
Abstract
Cushing’s disease (CD), a pituitary corticotroph-producing adenoma, is the most common cause of endogenous Cushing’s syndrome, a rare disorder characterized by chronic exposure to excess glucocorticoids leading to multisystemic comorbidities and increased mortality. The low incidence of CD makes it difficult to get accurate and exhaustive data on many aspects of the disease, like clinical features, diagnosis, management, and long-term outcome. Clinical presentation can be unspecific and highly variable, so that diagnosis is often challenging. Pituitary surgery is the first-line treatment, but remission is not always achieved. Radiotherapy, medical therapy, or lastly bilateral adrenalectomy might be necessary as additional therapeutic options. Although normalization of hypercortisolism significantly improves the glucocorticoid (GC)-related comorbidities (i.e., cardiovascular risk, osteopenia, and psychopathology), some of them are not completely reversible at long-term follow-up. A multidisciplinary and individualized approach is essential to choose the best approach to control hypercortisolism and treat comorbidities. Recurrence may occur even years after initial remission. All patients with CD should have ongoing surveillance to identify recurrences, need for additional treatment, and therapy for residual comorbidities. This chapter will review the main aspects on the diagnosis and management of CD.
The chapter has been endorsed by Prof. Maya Lodish, , University of California, San Francisco, California, USA
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cushing’s disease
- Hypercortisolism
- Pituitary corticotroph adenoma
- Radiation
- Medical therapy
- Transsphenoidal surgery
- Morbidity
- Mortality
- Quality of life
7.1 Introduction
Cushing’s syndrome (CS) constitutes a group of signs and symptoms due to extended and inappropriately high exposure to excess glucocorticoids (GCs). Iatrogenic corticosteroid administration is the most common cause of CS, and pituitary corticotroph adenoma (Cushing’s disease; CD) is the most frequent cause of endogenous excessive GC secretion. CD is a rare and severe disease, and chronic exposure to high GC levels has been associated with increased multisystemic morbidity and mortality. Therefore, an early diagnosis and treatment are mandatory to avoid long-term complications. Although most of the related comorbidities improve after initial therapy of hypercortisolism, many of them are not completely reversible and a lifelong follow-up is necessary to control comorbidities and rule out potential recurrences. Therefore, prompt identification and treatment of hypercortisolism and its comorbidities are important to reduce mortality. This chapter addresses epidemiology, diagnoses, treatment, outcomes, and follow-up of Cushing’s disease.
7.2 Epidemiology
Cushing’s syndrome is considered a rare disorder with an estimated incidence between 0.5 and 3.0/106 people/year; however, accurate data on prevalence and its incidence in the general population are scarce [1]. Only countries with national registries or databases provide information on its incidence and prevalence (i.e., incidence ranged from 1.2 to 1.7/106 inhabitants/year in Denmark [2]; standardized incidence rate (SIR) of 0.8/105/year in Iceland [3]; and SIR of 0.07/105/year in Malta [4]). The most frequent cause of endogenous hypercortisolism is an ACTH-producing pituitary adenoma, namely Cushing’s disease (CD). The incidence according to an 11-year period population-based study in Denmark is 1.2–1.7/106/year for CD, followed by an incidence of 0.6/106/year for adrenal adenomas [2]. Other types of CS are extremely rare. Some data indicate a high proportion of subclinical CS in risk populations such as patients with uncontrolled type 2 diabetes or early-onset osteoporosis [5]. CS is more frequent in females with an estimated female:male ratio of 3:1 and median age at diagnosis of around 40 years [1, 6]. CS is associated with increased mortality (SMR of 3.7 (2.3–5.3)); mortality rates are increased during the first year after diagnosis and in those patients with persistent hypercortisolism after initial therapy [2]. Although mortality risk decreases with remission of hypercortisolism, it still remains increased compared to general population, with circulatory diseases being the most important cause of mortality [7, 8].
7.3 Pathogenesis
Table 7.1 summarizes the different causes of CS. In adult population, the most frequent cause of endogenous CS is an adrenocorticotropic (ACTH)-dependent CS; a corticotrope pituitary adenoma (CD) is the cause of about 70% of ACTH-dependent CS, followed by an ectopic (extrapituitary) ACTH-secreting tumor [1]. CD is characterized by an excessive production of ACTH due to an ACTH-producing pituitary adenoma (90% of which are microadenomas), which induces high levels of cortisol and androgens, often accompanied by diffuse adrenal hyperplasia [9].
The molecular pathophysiology of CD is not completely understood. However, the identification of some molecular genetic abnormalities has provided new insight into the pathogenesis of pituitary ACTH-producing adenomas. Most pituitary tumors are sporadic as a consequence of monoclonal expansion of a mutated cell. CD rarely occurs, although cases have been described, in the context of germline mutations like multiple endocrine neoplasia (MEN) type 1, aryl hydrocarbon receptor-interacting protein (AIP), and cyclin-dependent kinase inhibitor (CDKN1B) genes in patients with MEN1 or MEN4 features and in patients with succinate dehydrogenase subunit mutations [10]. In contrast, somatic mutations of MEN1 or AIP have not been found in sporadic pituitary adenomas or CD. Instead, very rare mutations of the GC receptor gene or proteins related to GC8 receptor functions have been found in CD [11]. Inactivation of the ubiquitin-specific peptidase 8 gene (USP8), present in 35–62% of the corticotroph adenomas, is responsible for the increased expression of EGFR and ultimately ACTH synthesis. Epidermal growth factor receptor (EGFR) and pituitary-transforming gene (PTTG) can be overexpressed and play a causal role in the development of CD and defects in this pathway may also be a key to therapeutic targets. In animal models, a PTTG inhibitor (R-roscovitine) or an inhibitor of the EGFR inhibitor (gefitinib) inhibited corticotroph tumor growth and features of hypercortisolism [10], nevertheless, the translation of these findings in humans is still unknown.
7.4 Clinical Presentation
Manifestations of hypercortisolism are systemic and symptoms at clinical presentation may be unspecific and highly variable, and the diagnosis of the disease is often delayed up to several months or years. There is no pathognomonic sign or symptom. Table 7.2 summarizes the most frequent clinical presentation according to different series [1]. The most common features at diagnosis are weight gain, hypertension, skin abnormalities, myopathy, and menstrual irregularities. Hypertension and hirsutism present more frequently in ectopic CS than in other causes.
Several signs and symptoms are more suggestive and specific of hypercortisolism due to catabolic effects of cortisol on skeletal muscles, skin, and connective tissue, including skin atrophy, easy bruising with no trauma, proximal muscle weakness, or unexplained osteoporosis. Increased and impaired distribution of fat depots is one of the most precocious signs, with abnormally increased fat in the trunk, face, and neck. Wide reddish-purple striae of more than 1 cm on the abdomen and limbs and proximal muscle weakness are characteristic features seen in CS. Lower bone mineral density and a high prevalence of fractures are common in CS. Hirsutism, menstrual irregularities, obesity, diabetes, and low libido may also be present, but are also frequent in the general population. Psychopathology (emotional lability, cognitive disturbances, anxiety, depression) is very common. Metabolic syndrome features (dyslipidemia, impaired glucose metabolism, hypertension) together with an hypercoagulability state lead to an increased cardiovascular risk.
Usually, progression of symptomatology is slow, except for ectopic ACTH CS due to a paraneoplastic manifestation of a malignant tumor, where clinical presentation is usually rapid with prominent myopathy, edema, hyperpigmentation, and hypokalemia.
