
Abstract
Bronchiectasis is characterized by chronic, irreversible dilatation of the bronchi with thickening of the airway walls linked to chronic infection and inflammation that cause the associated loss of supportive tissue structures. Microbes are central protagonists in bronchiectasis, underpinning pathogenesis, and guiding treatment decisions. Historically, culture-based approaches have provided the basis for investigation of this ‘infective’ disease; however, emerging microbiome research now implicates more complex and heterogeneous microbial consortia, which may provide scope for improved stratification and targeted therapy. Such advances are necessary for this disease where licenced therapies remain lacking. As such, the stratification of patients based on detailed assessment of the microbiome is a highly appropriate approach. Emergent sequencing and analytical technologies now allow an unprecedented and expanding assessment of the microbiome incorporating bacteria, fungi and viruses and provide scope for novel diagnostic and treatment modalities for this disease of increasing global prevalence.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Aetiopathogenesis
Bronchiectasis is characterized by chronic, irreversible dilatation of the bronchi with thickening of the airway walls linked to degradation of bronchial elastin and supportive tissue structures. Patients experience chronic cough and recurrent respiratory infections associated with pulmonary exacerbations, and increased inflammation, that leads to airway damage, dyspnea and lung function decline [1, 2]. The gold standard for its confirmatory diagnosis is high-resolution tomography (HRCT) which can further delineate morphological subtypes. Bronchiectasis can be cylindrical, common and characterized by smooth tubular bronchi and mild disease; varicose, non-uniform dilation; or cystic, associated with more severe disease and complete loss of bronchial morphology [2, 3]. While the largest airways become visibly dilated, patients exhibit airflow limitation due to impaired drainage of bronchial secretions and obstruction in the small and medium airways caused largely by infectious and inflammatory insults [2, 4, 5]. Mucus inspissation and impaired mucociliary clearance, among other factors, support microbial colonization of the lung, a central tenant of current aetiopathogenic models [4, 6]. Clinically, patients present with cough and chronic sputum production, antecedent to confirmatory HRCT diagnosis, while other associated symptoms include malaise, chest discomfort, haemoptysis and weight loss [1, 2, 7]. Post-infectious bronchiectasis represents a key aetiology (after idiopathic disease) followed by immunodeficiency, ciliary disorders and obstructive lung disease, although estimates of each vary internationally [2, 8]. The vicious cycle hypothesis, first described by Cole, proposes that trigger factors, underpinned by genetic susceptibility or defects in host defence, set in motion a self-perpetuating cycle of infection, inflammation and impaired mucociliary clearance leading to progressive bronchial wall dilatation and destruction [4]. Paediatric bronchiectasis exhibits a distinct presentation compared to adults with a predominance of specific aetiologies and clinical manifestations including primary and secondary immunodeficiency, ciliary dyskinesia, congenital malformations, bronchiolitis obliterans and skeletal disease [8]. Airway insults from recurrent childhood infection further predisposes to the development of bronchiectasis [9]. It is noteworthy that the occurrence of bronchiectasis peaks at the extremes of age (i.e. in children 75 years) as these life stages are accompanied by significant shifts in both the microbiome and underlying immune status, which, in turn, may influence disease trajectory while providing scope for intervention [5, 10, 11].
