
Abstract
Over the past decades, glucocorticoids booked its place as one of the most commonly prescribed classes of drugs for several medical conditions. Although the adverse skeletal effects of glucocorticoids have been recognized for decades, attention to this side effect has gained increased attention because of the widespread long-term clinical use of glucocorticoids. Glucocorticoid-induced osteoporosis has been considered the most common cause of secondary osteoporosis. For its negative impact on the musculoskeletal health and its subsequent significantly elevated fracture risk, several international societies have published recommendations for the prevention and management of glucocorticoid-induced osteoporosis. Glucocorticoids effect on bone is both direct and indirect, including a receptor activator of nuclear factor-κB ligand (RANKL)–induced increase in osteoclasts, osteocyte apoptosis, and decreased recruitment and accelerated osteoblast apoptosis. In addition, glucocorticoids increase the risk for falls, which has been attributed to the reduction of muscle mass and a decrease in calcium resorption. On the other hand, the fracture risk rapidly decreases after glucocorticoid discontinuation. This chapter will review the epidemiology and pathophysiology of glucocorticoid-induced osteoporosis. This will be followed by discussing the effect of glucocorticoids on the musculoskeletal health, namely bones and muscles, clinical correlations of glucocorticoid-induced osteoporosis, risk stratification, screening and assessment, and glucocorticoid associated changes in bone mineral density and bone architecture. The chapter will then discuss the monitoring of BMD and fracture risk assessment as well as management of the glucocorticoid-induced osteoporosis. The chapter will conclude with an algorithm for assessment and management of glucocorticoid-induced osteoporosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glucocorticoids
- Glucocorticoid-induced osteoporosis
- GIO
- Steroids
- Osteoporosis
- Steroid-induced osteoporosis
- BMD
- Sarcopenia
Introduction
Much has been learned since 1932 when Harvey Cushing described the set of symptoms that denoted hypersecretion of ACTH most often due to a pituitary adenoma. A set of symptoms prominently featured include truncal obesity, a rounded face, increased fat around the neck, and peripheral muscle wasting, weakened bones, leading to vertebral compression fractures, striae on the abdomen and buttocks, hirsutism, fatigue, muscle weakness, fatigue, fluid retention, hypertension, and hyperglycemia [1]. The original publication by Cushing [2], as it turns out, was the first description of the effects of endogenous glucocorticoids on bone and muscle, a description that became identified with the effects of exogenous glucocorticoids, or steroid medication, for a variety of chronic inflammatory and neoplastic diseases.
Over the past decades, glucocorticoids booked its place as one of the most commonly prescribed classes of drugs for several medical conditions and in states of hypocortisolism [3]. Although the adverse skeletal effects of glucocorticoids have been recognized for decades, attention to this side effect has gained increased attention because of the widespread long-term clinical use of glucocorticoids in a variety of disorders including autoimmune, pulmonary, and gastrointestinal diseases, malignancies, and in patients receiving organ transplants. The problem has become so widely appreciated that guidelines for the prevention and treatment for glucocorticoid-induced osteoporosis (GIO) have been established in many countries based upon recommendations by expert scientific organizations [4].
The bone loss secondary to initiation of glucocorticoids is early and rapid, and the bone mineral loss correlates well with cumulative dose and duration. Fracture risk is increased even with a daily dose of prednisolone that is <5 mg/day [5]. Parenteral, oral, and even long-term inhaled glucocorticoids are associated with a significant bone loss. The rate of bone loss is noted to be more than 10% in the first year of therapy and thereafter tends to stabilize at 2–3% every year [6]. It predominantly involves trabecular bone, thus increasing the risk of vertebral fractures. Later, cortical bone is also involved (e.g., femoral neck) [7]. About 20% of those treated with glucocorticoids will have a fragility fracture within the first year of treatment [8]. Postmenopausal women and elderly men are at a higher risk for developing glucocorticoid-induced bone loss and fractures [9]. Similarly, the catabolic effects of glucocorticoids on skeletal muscles have been well known for many years. Administration of high doses of glucocorticoids to animals causes not only decreased muscle mass but also muscle dysfunction characterized by reduced force and weakness [10].
Even though the negative effect of glucocorticoid therapy on both bone and muscle health has been recognized and well documented, more than half of those on treatment do not receive bone mineral density (BMD) assessment or the recommended preventative therapy for osteoporosis [11]. A number of guidelines have already been published highlighting the importance of considering preventive measures for patients receiving glucocorticoid treatment on a long-term basis, or until the steroid therapy course is stopped. This chapter will review the epidemiology and pathophysiology of glucocorticoid-induced osteoporosis. This will be followed by discussing the effect of glucocorticoids on the musculoskeletal health, namely bones and muscles, clinical correlations of glucocorticoid-induced osteoporosis, risk stratification, screening and assessment and glucocorticoid-associated changes in bone mineral density and bone architecture. The chapter will then discuss the monitoring of BMD and fracture risk assessment as well as management of the glucocorticoid-induced osteoporosis. The chapter will conclude with an algorithm for assessment and management of glucocorticoid-induced osteoporosis.
Epidemiology
Epidemiologic studies provide valuable information about the use of glucocorticoids and its negative impact on musculoskeletal system. The prevalence of use of oral glucocorticoids in the community population ranges between 0.5% and 0.9%, rising to 2.7% in adults and older adults aged ≥50 years [12,13,14]. The prevalence has been reported to be similar in men and women. In the Global Longitudinal Study of Osteoporosis in Women (GLOW), conducted in 10 countries, 4.6% of 60,393 postmenopausal women were receiving glucocorticoids at baseline visit [15, 16]. The most frequent indications for oral glucocorticoids are inflammatory rheumatic disorders (rheumatoid arthritis, systemic lupus erythematosus, polymyalgia rheumatica, and temporal arteritis), lung disorders (asthma and chronic obstructive lung diseases), and organ transplantation.
Of the multiple side effects that can occur with glucocorticoid therapy, glucocorticoid-induced osteoporosis (GIO) has been identified the most prevalent form of bone affection. Examining the prevalence of GIO revealed that at least 50% of people receiving long-term glucocorticoid therapy are estimated to develop osteoporosis [17]. Furthermore, osteoporotic fractures are one of the most devastating glucocorticoid musculoskeletal complication, affecting 30–50% of patients [18,19,20,21]. Furthermore, secondary causes of osteoporosis, such as that induced by glucocorticoids, are particularly common in pre- or perimenopausal women. In a clinical study of pre- and postmenopausal women attending a specialty osteoporosis clinic (n = 384 patients), a secondary cause for osteoporosis was established in 8.6% of cases, and 21% of these were attributed to glucocorticoids, all of which were in premenopausal women [22]. Khosla and colleagues reported GIO in over 50% of patients aged 20–44 years with an established diagnosis of secondary osteoporosis [23].
In concordance, the risk of fractures is increased by twofold in patients treated with glucocorticoids, and the risk of vertebral fractures is even higher. In a study comparing 244, 235 oral glucocorticoids users and 244, 235 controls, the risk of hip fracture is 1.6, and that of vertebral fracture is 2.6; these numbers have been reproduced in many studies [24,25,26]. The global prevalence of fractures in patients receiving long-term glucocorticoids has been reported to be 30–50%. In 551 patients receiving long-term glucocorticoids, the prevalence of vertebral fractures was 37%, with 14% of patients having two or more asymptomatic vertebral fractures; 48% of patients aged ≥70 years and 30% of those aged <60 years had at least one vertebral fracture [27]. The prevalence increases with age, a key point for preventive strategies.
The highly cited study [8] from the United Kingdom General Practice Research Database demonstrated that a dose of about 5 mg of prednisone equivalent led to a measurable increase in fracture risk after only 3 months of glucocorticoid treatment. More recently [28], a report suggested that even 1 month of systemic glucocorticoid therapy was associated with increased fracture risk. If this is confirmed by other studies, it strengthens the need for clinicians to consider fracture risk at the time of prescribing prednisone and similar drugs. Indeed, it has been shown that on the first day of prednisone therapy, bone formation markers will be suppressed [29]. Even injections into joints can affect bone turnover markers for 2 weeks or more [30].
Pathophysiology
Mechanism of Action of Glucocorticoids
Glucocorticoids are a class of corticosteroids, which are a class of steroid hormones. Glucocorticoids are corticosteroids that bind to the cytosolic glucocorticoid receptor, which is present in almost every vertebrate animal cell [31]. The name “glucocorticoid” is a portmanteau (glucose + cortex + steroid) and is composed of its role in regulation of glucose metabolism, synthesis in the adrenal cortex, and its steroidal structure. A less common synonym is glucocorticosteroid. Glucocorticoids are distinguished from mineralocorticoids and sex steroids by their specific receptors, target cells, and effects. In technical terms, “corticosteroid” refers to both glucocorticoids and mineralocorticoids (as both are mimics of hormones produced by the adrenal cortex). Glucocorticoids are chiefly produced in the zona fasciculata of the adrenal cortex, whereas mineralocorticoids are synthesized in the zona glomerulosa. Glucocorticoids are part of the feedback mechanism in the immune system, which reduces certain aspects of immune function, such as inflammation. They are therefore used in medicine to treat diseases caused by an overactive immune system, such as allergies, asthma, autoimmune diseases, and sepsis.
Upon activation of the glucocorticoid receptor, the complex translocates to the nucleus, where it binds glucocorticoid response elements of an array of genes (Fig. 31.1). This in turn modulates transcription of target genes, resulting in expression of enzymes crucial for normal cellular functions such as gluconeogenesis and anti-inflammation. As such, hydrocortisone is one of the most important hormones in the human body. Noncanonical pathways play less important roles, but these nongenomic effects have also been shown to contribute to various mechanisms of downregulating inflammatory markers [32].

Molecular effects of glucocorticoids excess on bones: GC’s effects occur by four mechanisms: the classic genomic (the most important), which involves the cytosolic GC receptors (cGCR) and is divided into two processes, transrepression, and transactivation; nongenomic secondary, which is also initiated with cGCR; nongenomic executed by membrane receptors (mCGR); and nonspecific nongenomic, resulting from interactions with cell membranes (including the organelles). Glucocorticoids enter cells and become activated by 11β-HSD1 or occasionally deactivated by 11β-HSD2. The activated glucocorticoids bind to a cytoplasmic protein complex containing heat shock proteins and the glucocorticoids receptor. Complexes with Hsp70 and Hsp40 render the glucocorticoids receptor as a low-affinity receptor; however, complexes with Hsp90 give rise to a high ligand affinity of the glucocorticoids receptor. Upon ligand binding, the chaperone FKBP51 is exchanged for FKBP52, thereby allowing shuttling of the complex into the nucleus and into contact with the chromatin
At physiologic concentrations, endogenous glucocorticoids may have a role in promoting osteogenesis [33], while excess glucocorticoids increase osteoclastogenesis and suppress osteblastogenesis in cell culture, murine, and human models [7, 34]. Local metabolism of glucocorticoids in bone cells is controlled by a pair of complementary enzymes, 11β-hydroxysteroid dehydrogenase types 1 and 2 (11β-HSD1, 11β-HSD2), which respectively activate or deactivate glucocorticoid action by metabolizing the interconversion of biologically active or inert forms (Fig. 31.2) [35, 36]. Additional pathways of glucocorticoid action are thought to be multiple, including inducing proapoptotic molecules in osteoblasts and osteocytes and through antagonizing the osteoblastogenic Wnt pathway [34, 37, 38]. There continues to be active work into uncovering pathways of glucocorticoid mechanism at the cellular level [39], including suggestions of enhancing osteoblast activity through heat shock protein 90 [40].

As glucocorticoids enter cells it become activated by 11β-HSD1, thus inactive forms of corticosteroids (cortisone and prednisone) are converted into active forms (cortisol and prednisolone). 11β-HSD1 is also expressed in osteoblasts, which increase with age, which in turn favors the greater concentration of glucocorticoids in these cells. Increased expression of 11β-HSD1 enzyme is considered a risk factor for GC-induced osteoporosis. In its active form, the glucocorticoid binds with a receiver (GRα or GRβ), a member of the nuclear receptor super family, and migrates from the cytoplasm to the nucleus, where it can bind to glucocorticoid response elements (GREs), and to other transcription factors. The other side of the HSD system equation is composed of GC-inactivating enzyme and 11β-HSD2. The sensitivity of different types of glucocorticoids to this enzyme varies, and dexamethasone, by having a fluorine atom at the 9α position of the B ring, with site of HSD2 blocked, is the steroid which is most resistant to such inactivation, and therefore is what causes osteoporosis the most
Glucocorticoid therapy affects all three bone cell lines—osteoblasts, osteoclasts, and osteocytes (Fig. 31.3). The predominant action is by suppression of osteoblastic activity, resulting in inhibition of bone formation. Direct effects of glucocorticoids on bone formation are mediated largely through upregulation of peroxisome proliferator-activated receptor gamma receptor 2 (PPARγ2) [41] and effects on the Wnt/β-catenin signaling pathway [42, 43]. The former mechanism favors the differentiation of pluripotent precursor cells to adipocytes in preference to osteoblasts, resulting in decreased numbers of osteoblasts. On another front, the expression of dickkopf-related protein and sclerostin and the inhibitors of WNT signaling pathway are upregulated. Increased expression of sclerostin, which binds to the co-receptors for frizzled, Lrp4 and Lrp5, results in inhibition of Wnt signaling pathway leading to reduced differentiation of osteoblast precursors to mature osteoblasts and increased osteoblast and osteocyte apoptosis. The importance of sclerostin in mediating the effects of glucocorticoids on bone formation is emphasized by the demonstration, in mice with sclerostin deficiency, that bone integrity is maintained in the presence of glucocorticoid excess [44]. Furthermore, in a mouse model of glucocorticoid-induced osteoporosis, treatment with an antibody to sclerostin prevented the reduction in bone mass and strength [45].