Several physiological and pathological situations can produce an overactivity of the hypothalamic–pituitary–adrenal axis (Table 7.3). Hypercortisolism without signs or symptoms of CS might be found in physical stress (surgery, pain), anorexia nervosa, malnutrition, cortisol-binding globulin elevation, or hypothalamic amenorrhea; hypercortisolism with mild signs/symptoms of CS might be seen in psychiatric disorders (depression, bipolar disorder), alcoholism, pregnancy, morbid obesity, or poor controlled diabetes mellitus.
7.5 Diagnosis
In this first step of diagnosing CS, hypercortisolism needs to be confirmed using screening tools. Once CS is diagnosed, the cause must be identified to determine the specific etiology in order to guide treatment decisions.
7.5.1 Establishing the Diagnosis of Cushing’s Syndrome
Screening is recommended in individuals with a high risk of CS, after excluding current or recent use of any type of exogenous GC (oral, inhaled, topic, rectal, parenteral) and high dose of progestogens [12, 13]. In children with decreasing height percentile together with progressive obesity, suspicion should be raised for CS. In addition, in young adults with hypertension or osteopenia, in patients with adrenal incidentaloma, and in women with menstrual irregularities, screening for CS may be indicated, especially if patients have additional features of the syndrome. Some studies have revealed a higher prevalence of “occult CS” in patients with poorly controlled T2DM or early-onset osteoporosis; however, there is no strong evidence to support a routine screening for occult CS in these patients based on the data available currently [14,15,16,17].
Specificity and sensitivity of screening tools vary enormously across studies, and it is often necessary to perform several tests before reaching a diagnosis. Specificity is not optimal, and false positives may occur. Moreover, the use of screening tools and their variability vary across countries [18]. Importantly, it is important to recognize the impact of physiological stress on cortisol levels when interpreting results. Recommended screening tests are outlined in Fig. 7.1:
-
24-hour urinary free cortisol (UFC; at least two measurements) measures the cortisol that is not bound to cortisol-binding globulin (CBG) and thus filtered unchanged by the kidney. Therefore, UFC is not affected by medications or conditions that alter CBG. Concomitant measurement of urine creatinine helps to identify if collection is complete and to interpret collections with excessive volume (false positives with a 24-h diuresis of >5 l/day) [19]. False negatives may be seen in chronic kidney disease, if GFR < 60 ml/min and in cases with mild CS, in whom late-night salivary cortisol (LNSC) might be more useful [13, 20]. Importantly, the use of carbamazepine and fenofibrate or licorice can increase UFC levels.
-
1 mg overnight dexamethasone suppression test (DST; alternatively, 2 mg/day for 48 h). In healthy individuals, the administration of supraphysiological GC doses suppresses ACTH and cortisol secretion, while in CD patients there is a failure of this suppression when low doses of dexamethasone are given. Variable absorption, use of concomitant drugs, and renal or liver failure may influence the results of DST. Some drugs enhance hepatic enzymatic clearance of dexamethasone, reducing plasma dexamethasone concentration, and ultimately causing false-positive results of the DST, as well as alcohol abuse (2 weeks of alcohol abstinence is recommended to reduce false-positive results). On the contrary, other agents impair dexamethasone metabolism, increasing plasma dexamethasone levels and causing false-positive results in DST (Table 7.4). Some authors advocate for simultaneous measurement of cortisol and dexamethasone concentrations to ensure appropriate plasma dexamethasone levels (>3.3 nmol/l (>0.13 μg/dl)); however, dexamethasone measurement is not available in some countries [21]. The recommended serum cortisol cutoff for suppression is 50 nmol/l (1.8 μg/dl) to maximize the sensitivity as a screening tool.
-
Late-night salivary cortisol (LNSC; two measurements) is a useful tool to assess the loss of circadian rhythmicity (the absence of LNSC nadir), a consistent abnormality in patients with CS. Salivary cortisol concentrations are in equilibrium with serum active free cortisol, there is a good correlation in cortisol concentrations between both specimens, and salivary cortisol levels are not affected by the rate of saliva production [22]. It is important to note that circadian rhythm might be altered in shift workers, smokers, patients with depression, or in those who are critically ill. Also, false-positive results are commonly seen in patients with obesity or diabetes [23].

Algorithm for testing patients when Cushing’s syndrome (CS) is suspected and for the differential diagnoses of different etiologies once Cushing’s syndrome is confirmed. GC glucocorticoids, UFC urinary free cortisol, DST dexamethasone suppression test, LNSC late-night salivary cortisol, Dex-CRH dexamethasone–corticotropin-releasing hormone test, ACTH adrenocorticotropic hormone, CD Cushing’s disease, BIPSS bilateral inferior petrosal sinus sampling, CT computed tomography
To optimize sensitivity of screening tests, it is recommended to use the upper limit of the reference range for UFC and LNSC and a fasting cortisol lower than 50 nmol/L (1.8 μg/dl) following DST. Two abnormal screening tests are enough to confirm hypercortisolism in individuals with high pretest probability of having CS. For individuals with low pretest probability of having CS, or in those patients in whom cyclical CS is suspected, it is recommended to perform additional testing. In these cases, serial LNSC might be useful to follow the progression. Postponing additional testing to allow progression of biochemical and clinical features might be useful in some cases. Subjects with abnormal results in these tests should be referred to an endocrinologist for further testing [13]; performance and interpretation of subsequent testing require considerable expertise. Assay accuracy and normal ranges differ widely, so it is essential to interpret results in the context of the assay used and with appropriate normal ranges.
It is important to differentiate between endogenous pathological hypercortisolism (CS) and pseudo-Cushing states (alcoholism, depression, anorexia nervosa, obesity). Pseudo-Cushing seems to be mediated via increased hypothalamic secretion of CRH, instead of CRH suppression observed in CS. Commonly, pseudo-Cushing patients have biochemical hypercortisolism but minimum features of CS and no presence of tumor (Table 7.3).
UFC is recommended rather than DST in pregnancy, as well as in situations that can increase/decrease CBG (oral contraceptive pill use, critically ill, or nephrotic patients) or when concomitant use of antiepileptic drugs occurs, as these medications may enhance dexamethasone clearance. However, during pregnancy, only UFC values greater than 3 times the upper limit of normal can be considered for further testing for CS, since UFC excretion physiologically increases up to threefold during pregnancy. In contrast, DST rather than UFC is recommended in severe renal failure and adrenal incidentaloma for initial testing. If cyclical CS is suspected, LNSC or UFC rather than DST is recommended [13].
If further evaluation to rule out a possible non-Cushing’s hypercortisolism is required, a dexamethasone-suppressed CRH stimulation test might be useful in specific situations. This test involves a 48-h 2 mg/d DST followed by the administration of CRH (1 μg/kg iv) 2 hours after the last dose of dexamethasone is administered and cortisol measured 15 min later. However, the optimal cutoff for diagnosis needs further clarification and CRH is not available in some countries. The hypercortisolism of pseudo-Cushing’s states is thought to be mediated through increased hypothalamic secretion of CRH in the setting of an HPA axis that is otherwise appropriately restrained by negative feedback from cortisol. In contrast, the hypercortisolism of true CS suppresses hypothalamic CRH secretion and is less responsive to the negative feedback of exogenous CS. Therefore, in comparison with true CS, patients with pseudo-CS states show a greater suppression of cortisol production by exogenous GC and a diminished response to CRH injection [24]. Measurement of midnight serum cortisol while an individual is asleep (>50 nmol/l) provides high sensitivity for the diagnosis of CS, and it is useful to exclude CS when cortisol <50 nmol/l, but this test requires inpatient admission and this approach may not be possible in some clinical practice settings.