The Role of Infection
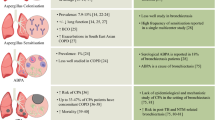
Infection is a hallmark of bronchiectasis as both a cause and consequence of disease. Culture-based studies have played a crucial role in our understanding of the microbiology of the bronchiectatic airway, where Haemophilus influenzae, Pseudomonas aeruginosa, Streptococcus pneumoniae, Moraxella catarrhalis and Staphylococcus aureus are frequently identified [2, 8, 12, 13]. Mycobacterium tuberculosis is also implicated as an important post-infective aetiology, particularly in Asia [8, 14], while infection by non-tuberculous mycobacteria (NTM) is associated with a worsening of pre-existing bronchiectasis and increased risk of fungal colonization by Aspergillus fumigatus [15,16,17]. Fungi are also important aetiological agents with heightened sensitization and the development of allergic bronchopulmonary aspergillosis (ABPA) increasingly recognized as negative prognostic indicators, while fungal genera including Candida, Penicillium, Cryptococcus, Clavispora and Scedosporium have been highlighted in more recent culture-independent studies of the bronchiectasis airway [8, 17,18,19,20]. The precise role of viruses in bronchiectasis is not well-established, with few large-scale and prospective studies available; however, several established respiratory viruses including coronavirus, rhinovirus and influenza have been commonly detected in bronchiectasis patients [2, 8, 21, 22]. While increasing evidence also supports a role for the human T-cell lymphotropic virus type 1 (HTLV-1) in acquired immunodeficiency linked to disease risk, conclusive mechanistic studies are lacking [8, 23]. Likewise, oropharyngeal species – generally reported as ‘contaminant’ microbes – may play insidious ‘pathobiont’ roles inciting deleterious immune responses directly or through their influence on overtly pathogenic species through microbial interactions [24, 25]. As such, the bronchiectasis microbiome may be best defined as a dynamic inter-kingdom network with an underlying and dysregulated production of cytokines, elastases and matrix metalloproteinases (MMPs) that, in turn, damage the structural integrity of the lung, leading to visible distortion of the airway [26,27,28]. Inciting microbial insults elicit a largely neutrophilic cellular immune response with increased macrophage recruitment, while a small but significant subset of patients exhibit eosinophil-dominant disease linked predominantly to environmental triggers [29]. The observed cytokine responses are heterogenous and reflect the nature of the underlying infectious triggers within the bronchiectasis airway and are typically characterized by elevated levels of IL-1β, IL-8, leukotriene (LT)B4, CXCL2 and TNFα [29]. These inflammatory profiles sustain the release of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) from the endothelium, leading to further neutrophil and eosinophil recruitment to the airways [29, 30]. The role of the neutrophil remains central to bronchiectasis and has been elegantly dissected in several studies which collectively illustrate its importance in the pathogenic process. The neutrophil, as a major airway inflammatory cell, produces serine proteases such as neutrophil elastase, which itself is described to represent a negative prognostic indicator in bronchiectasis which correlates to reduced microbial diversity and the presence of P. aeruginosa. Specifically, neutrophil elastase is highly expressed in bronchiectasis and associates with exacerbations, radiological extent of disease and lung function [26]. While active in bronchiectasis, the neutrophil itself is functionally compromised, leading to impaired bacterial phagocytosis and killing despite a prolonged viability and delayed apoptosis [31, 32]. Alteration of the sputum proteome in bronchiectasis patients infected by P. aeruginosa reveals the upregulation of pregnancy zone protein (PZP) associated release of neutrophil extracellular traps (NETs) tying airway infection to NET formation, disease severity and pathogenesis [33, 34]. Such detailed mechanistic study of the neutrophil has paved the way for clinical application in bronchiectasis evidenced by the success of a phase II clinical trial of the dipeptidyl peptidase 1 inhibitor (DPP-1) Brensocatib – an inhibitor of neutrophilic serine protease activation. This opens a new and urgently needed avenue towards clinical translation [35, 36]. The dysregulation of host neutrophilic function in bronchiectasis is also notable for its association with shifts in microbiome composition, whereby neutrophil-associated PZP levels predict a dysbiotic predominance of Proteobacteria including Pseudomonas, Enterobacteriaceae, Stenotrophomonas and Moraxella further illustrating the close relationships between microbiome profile and disease pathogenesis [33]. This particular association has been independently corroborated in subsequent microbiome studies and correlated to neutrophil elastase levels, which in turn associated with a decreased microbial diversity and increased P. aeruginosa abundance [37]. The salient features of bronchiectasis pathogenesis as currently understood are detailed in Fig. 7.1.