Role of Underlying Inflammation
In the general population, even mild elevations of C-reactive protein within the normal range increase nontraumatic fracture risk [46]. While some studies revealed that variations within the low levels of inflammatory markers and cytokines predict bone loss, other studies reported that elevated inflammatory markers are prognostic for fractures [47, 48].
Independently, it was reported that rheumatoid arthritis (RA) doubles the risk of hip and vertebral fractures, regardless of the use of glucocorticoids, and disease activity is consistently associated with low BMD [49]. In a prospective study of patients with early RA conducted at a time when biotherapies were not available, high bone loss was observed, mainly in patients with persistent inflammation during follow-up (supported by persistent high CRP) [50]. In ankylosing spondylitis, an inflammatory disease in which glucocorticoids are not used, there is bone loss and an increased risk of vertebral fractures, driven by inflammation [51, 52].
There is a strong biological rationale for these clinical observations. Osteoclastogenesis is under the control of RANK-ligand, which is produced not only by osteocytes in normal bone remodeling but also by lymphocytes and fibroblasts in other situations, such as estrogen deficiency and inflammation [53]. Osteoclastogenesis can be enhanced by a number of cytokines, the main pathway being driven by T-helper 17 cells subpopulation (i.e., interleukin IL-6 and IL-23) [54,55,56,57,58]. Tumor necrosis factor α (TNF-α) transgenic mice are models of osteoporosis with dramatic decrease in bone mass and deterioration of bone microarchitecture. Moreover, an over expression of sclerostin has been observed in these models, with a consequence of inflammation-related decrease in bone formation [59]. Finally, autoimmunity has a role in bone remodeling, as antibodies against citrullinated proteins (ACPAs) can increase osteoclast numbers and activity through citrullinated vimentin located at the surface of precursors and cells (through a TNF-α local effect) [60].
All these clinical observations and biological studies show that inflammation has a deleterious effect on bone remodeling, inducing an increase in resorption and a decrease in formation, before any effect of glucocorticoids themselves.
Effect on the Bone
Direct Effect on the Bone
The predominant effect of glucocorticoids on bone is the impairment in bone formation, with an additional early but transient increase in bone resorption. The initial increase in remodeling rate is accompanied by reduced bone formation at the level of the individual bone multicellular unit (BMU), and this combination of increased bone turnover and a negative remodeling balance results in rapid bone loss [61,62,63,64,65]. Subsequently, the decrease in bone formation, both at tissue and BMU levels, predominates leading to a low turnover state. The evidence that this is a direct effect, independent of the inflammation effect, comes from studies conducted in healthy volunteers where prednisone 5 mg daily is enough to rapidly and significantly decrease serum P1NP and osteocalcin, which are specific markers of bone formation; the changes are reversed after discontinuation of the prednisone [66].
Glucocorticoids also have direct effects on bone resorption, increasing the production of macrophage colony stimulating factor (M-CSF) and RANKL and decreasing production of osteoprotegerin (OPG) by osteoblastic cells and osteocytes, resulting in an increase both in the number and activity of osteoclasts [67, 68]. As a consequence, a prolonged lifespan of osteoclasts is observed (contrasting with the decrease in the lifespan of osteoblasts). This effect diminishes with time, possibly as a result of the reduction in number of osteoblasts and osteocytes. Therefore, it can be said that much of the glucocorticoid-related bone loss is caused by the reduced bone formation, which persists throughout glucocorticoid administration. Finally, there is some evidence from animal models that glucocorticoids affect osteocyte morphology and mineralization [69].
Indirect Effects on Bone
Other mechanism that may contribute to glucocorticoid-induced bone loss is that occur through indirect effects on bone. The first mechanism is that attributed to the effects of glucocorticoids on calcium metabolism. Glucocorticoids cause decrease of gastrointestinal absorption of calcium and induction of renal calcium loss. A secondary hyperparathyroidism state has been suggested as a determinant of bone effects. However, there is no evidence for elevated endogenous levels of parathyroid hormone in these patients, and histological features are not those related to an increased parathyroid hormone secretion [70]. Glucocorticoids also reduce production of sex steroid hormones inducing a state of hypogonadism. In addition, the bone loss has been linked to reduced physical activity and reduced production of growth hormone, insulin-like growth factor 1 (IGF1), and IGF1-binding protein (IGF-BP) [71]. Furthermore, as mentioned earlier, the underlying diseases for which glucocorticoid therapy is administered are often associated with increased inflammation, which contributes to bone loss through increased production of pro-inflammatory, pro-resorptive cytokines. While glucocorticoids suppress inflammation and hence should mitigate the adverse effects of inflammation, disease relapse despite therapy is associated with episodes of increased bone resorption.
Effect on the Muscles
The clinical side effects of steroid medications on bone and muscle have been the subject of numerous reviews and textbook descriptions. Glucocorticoids are known to regulate protein metabolism in skeletal muscle, producing a catabolic effect, which is opposite to that of insulin. In many catabolic diseases, such as sepsis, starvation, and cancer cachexia, endogenous glucocorticoids are elevated contributing to the loss of muscle mass and function.
Similar to bone, skeletal muscle homeostasis is disrupted by glucocorticoids. Glucocorticoids not only decrease muscle anabolism by inhibiting amino acid transport into muscle [72] but also increase muscle catabolism by altering three major pathways: the myostatin signaling pathway, the IGF-1–PI3K–Akt pathway, and the NF-κB pathway [73]. The result is a shift in net skeletal muscle protein balance toward proteolysis. There is also evidence that glucocorticoids inhibit muscle regeneration by interfering with myogenic differentiation [74] and/or immune responses that detect injury and trigger repair [73]. Furthermore, Schakman et al. demonstrated that glucocorticoids, but not IGF-I or TNF-α–NF-κB, play a key role in inducing proteolysis in acute inflammatory states via the autophagy and the ubiquitin–proteasome pathways [75]. Patients at risk include elderly patients, those with poor nutritional intake and those not participating in exercise that could counterbalance the negative metabolic effects of glucocorticoids [76].
In addition, glucocorticoids are associated with glucocorticoid-induced myopathy and critical illness myopathy (CIM). Glucocorticoid-induced myopathy is typically associated with the use of glucocorticoids in high doses, and, in particular, fluorinated glucocorticoid preparations. Patients develop insidious pain-free proximal muscle weakness that primarily affects the lower extremities several weeks or years into glucocorticoid therapy. Throughout its course, serum muscle enzyme levels generally remain in the normal range or mildly elevated [15, 77]. On histopathology, glucocorticoid-induced myopathy is characterized by preferential loss and atrophy of type II muscle fibers [78]. Muscle weakness usually begins to ameliorate within 3–4 weeks after glucocorticoid cessation but may last for up to 6 weeks.
On the other hand, critical illness myopathy (CIM) has been linked to treatment with high doses of glucocorticoids and neuromuscular blocking agents. Other risk factors such as renal failure, hyperglycemia, and increased severity of the underlying disease are also important [79]. It has been estimated that at least a third of the intensive care unit patients treated for status asthmaticus develop critical illness myopathy (CIM) [80]. Patients typically present with acute-onset, diffuse, flaccid muscle weakness that generally affects all limb muscles, neck flexors, and often the diaphragm and facial muscles [79]. Serum creatinine kinase levels generally increase 10- to 100-fold higher than normal, peaking at day 3–4 and normalizing after 10 days [81]. The diagnosis is made on the basis of distinctive electrophysiological activity and muscle/nerve biopsies analyses. Electrodiagnostic testing reveals low amplitude, short duration and polyphasic motor unit potentials, low amplitude compound action potentials, and fibrillation and sharp wave potentials [82]. On histopathology, critical illness myopathy (CIM) is characterized by varying degrees of muscle fiber necrosis and regeneration, no lymphocytic inflammation, preferential atrophy of type II fibers, and loss of thick myosin filaments [83].
Clinical Correlations of Glucocorticoid-Induced Osteoporosis
Owing to a much lower mechanical bone strength than would appear to be the case from observing BMD and a significant increase in fracture risk associated with the use of glucocorticoids, patients taking steroids should be assessed with care, and consideration should be given to several factors that make glucocorticoid-induced osteoporosis (GIO) a unique form of bone loss (Fig. 31.4). These factors can be summarized as follows:

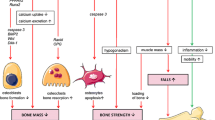
Pathophysiology of glucocorticoid-induced osteoporosis: Excessive amounts of systemic glucocorticoids lead to clinically significant adverse effects on the musculoskeletal system by inducing a state of inappropriate bone remodeling through direct and indirect mechanisms and muscle atrophy that contributes to osteoporosis and fractures. Early bone loss is driven by changes in hormone levels mainly estrogen and parathyroid hormone which stimulate receptor activator of nuclear factor-κB ligand (RANKL)–induced osteoclastogenesis. Osteocyte and osteoblast apoptosis prevents effective mechanosensing and new bone formation

Risk Factors for fractures in patients receiving glucocorticoids: Patients taking steroids should be assessed with care, and consideration should be given to several factors that make glucocorticoid-induced osteoporosis (GIO) a unique form of bone loss
GIO vs postmenopausal osteoporosis:
The primary difference between GIO and postmenopausal osteoporosis is the suppression of osteoblastic activity, leading to decreased bone formation. In GIO, following an early phase consists of rapid loss of bone mineral density due mostly to excessive bone resorption and impaired bone formation usually manifests and is more progressive with long-term therapy. Trabecular bone loss predominates, with most marked changes not only in the lumbar spine but also in the femoral neck and other sites. The effect on bone is dose dependent, with relative risk significantly increased at daily doses higher than 2.5 mg prednisolone equivalent [25]. It is also dependent on duration, with fracture risk returning to baseline months after cessation of therapy. However, changes to bone can occur sooner than many physicians realize. One study found that a 40 mg or higher prednisone equivalent a day can result in substantial BMD loss at the lumbar spine in just 2 months [84]. Meta-analyses indicate that exposure to steroid use is associated with a relative risk of fracture of about 1.6–1.98 and is independent of gender [26, 85].
Fractures most often occur at sites enriched with trabecular (cancellous) bone, such as the lumbar spine and femoral neck. Vertebral fractures may be asymptomatic and detected only by radiographic imaging. While much of the knowledge of GIO is extrapolated from experience with postmenopausal osteoporosis, it is important to note that fractures tend to occur at higher bone mineral density in glucocorticoid- induced osteoporosis [86].
Phases of bone loss associated with glucocorticoids:
GC-induced bone loss has a rapid with, as mentioned earlier, early phase of excessive bone resorption and, a slower, later phase marked by inadequate bone formation [42, 87, 88].Within the first few days of treatment, glucocorticoids transiently increase osteoclast numbers due to an antiapoptotic effect on mature osteoclasts, which probably results in early loss of bone [87]. Bone resorption may also be stimulated by higher doses of glucocorticoids [89]. In the second phase, chronic glucocorticoids excess suppresses remodeling by downregulating osteoblastogenesis and osteoclastogenesis and is characterized by depressed bone formation and turnover [88]. Studies have demonstrated that bone resorption decreases after 4 weeks of prednisolone administration to normal or below normal levels [90]. The decrease in bone formation and turnover in GIO is in contrast to the increase in bone resorption, and turnover that characterizes osteoporosis caused by a loss of sex steroids (i.e., in postmenopausal women).
The risk of fracture rapidly decreases when glucocorticoids are discontinued. A prospective study showed clinically significant improvement in bone mineral density at the lumbar spine within 6 months after discontinuation of glucocorticoids [91]. A large retrospective study showed an increased risk of a major osteoporotic fracture among patients with recent prolonged glucocorticoid use but not among those with intermittent or past use of these agents [92].
Differential sensitivity to glucocorticoid:
There is great variability of glucocorticoid linked side effects among individuals , including bone loss, for largely unknown reasons. Attention has been paid to the 11 β-hydroxysteroid dehydrogenase (11β-HSD) system, which is a pre-receptor modulator of glucocorticoid action. This system catalyzes the interconversion of active/inactive cortisone, and the 11β-HSD enzyme amplifies glucocorticoid signaling in osteoblasts. Interestingly, 11β-HSD, widely expressed in glucocorticoid target tissues including bone, can be modulated and amplified by pro-inflammatory cytokines [93, 94], age, and glucocorticoid administration itself, suggesting that the mechanism could be a key regulator of the effects of glucocorticoids on bone. Individual glucocorticoid sensitivity can also be regulated by polymorphisms in the glucocorticoid receptor gene [95].