7.5.2 Establishing the Etiology of CS
The first step to localize the source of hypercortisolism is the measurement of plasma ACTH (at least twice), although most of the commercially available ACTH assays are not always accurate, especially in the low range. To avoid falsely low results, samples should be collected in an ice bath and processed immediately. Measurement of ACTH can be done at any time of the day because normal circadian rhythm is lost; however, peak values are higher in the morning. In a patient with endogenous hypercortisolism, ACTH concentrations <1.1 pmol/l (< 5 pg/ml) suggest an ACTH-independent origin (an adrenal cause), while ACTH concentrations >4.4 pmol/l (20 pg/ml) suggest an ACTH-dependent origin (mostly pituitary or less frequently an ectopic source). Intermediate values require further evaluation, with a CRH stimulation test that might unmask ACTH responsiveness, or else repeating test over time to either confirm or rule out the diagnosis. The concept that underlies the use of CRH stimulation in CS is that pituitary tumors (CD) usually respond to CRH with an increase in ACTH and cortisol levels, while ectopic ACTH-secreting tumors usually do not. A mean cortisol increase at 30 and 45 minutes of >20% and a mean ACTH increase at 15 and 30 min of >35% over their respective mean baseline values are thought to be consistent with CD [25, 26]. An increase greater than 50% in ACTH levels and/or 20% in cortisol levels following 100 μg of intravenous CRH administration is highly suggestive of CD (Fig. 7.1) [27].
No single best approach to test patients with ACTH-dependent CS exists. The evaluation of CS requires a systematic approach. After biochemical testing has confirmed a diagnosis of ACTH-dependent hypercortisolism, the source of ACTH excess has to be determined. The choice of diagnostic tests used to distinguish a pituitary vs ectopic source varies by institution and depends on the availability of IPSS, cost considerations, patient preference, and availability of CRH and sensitive MRI technology. The most accurate method to discriminate between pituitary and non-pituitary sources of ACTH is to evaluate the central-to-peripheral ACTH gradient via bilateral inferior petrosal sinus sampling (BIPSS). Corticotroph tumors have a clear setup in the petrosal samples, while ectopic ACTH-producing tumors do not. BIPSS is considered the gold standard test to differentiate between a pituitary and an ectopic source of ACTH (sensitivity and specificity of 95%), and it is recommended for patients with pituitary lesions <6 mm or in those with discordant noninvasive tests [28, 29]. BIPSS is the best way to document a central-to-peripheral ACTH gradient in the blood draining the tumor. Despite the high sensitivity and specificity of BIPSS, false-positive results can occur in ectopic ACTH production with cyclical or mild hypercortisolism without suppression of normal corticotropes, and in CRH-producing tumors. It is recommended to perform BIPSS only in cases with documented hypercortisolism (at least twofold increase in UFC in the 6–8 weeks prior to BIPSS) to ensure that normal corticotroph cells are suppressed. Inability to cannulate veins or abnormal venous drainage might cause false-positive results. Concomitant measurement of prolactin can confirm successful catheterization. CRH is not available in some countries, and desmopressin administration has been shown to offer similar results to CRH stimulation in some studies. BIPSS has limited value in identifying intrapituitary localization of the tumor. If MRI is negative, the gradient of BIPSS might be helpful to choose the side for the initial surgical approach; however, if tumor is not found, the other side of the pituitary gland should be explored. Alternatively, bilateral internal jugular venous sampling has been proposed, since it is more simple and safe and it does not require specialized expertise; however, sensitivity is lower than that of BIPSS [30, 31].
Noninvasive tests to assess the etiology of CD:
-
High-dose dexamethasone suppression test (HDDST), CRH, or desmopressin stimulation tests are noninvasive tests that might contribute to localization of the source of CS when tumor is not seen or is very small on an MRI, or when BIPSS is not available. The HDDST relies on the concept that pituitary corticotroph tumor cells retain sensitivity (albeit impaired) to glucocorticoids, but tumor cells in ectopic ACTH secretion (EAS) do not. HDDST (2 mg every 6 h for 48 h) is used to distinguish between pituitary CD and EAS. Accuracy of these tests is inferior to that of BIPSS. In general, CD adenomas maintain sensitivity to CRH or desmopressin stimulation and are resistant to negative feedback regulation by GC (HDDST), while malignant tumors (ectopic ACTH source) do not. However, some benign carcinoid tumors may respond equally to what is observed in CD. Similarly, some ectopic ACTH-secreting tumors might express vasopressin receptors and respond to desmopressin like CD tumors; in these cases, desmopressin test is not helpful in differentiating the source of ACTH. Actually, it is not recommended to use the desmopressin test routinely until more data validating the test become available [13]. Discordant results of these tests are reported in up to 60% of patients, BIPSS being the most recommended one.
Additional testing might be helpful to delineate the source of ACTH, as well as to evaluate comorbidities of CS. Chromogranin A, 5-hydroxy-indoleacetic acid, calcitonin, and gastrin might point to ectopic ACTH tumors. Metabolic alkalosis and hypokalemia as a result of severe hypercortisolism (UFC > 1500 μg/24 h) are common in ectopic ACTH sources, but it can be also present in 10% of CD. Dehydroepiandrosterone sulfate is normal or increased in ACTH-dependent causes, but is decreased in cases of adrenal origin [32].
7.5.3 Localization of the ACTH Source
Once ACTH-dependent CS is confirmed and noninvasive tests or BIPSS suggest a pituitary origin, pituitary imaging is mandatory. T1 gadolinium contrast MRI identifies pituitary tumors in around 50% of patients with CD. Importantly, roughly 10% of healthy individuals have incidental pituitary lesions up to 6 mm; therefore, a lesion smaller than 6 mm does not always identify CD as a cause of CS (Fig. 7.1). Sensitivity of MRI can be improved up to 80% using more advanced techniques (spoiled gradient recalled acquisition or dynamic MRI sequences) [33]. A cranial CT scan might be requested by the surgeon since it provides better information on bone structures. It has recently been reported that pituitary adenomas not visible via MRI may be detectable by CRH-stimulated 18-F-fluoro-deoxy-glucose PET imaging [34].
7.6 Management
CD requires a multidisciplinary and individualized management strategy. The main goals of therapy are to reverse clinical features, normalize cortisol levels, minimalize morbidity, and reach long-term remission without recurrences. The best treatment options involve a multidisciplinary team, including an endocrinologist experienced in the management of CD. Surgery is considered the first therapeutic option. When surgery is not possible or non-curative, the choice for second-line therapy might depend on several factors, i.e., patient preferences, urgency to treat, location and size of remnant tumor, drug interactions and side effects, and cost and availability of medical therapies.
7.6.1 Surgical Treatment
Selective pituitary adenomectomy using transsphenoidal surgery (TSS; microscopic or endoscopic techniques) by an experienced neurosurgeon remains the first-line treatment for CD. If surgery is successful (pathological confirmation of the adenoma or biochemical demonstration of remission after resection), the patient is cured. Ideally, the whole pituitary tumor is removed and normal pituitary tissue is left. However, pituitary deficiencies as a consequence of surgery might be present after initial therapy and an interval of hypoadrenalism is common since normal corticotroph cells have been suppressed by longstanding hypercortisolism. Reasons for surgery failure include lack of experience of the surgeon, diffuse corticotroph hyperplasia (very rare), invasive tumor that cannot be resected, or adenomas arising in unusual sites (parasellar, pituitary stalk, neurohypophysis). On the contrary, a well-defined, noninvasive (to the cavernous sinus), and well-visualized tumor on MRI, histological confirmation of an ACTH-secreting tumor, low postoperative cortisol levels, and long-lasting adrenal insufficiency are favorable prognostic factors [35].
If surgery is not successful the first time, repeated TSS, medical therapy, and radiotherapy are potential second-line therapies. As a final resource, surgical or medical adrenalectomy can be performed if hypercortisolism is still uncontrolled. The choice of a second-line treatment must be discussed with the patient. Repeated surgery as soon as active and persistent hypercortisolism is confirmed might be undertaken especially if the pituitary tumor is visualized; nevertheless, overall successful rates are lower than for the first surgery and carries a high risk of pituitary insufficiencies [36].
