrell G, Mather T, Catlett R, Blick K, Esmon CT. C4b-binding protein exacerbates the host response to Escherichia coli. Blood. 1991;78:357–63.
Liaw PCY, Ferrell G, Esmon CT. A monoclonal antibody against activated protein C allows rapid detection of activated protein C in plasma and reveals a calcium ion dependent epitope involved in factor Va inactivation. J Thromb Haemost. 2003;1:662–70.
Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012;255:379–85.
Butenas S, van’t Veer C, Mann KG. “Normal” thrombin generation. Blood. 1999;94:2169–78.
Grottke O, Braunschweig T, Spronk HMH, Esch S, Rieg AD, van Oerle R, ten Cate H, Fitzner C, Tolba R, Rossaint R. Increasing concentrations of prothrombin complex concentrate induce disseminated intravascular coagulation in a pig model of coagulopathy with blunt liver injury. Blood. 2011;118:1943–51.
Miller RS, Weatherford DA, Stein D, Crane MM, Stein M. Antithrombin III and trauma patients: factors that determine low levels. J Trauma. 1994;37:442–5.
Liener UC, Brückner UB, Strecker W, Steinback G, Kinzl L, Gebhard F. Trauma severity-dependent changes in ATIII activity. Shock. 2001;15:344–7.
Owings JT, Bagley M, Gosselin R, Romac D, Disbrow E. Effect of critical injury on plasma antithrombin activity: low antithrombin levels are associated with thromboembolic complications. J Trauma. 1996;41:396–406.
Yanagida Y, Gando S, Hayakawa M, Sawamura A, Uegaki S, Kubota N, Homma T, Ono Y, Honma Y, Wada T, Jesmin S. Normal prothrombinase activity, increased systemic thrombin generation, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma-shock. Surgery. 2013;154:48–57.
Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009;49:2652–60.
Chandler WL. Procoagulant activity in trauma patients. Am J Clin Pathol. 2010;134:90–6.
Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79:1111–5.
Nakashima M, Uematsu T, Umemura K, Maruyama I, Tsuruta K. A novel recombinant human soluble thrombomodulin, ART-123, activates the protein C pathway in healthy male volunteers. J Clin Pharmacol. 1998;38:540–4.
Mohri M, Sata M, Gomi K, Maruyama Y, Osame M, Maruyama I. Abnormalities in the protein C anticoagulant pathway detected by a novel assay using human thrombomodulin. Lupus. 1997;6:590–6.
Giles AR, Nesheim ME, Mann KG. Studies of factors V and VIII:C in an animal model of disseminated intravascular coagulation. J Clin Invest. 1984;74:2219–25.
Wyshock EG, Sufferendini AF, Parrillo JE, Colman RE. Cofactors V and VIII after endotoxin administration to human volunteers. Thromb Res. 1995;80:377–89.
Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardivasc Med. 2005;15:302–8.
Terraube V, O’Donnell JS, Jenkins PV. Factor VIII and von Willebrand factor interaction: biological, clinical and therapeutic importance. Haemophilia. 2010;16:3–13.
Clarke BJ, Sridhara S, Woskowska Z, Blajchman MA. Consumption of plasma factor VII in a rabbit model of non-overt disseminated intravascular coagulation. Thromb Res. 2003;108:329–34.
Gando I, Makise H, Tedo I. Variation in wound healing factors in trauma patients. Jpn J Surg. 1990;91:17–22.
Risberg B. Fibrinolysis in trauma. Eur Surg Res. 1978;10:373–81.
Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, Khan S, De’ath HD, Allard S, Hart DP, Pasi KJ, Hunt BJ, Stanworth S, Maccallums PK, Brohi K. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11:307–14.
Stump DC, Taylor FBJ, Nesheim ME, Giles AR, Dzik WH, Bovill EG. Pathologic fibrinolysis as a cause of clinical bleeding. Semin Thromb Hemost. 1990;16:260–73.
Levi M, ten Cate H, van der Poll T, van Daventer SJH. Pathogenesis of disseminated intravascular coagulation in sepsis. JAMA. 1993;270:975–9.
Gando S, Kameue T, Nanzaki S, Nakanishi Y. Massive fibrin formation with consecutive impairment of fibrinolysis in patients with out-of-hospital cardiac arrest. Thromb Hemost. 1997;77:278–82.
Hayakawa M, Gando S, Ieko M, Honma Y, Homma T, Yanagida Y, Kubota N, Uegaki S, Sawamura A, Asakura H. Massive amount of tissue factor induce fibrinogenolysis without tissue hypoperfusion in rats. Shock. 2013;39:514–9.