Time effect:
The increase in GIO and its consequent fracture risk is immediate, as early as 3 months after the initiation of therapy and reverses sharply after discontinuation of glucocorticoids. This cannot be explained by BMD changes, but can be related to the added effects of glucocorticoids on bone remodeling previously uncoupled by the inflammation itself, and the dramatic effect on bone strength through induced apoptosis of osteocytes. Data also suggest a rapid increase in rate of falls after start of oral glucocorticoids [25]. Thus, primary prevention, after careful assessment of the fracture risk, is recommended in high-risk patients.
Dose effect:
In epidemiological studies, the increased risk of fractures is observed even at low doses of prednisone, that is, 2.5–5 mg per day. The appropriate care of patients receiving such low doses has been advised in recent guidelines, which will be discussed later in this chapter. There is a dose-dependent increase in fracture incidence. Interestingly, the fracture risk is related to the current daily dose, more than to the cumulative dose [96]; this may be attributed to the difficulty of an accurate calculation of this cumulative dose.
Prior versus current glucocorticoids use:
Ever use of glucocorticoids is associated with an increased risk of hip fracture, and this justifies the assessment of osteoporosis and fracture risk in all patients. However, the risk is mainly associated with recent and prolonged glucocorticoid use, more than to remote or short courses [97].
Role of underlying disease:
Persistent inflammation is associated with bone loss as shown in longitudinal studies in patients with active RA or ankylosing spondylitis (SpA). In contrast, prospective open studies show that complete control of inflammation (in parallel with clinical improvement and thus increased mobility) is accompanied by the absence of bone loss [98]. This is not only expected in spondyloarthritis in the absence of glucocorticoids but is also observed in RA of the hand, spine, and hip and in patients receiving low doses of glucocorticoids [98,99,100]. In the BeSt study, conducted in patients with recent-onset active RA, bone loss was limited in all treated groups, including in the group initially treated with high-dose prednisone [101]. Thus, the concept that a high level of inflammation is more deleterious for bone than a low dose of glucocorticoids, controlling this inflammation is relevant as far as surrogate markers (BMD, biological parameters) are concerned. However, there is no evidence for a reduction in fracture risk with such a strategy [102], and further epidemiological studies are important in this aspect.
Role of patient characteristics:
Age , female gender, low BMI, history of falls and previous fractures, duration of menopause, and smoking are associated with fracture risk in patients with glucocorticoids, similarly to how they are in primary osteoporosis. It has been reported that the prevalence of non-vertebral fractures is a strong determinant of the risk of having vertebral fractures in patients with RA [103], implying that the individual’s skeleton is already of inadequate strength to withstand the trauma of daily living. Beyond glucocorticoids use, these risk factors must be assessed in all patients, and all causes of secondary osteoporosis are added risk factors of fractures in patients on glucocorticoids therapy [104].
Glucocorticoids Pharmacologic Preparation
Most common forms of administered glucocorticoids are oral preparations. Intravenous, inhaled, injected, and transdermal preparations are also frequently encountered. However, side effects, including to bone, are not limited to oral or intravenous administrations. Injected glucocorticoids, particularly when repeated, and topical therapy may both lead to systemic effects.
Systemic (Tablets/Injections):
Glucocorticoids are transported in the bloodstream bound to corticosteroid-binding globulin and albumin, in equilibrium with the biologically active free form that binds to the glucocorticoid receptor. Individual sensitivity to glucocorticoid signaling is also affected by genetic variability of the glucocorticoid receptor, with alternative splicing and polymorphisms identified [105, 106]. Furthermore, pharmaceutical preparations have differences in absorption, transport, and target affinity, and thus a varied range of potencies and duration of effects. Glucocorticoid potency ranges from lower potency such as with cortisone and prednisone, to higher potency, such as with dexamethasone and betamethasone. In general, when oral cortisol (hydrocortisone) is used as a baseline, prednisone and prednisolone have about four times the potency, while methylprednisolone and triamcinolone are about five times the potency, and dexamethasone and betamethasone are about 25 times the potency.
Inhaled glucocorticoids:
For almost two decades, inhaled glucocorticoids have been used widely in the management of chronic lung disease, mainly asthma [107]. However, the effect of inhaled glucocorticoids on bone and whether their use leads to GIO is somewhat controversial. In a meta-analysis by Richy and colleagues, inhaled glucocorticoids were associated with a 1.2–1.8-times increased risk of vertebral fracture and a 1.6-times increased risk of hip fracture [108]. This meta-analysis also demonstrated that inhaled glucocorticoids were associated with lower bone density at the spine and hip and lower levels of bone formation markers (osteocalcin and procollagen type 1 C-terminal propeptide). Vestergaard and colleagues found an increased risk of any fracture (adjusted for comorbid diseases, but not respiratory severity) associated with inhaled glucocorticoids only for daily dosages above 7.5 mg of prednisolone equivalents (equivalent to 1875 μg of budesonide/day) [109].
Fujita and colleagues examined lumbar BMD (and biochemical markers) in inhaled glucocorticoids users with no oral glucocorticoids for at least 1 year and found significant lower BMD and serum osteocalcin among the inhaled glucocorticoids users versus controls in the postmenopausal group only [110]. Wong and colleagues found a negative relationship between total cumulative dose of inhaled glucocorticoids and BMD in asthma patients [111].
Risk Stratification, Screening, and Assessment
Management of patients who start/remain on glucocorticoids is based on risk assessment and prevention of osteoporosis. The rate of screening for bone disease traditionally varies according to the medical specialty of the provider prescribing the chronic glucocorticoid therapy [112], though it can be said that awareness has increased in recent years [113]. There are several recommendations for identifying high risk of fracture in patients on glucocorticoids (Table 31.1). Screening for fracture risk should be performed soon after the initiation of glucocorticoid treatment. Currently, tools to estimate the risk of fracture among patients who are younger than 40 years of age are lacking. The risk of fracture increases, and the time to fracture decreases considerably with increasing age among patients who receive glucocorticoids [114]. The risk of fracture among patients of ages ≥40 can be estimated with the use of bone mineral density (BMD) testing and the fracture risk assessment tool (FRAX). Table 31.1 summarizes the main clinical risk factors for glucocorticoid-induced osteoporosis (GIO).
The 2017 ACR guidelines [115] allow clinicians to evaluate fracture risk in adult glucocorticoid users of all ages. Under these guidelines, adults <40 years of age are classified as low risk unless they have prevalent fragility fracture, or are high-dose steroid users with extremely low BMD or rapid BMD loss. This risk stratification algorithm flags high-risk young adults for more aggressive care, while exempting most young people from monitoring and treatment of little benefit to them. However, it does not account for clinical GIO risk factors such as malnutrition, low body weight, thyroid and parathyroid disease, family history of hip fracture, and alcohol and tobacco use. These are common comorbidities among young patients with inflammatory conditions like inflammatory bowel disease and rheumatoid arthritis, and substantially impact individual fracture risk. Adults ≥40 years of age are risk stratified using FRAX scores, an established method that incorporates clinical GIO risk factors. However, this method has specific limitations when applied to glucocorticoid users. FRAX equations do not adequately adjust for high-dose or prolonged glucocorticoid exposure, cannot assess the BMD-independent effects of glucocorticoids on bone [26], and rely on hip BMD when glucocorticoids cause disproportionate loss of trabecular BMD, best measured at the spine.
The guideline authors address these issues by recommending annual fracture risk assessment for all patients that evaluates glucocorticoid dose, duration, and exposure pattern, screens for fall risk, frailty, and the clinical risk factors as described and assesses body mass index, muscle strength, and signs of occult fracture. The authors recommend that patients with concerning findings on this assessment undergo serial BMD testing regardless of their original fracture risk classification. The guidelines also endorse the Fracture Risk Calculator, an alternative to FRAX that incorporates spine BMD, for patients with discordant hip and spine BMD measurements (available at: https://riskcalculator.fore.org/).
Glucocorticoid-Induced Changes in BMD and Bone Microarchitecture
In postmenopausal osteoporosis, the risk of osteoporotic fractures has been shown to be doubled for each standard deviation decrease in BMD [116], but this may underestimate the fracture risk for patients treated with glucocorticoids. In glucocorticoid-treated asthmatic patients with vertebral fractures, Luengo et al. [117] found that BMD was higher compared with a group with postmenopausal osteoporosis and fractures. Similarly, Peel et al. [118] found that steroid-treated patients with RA had a 6.2-fold increased risk of vertebral fractures with only a 0.8–1.5 S.D. decrease in lumbar spine BMD. Therefore, in addition to BMD, the decision to start treatment may also depend on assessment of clinical risk factors.
Increased rates of bone loss in the hip, spine, and radius are well documented in individuals treated with glucocorticoids. Earlier studies revealed that the BMD loss is an immediate consequence of the introduction of glucocorticoids therapy and affects the trabecular bone (i.e., spine) more than it does the cortical bone (i.e., femur). According to a meta-analysis of 56 cross-sectional studies and 10 longitudinal studies, bone loss assessed by dual-energy X-ray absorptiometry can be 5–15% during the first year of treatment [24]. The main determinant of BMD at any time is the cumulative dose. The increased rate of bone loss persists in chronic glucocorticoids users, but at slower rate.
Data obtained from assessment of bone microarchitecture using high-resolution peripheral computed tomography (HRpQCT) are sparse. In a cross-sectional study of 30 postmenopausal women who had received oral glucocorticoids for longer than 3 months, despite similar areal BMD values to 60 control subjects, significantly lower total, cortical, and trabecular volumetric BMD, thinner cortices, increased trabecular separation, and reduced trabecular number were reported in the radius and tibia; whole bone stiffness, assessed using finite element analysis, was also significantly reduced in comparison with the controls [119]. Although the patients and controls were generally well matched, however, bisphosphonate use was significantly more common in the former (100% vs. 8.6%), so definite attribution of the observed differences to glucocorticoid therapy cannot be made.
Trabecular bone score (TBS) provides an indirect index of trabecular bone architecture that can be obtained from DXA images of the lumbar spine and has predictive value for fracture independent of BMD [120]. In 64 postmenopausal women who had taken prednisolone in a dose of ≥5 mg daily for >3 months, TBS was significantly lower than in a group of non-glucocorticoid-treated controls, although lumbar spine BMD T-scores were not significantly different [121]. Similar findings have been reported in 416 individuals on long-term glucocorticoids (≥5 mg daily for 3 months), the decrease in TBS being most marked in men and in individuals with fracture [122]. These findings indicate that glucocorticoids have adverse effects on spine bone microarchitecture that are independent of BMD and which may contribute to increased fracture risk.
Monitoring BMD Changes/Response to Therapy
It is recommended for all adults >40 years of age, and for adults <40 years of age with prevalent osteoporotic fracture or other osteoporosis risk factors, to arrange for BMD testing at the start of glucocorticoid treatment. For adults >40 years of age, serial BMD monitoring is recommended every 1–3 years for those not on anti-osteoporotic treatment, and every 2–3 years during treatment for those taking “very high dose glucocorticoids” (>30 mg/day prednisone equivalent with >5 g annual cumulative exposure), poor medication response, adherence or absorption, or other risk factors for bone loss. BMD assessment should be carried out every 2–3 years after completing treatment. For adults <40 years of age, BMD monitoring every 2–3 years regardless of treatment is recommended for those with a moderate to high fracture risk, very high dose glucocorticoid exposure, or other risk factors [123].
Fracture Risk Assessment in Individuals Treated with Steroids
The WHO fracture risk assessment tool (FRAX) (http://www.shef.ac.uk/FRAX) algorithm has been developed to estimate the 10-year risk of hip and other major fractures (clinical spine, humerus, or wrist fracture) based on clinical risk factors, with or without BMD. The risk factors included in FRAX are: age, sex, body mass index (BMI), personal history of fracture, parental history of hip fracture, current smoking, alcohol intake, glucocorticoid use, rheumatoid arthritis, and other causes of secondary osteoporosis and femoral neck (not spine) BMD. These clinical risk factors are largely independent of BMD and can thus improve the fracture risk assessment. FRAX cannot be used in premenopausal women, men aged <40 years and in subjects previously treated with anti-osteoporotic drugs.