Surgical complications are more likely to occur in macroadenomas or extensive pituitary exploration. Anterior pituitary deficiencies are observed in about 20 to 25% of the cases, as well as transient central diabetes insipidus. Permanent diabetes insipidus is less frequent. Symptomatic hyponatremia might occur between 1 and 10 days after surgery, with a maximum antidiuresis around day 5, in about 8% of the patients [37]. It is recommended to measure serum sodium several times during the first days after surgery. Other complications include venous thrombosis, hemorrhage, and infections [38]. Since a hypercoagulability state exists in CS, perioperative prophylaxis a few days prior to surgery is recommended [39]. Within 2 weeks of surgery, measurement of free T4 and prolactin may help to identify hypopituitarism, when compared to preoperative values.
7.6.2 Radiotherapy
Conventional fractionated photon beam radiotherapy (1.7–2 Gy daily for a total dose of 45 Gy over 6 weeks) or stereotactic radiosurgery (single-dose radiation, including gamma knife, linear accelerator, and proton beam) control hypercortisolemia in up to 80% of patients within 3–5 years [40], but results vary across series [41]. Radiotherapy is indicated in persistent CD after surgery and when local invasion precludes a surgical cure. Radiosurgery may provide a faster biochemical control and less risk of radiation damage to the surrounding structures than conventional radiation therapy, but there are no direct comparative studies. Since medical therapy is effective in normalizing cortisol, it is recommended before administering radiotherapy, since these agents will be required while waiting for the effects of radiation. Long-term follow-up is mandatory to detect both relapse and pituitary insufficiencies. The risk of a second neoplasia (most frequently meningiomas) is estimated in 2% of the patients at 20 years, but further studies are required to confirm these data [42]. Optic neuropathy and other cranial neuropathies may also occur (2%). Measuring UFC without concomitant glucocorticoid supplementation at 6–12 months after radiation therapy is necessary to assess if adrenal insufficiency (AI) has developed. In addition, patients should be counseled on symptoms of AI and to alert their physician if they develop symptoms so that more frequent testing may be undertaken. Diurnal rhythm is not necessarily achieved following radiotherapy, so LNSC is not a good tool to assess remission.
7.6.3 Bilateral Adrenalectomy
In general, bilateral adrenalectomy is considered the last treatment option: It provides definitive and immediate control of hypercortisolism, resulting in permanent hypocortisolism that requires lifelong GC and mineralocorticoid replacement therapy and careful education of the patient. Furthermore, regular pituitary MRI and assessment of hyperpigmentation and ACTH levels are mandatory because of the risk of developing Nelson’s syndrome (corticotroph tumor progression) described in up to 25% of patients [43]. If remission is not achieved after the second surgery, the decision of pituitary radiotherapy or bilateral adrenalectomy must be individualized and requires consideration of the pituitary status after the second surgery. Another factor that needs to be taken into account includes patient tolerance to medical therapy while awaiting the effects of radiotherapy. Finally, women who wish to maintain fertility without the need for ovulation induction may opt for adrenalectomy instead of pituitary radiotherapy [44].
7.6.4 Medical Therapy
Medical therapy is indicated in preoperative patients with severe hypercortisolism and associated clinical manifestations, as well as in patients awaiting a response after radiotherapy, and/or when palliative treatment is needed. Hypoadrenalism may occur when treating with these agents; therefore, the possibility of adrenal insufficiency must be addressed if suspected and interrupting the medication may be indicated. Some of these agents affect CYP3A4 leading to significant drug–drug interactions; thus, reviewing all other medications taken by a patient is necessary before starting the treatment. Follow-up for individuals on medical therapy should include UFC and clinical features, aiming for normalization of both [45]. Medical treatments include steroidogenesis inhibitors, agents that modulate ACTH secretion or GC receptor antagonists (Table 7.5).
7.6.4.1 Steroidogenesis Inhibitors
Steroidogenesis inhibitors are effective at blocking the hypercortisolemia despite the fact that they do not treat the underlying tumor. The most commonly used steroidogenesis inhibitors include metyrapone, ketoconazole, mitotane, and etomidate.
-
Metyrapone inhibits 11-β hydroxylase (conversion of 11-deoxycortisol to cortisol) and inhibits aldosterone biosynthesis with accumulation of aldosterone precursors that have a weak mineralocorticoid activity. It has a rapid onset of action, and it controls hypercortisolism in 50–75% of the patients with CS [46]. The risk of hirsutism and acne development due to accumulation of androgenic precursors may be metyrapone less desirable as initial therapy in females. Monitoring for edema, hypokalemia, and hypertension is recommended due to the accumulation of mineralocorticoid precursors.
-
Ketoconazole is an imidazole derivative that inhibits steroidogenesis by blocking side-chain cleavage, and to a lesser degree 17,20-desmolase, and 11-β hydroxylase enzymes. It has a rapid onset of action and an acidic environment is required to maximize the absorption; therefore, concomitant use of proton-pump inhibitors is contraindicated. Efficacy reported varies across series; on average, it normalizes UFC levels in 60% of the patients with CD [47]. Liver function should be monitored; mild elevation of liver enzymes, up to threefold, is not a contraindication. Hepatotoxicity is usually mild and resolves after drug withdrawal. The possibility of development of hypogonadism in men may favor the use of metyrapone as initial medical therapy in men. Recently, ketoconazole was withdrawn from the market due to hepatic dyscrasia for the treatment of fungal infections by the European Medicines Agency; however, it is indicated to control hypercortisolism.
Combination therapy with both ketoconazole and metyrapone can be used to control severe hypercortisolemia.
-
Mitotane has a specific adrenolytic action and acts by inhibition of CYP11A1, P450 side-chain cleavage. Mitotane provides a long-term suppression of hypercortisolism in patients with CD. However, its onset of action is slow (weeks or months) and potential adverse events may be serious (gastrointestinal and neurological); careful monitoring of drug levels is required and only available in few centers [48]. However, monitoring circulating levels of mitotane is made available by the manufacturer in many countries. Mitotane increases CBG and plasma total cortisol levels; thus, biochemical monitoring relies on UFC or salivary cortisol. Hypoadrenalism is common and requires concomitant supplementation with higher doses of hydrocortisone than in other causes of hypoadrenalism, because mitotane activates CYP3A4 and increases hydrocortisone clearance.
-
Etomidate is an intravenous anesthetic and an imidazole derivative used when rapid control of cortisol levels is required, and oral agents cannot be taken. Etomidate may be useful in emergency settings with unmanageable symptoms (respiratory failure, psychosis). Monitoring cortisol levels every 4–6 h is required to titrate the infusion rate.
-
Osilodrostat (LCI699), an oral 11 β-hydroxylase inhibitor, has recently been authorized for its use in Cushing’s syndrome by the European Medicine Agency (January 2020). Osilodrostat was effective at lowering the levels of cortisol in at least 80% of the patients compared to placebo, and it was in general well-tolerated and more convenient since it can be given orally twice a day [49].
-
Levoketoconazole (COR-003), a ketoconazole stereoisomer, is under investigation. Preliminary results show promising results in the treatment of CD patients; however, it is still being evaluated in multicenter trials [50].