Murakami H, Gando S, Hayakawa M, Sawamura A, Sugano M, Kubota N, Uegaki S, Jesmin S. Disseminated intravascular coagulation (DIC) at an early phase of trauma continuously proceeds to DIC at a late phase of trauma. Clin Appl Thromb Hemost. 2012;18:364–9.
Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med. 2010;38(Suppl):S35–42.
Bergentz SE, Leandoer L. Disseminated intravascular coagulation in shock. Ann Chir Gynecol Fenn. 1971;197(60):175–9.
Turpini R, Stefanini M. The nature and mechanism of the hemostatic breakdown in the course of experimental hemorrhagic shock. J Clin Invest. 1959;38:53–65.
Borgström S, Gelin LE, Zederfeldt B. The formation of vein thrombi following tissue injury. An experimental study in rabbits. Act Chir Scand. 1959;247(Suppl):1–36.
Allardyce B, Hamit HF, Matsumoto T, Moseley RV. Pulmonary vascular changes in hypovolemic shock: radiography of the pulmonary microcirculation and the possible role of platelet embolism in increasing vascular resistance. J Trauma. 1999;9:403–11.
Lungqvist U, Bergentz SE, Lewis DH. The distribution of platelets, fibrin and erythrocytes in various organs following experimental trauma. Eur Surg Res. 1971;3:293–300.
Leandoer L, Bergentz SE. Haemorrhagic shock in the dog. The formation of thromboemboli during antifibrinolytic therapy. Eur Surg Res. 1970;2:341–7.
Avikainen V, Eklund B. Disseminated intravascular coagulation after inhibition of fibrinolysis with tranexamic acid (AMCA) and proteinase inhibitor trasylol in experimental traumatic and haemorrhagic shock. Ann Chir Gynecol Fenn. 1974;63:226–34.
Hardaway RM. The significance of coagulative and thrombotic changes after haemorrhage and injury. J Clin Pathol (R Coll Pathol). 1970;4(Suppl):110–20.
Nuytinck HKS, Offermans XJMW, Kubat K, Goris RJA. Whole-body inflammation in trauma patients. An autopsy study. Arch Surg. 1988;123:1519–24.
Kaufman HH, Hui KS, Mattson JC, Borit A, Childs TL, Hoots WK, Bernstein DP, Makekla ME, Wnger KA, Kahan BD, Gildenberg PL. Clinicopathological correlations of disseminated intravascular coagulation in patients with head injury. Neurosurgery. 1984;15:34–42.
Stein SC, Smith DH. Coagulopathy in traumatic brain injury. Neurocrit Care. 2004;1:479–88.
Gando S, Nanzaki S, Kemmotsu O. Coagulofibriolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070–7.
Stein SC, Chen XH, Sinson GP, Smith DH. Intravascular coagulation: a major secondary insult in nonfatal traumatic brain injury. J Neurosurg. 2002;97:1373–7.
Stein SC, Graham DI, Chen XH, Smith DH. Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery. 2004;54:687–91.
Schwarzmaier SM, Kim SW, Trabold R, Plesnila N. Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J Neurotrauma. 2010;27:121–30.
Bergentz SE, Nilsson IM. Effect of trauma on coagulation and fibrinolysis in dogs. Acta Chir Scand. 1961;122:21–9.
Riseborough E, Herndon JH. Alterations in pulmonary function, coagulation and fat metabolism in patients with fractures of the lower limbs. Clin Orthopedic Relat Res. 1976;115:248–67.
Shier MR, Wilson RF. Fat embolism syndrome: traumatic coagulopathy with respiratory distress. Surg Annu. 1980;12:139–68.
Saldeen T. The importance of intravascular coagulation and inhibition of the fibrinolytic system in experimental fat embolism. J Trauma. 1970;10:287–98.
Saldeen T. Fat embolism and signs of intravascular coagulation in a posttraumatic autopsy material. J Trauma. 1970;10:273–86.
Kao SJ, Yeh DYW, Chen HI. Clinical and pathological features of fat embolism with acute respiratory distress syndrome. Clin Sci. 2007;113:279–85.
Serck-Hanssen A. Posttraumatic fat embolism. Red cell aggregation, hyaline microthrombi and platelet aggregates in 5 fatal cases. Acta Path Microbiol Scand. 1965;65:31–45.
Arai F, Kita T, Nakai T, Hori T, Maki N, Kakiuchi M, Sasaki S. Histopathologic features of fat embolism in fulminant fat embolism syndrome. Anesthesiology. 2007;107:509–11.