One of the limitations of FRAX is that use of oral GCs is recorded as a dichotomous risk factor and does not take into account the dose of glucocorticoid and the duration of use. Moreover, FRAX does not take into account the difference in risk between prior and current use [97]. FRAX assumes an average dose of prednisolone (2.5–7.5 mg/day or its equivalent) and may underestimate fracture risk in patients taking higher doses and may overestimate risk in those taking lower doses. Moreover, the predictive value of FRAX has been mainly validated for non-vertebral fractures although the principal risk in glucocorticoids users is for vertebral fractures. Adjustment of FRAX has been proposed for postmenopausal women and men aged ≥50 years with lower or higher doses than 2.5–7.5 mg/day: a factor of 0.8 for low-dose exposure and 1.15 for high-dose exposure for major osteoporotic fractures and 0.65 and 1.20 for hip fracture probability [124]. For very high doses of glucocorticoids, greater upward adjustment of fracture probability may be required. Moderate risk was defined as a 10-yearr major osteoporosis fracture risk of 10–19% and a hip fracture risk of 1.1–2.9%, with both doses adjusted. Pharmacologic therapy was suggested for these two groups. Low-risk patients were defined to have a major osteoporosis fracture risk of <10% and a hip fracture risk of ≤1% in 10 years. These patients can be treated conservatively with adequate dietary calcium and vitamin D, with supplements of the latter if necessary.
Using data from the UK General Research Practice Database, Kanis et al. [125] have provided adjustments that can be incorporated into the FRAX calculations to adjust for different doses of glucocorticoids (Table 31.2). For daily doses of over 7.5 mg daily of prednisolone or equivalent, greater upward adjustment of fracture probability may be required. It should be noted that the duration of glucocorticoid therapy and cumulative dose are not accommodated within the FRAX algorithm. In addition, the use of total hip BMD in FRAX may result in underestimation of fracture risk in patients with differentially low spine BMD, although a correction for this discordance has been proposed [40, 41, 126, 127]. A final caveat is that the response to treatment in glucocorticoid-treated individuals selected on the basis of FRAX-derived fracture probability has not been documented.
FRAX assessment has already been included in some guidelines at different steps of the treatment decision. The American College of Rheumatology (ACR) has created guidelines addressing management of GIO, last updated in 2017 [115]. Adult patients are risk stratified by age and their fracture risk as well as the steroid therapy dose. In concordance, the International Osteoporosis Foundation (IOF)–European Calcified Tissue Society [129] recommendations advised that a treatment decision for postmenopausal women and for men aged ≥50 years exposed to oral glucocorticoids for ≥3 months should be based on fracture risk assessment with FRAX adjusted for glucocorticoid use (with or without BMD testing). Treatment can be considered directly (without FRAX assessment) if patients are at high risk defined by one of the following criteria: prevalent fracture, age ≥70 years, and exposure to a glucocorticoid dose ≥7.5 mg per day or low BMD (T ≤ −2.5).
Glucocorticoid-Induced Osteoporosis in Children and Adolescents
Children represent a different approach as bone development is still critical. In children, weight gain, growth retardation, and Cushingoid features are the most frequent adverse reaction to chronic oral glucocorticoid use, but a recent meta-analysis that included a total of 6817 children noted a 21% incidence of decreased bone mineral density [130]. Growth retardation, whether directly or through suppression of the hypothalamic–pituitary axis or of sex steroid hormone production, puts children at considerable risk of osteoporosis in adulthood, as peak bone mass is achieved in late adolescence and early adulthood. While there are few prospective randomized controlled trials, there is general consensus for ensuring adequate calcium and vitamin D intake and for avoidance of further pharmacologic means such as bisphosphonates. Bisphosphonate use should only be considered, very carefully, for children who have an osteoporotic fracture who are still continuing long-term glucocorticoid use.
For inhaled steroid use in children, commonly prescribed for asthma, most studies have not reported significant effects of inhaled steroid on bone markers [131,132,133], although a few have identified decreased BMD with high dose inhaled corticosteroids [134]. On the other hand, a point of argument has been raised, as better control of asthma leads to greater physical activity, which is beneficial to bone development. It is now well-accepted that no bone-specific monitoring or pharmacologic treatment is needed in intermittent or routine inhaled corticosteroid use for asthma, though “periodic” evaluations of bone density may be advised in long-term, high-dose therapies [135].
Management of Glucocorticoid-Induced Osteoporosis
In a study of a large managed care population in the United States, Saag and colleagues [136] found low rates of preventative interventions in individuals on long-term glucocorticoid therapy. Postmenopausal women were the most likely to receive recommended interventions, yet only approximately 50% were treated with anti-osteoporotic medication. In total, 19% of postmenopausal women underwent bone mass measurements. This number dropped to <6% in women under 50 years of age as well as in men. The study also found that rheumatologists were three to four times more likely to initiate the above interventions than internists or family practitioners. Interventions carried out aiming at improving physician management of GIO have largely been unsuccessful. When physicians were randomized to receive a web-based GIO intervention (including a personalized performance-audit and feedback) versus control intervention, there was no significant increase in BMD testing (19% versus 21%) or prescription of antiosteoporotic medications (32% versus 29%) in the year following the intervention [137].
There is a mismatch between BMD data and fracture data in patients receiving glucocorticoids because of the disparity related to the alteration of bone quality. At similar levels of BMD, postmenopausal women taking glucocorticoids have considerably higher risk of fracture than controls not using glucocorticoids [96]. There is a debate on the appropriate T-score threshold to be considered a risk and as an indication for treatment in patients with glucocorticoids: the same diagnostic criterion as in postmenopausal women has been suggested (T ≤ −2.5), but a higher threshold (i.e., T ≤ −1.5) has been proposed for intervention, because bone loss can be 10% or more in some individuals over the first year of glucocorticoids use [138].
There is no means to provide an evidence-based threshold for treatment decisions. A practical approach is to recommend a BMD measurement in glucocorticoids users (optimally at the initiation of treatment) and to consider that patients with T ≤ −2.5 as those who should receive the highest priority for treatment [139]. However, beyond the BMD, a more comprehensive approach of the risk and clinical judgment is recommended. This will be discussed in further details later in this chapter.
Management of steroid-induced osteoporosis can be stratified into general measures and pharmacological measures.
General Measures
At the initiation of glucocorticoid treatment, clinical assessment should be carried out to assess for: measurement of the patient’s height, as height loss in the follow-up could be related to asymptomatic vertebral fractures. Biological tests are performed to screen for other causes of bone diseases. There is no indication for assessment of biochemical markers of bone remodeling either at baseline or during follow-up, as bone turnover is not reliable for interpretation on individual basis among and is consistently low in glucocorticoid users [104].
A number of life-style measures may mitigate the harmful skeletal effects of glucocorticoids, although the evidence base for this approach is weak and requires extrapolation from studies in non-glucocorticoid-treated individuals. The risk of falling should be assessed in particular in elderly patients, patients with painful joints of the lower limbs, and patients with massive doses of glucocorticoids. Fall risk should be assessed at baseline and preventive measures instituted wherever appropriate. Exercise, tailored to the individual patient, and good nutrition with adequate dietary calcium intake should be advocated with avoidance of smoking and alcohol abuse. Maintenance of an adequate vitamin D status should also be advised.
As the daily dose of glucocorticoids is a determinant of fracture risk, attention should be paid to keep the dose of glucocorticoids to a minimum. This must be constantly reviewed by considering both the reduction of the dose to the minimally active, considering alternative administration such as intra-articular injections., or the use of steroid-sparing drugs such as methotrexate or azathioprine or alternative routes of administration (e.g., inhaled or topical) where appropriate. Topical therapy (such as inhaled glucocorticoids or glucocorticoid enemas for asthma or bowel disease, respectively) is preferred over enteral or parenteral glucocorticoids whenever possible. Nonsteroidal therapies should be used when possible to maintain remission, once achieved. However, it is also important to maintain suppression of the underlying disease, since this will prevent the adverse skeletal effects of inflammation and other effects of increased disease activity.
Nutrition/Calcium and Vitamin D Supplementation
Attention to nutrition must be paid to prevent protein and calcium intake deficiencies. Calcium and vitamin D have been used for decades in GIO, although there are controversies about their effect on BMD. In 66 patients with RA receiving prednisone, 1000 mg/day of calcium carbonate and 500 IU/day of vitamin D3 induced a positive change of 0.63% per year at the lumbar spine, versus a decrease of 1.31% per year in the placebo group; there was no effect at the femoral neck [140]. No benefit was observed in another study with a 3-year follow-up [141].
However, it is reasonable to consider that any deficiency in calcium and vitamin D could be deleterious in patients beginning or receiving glucocorticoids. For calcium, the recommendation is to have an intake of 1000–1500 mg/day, and supplementation should be prescribed only to patients whose dietary intake does not provide this adequate quantity. Glucocorticoid-treated patients may seldom be outdoors and thus exposed more than the general population to vitamin D deficiency. Vitamin D Supplementation is considered adequate in the range of 800–2000 IU per day. There is no evidence of an advantage using calcitriol or alfacalcidol, as there is a large variability of outcomes with these vitamin D metabolites over plain vitamin D [104].
Pharmacologic
Anti-resorptives and teriparatide have been assessed in prevention and treatment of GIO (Fig. 31.5). There are a number of issues regarding their efficacy. In contrast to BMD, which was considered as the main end point, fracture incidence has not been a primary end point of any study. Furthermore, the duration of the studies tends to be short (1 year on average), and the number of men and premenopausal women in these studies is low. Thus, the efficacy on fracture(s) prevention in patients treated with glucocorticoids is mainly based on bridging data between the short-term change in BMD, and the long-term change in BMD and reduction of fracture risk in postmenopausal patients diagnosed with osteoporosis.

Management of glucocorticoid-induced osteoporosis: counteracting the negative impact of glucocorticoids on the bone: Mechanism of action of the antiresorptive (bisphosphonates and denosumab) or anabolic agents (teriparatide, abaloparatide, and romosozumab) on bone
In addition, there is inevitable heterogeneity in glucocorticoid-treated trial populations, with respect to age, underlying disease, comorbidities and co-medications, dose and duration of glucocorticoid therapy, and the timing of bone protective therapy. Furthermore, the duration of most treatment studies has been relatively short and this, combined with smaller trial populations, reduces the strength of the safety database [142].
Antiresorptive Agents
Bisphosphonates
Bisphosphonates are the most popular anti-osteoporotic medication. Alendronate (oral 5 or 10 mg once daily, or 70 mg once weekly), risedronate (oral 5 mg daily or 35 mg one weekly), and zoledronate (intravenous infusion 5 mg once yearly) are all approved for this indication. All have been shown to have beneficial effects on lumbar spine and hip BMD in people treated with glucocorticoids [143,144,145,146,147,148,149,150], and for alendronate and risedronate, there is also evidence from post hoc analyses for a reduction in the rate of vertebral fractures [148, 149].
Alendronate was assessed in a placebo-controlled study in 477 men and women over 48 weeks. There was a 2.1% and 2.9% increase at the lumbar spine in the 5 and 10 mg alendronate groups, respectively, and a 0.4% decrease in the placebo group. At the femoral neck the changes were +1, +1.2, and −1.2%, respectively. Interestingly the decrease of BMD in the placebo group (receiving calcium and vitamin D) was driven by the duration of glucocorticoids: −2.9, −1.4, +0.8 in patients receiving GCs for less than 4 months, 4–12 months, and more than 12 months, respectively [151]. In a follow-up study in a second year , performed in 208 out of the 477 patients, there were fewer patients with new vertebral fractures in the treated group (0.7%) than in the placebo group (6.8%) [152].
Two 1-year studies were performed with risedronate, one for prevention in patients beginning glucocorticoids and one for treatment of GIO in patients chronically treated with glucocorticoids. Data from pooling these two studies suggest a reduction of fractures in the first year of therapy: 16% of placebo patients and 5% of those on risedronate 5 mg/day [153,154,155].
In a comparative double blind randomized study, zoledronic acid (1 injection) induced a higher BMD increase than risedronate (daily) in treatment (+4.06 vs +2.71%) and prevention (+2.6 vs 0.6%) subgroups over 1 year at the lumbar spine [156].
The number of non-vertebral and hip fractures has been insufficient in individual trials to assess an impact of bisphosphonates. However, data from cohort studies provide some evidence for efficacy at these sites. In an observational cohort study of women aged >65 years taking alendronate or risedronate, Thomas et al. [157] studied the baseline incidence of clinical fractures in the first 3 months after starting glucocorticoid therapy and the fracture incidence in the following 12 months. Compared with the baseline incidence, both clinical vertebral and non-vertebral fracture incidence were significantly lower. Treatment within the first 90 days of glucocorticoid use was associated with a significant reduction in clinical fractures (including vertebral) of 48% at 1 year and 32% at 3 years when compared with nonuse. Finally, in three matched cohorts derived from healthcare administrative data from Ontario, Canada, Amiche et al. [158] reported that in individuals initiating long-term glucocorticoids, therapy within the first 6 months with alendronate or risedronate was associated with a decrease in incident hip fracture (alendronate 0.49 (0.34–0.69), risedronate 0.58 (0.36–0.90). The results confirmed a reduction in vertebral fracture risk with etidronate, alendronate, and risedronate, but no decrease in risk of forearm or humerus fractures for any bisphosphonate. The analysis was limited to oral bisphosphonates, and zoledronic acid was not considered. Overall, therefore, these studies would be consistent with a beneficial effect of bisphosphonates both on vertebral and non-vertebral fracture, including hip fracture.