7.6.4.2 Another Modality for Treating CS
Another modality for treating CS includes the class of medications that are GC receptor antagonists. One such drug is mifepristone (RU486), which is both a GC receptor antagonist and an anti-progestin. Few experiences with CD are reported, and the assessment of its efficacy without a biochemical marker makes its use challenging and restricts the ability to assess overtreatment or undertreatment. Cortisol levels remain unchanged or increased while on mifepristone and are not useful to guide efficacy of the therapy. Therefore, dose adjustment is based on clinical symptoms, which have been shown to improve in a significant number of patients (diabetes in 60%, hypertension in 40%, and at least one of the following clinical parameters: weight, depression, quality of life (QoL), or clinical appearance in 87%) [51]. Mifepristone is a second-line therapy in CS relegated for patients with diabetes who failed surgery or those with CS who are not surgical candidates and have persistent disease. CORT125134 is a new selective GC receptor antagonist under investigation.
7.6.4.3 Tumor-Directed Therapeutic Agents
Tumor-directed therapeutic agents act directly on corticotroph cells, inhibiting ACTH production
-
Dopamine D2 receptor agonists (bromocriptine, cabergoline) may be effective in a subset of CD patients. Usually, higher doses of cabergoline than the ones used for hyperprolactinemia are required (2–3 mg/week). Efficacy is limited; small studies showed normalization of UFC in 30–40% of patients [52]. Clinical trials are needed to evaluate combination therapy with cabergoline.
-
Pasireotide (SOM230) is a somatostatin receptor (SST) agonist with higher and broader affinity for SST 1, 2, 3, and 5 than octreotide or lanreotide. It has been demonstrated to be useful as a tumor-directed medical therapy for CD, since corticotroph tumors have a high expression of SST5. It is approved for patients who have failed surgery or for those with contraindications to surgery. The main side effects of pasireotide include gastrointestinal upset and hyperglycemia. Long-acting pasireotide (monthly administration) has been developed, with a similar safety profile and efficacy to that of twice-daily formulations, normalizing UFC in about 40% of CD patients [53].
Potential new drugs are under investigation as a tumor-directed targets, such as retinoic acid, silibinin, and roscovitine, whose efficacy and safety are still unclear [54].
7.6.5 Perioperative Management
GC replacement before surgery for Cushing’s syndrome is not necessary unless the patient has been treated with adrenal enzyme inhibitors. Regimens differ across centers, and no study comparisons between one approach and the other have been done. A typical regimen is the administration of GC during the 24 h prior to transsphenoidal surgery [55].
Almost all patients require GC replacement after transsphenoidal surgery for an ACTH-producing adenoma until the HPA axis is recovered [55]. Some patients may have a “GC withdrawal syndrome” despite the use of physiologic GC replacement therapy, and they should be warned that this is expected after “successful surgery.” Occasionally transient supraphysiological doses of GC are needed [56].
7.7 Follow-Up
Initial remission rates are achieved in 80–90% of the patients with microadenomas and 60% of patients with macroadenomas with an experienced neurosurgeon. However, 20% of the patients present late recurrences even 10–15 years after initial therapy [2, 6]. Therefore, lifelong follow-up is mandatory for CD.
Measurement of morning serum cortisol during the first week after surgery is recommended to assess remission by withholding treatment with GCs at least 24 hours [44]. Hydrocortisone (<20 mg) 2–3 times daily is the preferred regimen of GC supplementation, since it has a shorter half-life and supraphysiological doses should be avoided to allow quicker recovery of HPA axis. Usually, GCs are maintained for a few days before withdrawal, being careful about the potential development of adrenal insufficiency. It is recommended to measure morning serum cortisol every 2–3 months, followed by an ACTH stimulation test if cortisol levels are >200 nmol/l (7.3 μg/dl). Alternatively, insulin-induced hypoglycemia can be done to assess recovery of the HPA axis within 6 weeks of surgery [45]. GC can be stopped if morning serum cortisol or cortisol response to ACTH stimulation test is >500 nmol/l (>18 μg/dl). If baseline cortisol is still <138 nmol/l (5 μg/dl), GC replacement should be continued while retesting the patient every 3–6 months. Recovery of the HPA axis may take up to 1 year after TSS. Once HPA axis function is recovered, assessment for possible recurrences should be performed annually or sooner if patients have clinical symptoms. Interestingly, LNSC seems to be one of the earliest biochemically detectable alterations of recurrence, preceding elevations of UFC [57].
There is no consensus on the criteria for “cure” after initial surgery for CD. Persistent elevations of UFC after the immediate postoperative period are indicative of lack of remission. While levels of cortisol at either end of the spectrum are more clear-cut, there are intermediate levels that may present a diagnostic dilemma. It is well accepted that serum cortisol levels <50 nmol/l (<1.8 μg/dl) or UFC levels below the normal range (<55 nmol/24 h, ideally <28 nmol/24 h) define remission and are associated with low recurrence rate of 10% at 10 years and high remission rates at long-term follow-up of 85–100%. On the contrary, persistent cortisol >138 nmol/l (>5 μg/dl) for up to 6 weeks requires further evaluation [58]. In patients with intermediate postoperative values of serum plasma cortisol (between 50 and 138 nmol/l), management and follow-up should be individualized; a postoperative cortisol cutoff <138 nmol/l (5 μg/dl) is associated with long-term remission rates of between 65 and 80%. Sometimes cortisol levels decline gradually over the months following surgery, indicating a progressive necrosis of the remaining tumor cells. Therefore, it is important to confirm that levels reached a nadir, before additional testing and therapy are prescribed in patients with persisting hypercortisolism immediately after surgery [59]. Young individuals (<25 years) and patients with macroadenomas are at a higher risk of recurrences [60]. Some patients might present a gradual decline of cortisol levels over the months following surgery, so follow-up is mandatory.
The true long-term recurrence rate is uncertain because of different criteria of remission and inadequate follow-up. The median interval to recurrence is about 40 months but varies across series and duration of follow-up. Postoperative serum cortisol levels or UFC might provide prognostic information. Long-term monitoring should be done annually for several years using LNSC, DST, and/or UFC or at any time that patients experience a return of symptomatology, to rule out recurrences. Documentation of recovery of a diurnal cortisol pattern might be helpful to support a remission state.
Testing for GH deficiency is advised at least 12 months after remission, because hypercortisolism affects GH axis [61] and a postoperative MRI scan is recommended beyond 3 months after surgery [45].
7.8 Prognosis
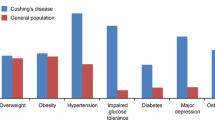
Although biochemical remission is associated with significant clinical improvement, some symptoms and comorbidities may not completely normalize [7]. Therefore, it is mandatory to monitor and provide additional treatment for cortisol-dependent comorbidities throughout the life of a CS survivor (Fig. 7.2).

Main clinical manifestation after remission of Cushing’s syndrome
7.8.1 Mortality
Higher mortality, mainly due to cardiovascular and infectious diseases, is observed within the first year after diagnosis and initial therapy, and in those patients whose initial cure was not obtained. Before the development of steroidogenesis inhibitors and surgical techniques, prognosis was poor with a mean survival of 4 years [62]. In patients who have persistent mild hypercortisolism despite therapy, SMR is increased 3.8- to 5.0-fold compared with the general population [2, 8]. Although more recent studies show that mortality rates have been dramatically reduced after successful normalization of cortisol, mortality is still increased up to 2–3 times in most of the series, while others reported similar SMR to that of age-matched populations [60, 63, 64].
7.8.2 Cardiovascular Risk and Metabolic Syndrome
Cardiovascular risk and metabolic syndrome may remain abnormal after years of cortisol normalization. Hypertension, glucose, and lipid metabolism improve after normalization of hypercortisolism, but a higher risk of diabetes, dyslipidemia, and hypertension remains years after remission compared to a healthy population or patients with nonfunctioning pituitary adenomas [7, 65]. Moreover, persistence of central obesity contributes to insulin resistance and an increased inflammatory and hypercoagulable state with an unfavorable adipokine profile (increased levels of TNF-alpha, interleukin-6, and leptin) contributing to persistence of increased cardiovascular risk. An increased risk of myocardial infarction, greater prevalence of coronary calcifications, left ventricular dysfunction, and cerebrovascular disease have also been reported in survivors of CS [7, 63,64,65,66].