Overview of the pathogenesis in bronchiectasis and the role of the microbiome. (1) Trigger factors include host genetics, defects in host defence and/or ciliary dysfunction which predispose to microbial colonization. (2) Colonization or net ‘immigration’ of microbes leading to the emergence of deleterious microbiome signatures defined by the presence of host response to specific bacteria, fungi and viruses (grey box). Environmental exposures to air-, surface- and device-associated microbes including pollution, and other geographically variable environmental factors may also contribute (yellow box). (3) Deleterious and impaired host responses directed towards the ‘elimination’ of microbes are triggered by pathogen-associated molecular patterns, virulence factors and allergens culminating in a dysfunctional and airway damaging immune response. A transition from acute to persistent infection is accompanied by a decreased diversity in the microbiome, altered composition and network configuration. (4) Chronic infection and inflammation cycles, leading to progressive airway damage and loss of structure with associated changes in radiological morphology. (5) Loss of structure and increasing dilatation of the bronchi, coupled to mucus plugging and impaired mucociliary clearance predisposing to an increased risk of subsequent infection and progressive clinical decline
The Case for Microbiome Research in Bronchiectasis
Building upon Cole’s initial hypothesis, a more complex and holistic picture has emerged as sophisticated multi-omic technologies are increasingly applied in bronchiectasis and other chronic respiratory diseases providing both opportunities and challenges [38]. Fine-structure analysis of disease progression permits a greater appreciation of pathogenic mechanisms, while improved patient stratification provides scope for personalized therapeutic approaches [36, 39]. Contemporary models such as the recently advanced ‘vicious vortex’ paradigm proposed by Flume et al. seek to capture the dynamic interactions embodied by each pathophysiologic step of the disease cycle as they promote persistent and progressive inflammation and airway damage over time [6]. This represents a major academic and clinical challenge in the setting of a highly heterogenous condition such as bronchiectasis, where arrival at the endpoint of structural airway damage may be reached through distinct endophenotypic routes [36, 39]. In this context, the microbiome represents a potentially important window into disease progression and pathogenesis with prognostic potential and scope for improved patient stratification in this heterogenous clinical setting (Fig. 7.1) [36, 40, 41]. The failure of high-profile multi-centre antibiotic clinical trials in bronchiectasis has further highlighted the need and potential for the integration of microbiome approaches into clinical trial design with the aim of improved patient selection and stratification including appropriately chosen clinical endpoints that address past inconsistencies resulting in a failure to replicate results across geographically distinct patient populations [8, 36, 42]. Early work, driven by culture-based assessment of resident microbial pathogens and later advanced by characterization of associated immune responses, has laid the foundation for our understanding of bronchiectasis and the potential therapeutic approaches for its clinical management [12]. Intriguingly, however, the introduction of antibiotics for bronchiectasis-associated microbiomes has not been met with the anticipated decline of this ‘infective’ condition, contradicting a simplistic model of bacterial overgrowth amenable to suppression by antibiotics. Therefore, early presumptions regarding amenability to antimicrobials to provide resolution has likely contributed to a neglect of this complex and poorly understood respiratory condition [2, 36]. A further feature contributing to delays in understanding the airway microbiome is the previous and incorrect assumption of lung sterility. As a consequence, the lung microbiome has received less attention compared to other anatomical sites (such as the gut) leading to significant gaps in understanding the lung microbiome – gaps now actively being closed in the context of data illustrating the presence and key functional role of the microbiome in the respiratory diseases including bronchiectasis [38, 43]. The time for a more detailed exploration of the microbiome in bronchiectasis has arrived and will likely provide a clearer understanding of disease pathogenesis, mechanisms of infection and a potential for therapeutic advancements in its management.
The Airway Microbiology in Bronchiectasis
Several studies have described airway colonization in bronchiectasis by distinct microbial entities, largely based on culture. Although prevalence varies, H. influenzae and P. aeruginosa represent the most common bacterial species identified followed by S. pneumoniae, M. catarrhalis, S. aureus and others including K. pneumoniae, while S. maltophilia and Achromobacter xylosoxidans are less frequently found [13, 44]. While extensively characterized, a significant bias exists towards western populations in the microbiological surveillance of the bronchiectasis airway, and the growing awareness of geographic differences most notably in Asian populations have been described warranting study [8, 45]. In comparison to western populations, Asian patients exhibit higher P. aeruginosa colonization rates (relative to H. influenzae), while K. pneumoniae is also more frequently isolated [45]. A comparative analysis of European, US and Indian registry data also reveal similar patterns with higher rates of P. aeruginosa and exacerbation risk associated with prior M. tuberculosis infection in Indians [14]. Given such disparities, other geographic and population-associated differences in microbiology and microbiome composition may exist contributing further to disease heterogeneity and therapeutic challenges [2, 8, 36]. Importantly, no microbial pathogen has been identified in up to 70% of bronchiectasis sputum cultures – even in the presence of clear and measurable inflammatory responses – further highlighting our incomplete understanding of pathogenesis and, perhaps importantly, the need for broader microbiome analysis unrestricted by selective culture-based methodologies [29, 41, 46]. Genetic analyses of the microbiome, aided by next-generation sequencing (NGS), are now emerging in bronchiectasis, supporting the extensive culture-based literature (Fig. 7.2). The derivation and interrogation of such data may ultimately allow for an integration of direct microbiome sequencing into clinical diagnostics and the selection of therapies, potentially offering more personalized and effective treatment approaches at the individual level in the future care of bronchiectasis.