The safety profile of bisphosphonates in glucocorticoid-induced osteoporosis has been less well studied than in postmenopausal osteoporosis because of the small number of participants included and shorter duration of the trials. Attention has been paid recently to osteonecrosis of the jaw and atypical femoral fractures such as side effect of long-term administration of antiresorptive drugs in osteoporosis; these events are very rare [159, 160] but glucocorticoids use is one of the identified risk factors. Buccal hygiene procedures should be implemented to prevent any local increased risk of infection. Whether these rare events can change the duration of antiresorptive treatments in long-term glucocorticoids users; this requires further studies. Because of comorbidities and co-medications, people taking glucocorticoids may be more susceptible to side effects [161, 162].
Bisphosphonates should be used cautiously in premenopausal women, as they cross the placenta; appropriate contraception must be used if necessary and preference given to a short bone half-life bisphosphonate [142].
Denosumab
Denosumab inhibits bone resorption by binding to RANKL and interfering with the development of osteoclasts. A non-inferiority trial comparing denosumab with risedronate in patients who were beginning to receive glucocorticoids and in those who had received these agents long-term showed superiority of denosumab with respect to increases in bone mineral density at the spine at 12 months and non-inferiority with respect to rates of fracture [163].
A systematic review and meta-analysis of four randomized-controlled trials evaluating the efficacy and safety of denosumab for the prevention and/or treatment of GIO [164] revealed that treatment with denosumab provided significantly greater increments in lumbar spine and total hip BMD, compared with bisphosphonate therapy or placebo. There was no difference in fracture incidence; however, the total number of reported fractures across trials was low, and the studies were not powered to detect fracture differences between treatment groups. In a previously published meta-analysis excluding GIO studies [165], denosumab likewise increased spine and hip BMD greater than that observed with bisphosphonate therapy. Fracture wise, denosumab was associated with significantly fewer fractures at 24 months, when compared with alendronate (risk ratio 0.51, 95% CI 0.27–0.97).
A third previously published meta-analysis of 11 studies using denosumab to treat postmenopausal women with osteoporosis indicated an increased risk of serious adverse events related to infections [166]. However, the meta-analysis carried out by Yanbeiy and Hansen [164] did not detect a difference in the frequency of infections between denosumab and control groups. Rates of adverse events and serious adverse events were also similar between denosumab and control groups. In summary, denosumab represents a reasonable therapeutic choice for patients with GIO.
Anabolic in the Management of Glucocorticoid-Induced Osteoporosis
The predominant role of reduced bone formation in glucocorticoid-induced osteoporosis provides a rationale for the use of anabolic agents in its treatment. In an active comparator controlled, randomized, double blind study the effects of 18-month treatment with subcutaneous teriparatide, 20 μg/day, or oral alendronate 10 mg/day, were compared in 428 men and women with glucocorticoid-induced osteoporosis [167]. Teriparatide therapy resulted in significantly greater increases in spine and hip BMD, and this was seen in both premenopausal and postmenopausal women and in men [167]. In another multicenter, randomized, double-blind study of teriparatide 20 microg/day versus alendronate 10 mg/day in patients with GIO (277 postmenopausal women, 67 premenopausal women, 83 men) [168], at 18 months, mean percent increases from baseline in lumbar spine BMD were significantly greater in the teriparatide versus alendronate group in postmenopausal women (7.8% versus 3.7%, p < 0.001), premenopausal women (7.0% versus 0.7%, p < 0.001), and men (7.3% versus 3.7%, p = 0.03). Radiographic vertebral fractures occurred in one teriparatide (one postmenopausal) and ten alendronate patients (six postmenopausal, four men), and nonvertebral fractures occurred in 12 teriparatide (nine postmenopausal, two premenopausal, one man) and eight alendronate patients (six postmenopausal, two men). The proportion of patients reporting adverse events in teriparatide versus alendronate groups was consistent across subgroups. The magnitude of increase in BMD was somewhat less than that seen in nonglucocorticoid-treated postmenopausal women in another study [169], possibly as a result of the opposing actions of intermittent PTH and glucocorticoids on osteoblastogenesis , and osteoblast and osteocyte apoptosis [170,171,172]. Although fracture was not a primary end-point of the study, in concordance with the results of the study carried out by Langdahl et al. [168], there were significantly fewer new vertebral fractures occurred in the patients treated with teriparatide when compared with those treated with alendronate (0.6% vs. 6.1%; p = 0.004). The incidence of non-vertebral fractures was similar in the two treatment groups. In a study carried out by Saag and colleagues [173], results after 36 months of treatment demonstrated a continued increase in spine and hip BMD in the teriparatide-treated group, with superiority over alendronate at the 24- and 36-month time points. A lower incidence of new vertebral fractures was also seen in the teriparatide group at 36 months (1.7% vs 7.7%, p = 0.007), with a similar incidence of non-vertebral fractures in the two groups. Interestingly, measurements of TBS in a subpopulation of this study demonstrated a significant increase after 36 months in teriparatide-treated patients, but no significant change in those treated with alendronate [122]. While the long duration of this study is unique among treatment trials for glucocorticoid-induced osteoporosis, it should be noted that the participant discontinuation rate at 36 months was 44%.
However, bone loss and fractures occur rapidly after teriparatide is discontinued; therefore, after discontinuation, an antiresorptive agent such as bisphosphonate or denosumab should be initiated. Initial treatment with an anabolic agent such as teriparatide or abaloparatide, followed by an antiresorptive agent, may be considered for treatment of severe osteoporosis (bone mineral density T score below −2.5 in patients with a history of fracture).With regard to safety, increased pre-dose serum calcium levels were significantly more common in the teriparatide than alendronate-treated group (21% vs. 7%), but no other concerns were identified [174].
Romosozumab
Romosozumab is a monoclonal antibody against sclerostin, a protein secreted by osteocytes that inhibits bone formation through regulation of osteoblasts. Through its mechanism of action through which it blocks the repressive effect of sclerostin on the Wnts pathway, romosozumab acts as a potent bone forming stimulator. Furthermore, since sclerostin promote the formation of osteoclasts through a RANKL-dependent mechanism, the inhibitory effect of romosozumab on bone resorption gives romosozumab the dual function effect on bone, i.e., stimulate bone formation and inhibit bone resorption, which gives promising positive implications for the management of GIO. Earlier studies on glucocorticoid-treated rats revealed that sclerostin-antibody treatment resulted in marked improvements in bone mass across the rats’ skeleton and in osteocyte viability, resulting in decreased bone fragility [175]. However, like abaloparatide, sclerostin antibodies have not been studied in patients on chronic steroids; however, they have shown benefit when administered subcutaneously in postmenopausal women and men. In postmenopausal women specifically, romosozumab increased BMD when compared with placebo and teriparatide and decreased the incidence of fractures when compared with placebo and alendronate [176,177,178,179,180]. There is currently an ongoing study to assess the efficacy of romosozumab versus denosumab for osteoporosis in long-term glucocorticoid users in an open randomized parallel group-controlled trial (https://www.smartpatients.com/trials/NCT04091243#locations).
Third-Line Agents
Treatment either with raloxifene (a selective estrogen-receptor modulator) in postmenopausal women should be reserved for patients in whom other treatments are contraindicated or in whom such treatments have failed. Raloxifene is approved by the Food and Drug Administration for the prevention and treatment of glucocorticoid-induced osteoporosis in postmenopausal women. One trial showed that in postmenopausal women who received glucocorticoids, raloxifene significantly increased absolute bone mineral density (measured in grams per square centimeter) at the lumbar spine by 1.3% from the baseline measure, as compared with calcium and vitamin D supplementation, which decreased the absolute bone mineral density [181].
However, there was no difference in bone mineral density at the femoral neck between the treatment groups, and trials assessing rates of fracture among patients who have received both glucocorticoids and raloxifene are lacking. Although raloxifene has been shown to reduce the risk of estrogen receptor-positive breast cancer [182], potential adverse effects include hot flashes, leg cramps, venous thromboembolism , and fatal stroke [183].
Follow-Up
In cases of a fracture occurring ≥18 months after initiation of oral bisphosphonate therapy or significant bone loss (≥10%/y) after 12-months of therapy, it is recommended to treat with oral bisphosphonate. Alternatively, an intravenous bisphosphonate can be considered, if absorption or adherence problems are suspected. Another class of osteoporosis medication (teriparatide and denosumab) can be prescribed in case of intolerability or lack of efficacy to first line of management [115].
For patients who have completed 5 years of oral bisphosphonate therapy and are expected to continue glucocorticoid treatment, further treatment for osteoporosis is recommended and may include continuing oral bisphosphonate for 7–10 years or switching to a different class of osteoporosis medication. When glucocorticoid therapy is discontinued, fracture risk should be reassessed. If the fracture risk is deemed to be low, it is recommenced to withhold osteoporosis therapy. Otherwise, treatment should be continued [104, 115].
Algorithm for Assessment and Management of GIO
Undertreatment of glucocorticoid-induced osteoporosis has been widely recognized [21, 184]. In a population-based study of adults age ≥20 years, rates of BMD testing and prescription of bone protective medication between 1998 and 2008 were studied in individuals prescribed systemic glucocorticoids for 90 days or longer [185]. Overall, in the first 6 months after initiation of glucocorticoid therapy, only 6% had BMD testing, 22% received therapy, and 25% had both interventions.
Undertreatment was greatest in younger people and men, and primary care physicians had lower prescription rates than rheumatologists. Similar results have been reported using information from a national public health-insurance database in France, with only 8% undergoing BMD testing and prescriptions of calcium ±vitamin D alone or together with bisphosphonates were issued in 18% and 12% respectively [186]. In a large cohort from Canada of men and women aged 66 years or over who were initiating long-term glucocorticoid therapy, Amiche et al. reported that only 13% were prescribed bone protective therapy [158]. The problem of undertreatment is compounded by poor persistence with bisphosphonate therapy, particularly in younger people, those with comorbidities, and those in whom BMD measurements have not been made [187].
According to the 2017 guidelines by the American College of Rheumatology [115], glucocorticoid-treated patients can be classified into the following fracture risk categories:
High Fracture Risk
-
All adults with prior osteoporotic fracture.
-
Men aged ≥50 years and postmenopausal women with hip or spine bone mineral density T-score ≤ −2.5.
-
Adults aged ≥40 years with a glucocorticoid-adjusted Fracture Risk Assessment Tool (FRAX) 10-year risk for major osteoporotic fracture or hip fracture of ≥20% or ≥3%, respectively.
Moderate Fracture Risk
-
Adults aged ≥40 years with a glucocorticoid-adjusted FRAX 10-year risk for major osteoporotic fracture of 10%–19% or risk for hip fracture of >1%–<3%.
-
Adults aged <40 years with a hip or spine bone mineral density Z-score of <−3 or rapid bone loss of ≥10% at the hip or spine over 1 year and glucocorticoid treatment at ≥7.5 mg/d for ≥6 months.
Low Fracture Risk
-
Adults aged ≥40 years with a glucocorticoid-adjusted FRAX 10-year risk for major osteoporotic fracture of <10% and risk for hip fracture of ≤1%.
-
Adults aged <40 years with none of the above risk factors other than glucocorticoid treatment.
Figure 31.6 shows an algorithm to for assessment and management of GIO. Stages of assessment and management can be split into three sections:

An algorithm for assessment and management of glucocorticoid-induced osteoporosis
Initial fracture risk assessment:
A clinical fracture risk assessment includes obtaining a history with full details of glucocorticoid use (dose, duration, and pattern of use), an evaluation for falls, fractures, frailty, and other osteoporosis risk factors (malnutrition, significant weight loss or low body weight, hypogonadism, secondary hyperparathyroidism, thyroid disease, family history of hip fracture, and history of alcohol use [at ≥3 units/day] or smoking) and other clinical comorbidities, in addition to a physical examination including measurement of weight and height (without shoes), testing of muscle strength, and assessment for other clinical findings of undiagnosed fracture (i.e., spinal tenderness, deformity, and reduced space between lower ribs and upper pelvis) as appropriate given the patient’s age. The risk of major osteoporotic fracture calculated with the FRAX tool (https://www.shef.ac.uk/FRAX/tool.jsp) should be increased by 1.15 and the risk of hip fracture by 1.2, if the prednisone dose is >7.5 mg/day (e.g., if the calculated hip fracture risk is 2.0%, increase to 2.4%). It is recognized that in some cases, bone mineral density (BMD) testing may not be available.
Reassessment of fracture risk:
A clinical fracture risk reassessment carried out as above. Very high dose of glucocorticoids treatment was defined as treatment
with prednisone ≥30 mg/day and a cumulative dose of >5 gm in the past year. Reliability of FRAX (https://www.shef.ac.uk/FRAX/tool.jsp) after osteoporosis treatment is debated, but FRAX calculation can be repeated in adults age ≥40 years who have not received treatment.