7.8.3 Psychopathology and Cognition
Even years after CD is cured, patients have an increased prevalence of psychopathology (anxiety disorders, major depression, maladaptive personality) compared to healthy individuals or patients treated for other types of pituitary diseases [7]. The use of screening tools in clinical practice to identify psychopathology promptly is recommended [67]. Cognitive impairment (short-term memory and attention deficits, as well as working memory and impaired decision making) is a common complaint after hypercortisolism [68]. Chronic hypercortisolism exposure causes structural changes in cerebral areas; specifically, structural brain abnormalities (more severe white matter lesions commonly associated with increased cardiovascular risk, smaller gray matter volumes in the anterior cingulate cortex, greater cerebellar volumes, hippocampal dysfunction) were found in patients in long-term remission of their CS compared to matched controls [69,70,71,72]. Fluoxetine has been suggested as a neuroprotectant and antidepressant for these patients, but no prospective studies are available yet [73]. Data on long-term recovery regarding cognitive impairment are scarce.
7.8.4 Bone Metabolism
Factors involved in higher risk of osteoporosis in CD are multiple: direct effects of GC on bone cells, GC-induced hypogonadism, hypopituitarism secondary to surgery, hydrocortisone replacement therapy, or reduced bone strain due to muscle atrophy [74]. Approximately 40% of CS patients present with fractures, particularly vertebral fractures. Although there is a decrease in bone mineral density (BMD) and increased fracture risk during hypercortisolism and before diagnosis, studies report BMD normalization after several years of cure [75, 76]. Meanwhile, in severe osteopenia, alendronate might be useful to induce a more rapid improvement of BMD or to prevent further bone loss in persistent postsurgical hypercortisolism [77]. Time of excess GC exposure and duration of GC replacement after surgery were found to be predictors of low BMD in CS. A detailed fracture assessment is recommended, evaluating BMD at the spine and hip, adequate calcium and vitamin D supplementation, and individualized long-term follow-up accordingly.
7.8.5 Quality of Life
Because of all comorbidities associated with CS despite cure, health-related quality of life is likely to be affected long term. Perceived QoL is impaired especially in patients with CD [7, 78], even independently of the disease control or adequately replaced hypopituitarism [2], compared to healthy individuals or other pituitary tumors. Although QoL improves in patients in remission, long-term residual impairment in physical and social functioning is commonly reported. Altogether, it has a significant impact, with social and economic consequences, since inability to return to work or sick days is common [73, 79].
7.8.6 Other Cortisol-Related Comorbidities
CS patients have a significant increased risk of venous thromboembolic events and impaired coagulation profile (activation of coagulation cascades and impaired fibrinolysis) compared to the general population, especially in the postoperative period [80]. Muscle myopathy may persist long-term after cure; aerobic and resistance exercises might be effective in attenuating GC-induced muscle atrophy. Increased rate of nephrolithiasis in both active and cured CD compared to healthy controls has been reported, but the pathogenic mechanisms in CS are not yet elucidated.
Importantly, resolution of hypercortisolism is associated with new onset or exacerbation of pre-existing autoimmune or inflammatory diseases (psoriasis, rheumatoid arthritis, ulcerative colitis, etc.), likely due to the suppressive effects of hypercortisolism on the immune system [81]. The most common autoimmune disease is autoimmune thyroiditis; therefore, patients with positive antibodies should be followed after remission of hypercortisolism to identify precociously the onset of a hypothyroidism.
7.9 Summary and Conclusions
CD is a rare disorder characterized by chronic exposure to excess glucocorticoids leading to multisystemic comorbidities and increased mortality. Although clinical presentation is often nonspecific and hypercortisolism might not be suspected, a prompt diagnosis is mandatory to reduce long-term comorbidities due to hypercortisolism. Pituitary surgery can be curative in most of the cases but not all. Radiotherapy and medical therapy can be used as additional therapeutic options. Fortunately, new medical therapies are under development with promising results. A multidisciplinary and individualized approach is essential to choose the best treatment approach, minimize complications, and manage cortisol-related comorbidities, since some of them persist even after hormonal normalization. A lifelong surveillance is advisable to identify recurrences and treatment of persistent comorbidities.
References
Valassi E, Santos A, Yaneva M, Toth M, Strasburger CJ, Chanson P, et al. The European Registry on Cushing's syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur J Endocrinol. 2011;165(3):383–92.
Lindholm J, Juul S, Jorgensen JO, Astrup J, Bjerre P, Feldt-Rasmussen U, et al. Incidence and late prognosis of cushing's syndrome: a population-based study. J Clin Endocrinol Metab. 2001;86(1):117–23.
Agustsson TT, Baldvinsdottir T, Jonasson JG, Olafsdottir E, Steinthorsdottir V, Sigurdsson G, et al. The epidemiology of pituitary adenomas in Iceland, 1955-2012: a nationwide population-based study. Eur J Endocrinol. 2015;173(5):655–64.
Gruppetta M, Mercieca C, Vassallo J. Prevalence and incidence of pituitary adenomas: a population based study in Malta. Pituitary. 2013;16(4):545–53.
Leon-Justel A, Madrazo-Atutxa A, Alvarez-Rios AI, Infantes-Fontan R, Garcia-Arnes JA, Lillo-Munoz JA, et al. A probabilistic model for Cushing's syndrome screening in at-risk populations: a prospective multicenter study. J Clin Endocrinol Metab. 2016;101(10):3747–54.
Steffensen C, Bak AM, Rubeck KZ, Jorgensen JO. Epidemiology of Cushing's syndrome. Neuroendocrinology. 2010;92(Suppl 1):1–5.
Feelders RA, Pulgar SJ, Kempel A, Pereira AM. The burden of Cushing's disease: clinical and health-related quality of life aspects. Eur J Endocrinol. 2012;167(3):311–26.
Etxabe J, Vazquez JA. Morbidity and mortality in Cushing's disease: an epidemiological approach. Clin Endocrinol. 1994;40(4):479–84.
Sharma ST, Nieman LK, Feelders RA. Cushing's syndrome: epidemiology and developments in disease management. Clin Epidemiol. 2015;7:281–93.
Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing's syndrome. Lancet. 2015;386(9996):913–27.
Karl M, Lamberts SW, Koper JW, Katz DA, Huizenga NE, Kino T, et al. Cushing's disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians. 1996;108(4):296–307.
Mann M, Koller E, Murgo A, Malozowski S, Bacsanyi J, Leinung M. Glucocorticoid-like activity of megestrol. A summary of Food and Drug Administration experience and a review of the literature. Arch Intern Med. 1997;157(15):1651–6.
Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526–40.
Tabarin A, Perez P. Pros and cons of screening for occult Cushing syndrome. Nat Rev Endocrinol. 2011;7(8):445–55.
Catargi B, Rigalleau V, Poussin A, Ronci-Chaix N, Bex V, Vergnot V, et al. Occult Cushing's syndrome in type-2 diabetes. J Clin Endocrinol Metab. 2003;88(12):5808–13.
Reimondo G, Pia A, Allasino B, Tassone F, Bovio S, Borretta G, et al. Screening of Cushing's syndrome in adult patients with newly diagnosed diabetes mellitus. Clin Endocrinol. 2007;67(2):225–9.
Chiodini I, Vainicher CE, Morelli V, Palmieri S, Cairoli E, Salcuni AS, et al. Mechanisms in endocrinology: endogenous subclinical hypercortisolism and bone: a clinical review. Eur J Endocrinol. 2016;175(6):R265–R82.