Timeline of culture-based and culture-independent microbiome research in bronchiectasis. A lollipop chart illustrates the growth of culture-based and culture-independent research on the bronchiectasis microbiome over time (2000–2020). Studies are indicated by coloured lollipops with stick height (y-axis, logarithmic) representing the number patients in each study. The type of analysis performed in each study is indicated in the right-hand legend (grey, culture based; red, bacteria 16S rRNA analysis; blue, fungal ITS analysis; purple, WGS metagenomic shotgun analysis)
The Bacteriome in Bronchiectasis
While the emergence of culture-independent lung microbiome analysis has its origins in cystic fibrosis (CF), it is only more recently that culture-independent approaches have been applied to (non-CF) bronchiectasis (Fig. 7.2) [41, 47]. Pioneering pyrosequencing efforts initially documented a limited shift in community composition during exacerbation and following antibiotic therapy with characterization of P. aeruginosa, H. influenzae, Prevotella and Veillonella as part of a complex community in a cross-sectional cohort of 40 patients [48]. Rogers et al. subsequently applied 16S rRNA sequencing to a larger cohort (n = 86) from a clinical trial of macrolide intervention (the BLESS study) highlighting this as a potentially informative analytical measure in respiratory trials [49]. The BLESS intervention (low-dose erythromycin – 400 mg twice daily) demonstrated success in reducing exacerbations among longitudinally sampled patients compared to the control arm with 16S rRNA analysis providing insight into microbiome composition as a correlate of the observed therapeutic response. This inclusion of microbiome analysis revealed granular changes in microbiome composition and allowed patient stratification according to the dominant organism, where a significantly worse outcome is observed in those with Pseudomonas or Veillonella-dominant profiles [40, 49]. The association of the commensal genus Veillonella with exacerbation further represents a novel perspective on this bacterial taxa and suggests a potential insidious role for this anaerobe within the complex bronchiectasis microbial community [40]. Microbiome profiles importantly were also predictive of the observed host immune response, with H. influenzae inducing increases in MMP2 and MMP8 compared to patients with Pseudomonas-dominant profiles, while both organisms induced significantly elevated levels of serum CRP, sputum IL-1β and IL-8. Taxonomic diversity exhibits negative correlation with clinical outcomes, and lower IL-1β and IL-8 for instance suggest better outcomes with greater microbial richness observed in the erythromycin treatment arm [28, 49]. In further corroboration of the observed microbial-host interactions in the BLESS cohort, loss-of-function variants are identified in the human FUT2 fucosyltransferase gene (responsible for coating mucosal surfaces with fucosylated glycans) and found to influence the composition of the microbiome while decreasing the risk of pulmonary exacerbations and P. aeruginosa colonization [24]. Overall, the targeted 16S analyses of the BLESS cohort underscored the stability of the bronchiectasis microbiome but noted the supplantation of H. influenzae by the more pathogenic P. aeruginosa strains. Such displacement, along with the observed increases in macrolide resistance, confers potentially undesirable long-term consequences of this therapeutic intervention, given the established negative association of P. aeruginosa with bronchiectasis [44]. Therefore, while the effect of erythromycin therapy is largely beneficial, this depends on the baseline microbiome composition and importantly did not significantly alter exacerbation rates in Pseudomonas-dominant patients, while its benefits in non-Pseudomonas-dominant patients came with a greater risk of subsequent Pseudomonas colonization. Further analysis, based on whole-genome metagenomic shotgun sequencing, has also highlighted the increased burden of macrolide resistance determinants in antibiotic-treated patients over time [50]. These observations clearly highlight the value of targeted and metagenomic sequencing of the microbiome as a key secondary outcome measure in clinical trials and administered therapeutics. A similar trend in antimicrobial resistance, through metagenomics, was observed in macrolide-treated severe asthma, further underscoring this phenomena in chronic respiratory disease [51]. Additional ultra-deep metagenomic analysis of respiratory specimens from non-diseased (healthy) subjects reveals the presence of a core macrolide resistome, which importantly remains consistent even across distinct respiratory disease states, including bronchiectasis [52]. Resistance genes are correlated to commensal species representing potential resistance reservoirs, detected in host sputum and on patient inhaler devices highlighting the potential of metagenomics for surveillance of resistance in the environment as well as within host respiratory microbiomes [52].