Pharmacologic treatment for adults:
Recommended doses of calcium and vitamin D are 1000–1200 mg/day and 600–800 IU/day (serum level ≥20 ng/ml), respectively. Lifestyle modifications include a balanced diet, maintaining weight in the recommended range, smoking cessation, regular weight-bearing and resistance training exercise, and limiting alcohol intake to 1–2 alcoholic beverages/day. Very high-dose glucocorticoid treatment was defined as treatment with prednisone ≥30 mg/day and a cumulative dose of >5 gm in the past year. The risk of major osteoporotic fracture calculated with the FRAX tool (https://www.shef.ac.uk/FRAX/tool.jsp) should be increased by 1.15 and the risk of hip fracture by 1.2, if the prednisone dose is >7.5 mg/day.
Table 31.3 shows a comparison of the main guidelines published regarding the treatment indications for glucocorticoid-induced osteoporosis.
In conclusion, there have been significant advances in our understanding of the mechanisms by which glucocorticoids affect bone and increase fracture risk. Yet, the clinical management of glucocorticoid-induced osteoporosis remains suboptimal. Use of glucocorticoid for a duration of over 3 months leads to decreased bone mineral density, primarily through osteoblast suppression, as well as through increasing osteoclast life span. Awareness of osteoporosis risk mandates the stratification of patients into low, moderate, and high risk categories. Fracture risk should be assessed in all patients at the initiation of prolonged glucocorticoids therapy. All patients should maintain adequate intake of calcium and vitamin D, while those in moderate and high risk categories should initiate bisphosphonate therapy, or if bisphosphonates are contraindicated, use of alternative agents such as teriparatide or denosumab.
References
National Endocrine and Metabolic Diseases Information Service. Cushing’s disease. National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Department of Health and Human Services; 2008. p. 1–10. NIH Publication 08–300.
Cushing H. The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bull Johns Hopkins Hosp. 1932;50:137–95.
Seibel MJ, Cooper MS, Zhou H. Glucocorticoid-induced osteoporosis: mechanisms, management, and future perspectives. Lancet Diabetes Endocrinol. 2013;1:59–70.
Compston JE. Emerging consensus on prevention and treatment of glucocorticoid-induced osteoporosis. Curr Rheumatol Rep. 2007;9:78–84.
Whittier X, Saag KG. Glucocorticoid-induced osteoporosis. Rheum Dis Clin N Am. 2016;42:177–89, x.
LoCascio V, Bonucci E, Imbimbo B, Ballanti P, Adami S, Milani S, et al. Bone loss in response to long-term glucocorticoid therapy. Bone Miner. 1990;8:39–51.
Canalis E, Mazziotti G, Giustina A, Bilezikian JP. Glucocorticoid-induced osteoporosis: pathophysiology and therapy. Osteoporos Int. 2007;18:1319–28.
Van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993–1000.
Tatsuno I, Sugiyama T, Suzuki S, Yoshida T, Tanaka T, Sueishi M, et al. Age dependence of early symptomatic vertebral fracture with high-dose glucocorticoid treatment for collagen vascular diseases. J Clin Endocrinol Metab. 2009;94:1671–7.
ShinYS FH, Khiroya R, IbebunjoCMartyn J. Prednisolone-induced muscle dysfunction is caused more by atrophy than by altered acetylcholine receptor expression. Anesth Analg. 2000;91:322–8.
Majumdar SR, Lix LM, Yogendran M, Morin SN, Metge CJ, Leslie WD. Population-based trends in osteoporosis management after new initiations of long-term systemic glucocorticoids (1998–2008). J Clin Endocrinol Metab. 2012;97:1236–42.
Soucy E, Bellamy N, Adachi JD, et al. A Canadian survey on the management of corticosteroid induced osteoporosis by rheumatologists. J Rheumatol. 2000;27:1506–12.
Fardet L, Petersen I, Nazareth I. Prevalence of long-term oral glucocorticoid prescriptions in the UK over the past 20 years. Rheumatology (Oxford). 2011;50:1982–90.
Overman RA, Yeh JY, Deal CL. Prevalence of oral glucocortidoid usage in the United States: a general population perspective. Arthr Care Res. 2013;65:294–8.
Díez-Pérez A, Hooven FH, Adachi JD, et al. Regional differences in treatment for osteoporosis. The Global Longitudinal Study of Osteoporosis in Women (GLOW). Bone. 2011;49:493–8.
Silvermann S, Curtis J, Saag K, et al. International management of bone health in glucocorticoid-exposed individuals in the observational GLOW study. Osteoporos Int. 2015;26:419–20.
Lukert BP, Raisz LG. Glucocorticoid induced osteoporosis: pathogenesis and management. Ann Intern Med. 1990;112(5):352–64.
Angeli A, Guglielmi G, Dovio A, Capelli G, de Feo D, Giannini S, et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone. 2006;39:253–9.
Mazziotti G, Angeli A, Bilezikian JP, Canalis E, Giustina A. Glucocorticoid-induced osteoporosis: an update. Trends Endocrinol Metab. 2006;17:144–9.
Shaker JL, Lukert BP. Osteoporosis associated with excess glucocorticoids. Endocrinol Metab Clin N Am. 2005;34:341–56.
Feldstein AC, Elmer PJ, Nichols GA, Herson M. Practice patterns in patients at risk for glucocorticoid-induced osteoporosis. Osteoporos Int. 2005;16:2168–74.
Cakir B, Odabasi E, Turan M, Guler S, Kutlu M. Secondary osteoporosis in women. A retrospective analysis. Arch Gynecol Obstet. 2002;266(4):214–7.
Khosla S, Lufkin EG, Hodgson SF, Fitzpatrick LA, Melton LJ III. Epidemiology and clinical features of osteoporosis in young individuals. Bone. 1994;15(5):551–5.
van Staa TP, Leufkens HG, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int. 2002;13:777–87.
Van Staa TP, Leufkens HG, Abenhaim L, et al. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15:993–1000.
Kanis JA, Johansson H, Oden A, et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res. 2004;19:893–9.
Angeli A, Guglielmi G, Dovio A, et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone. 2006;39:253–9.
Waljee AK, Rogers MAM, Lin P, et al. Short term use of oral corticosteroids and related harms among adults in the United States: population based cohort study. BMJ. 2017;357:j1415.
Godschalk MF, Downs RW. Effect of short-term glucocorticoids on serum osteocalcin in healthy young men. J Bone Miner Res. 1988;3:113–5.
Weitoft T, Larsson A, Saxne T, Ronnblom L. Changes of cartilage and bone markers after intra-articular glucocorticoid treatment with and without post-injection rest in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:1750–3.
Pelt AC. Glucocorticoids: effects, action mechanisms, and therapeutic uses. Hauppauge, NY: Nova Science; 2011. ISBN 978-1617287589.
Hofbauer LC, Rauner M. Minireview: live and let die: molecular effects of glucocorticoids on bone cells. Mol Endocrinol (Baltimore, MD). 2009;23:1525–31.
Kalak R, Zhou H, Street J, et al. Endogenous glucocorticoid signalling in osteoblasts is necessary to maintain normal bone structure in mice. Bone. 2009;45:61–7.
Komori T. Glucocorticoid signaling and bone biology. Horm Metab Res = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2016;48:755–63.
Sher LB, Harrison JR, Adams DJ, Kream BE. Impaired cortical bone acquisition and osteoblast differentiation in mice with osteoblast-targeted disruption of glucocorticoid signaling. Calcif Tissue Int. 2006;79:118–25. [PubMed: 16927049].
Sher LB, Woitge HW, Adams DJ, et al. Transgenic expression of 11beta-hydroxysteroid dehydrogenase type 2 in osteoblasts reveals an anabolic role for endogenous glucocorticoids in bone. Endocrinology. 2004;145:922–9. [PubMed: 14617568].
Sato AY, Richardson D, Cregor M, et al. Glucocorticoids induce bone and muscle atrophy by tissue-specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology. 2017;158:664–77.
Geurtzen K, Vernet A, Freidin A, et al. Immune suppressive and bone inhibitory effects of prednisolone in growing and regenerating zebrafish tissues. J Bone Miner Res. 2017;
Yang N, Baban B, Isales CM, Shi XM. Role of glucocorticoid-induced leucine zipper (GILZ) in inflammatory bone loss. PLoS One. 2017;12:e0181133.
Chen H, Xing J, Hu X, et al. Inhibition of heat shock protein 90 rescues glucocorticoid-induced bone loss through enhancing bone formation. J Steroid Biochem Mol Biol. 2017;171:236–46.
Wu Z, Bucher NLR, Farmer SR. Induction of peroxisome proliferator-activated receptor γ during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPh, C/EBPy, and glucocorticoids. Mol Cell Biol. 1996;16:4128–36.
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids—potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102(2):274–82.
Ohnaka K, Tanabe M, Kawate H, Nawata H, Takayanagi R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem Biophys Res Commun. 2005;329:177–81.
Sato AY, Cregor M, Delgado-Calle J, Condon KW, Allen MR, Peacock M, Plotkin LI, Bellido T. Protection from glucocorticoid-induced osteoporosis by anti-catabolic signaling in the absence of sost/sclerostin. J Bone Miner Res. 2016;31:1791–802.
Yao W, Dai W, Jiang L, Lay EY, Zhong Z, Ritchie RO, Li X, Ke H, Lane NE. Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporos Int. 2016;27(1):283–94.
Schett G, Kiechl S, Weger S, et al. High-sensitivity C-reactive protein and risk of non traumatic fractures in the Bruneck study. Arch Intern Med. 2006;166:2495–501.
Ding C, Parameswaran V, Udayan R, et al. Circulating levels of inflammatory markers predict change in bone mineral density and resorption in older adults: a longitudinal study. J Clin Endocrinol Metab. 2008;93:1952–8.
Cauley JA, Danielson ME, Boudreau RM, et al.; for the Health ABC study. Inflammatory markers and incident fracture risk in older men and women: the Health Aging and Body Composition Study. J Bone Miner Res. 2007;22:1088–95.
van Staa TP, Geusens P, Bijlsma JW, et al. Clinical assessment of the long-term risk of fracture in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:3104–12.
Gough AK, Lilley J, Eyre S, et al. Generalised bone loss in patients with early rheumatoid arthritis. Lancet. 1994;344:23–7.
Maillefert JF, Aho LS, El Maghraoui A, et al. Changes in bone density in patients with ankylosing spondylitis: a two-year follow-up study. Osteoporos Int. 2001;12:605–9.
Briot K, Durnez A, Paternotte S, et al. Bone oedema on MRI is highly associated with low bone mineral density in patients with early inflammatory back pain: results from the DESIR cohort. Ann Rheum Dis. 2013;72:1914–9.
Charactcharoenwitthaya N, Khosla S, Atkinson EJ, et al. Effect of blockade of TNF-α and interleukine-1 action on bone resorption in early postmenopausal women. J Bone Miner Res. 2007;22:724–9.
Kong YY, Feige U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9.
Zaiss MM, Axmann R, Zwerina J, et al. Treg cells suppress osteoclast formation. A new link between the immune system and bone. Arthritis Rheum. 2007;56:4104–12.
Lam J, Takeshita S, Barker JE, et al. TNFα induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–8.
Sato K, Suematsu A, Okamoto K, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82.
Axmann R, Böhm C, Krönke G, et al. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009;60:2747–56.
Chen XX, Baum W, Dwyer D, et al. Sclerostin inhibition reverses systemic, periarticular and local bone loss in arthritis. Ann Rheum Dis. 2013;72:1732–6.
Harre U, Georgess D, Bang H, et al. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. 2012;122:1791–802.
Bresssot C, Meunier PJ, Chapuy MC, Lejeune E, Edouard C, Darby AJ. Histomorphometric profile, pathophysiology and reversibility of corticosteroid-induced osteoporosis. Metab Bone Dis Relat Res. 1979;1:303–11.
Dempster DW, Arlot MA, Meunier PJ. Mean wall thickness and formation periods of trabecular bone packets in corticosteroid-induced osteoporosis. Calcif Tissue Int. 1983;35:410–7.
Dempster DW. Bone histomorphometry in glucocorticoid induced osteoporosis. J Bone Miner Res. 1989;4:137–47.
Chappard D, Legrand E, Basle MF, Fromont P, Racineux JL, Rebel A, Audran M. Altered trabecular architecture induced by corticosteroids: a bone histomorphometric study. J Bone Miner Res. 1996;11:676–85.
Dalle Carbonare L, Arlot ME, Chavassieux PM, Roux JP, Portero NR, Meunier PJ. Comparison of trabecular bone architecture and remodeling in glucocorticoid-induced and postmenopausal osteoporosis. J Bone Miner Res. 2001;16:97–103.
Ton FN, Gunawardene SC, Lee H, et al. Effects of low-dose prednisone on bone metabolism. J Bone Miner Res. 2005;20:464–70.
Swanson C, Lorentzon M, Conaway HH, Lerner UH. Glucocorticoid regulation of osteoclast differentiation and expression of receptor activator of nuclear factor-kappaB (NF-kappaB) ligand, osteoprotegerin, and receptor activator of NF-kappaB in mouse calvarial bones. Endocrinology. 2006;147(7):3613–22.