Valassi E, Franz H, Brue T, Feelders RA, Netea-Maier R, Tsagarakis S, et al. Diagnostic tests for Cushing's syndrome differ from published guidelines: data from ERCUSYN. Eur J Endocrinol. 2017;176(5):613–24.
Mericq MV, Cutler GB Jr. High fluid intake increases urine free cortisol excretion in normal subjects. J Clin Endocrinol Metab. 1998;83(2):682–4.
Chan KC, Lit LC, Law EL, Tai MH, Yung CU, Chan MH, et al. Diminished urinary free cortisol excretion in patients with moderate and severe renal impairment. Clin Chem. 2004;50(4):757–9.
Ueland GA, Methlie P, Kellmann R, Bjorgaas M, Asvold BO, Thorstensen K, et al. Simultaneous assay of cortisol and dexamethasone improved diagnostic accuracy of the dexamethasone suppression test. Eur J Endocrinol. 2017;176(6):705–13.
Dorn LD, Lucke JF, Loucks TL, Berga SL. Salivary cortisol reflects serum cortisol: analysis of circadian profiles. Ann Clin Biochem. 2007;44(Pt 3):281–4.
Liu H, Bravata DM, Cabaccan J, Raff H, Ryzen E. Elevated late-night salivary cortisol levels in elderly male type 2 diabetic veterans. Clin Endocrinol. 2005;63(6):642–9.
Yanovski JA, Cutler GB Jr, Chrousos GP, Nieman LK. Corticotropin-releasing hormone stimulation following low-dose dexamethasone administration. A new test to distinguish Cushing's syndrome from pseudo-Cushing's states. JAMA. 1993;269(17):2232–8.
Nieman LK, Oldfield EH, Wesley R, Chrousos GP, Loriaux DL, Cutler GB Jr. A simplified morning ovine corticotropin-releasing hormone stimulation test for the differential diagnosis of adrenocorticotropin-dependent Cushing's syndrome. J Clin Endocrinol Metab. 1993;77(5):1308–12.
Chrousos GP, Schulte HM, Oldfield EH, Gold PW, Cutler GB Jr, Loriaux DL. The corticotropin-releasing factor stimulation test. An aid in the evaluation of patients with Cushing's syndrome. N Engl J Med. 1984;310(10):622–6.
Arnaldi G, Angeli A, Atkinson AB, Bertagna X, Cavagnini F, Chrousos GP, et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2003;88(12):5593–602.
Nieman LK, Ilias I. Evaluation and treatment of Cushing's syndrome. Am J Med. 2005;118(12):1340–6.
Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing's syndrome. Lancet. 2006;367(9522):1605–17.
Swearingen B, Katznelson L, Miller K, Grinspoon S, Waltman A, Dorer DJ, et al. Diagnostic errors after inferior petrosal sinus sampling. J Clin Endocrinol Metab. 2004;89(8):3752–63.
Colao A, Faggiano A, Pivonello R, Pecori Giraldi F, Cavagnini F, Lombardi G, et al. Inferior petrosal sinus sampling in the differential diagnosis of Cushing's syndrome: results of an Italian multicenter study. Eur J Endocrinol. 2001;144(5):499–507.
Hong AR, Kim JH, Hong ES, Kim IK, Park KS, Ahn CH, et al. Limited diagnostic utility of plasma adrenocorticotropic hormone for differentiation between adrenal Cushing syndrome and Cushing disease. Endocrinol Metab (Seoul). 2015;30(3):297–304.
Grober Y, Grober H, Wintermark M, Jane JA, Oldfield EH. Comparison of MRI techniques for detecting microadenomas in Cushing's disease. J Neurosurg. 2018;128(4):1051–7.
Boyle J, Patronas NJ, Smirniotopoulos J, Herscovitch P, Dieckman W, Millo C, et al. CRH stimulation improves (18)F-FDG-PET detection of pituitary adenomas in Cushing's disease. Endocrine. 2019;
Bochicchio D, Losa M, Buchfelder M. Factors influencing the immediate and late outcome of Cushing's disease treated by transsphenoidal surgery: a retrospective study by the European Cushing's Disease Survey Group. J Clin Endocrinol Metab. 1995;80(11):3114–20.
Hofmann BM, Hlavac M, Kreutzer J, Grabenbauer G, Fahlbusch R. Surgical treatment of recurrent Cushing's disease. Neurosurgery. 2006;58(6):1108–18. discussion -18
Adams JR, Blevins LS Jr, Allen GS, Verity DK, Devin JK. Disorders of water metabolism following transsphenoidal pituitary surgery: a single institution's experience. Pituitary. 2006;9(2):93–9.
Prevedello DM, Pouratian N, Sherman J, Jane JA Jr, Vance ML, Lopes MB, et al. Management of Cushing's disease: outcome in patients with microadenoma detected on pituitary magnetic resonance imaging. J Neurosurg. 2008;109(4):751–9.
van der Pas R, de Bruin C, Leebeek FW, de Maat MP, Rijken DC, Pereira AM, et al. The hypercoagulable state in Cushing's disease is associated with increased levels of procoagulant factors and impaired fibrinolysis, but is not reversible after short-term biochemical remission induced by medical therapy. J Clin Endocrinol Metab. 2012;97(4):1303–10.
Petit JH, Biller BM, Yock TI, Swearingen B, Coen JJ, Chapman P, et al. Proton stereotactic radiotherapy for persistent adrenocorticotropin-producing adenomas. J Clin Endocrinol Metab. 2008;93(2):393–9.
Starke RM, Williams BJ, Vance ML, Sheehan JP. Radiation therapy and stereotactic radiosurgery for the treatment of Cushing's disease: an evidence-based review. Curr Opin Endocrinol Diabetes Obes. 2010;17(4):356–64.
Minniti G, Traish D, Ashley S, Gonsalves A, Brada M. Risk of second brain tumor after conservative surgery and radiotherapy for pituitary adenoma: update after an additional 10 years. J Clin Endocrinol Metab. 2005;90(2):800–4.
Assie G, Bahurel H, Coste J, Silvera S, Kujas M, Dugue MA, et al. Corticotroph tumor progression after adrenalectomy in Cushing's disease: a reappraisal of Nelson's syndrome. J Clin Endocrinol Metab. 2007;92(1):172–9.
Biller BM, Grossman AB, Stewart PM, Melmed S, Bertagna X, Bertherat J, et al. Treatment of adrenocorticotropin-dependent Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93(7):2454–62.
Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015;100(8):2807–31.
Ceccato F, Zilio M, Barbot M, Albiger N, Antonelli G, Plebani M, et al. Metyrapone treatment in Cushing's syndrome: a real-life study. Endocrine. 2018;62(3):701–11.
Castinetti F, Guignat L, Giraud P, Muller M, Kamenicky P, Drui D, et al. Ketoconazole in Cushing's disease: is it worth a try? J Clin Endocrinol Metab. 2014;99(5):1623–30.
Baudry C, Coste J, Bou Khalil R, Silvera S, Guignat L, Guibourdenche J, et al. Efficiency and tolerance of mitotane in Cushing's disease in 76 patients from a single center. Eur J Endocrinol. 2012;167(4):473–81.
Pivonello R, Fleseriu M, Newell-Price J, Bertagna X, Findling J, Shimatsu A, et al. Efficacy and safety of osilodrostat in patients with Cushing's disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol. 2020;8(9):748–61.
Feelders RA, Newell-Price J, Pivonello R, Nieman LK, Hofland LJ, Lacroix A. Advances in the medical treatment of Cushing's syndrome. Lancet Diabetes Endocrinol. 2019;7(4):300–12.