While baseline microbiomes appear predictive of clinical course and therapeutic response, stability throughout exacerbation and treatment has emerged as a recurrent finding among microbiome studies in bronchiectasis [41, 49, 53]. Indeed, individual microbiomes can persist for many years in longitudinally sampled patients [54]. There is, however, a generally consistent correlation between diversity measures of the microbiome and clinical outcome that likely reflects the increased dominance of particular pathogens, in agreement with the adapted island model of the lung microbiome, where reduced diversity departs from the healthy microbiome state [37, 43, 53, 55]. Notwithstanding this concept, the mere fact that bacterial microbiomes remain largely stable seems to contradict simplistic models of targeted antimicrobial elimination of pathogenic microbes as a basis for therapeutic efficacy. Furthermore, how antimicrobial agents such as macrolides, which have no activity against P. aeruginosa, offer the observed therapeutic benefit in bronchiectasis remains poorly understood. This may involve more indirect influences upon other microbial constituents and their associated community structure but extend to include the drug’s anti-inflammatory and immunomodulatory effects [56].
Non-tuberculous Mycobacteria
The role of non-tuberculous mycobacteria (NTM) is important and warrants particular mention given its incidence as a cause and consequence in bronchiectasis including its associated and significant challenges in detection by NGS methodologies [16, 57, 58]. Bronchiectasis and NTM infection are highly correlated to airway distortion, which in itself is thought to predispose to NTM colonization and disease progression in bronchiectasis [59]. Mycobacterium avium complex (MAC) is the most prevalent NTM detected although geographic variation exists [8]. Analysis of US registry data reveals a significant burden of MAC, followed by M. abscessus and M. chelonae in bronchiectasis who develop symptoms in later life and who are predominantly female [16]. As such, the presence of NTM potentially represents a sub-phenotype of bronchiectasis and a possible means of stratification towards microbiome-directed therapy. Importantly, however, recent work highlights the difficulty in a reliable detection of mycobacteria using targeted 16S rRNA sequence analysis whereby Mycobacterium spp. are frequently absent from 16S rRNA gene profiles in samples positive for mycobacterial cultures [57]. This discrepancy between culture-based and culture-independent analysis suggests a lack of sensitivity in culture-independent methods for the detection of mycobacteria. This likely reflects the relatively low copy number of 16S rRNA genes per genome that leads to an underrepresentation of these bacteria by targeted 16S analysis. Notwithstanding this technical limitation, Sulaiman et al. demonstrate the existence of distinct host phenotypes in NTM-positive bronchiectasis patients, where impaired IFN-γ and GM-CSF production is coupled to significant association with upper airway taxa and T-helper-17 cytokines [57]. These observations add credence to the concept of NTM infection as a potential endophenotype and treatable trait in bronchiectasis and is important given the association of NTM with susceptibility to fungal infection which leads to a complex microbiological picture in the airway and its associated treatment challenges [15, 39].
The Mycobiome in Bronchiectasis
Given the structural distortions of the airway observed in bronchiectasis, the risk of fungal exposure and subsequent colonization is increased. This results in an increased sensitization to fungal allergens and the occurrence of allergic bronchopulmonary aspergillosis (ABPA) [17]. As seen in other chronic respiratory disease states, fungal sensitization and allergy are associated with negative outcomes in bronchiectasis including poorer lung function and increased exacerbation [17, 60,61,62]. The immunology of fungal sensitization and its associated host response is extensively characterized in CF where Aspergillus fumigatus represents a key predominant airway fungal pathogen identified in association with Th2-driven response and antecedent to the emergence of ABPA that is accompanied by significant clinical symptoms and lung function decline [62]. While the host response to fungi in bronchiectasis remains lesser studied, marked increases in sensitization to Aspergillus antigens are noted as is the increased activity of the anti-fungal chitinase enzyme chitotriosidase (CHIT-1), both of which exhibit geographic variation [19, 63, 64]. While important, the diagnosis of fungal infection remains challenging due to the poor sensitivity and specificity associated with existing diagnostics and a lack of standardization between centres. This leads to delays in therapy and therefore adverse outcomes [62, 65]. The application of ITS amplicon sequencing has therefore been proposed and examined as an alternative detection method highlighting the complex fungal profiles seen in CF that appear distinct from non-CF bronchiectasis. In a head-to-head comparison, lower fungal diversity is observed in CF compared to non-CF bronchiectasis [18]. As fungal colonization, sensitization and ABPA all individually represent potentially ‘treatable traits’ in bronchiectasis, a detailed characterization of the airway mycobiome is