Hofbauer LC, Gori F, Riggs BL, Lacey DL, Dunstan CR, Spelsberg TC, Khosla S. Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblasts: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology. 1999;140:4382–9.
Lane NE, Yao W, Balooch M, Nalla RK, Balooch G, Habelitz S, Kinney JH, Bonewald LF. Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or oestrogen-deficient mice. J Bone Miner Res. 2006;21:466–76.
Rubin MR, Bilezikian JP. The role of parathyroid hormone in the pathogenesis of glucocorticoid-induced osteoporosis: a re-examination of the evidence. J Clin Endocrinol Metab. 2002;87:4033–41.
Mazziotti G, Formenti AM, Adler RA, Bilezikian JP, Grossman A, Sbardella E, Minisola S, Giustina A. Glucocorticoid induced osteoporosis: pathophysiological role of GH/IGF-I and PTH/VITAMIN D axes, treatment options and guidelines. Endocrine. 2016;54(3):603–11.
Hasselgren PO, Fischer JE. Counterregulatory hormones and mechanisms in amino acid metabolism with special reference to the catabolic response in skeletal muscle. Curr Opin Clin Nutr Metab Care. 1999;2(1):9–14.
Hanaoka BY, Peterson CA, Horbinski C, Crofford LJ. Implications of glucocorticoid therapy in idiopathic inflammatory myopathies. Nat Rev Rheumatol. 2012;8(8):448–57.
Schakman O, Gilson H, Kalista S, Thissen JP. Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res. 2009;72(Suppl. 1):36–41.
Schakman O, Dehoux M, Bouchuari S, et al. Role of IGF-I and TNF-α/NF-κB pathway in the induction of muscle atrogenes by acute inflammation. Am J Physiol Endocrinol Metab. 2012;303(6):E729–39.
Hanaoka B, Peterson C, Crofford L. Glucocorticoid effects on skeletal muscle: benefit and risk in patients with autoimmune inflammatory rheumatoid diseases. Expert Rev Clin Immunol. 2012;8(8):695–7.
Pereira RM, Freire de Carvalho J. Glucocorticoid-induced myopathy. Joint Bone Spine. 2011;78(1):41–4.
Dekhuijzen PN, Gayan-Ramirez G, Bisschop A, De Bock V, Dom R, Decramer M. Corticosteroid treatment and nutritional deprivation cause a different pattern of atrophy in rat diaphragm. J Appl Physiol. 1995;78(2):629–37.
Lacomis D, Zochodne DW, Bird SJ. Critical illness myopathy. Muscle Nerve. 2000;23(12):1785–8.
Douglass JA, Tuxen DV, Horne M, et al. Myopathy in severe asthma. Am Rev Respir Dis. 1992;146(2):517–9.
Dhand UK. Clinical approach to the weak patient in the intensive care unit. Respir Care. 2006;51(9):1024–40.
Latronico N, Bolton CF. Critical illness polyneuropathy and myopathy: a major cause of muscle weakness and paralysis. Lancet Neurol. 2011;10(10):931–41.
Lacomis D, Giuliani MJ, Van Cott A, Kramer DJ. Acute myopathy of intensive care: clinical, electromyographic, and pathological aspects. Ann Neurol. 1996;40(4):645–54.
Trabecular bone mineral density and lean body mass. Osteoporos Int. 2006; 17:105–108.
Steinbuch M, Youket TE, Cohen S. Oral glucocorticoid use is associated with an increased risk of fracture. Osteoporos Int. 2004;15:323–8.
Van Staa TP, Laan RF, Barton IP, et al. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;48:3224–9.
O’Brien CA, Jia D, Plotkin LI, et al. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145(4):1835–41.
Weinstein RS, Jia D, Powers CC, et al. The skeletal effects of glucocorticoid excess override those of orchidectomy in mice. Endocrinology. 2004;145(4):1980–7.
Ton FN, Gunawardene SC, Lee H, Neer RM. Effects of low-dose prednisone on bone metabolism. J Bone Miner Res. 2005;20(3):464–70.
Weinstein RS. Glucocorticoid-induced osteoporosis. Rev Endocr Metab Disord. 2001;2(1):65–73.
Laan RF, van Riel PL, van de Putte LB, van Erning LJ, van’t Hof MA, Lemmens JA. Low-dose prednisone induces rapid reversible axial bone loss in patients with rheumatoid arthritis: a randomized, controlled study. Ann Intern Med. 1993;119:963–8.
Majumdar SR, Morin SN, Lix LM, Leslie WD. Influence of recency and duration of glucocorticoid use on bone mineral density and risk of fractures: population based cohort study. Osteoporos Int. 2013;24:2493–8.
Cooper MS, Blumsohn A, Goddard PE, et al. 11 beta-hydroxysteroid dehydrogenase type 1 activity predicts the effects of glucocorticoids on bone. J Clin Endocrinol Metab. 2003;88:3874–7.
Cooper MS, Bujalska I, Rabbitt E, et al. Modulation of 11beta-hydroxisteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts; an autocrine switch form glucocorticoid inactivation to activation. J Bone Miner Res. 2001;16:1037–44.
Russcher H, Smit P, van den Akker ELT, et al. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid-regulated gene expression. J Clin Endocrinol Metab. 2005;90:5804–10.
Van Staa TP, Laan RF, Barton IP, et al. Bone density threshold and other predictors of vertebral fracture in patients receiving oral glucocorticoid therapy. Arthritis Rheum. 2003;11:3224–9.
Majumdar SR, Morin SN, Lix LM, et al. Influence of recency and duration of glucocorticoid use on bone mineral density and risk of fractures: population-based cohort study. Osteoporos Int. 2013;24:2493–8.
Wijbrandts CA, Klaasen R, Dijkgraaf MGW, et al. Bone mineral density in rheumatoid arthritis patients 1 year after adalimumab therapy: arrest of bone loss. Ann Rheum Dis. 2009;68:373–6.
Haugeberg G, Conaghan PG, Quinn M, et al. Bone loss in patients with active early rheumatoid arthritis: infliximab and methotrexate compared with methotrexate treatment alone. Explorative analysis from a 12-month randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2009;68:1898–901.
Haugeberg G, Strand A, Kvien TK, et al. Reduced loss of hand bone density with prednisolone in early rheumatoid arthritis: results from a randomized placebo controlled trial. Arch Intern Med. 2005;165:1293–7.
Güler-Yüksel M, Bijsterbosch J, Goekoop-Ruiterman YP, et al. Changes in bone mineral density in patients with recent onset, active rheumatoid arthritis. Ann Rheum Dis. 2008;67:823–8.
Kawai VK, Grijalva CG, Arbogast PG, et al. Initiation of tumor necrosis factor α antagonists and risk of fractures in patients with selected rheumatic and autoimmune diseases. Arthritis Care Res. 2013;65:1085–94.
Ghazi M, Kolta S, Briot K, et al. Prevalence of vertebral fractures in patients with rheumatoid arthritis: revisiting the role of glucocorticoids. Osteoporos Int. 2012;23:581–7.
Briot K, Roux C. Glucocorticoid-induced osteoporosis. RMD Open. 2015;1:e000014.
Song QQ, Xie WY, Tang YJ, et al. Genetic variation in the glucocorticoid pathway involved in inter individual differences in the glucocorticoid treatment. Pharmacogenomics. 2017;18:293–316.
Whirledge SD, Jewell CM, Barber LM, et al. Generating diversity in human glucocorticoid signaling through a racially diverse polymorphism in the beta isoform of the glucocorticoid receptor. Lab Invest. 2017;
Aris R, Donohue JF, Ontjes D. Inhaled corticosteroids and fracture risk: having our cake and eating it too. Chest. 2005;127(1):5–7.
Richy F, Bousquet J, Ehrlich GE, et al. Inhaled corticosteroids effects on bone in asthmatic and COPD patients: a quantitative systematic review. Osteoporos Int. 2003;14(3):179–90.
Vestergaard P, Rejnmark L, Mosekilde L. Fracture risk associated with systemic and topical corticosteroids. J Intern Med. 2005;257(4):374–84.
Fujita K, Kasayama S, Hashimoto J, et al. Inhaled corticosteroids reduce bone mineral density in early postmenopausal but not premenopausal asthmatic women. J Bone Miner Res. 2001;16(4):782–7.
Wong CA, Walsh LJ, Smith CJ, et al. Inhaled corticosteroid use and bone mineral density in patients with asthma. Lancet. 2000;355(9213):1399–403.
Curtis JR, Westfall AO, Allison JJ, et al. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum. 2005;52:2485–94.
Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70.
Tatsuno I, Sugiyama T, Suzuki S, et al. Age dependence of early symptomatic vertebral fracture with high-dose glucocorticoid treatment for collagen vascular diseases. J Clin Endocrinol Metab. 2009;94:1671–7.
Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, Humphrey MB, Lane NE, Magrey M, Miller M, Morrison L, Rao M, Byun Robinson A, Saha S, Wolver S, Bannuru RR, Vaysbrot E, Osani M, Turgunbaev M, Miller AS, McAlindon T. American college of rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Care Res. 2017;69(8):1095–110.
Kanis JA, Delmas P, Burckhardt P, Cooper C, Torgerson D. Guidelines for diagnosis and management of osteoporosis. Osteoporos Int. 1997;7:390–406.
Luengo M, Picado C, Rio LD, Guañabens N, Montserrat JM, Setoain J. Vertebral fractures in steroid dependent asthma and involutional osteoporosis: a comparative study. Thorax. 1991;46:803–6.
Peel NFA, Moore DJ, Barrington NA, Bax DE, Eastell R. Risk of vertebral fracture and relationship to bone mineral density in steroid treated rheumatoid arthritis. Ann Rheum Dis. 1995;54:801–6.
Sutter S, Nishiyama KK, Kepley A, Zhou B, Wang J, McMahon DJ, Guo XE, Stein EM. Abnormalities in cortical bone, trabecular plates, and stiffness in postmenopausal women treated with glucocorticoids. J Clin Endocrinol Metab. 2014;99(11):4231–40.
McCloskey EV, Odén A, Harvey NC, Leslie WD, Hans D, Johansson H, Barkmann R, Boutroy S, Brown J, Chapurlat R, Elders PJ, Fujita Y, Glüer CC, Goltzman D, Iki M, Karlsson M, Kindmark A, Kotowicz M, Kurumatani N, Kwok T, Lamy O, Leung J, Lippuner K, Ljunggren Ö, Lorentzon M, Mellström D, Merlijn T, Oei L, Ohlsson C, Pasco JA, Rivadeneira F, Rosengren B, Sornay-Rendu E, Szulc P, Tamaki J, Kanis JA. A meta-analysis of trabecular bone score in fracture risk prediction and its relationship to FRAX. J Bone Miner Res. 2016;31(5):940–8.
Paggiosi MA, Peel NF, Eastell R. The impact of glucocorticoid therapy on trabecular bone score in older women. Osteoporos Int. 2015;26(6):1773–80.
Saag KG, Agnusdei D, Hans D, Kohlmeier LA, Krohn KD, Leib ES, MacLaughlin EJ, Alam J, Simonelli C, Taylor KA, Marcus R. Trabecular bone score in patients with chronic glucocorticoid therapy-induced osteoporosis treated with alendronate or teriparatide. Arthritis Rheumatol. 2016;68(9):2122–8.
Wallace BS, Curtis KG, Waljee JR, Akbar K, et al. Just the FRAX: management of glucocorticoid-induced osteoporosis. Gastroenterology. 2018;154(3):748–50.
Leib ES, Saag KG, Adachi JD, et al.; FRAX(®) Position Development Conference Members. Official Positions for FRAX(®) clinical.
Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int. 2011;22(3):809–16.
Leslie WD, Lix LM, Johansson H, Oden A, McCloskey E, Kanis JA. Spine-hip discordance and fracture risk assessment: a physician-friendly FRAX enhancement. Osteoporos Int. 2011;22(3):839–47.
Johansson H, Kanis JA, Odén A, Leslie WD, Fujiwara S, Glüer CC, Kroger H, LaCroix AZ, Lau E, Melton LJ 3rd, Eisman JA, O’Neill TW, Goltzman D, Reid DM, McCloskey E. Impact of femoral neck and lumbar spine BMD discordances on FRAX probabilities in women: a meta-analysis of international cohorts. Calcif Tissue Int. 2014;95(5):428–35.
Kanis JA, Johansson H, Oden A, Johnell O, de Laet C, Melton LJ III, Tenenhouse A, Reeve J, Silman AJ, Pols HA, Eisman JA, McCloskey EV, Mellstrom D. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res. 2004;19:893–9.
Lekamwasam S, Adachi JD, Agnusdei D, et al.; Joint IOF-ECTS GIO Guidelines Working Group. A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int. 2012; 23:2257–76.
Aljebab F, Choonara I, Conroy S. Systematic review of the toxicity of long-course oral corticosteroids in children. PLoS One. 2017;12:e0170259.
Roux C, Kolta S, Desfougeres JL, et al. Long-term safety of fluticasone propionate and nedocromil sodium on bone in children with asthma. Pediatrics. 2003;111:e706–13.