Fleseriu M, Biller BM, Findling JW, Molitch ME, Schteingart DE, Gross C, et al. Mifepristone, a glucocorticoid receptor antagonist, produces clinical and metabolic benefits in patients with Cushing's syndrome. J Clin Endocrinol Metab. 2012;97(6):2039–49.
Pivonello R, De Martino MC, Cappabianca P, De Leo M, Faggiano A, Lombardi G, et al. The medical treatment of Cushing's disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. 2009;94(1):223–30.
Lacroix A, Gu F, Gallardo W, Pivonello R, Yu Y, Witek P, et al. Efficacy and safety of once-monthly pasireotide in Cushing's disease: a 12 month clinical trial. Lancet Diabetes Endocrinol. 2018;6(1):17–26.
Theodoropoulou M, Reincke M. Tumor-directed therapeutic targets in Cushing disease. J Clin Endocrinol Metab. 2019;104(3):925–33.
Prete A, Corsello SM, Salvatori R. Current best practice in the management of patients after pituitary surgery. Ther Adv Endocrinol Metab. 2017;8(3):33–48.
Hochberg Z, Pacak K, Chrousos GP. Endocrine withdrawal syndromes. Endocr Rev. 2003;24(4):523–38.
Danet-Lamasou M, Asselineau J, Perez P, Vivot A, Nunes ML, Loiseau H, et al. Accuracy of repeated measurements of late-night salivary cortisol to screen for early-stage recurrence of Cushing's disease following pituitary surgery. Clin Endocrinol. 2015;82(2):260–6.
Hammer GD, Tyrrell JB, Lamborn KR, Applebury CB, Hannegan ET, Bell S, et al. Transsphenoidal microsurgery for Cushing's disease: initial outcome and long-term results. J Clin Endocrinol Metab. 2004;89(12):6348–57.
Valassi E, Biller BM, Swearingen B, Pecori Giraldi F, Losa M, Mortini P, et al. Delayed remission after transsphenoidal surgery in patients with Cushing's disease. J Clin Endocrinol Metab. 2010;95(2):601–10.
Swearingen B, Biller BM, Barker FG 2nd, Katznelson L, Grinspoon S, Klibanski A, et al. Long-term mortality after transsphenoidal surgery for Cushing disease. Ann Intern Med. 1999;130(10):821–4.
Tzanela M, Karavitaki N, Stylianidou C, Tsagarakis S, Thalassinos NC. Assessment of GH reserve before and after successful treatment of adult patients with Cushing's syndrome. Clin Endocrinol. 2004;60(3):309–14.
Plotz CM, Knowlton AI, Ragan C. The natural history of Cushing's syndrome. Am J Med. 1952;13(5):597–614.
Dekkers OM, Horvath-Puho E, Jorgensen JO, Cannegieter SC, Ehrenstein V, Vandenbroucke JP, et al. Multisystem morbidity and mortality in Cushing's syndrome: a cohort study. J Clin Endocrinol Metab. 2013;98(6):2277–84.
Clayton RN, Raskauskiene D, Reulen RC, Jones PW. Mortality and morbidity in Cushing's disease over 50 years in Stoke-on-Trent, UK: audit and meta-analysis of literature. J Clin Endocrinol Metab. 2011;96(3):632–42.
Webb SM, Mo D, Lamberts SW, Melmed S, Cavagnini F, Pecori Giraldi F, et al. Metabolic, cardiovascular, and cerebrovascular outcomes in growth hormone-deficient subjects with previous cushing's disease or non-functioning pituitary adenoma. J Clin Endocrinol Metab. 2010;95(2):630–8.
Barahona MJ, Resmini E, Vilades D, Pons-Llado G, Leta R, Puig T, et al. Coronary artery disease detected by multislice computed tomography in patients after long-term cure of Cushing's syndrome. J Clin Endocrinol Metab. 2013;98(3):1093–9.
Santos A, Resmini E, Pascual JC, Crespo I, Webb SM. Psychiatric symptoms in patients with Cushing's syndrome: prevalence. Diagn Manage Drugs. 2017;77(8):829–42.
Tiemensma J, Kokshoorn NE, Biermasz NR, Keijser BJ, Wassenaar MJ, Middelkoop HA, et al. Subtle cognitive impairments in patients with long-term cure of Cushing's disease. J Clin Endocrinol Metab. 2010;95(6):2699–714.
Santos A, Resmini E, Gomez-Anson B, Crespo I, Granell E, Valassi E, et al. Cardiovascular risk and white matter lesions after endocrine control of Cushing's syndrome. Eur J Endocrinol. 2015;173(6):765–75.
Andela CD, van der Werff SJ, Pannekoek JN, van den Berg SM, Meijer OC, van Buchem MA, et al. Smaller grey matter volumes in the anterior cingulate cortex and greater cerebellar volumes in patients with long-term remission of Cushing's disease: a case-control study. Eur J Endocrinol. 2013;169(6):811–9.
Resmini E, Santos A, Gomez-Anson B, Lopez-Mourelo O, Pires P, Vives-Gilabert Y, et al. Hippocampal dysfunction in cured Cushing's syndrome patients, detected by (1) H-MR-spectroscopy. Clin Endocrinol. 2013;79(5):700–7.
Bourdeau I, Bard C, Noel B, Leclerc I, Cordeau MP, Belair M, et al. Loss of brain volume in endogenous Cushing's syndrome and its reversibility after correction of hypercortisolism. J Clin Endocrinol Metab. 2002;87(5):1949–54.
Santos A, Crespo I, Aulinas A, Resmini E, Valassi E, Webb SM. Quality of life in Cushing's syndrome. Pituitary. 2015;18(2):195–200.
Toth M, Grossman A. Glucocorticoid-induced osteoporosis: lessons from Cushing's syndrome. Clin Endocrinol. 2013;79(1):1–11.
Kristo C, Jemtland R, Ueland T, Godang K, Bollerslev J. Restoration of the coupling process and normalization of bone mass following successful treatment of endogenous Cushing's syndrome: a prospective, long-term study. Eur J Endocrinol. 2006;154(1):109–18.
Vestergaard P, Lindholm J, Jorgensen JO, Hagen C, Hoeck HC, Laurberg P, et al. Increased risk of osteoporotic fractures in patients with Cushing's syndrome. Eur J Endocrinol. 2002;146(1):51–6.
Mancini T, Doga M, Mazziotti G, Giustina A. Cushing's syndrome and bone. Pituitary. 2004;7(4):249–52.
Valassi E, Feelders R, Maiter D, Chanson P, Yaneva M, Reincke M, et al. Worse health-related quality of life at long-term follow-up in patients with Cushing's disease than patients with cortisol producing adenoma. Data from the ERCUSYN. Clin Endocrinol (Oxf). 2018;88(6):787–98.
van Aken MO, Pereira AM, Biermasz NR, van Thiel SW, Hoftijzer HC, Smit JW, et al. Quality of life in patients after long-term biochemical cure of Cushing's disease. J Clin Endocrinol Metab. 2005;90(6):3279–86.
Wagner J, Langlois F, Lim DST, McCartney S, Fleseriu M. Hypercoagulability and risk of venous thromboembolic events in endogenous Cushing's syndrome: a systematic meta-analysis. Front Endocrinol. 2018;9:805.
Pivonello R, Isidori AM, De Martino MC, Newell-Price J, Biller BM, Colao A. Complications of Cushing's syndrome: state of the art. Lancet Diabetes Endocrinol. 2016;4(7):611–29.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Aulinas, A., Webb, S.M. (2022). Cushing’s Disease. In: Tamagno, G., Gahete, M.D. (eds) Pituitary Adenomas. Springer, Cham. https://doi.org/10.1007/978-3-030-90475-3_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-90475-3_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-90474-6
Online ISBN: 978-3-030-90475-3
eBook Packages: MedicineMedicine (R0)