Noble RL, Collip JB. A quantitative method for the production of experimental traumatic shock without hemorrhage in unanesthetized animals. Quart J Exper Physiol. 1942;31:187–99.
Tanabe K, Yoshitake J. A study on coagulation and fibrinolytic dynamics in experimental traumatic shock. Masui (Jpn J Anesthesiol). 1981;30:826–31.
Kugimiya H. A pathophysiological and biochemical study on the experimental traumatic shock in rats. A relationship between coagulation/fibrinolytic system and DIC. Masui (Jpn J Anesthesiol). 1982;31:75–84.
Armstead VE, Opetanova IL, Minchenko AG, Lefer AM. Tissue factor expression in vital organs during murine traumatic shock. Anesthesiology. 1999;91:1844–52.
Hayakawa M, Gando S, Ono Y, Wada T, Yanagida Y, Sawamura A, Ieko M. Noble-Collip drum induces disseminated intravascular coagulation but not acute coagulopathy of trauma-shock. Shock. 2015;43:261–7.
Parr MJ, Bouillon B, Brohi K, Dutton RP, Hauser CJ, Hess JR, Holcomb JB, Kluger Y, Mackway-Jones K, Rizoli SB, Yukioka T, Hoyt DB. Traumatic coagulopathy: where are the good experimental models? J Trauma. 2008;65:766–71.
Wada H, Thachil J, Di Nisio M, Mathew P, Kurosawa S, Gando S, Kim HK, Nielsen JD, Dempfle CE, Levi M, Toh CH. Guidance for diagnosis and treatment of disseminated intravascular coagulation from harmonization of the recommendations from three guidelines. J Thromb Haemost. 2013;11:761–7.
Gando S, Wada H, Kim HK, Kurosawa S, Nielsen JD, Thachil J, Toh CH. Comparison of disseminated intravascular coagulation in trauma with coagulopathy of trauma/acute coagulopathy of trauma-shock. J Thromb Haemost. 2012;10:2593–5.
Jansen JO, Thomas R, Loudon MA, Brooks A. Damage control resuscitation for patients with major trauma. BMJ. 2009;338:b1778.
Spahn DR, Bouillon B, Cerny V, Coats TJ, Duranteau J, Fernández-Mondéjar E, Filipescu D, Hunt BJ, Komadina R, Nardi G, Neugebauer E, Ozier Y, Riddez L, Schultz A, Vincent JL, Rossaint R. Management of bleeding and coagulopathy following major trauma: an updated European guideline. Crit Care. 2013;17:R76.
CRASH-2 collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant hemorrhage (CRASH-2): a randomized, placebo-controlled trial. Lancet. 2010;376:23–32.
CRASH-2 collaborators, The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomized controlled trial. Lancet. 2011;377:1096–101.
Pusateri A, Weiskopf R, Bebarta V, Butler F, Cestero RF, Chaudy IH, Deal V, Doriac WC, Gerhardt RT, Given MB, Hansen DR, Hoots WK, Klein HG, MacDonald VW, Mattox KL, Michael RA, Mogford J, Montcalm-Smith EA, Niemeyer DM, Prusaczyk WK, Rappold JF, Rassmussen T, Rentas F, Ross J, Thompson C, Tucker LD, US DoD Hemorrhage and Resuscitation Research and Development Committee. Tranexamic acid and trauma: current status and knowledge gaps with recommended research priorities. Shock. 2013;39:121–6.
Naplitano LM, Cohen MJ, Cotton RA, Schreiber MA, Moore EE. Tranexamic acid in trauma: how should we use it? J Trauma Acute Care Surg. 2013;74:1575–86.
Roberts I, Prieto-Merino D. Applying results from clinical trials: tranexamic acid in trauma patients. J Intensive Care. 2014;2:56.
Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma. 2003;54:1127–30.
Hess J, Brohi K, Dutton RP, Hauser CJ, Holcomb JB, Mackway-Jones K, Parr MJ, Rizoli SB, Yukioka T, Hoyt DB, Bouillon B. The coagulopathy of trauma: a review of mechanisms. J Trauma. 2008;65:748–54.
Gando S. Hemostasis and thrombosis in trauma patients. Semin Thromb Haemost. 2015;41:26–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gando, S. (2016). Disseminated Intravascular Coagulation. In: Gonzalez, E., Moore, H., Moore, E. (eds) Trauma Induced Coagulopathy. Springer, Cham. https://doi.org/10.1007/978-3-319-28308-1_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-28308-1_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28306-7
Online ISBN: 978-3-319-28308-1
eBook Packages: MedicineMedicine (R0)