Szefler S, Weiss S, Tonascia J, et al. Long-term effects of budesonide or nedocromil in children with asthma. N Engl J Med. 2000;343:1054–63.
Zieck SE, George J, Blakeley BA, et al. Asthma, bones and corticosteroids: are inhaled corticosteroids associated with fractures in children with asthma? J Paediatr Child Health. 2017;53:771–7.
Skoner DP. Inhaled corticosteroids: effects on growth and bone health. Ann Allergy Asthma Immunol. 2016;117:595–600.
Wolfgram PM, Allen DB. Effects of inhaled corticosteroids on growth, bone metabolism, and adrenal function. Adv Pediatr. 2017;64:331–45.
Saag KG, Gehlbach SH, Curtis JR, Youket TE, Worley K. Trends in prevention of glucocorticoid-induced osteoporosis. J Rheumatol. 2006;33:1651–7.
Curtis JR, Westfall AO, Allison J, Becker A, Melton ME, Freeman A, et al. Challenges in improving the quality of osteoporosis care for long-term glucocorticoid users: a prospective randomized trial. Arch Intern Med. 2007;167:591–6.
Selby PL, Halsey JP, Adams KRH, et al. Corticosteroids do not alter the threshold for vertebral fracture. J Bone Miner Res. 2000;15:952–6.
Briot K, Cortet B, Roux C, et al. 2014 update of recommendations on the prevention and treatment of glucocorticoid-induced osteoporosis. Joint Bone Spine. 2014;81:493–501.
Buckley LM, Leib ES, Cartularo KS, et al. Calcium and vitamin D3 supplementation prevents bone loss in the spine secondary to low-dose corticosteroids in patients with rheumatoid arthritis. Ann Intern Med. 1996;125:961–8.
Adachi J, Bensen WG, Bianchi F, et al. Vitamin D and calcium in the prevention of corticosteroid induced osteoporosis: a 3 year follow-up. J Rheumatol. 1996;23:995–1000.
Compston J. Glucocorticoid-induced osteoporosis: an update. Endocrine. 2018;61(1):7–16.
Saag KG, Emkey R, Schnitzer TJ, Brown JP, Hawkins F, Goemaere S, Thamsborg G, Liberman UA, Delmas PD, Malice MP, Czachur M, Daifotis AG. Alendronate for the prevention and treatment of glucocorticoid induced osteoporosis. Glucocorticoid-Induced Osteoporosis Intervention Study Group. N Engl J Med. 1998;339:292–9.
Adachi JD, Saag KG, Delmas PD, Liberman UA, Emkey RD, Seeman E, Lane NE, Kaufman JM, Poubelle PE, Hawkins F, Correa-Rotter R, Menkes CJ, Rodriguez-Portales JA, Schnitzer TJ, Block JA, Wing J, McIlwain HH, Westhovens R, Brown J, Melo-Gomes JA, Gruber BL, Yanover MJ, Leite MO, Siminoski KG, Nevitt MC, Sharp JT, Malice MP, Dumortier T, Czachur M, Carofano W, Daifotis A. Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo-controlled extension trial. Arthritis Rheum. 2001;44:202–11.
Stoch SA, Saag KG, Greenwald M, Sebba AI, Cohen S, Verbruggen N, Giezek H, West J, Schnitzer TJ. Once-weekly oral alendronate 70 mg in patients with glucocorticoid-induced bone loss: a 12-month randomized, placebo-controlled clinical trial. J Rheumatol. 2009;36:1705–14.
Cohen S, Levy RM, Keller M, Boling E, Emkey RD, Greenwald M, Zizic TM, Wallach S, Sewell KL, Lukert BP, Axelrod DW, Chines AA. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel group study. Arthritis Rheum. 1999;42:2309–18.
Reid DM, Hughes RA, Laan RF, Sacco-Gibson NA, Wenderoth DH, Adami S, Eusebio RA, Devogelaer JP. Efficacy and safety of daily risedronate in the treatment of corticosteroid induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res. 2000;15:1006–13.
Wallach S, Cohen S, Reid DM, Hughes RA, Hosking DJ, Laan RF, Doherty SM, Maricic M, Rosen C, Brown J, Barton I, Chines AA. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int. 2000;67:277–85.
Reid DM, Adami S, Devogelaer JP, Chines AA. Risedronate increases bone density and reduces vertebral fracture risk within one year in men on corticosteroid therapy. Calcif Tissue Int. 2001;69:242–7.
Reid DM, Devogelaer JP, Saag K, Roux C, Lau CS, Reginster JY, Papanastasiou P, Ferreira A, Hartl F, Fashola T, Mesenbrink P, Sambrook PN, HORIZON investigators. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373:1253–63.
Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. N Engl J Med. 1999;339:292–9.
Adachi JD, Saag KG, Delmas PD, et al. Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo controlled extension trial. Arthritis Rheum. 2001;44:202–11.
Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 1999;42:2309–18.
Reid DM, Hughes RA, Laan RF, et al. Efficacy and safety of daily risedronate in the treatment of corticosteroid-induced osteoporosis in men and women: a randomized trial. European Corticosteroid-Induced Osteoporosis Treatment Study. J Bone Miner Res. 2000;15:1006–13.
Wallach S, Cohen S, Reid DM, et al. Effects of risedronate treatment on bone density and vertebral fracture in patients on corticosteroid therapy. Calcif Tissue Int. 2000;67:277–85.
Reid DM, Devogelaer JP, Saag K, et al. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373:1253–63.
Thomas T, Horlait S, Ringe JD, Abelson A, Gold DT, Atlan P, Lange JL. Oral bisphosphonates reduce the risk of clinical fractures in glucocorticoid-induced osteoporosis in clinical practice. Osteoporos Int. 2013;24(1):263–9.
Amiche MA, Lévesque LE, Gomes T, Adachi JD, Cadarette SM. Effectiveness of oral bisphosphonates in reducing fracture risk among oral glucocorticoid users: three matched cohort analyses. J Bone Miner Res. 2017;33:419–29.
Barasch A, Cunha-Cruz J, Curro FA, et al. Risk factors for osteonecrosis of the jaws: a case-control study from the CONDOR dental PRRN. J Dent Res. 2011;90:439–44.
Feldstein AC, Black D, Perrin N, et al. Incidence and demography of femur fractures with and without atypical features. J Bone Miner Res. 2012;27:977–86.
Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster DW, Ebeling PR, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O’Keefe R, Papapoulos S, Howe TS, van der Meulen MC, Weinstein RS, Whyte MP. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2014;29(1):1–23.
Koh JH, Myong JP, Yoo J, Lim YW, Lee J, Kwok SK, Park SH, Ju JH. Predisposing factors associated with atypical femur fracture among postmenopausal Korean women receiving bisphosphonate therapy: 8 years’ experience in a single center. Osteoporos Int. 2017;28(11):3251–9.
Saag KG, Wagman RB, Geusens P, et al. Denosumab versus risedronate in glucocorticoid- induced osteoporosis: a multicentre, randomised, double-blind, active controlled, double-dummy, non-inferiority study. Lancet Diabetes Endocrinol. 2018;6:445–54.
Yanbeiy ZA, Hansen KE. Denosumab in the treatment of glucocorticoid-induced osteoporosis: a systematic review and meta-analysis. Drug Des Devel Ther. 2019;13:2843–52.
Lyu H, Jundi B, Xu C, et al. Comparison of denosumab and bisphosphonates in patients with osteoporosis: a meta-analysis of randomized controlled trials. J Clin Endocrinol Metab. 2019;104(5):1753–65. https://doi.org/10.1210/jc.2018-02236.
Zhou Z, Chen C, Zhang J, et al. Safety of denosumab in postmenopausal women with osteoporosis or low bone mineral density: a meta-analysis. Int J Clin Exp Pathol. 2014;7(5):2113–22.
Saag KG, Shane E, Boonen S, Marín F, Donley DW, Taylor KA, Dalsky GP, Marcus R. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med. 2007;357:2028–39.
Langdahl BL, Marin F, Shane E, Dobnig H, Zanchetta JR, Maricic M, Krohn K, See K, Warner MR. Teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: an analysis by gender and menopausal status. Osteoporos Int. 2009;20(12):2095–104.
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41.
Oxlund H, Ortoft G, Thomsen JS, Danielsen CC, Ejersted C, Andreassen TT. The anabolic effect of PTH on bone is attenuated by simultaneous glucocorticoid treatment. Bone. 2006;39:244–52.
Nishida S, Yamaguchi A, Tanizawa T, Endo N, Mashiba T, Uchiyama Y, Suda T, Yoshiki S, Takahashi HE. Increased bone formation by intermittent parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone. 1994;15:717–23.
Jilka RL, Weinstein RS, Bellido T, Roberson P, Parfitt AM, Manolagas SC. Increased bone formation by prevention of osteoblast apoptosis by parathyroid hormone. J Clin Invest. 1999;104:439–46.
Saag KG, Zanchetta JR, Devogelaer JP, Adler RA, Eastell R, See K, Krege JH, Krohn K, Warner MR. Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double blind, controlled trial. Arthritis Rheum. 2009;60:3346–55.
Glüer CC, Marin F, Ringe JD, Hawkins F, Möricke R, Papaioannu N, Farahmand P, Minisola S, Martínez G, Nolla JM, Niedhart C, Guañabens N, Nuti R, Martín-Mola E, Thomasius F, Kapetanos G, Peña J, Graeff C, Petto H, Sanz B, Reisinger A, Zysset PK. Comparative effects of teriparatide and risedronate in glucocorticoid-induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J Bone Miner Res. 2013;28(6):1355–68.
Achiou Z, Toumi H, Touvier J, et al. Sclerostin antibody and interval treadmill training effects in a rodent model of glucocorticoid-induced osteopenia. Bone. 2015;81:691–701.
Cosman F, Crittenden DB, Ferrari S, et al. FRAME study: the foundation effect of building bone with 1 year of romosozumab leads to continued lower fracture risk after transition to denosumab. J Bone Miner Res. 2018;33(7):1219–26.
Genant HK, Engelke K, Bolognese MA, et al. Effects of romosozumab compared with teriparatide on bone density and mass at the spine and hip in postmenopausal women with low bone mass. J Bone Miner Res. 2017;32(1):181–7.
Langdahl BL, Libanati C, Crittenden DB, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet. 2017;390(10102):1585–94.
Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532–43.
Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377(15):1417–27.
Mok CC, Ying KY, To CH, et al. Raloxifene for prevention of glucocorticoid induced bone loss: a 12-month randomised double-blinded placebo-controlled trial. Ann Rheum Dis. 2011;70:778–84.
Cummings SR, Eckert S, Krueger KA, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA. 1999;281:2189–97.
Barrett-Connor E, Mosca L, Collins P, et al. Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med. 2006;355:125–37.
Curtis JR, Westfall AO, Allison JJ, Becker A, Casebeer L, Freeman A, Spettell CM, Weissman NW, Wilke S, Saag KG. Longitudinal patterns in the prevention of osteoporosis in glucocorticoid-treated patients. Arthritis Rheum. 2005;52:2485–94.
Majumdar SR, Lix LM, Morin SN, Yogendran M, Metge CJ, Leslie WD. The disconnect between better quality of glucocorticoid-induced osteoporosis preventive care and better outcomes: a population-based cohort study. J Rheumatol. 2013;40(10):1736–41.
Trijau S, de Lamotte G, Pradel V, Natali F, Allaria-Lapierre V, Coudert H, Pham T, Sciortino V, Lafforgue P. Osteoporosis prevention among chronic glucocorticoid users: results from a public health insurance database. RMD Open. 2016;2:e000249.
Curtis JR, Westfall AO, Allison JJ, Freeman A, Saag KG. Channeling and adherence with alendronate and risedronate among chronic glucocorticoid users. Osteoporos Int. 2006;17(8):1268–74.
Lekamwasam S, Adachi JD, Agnusdei D, et al. A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int. 2012;23:2257–76.
Lekamwasam S, Adachi JD, Agnusdei D, et al. An appendix to the 2012 IOF-ECTS guidelines for the management of glucocorticoid-induced osteoporosis. Arch Osteoporos. 2012;7:25–30.
Suzuki Y, Nawata H, Soen S, et al. Guidelines on the management and treatment of glucocorticoid-induced osteoporosis of the Japanese Society for Bone and mineral research: 2014 update. J Bone Miner Metab. 2014;32:337–50.
Compston J, Cooper A, Cooper C, et al.; National Osteoporosis Guideline Group (NOGG). UK clinical guideline for the prevention and treatment of osteoporosis. Arch Osteoporos. 2017;12:43.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
El Miedany, Y. (2022). Glucocorticoids and Musculoskeletal Health. In: El Miedany, Y. (eds) New Horizons in Osteoporosis Management. Springer, Cham. https://doi.org/10.1007/978-3-030-87950-1_31
Download citation
DOI: https://doi.org/10.1007/978-3-030-87950-1_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-87949-5
Online ISBN: 978-3-030-87950-1
eBook Packages: MedicineMedicine (R0)