worthy despite the inherent technical challenges related to mycobiome analysis [39, 66, 67]. In the largest investigation of the bronchiectasis mycobiome performed to date, the Cohort of Asian and Matched European Bronchiectasis (CAMEB) compared mycobiome profiles generated by ITS amplicon sequencing in patients from Asian and European bronchiectasis cohorts ‘matched’ for age, sex and disease severity [19]. This important work provided the first broad insight into the airway mycobiome in bronchiectasis and identified host responses related to fungal presence that associate negatively with clinical outcomes. This study illustrated the higher abundance of potentially pathogenic taxa including Aspergillus, Penicillium and Cryptococcus in bronchiectasis (compared to healthy controls) and the presence of an unfavourable allergic sensitization and immune response profile associated with Aspergillus. The study design further allowed comparisons between the Asian (Singaporean and Malaysian) and European (Scottish) patients, matched by age, sex and Bronchiectasis Severity Index (BSI) score. This permitted a clear assessment of geographic differences in the mycobiome while controlling for disease severity. Differences in mycobiome profiles were identified including increased relative abundances of Simplicillium, Trichosporon and Aspergillus in the Asians, while a higher abundance of Wickerhamomyces, Clavispora and Cryptococcus distinguished Europeans. Candida was frequently observed across both cohorts at comparable frequency, while the Scottish cohort exhibited a higher prevalence of Saccharomyces, Penicillium, Cryptococcus, Clavispora and Botrytis. Further analysis using a validated qPCR method that included quantification of the various Aspergillus species present in the airway revealed a predominance of A. terreus in the Scottish patient group, while A. fumigatus conidial burden was greatest in Asians [19, 68]. While these findings cannot be generalized beyond the local regions studied, they do illustrate the geographic variation observed in bronchiectasis and highlight both the similarities and differences that can be uncovered when appropriately designed populations are compared [8, 19]. Further stratification of the CAMEB study participants by an immunological classification system that accounted for fungal presence and associated host biomarkers including Aspergillus-specific IgE and IgG and sputum galactomannan identified high frequencies of fungal sensitization and ABPA. These Aspergillus-associated disease states further revealed a clear association with disease severity, exacerbation frequency and lung function decline, particularly in those with serological ABPA (sABPA) [19]. This important, novel and clinically relevant observation was next further investigated using an extended panel of fungal allergens revealing a remarkable and very high level of sensitization among bronchiectasis patients [20]. Sensitization level and the occurrence of poly-sensitization was linked to poorer lung function but not exacerbations; however, assessment of the host airway immune response allowed a clustering of patients according to “immunoallertypes”: one fungal driven and proinflammatory and a second characterized by sensitization to house dust mite coupled to chemokine dominance. Critically, the fungal patient cluster demonstrates greater disease severity and poorer lung function [20]. Current works characterizing the mycobiome and the sensitization response in bronchiectasis have thus far revealed that combining immune profiling with patient clustering reveals novel disease endophenotypes that potentially may be amenable to tailored and personalized bronchiectasis therapy.
The Virome in Bronchiectasis
The virome represents the most challenging and therefore least well-described aspect of the human microbiome and has yet to be clearly defined in the lung [38]. In bronchiectasis, the role of viruses remains unclear; however, they have been detected in the airway, and emerging evidence supports a potential role in disease [21, 22, 69]. The virome may be considered from a number of perspectives in bronchiectasis: (1) the role of common respiratory viruses and their impact on health status, (2) acquired immunodeficiency associated with viral infection and (3) bacteriophages and their influence on the bacterial hosts, including as potential facilitators of horizontal gene transfer. The first two areas have been addressed in several existing studies, while the third represents a novel area in bronchiectasis yet to be meaningfully addressed by research. Considering what is known in other chronic respiratory disease states, viruses are considered important triggers of exacerbation. Gao et al. assessed the presence of viruses at exacerbation in a cohort of bronchiectasis patients from Guangzhou, China, representing the first large-scale prospective study determining the incidence and clinical impact of viral infection. Common viruses documented included coronavirus, rhinovirus and influenza A and B viruses. While systemic and lower airway symptoms were not significantly different between virus-positive and virus-negative exacerbations, several systemic and airway inflammatory markers (serum IL-6 and TNF-α; sputum IL-1β and TNF-α) distinguished virus-positive patients [22]. In subsequent work from Australia, a high frequency of stable bronchiectasis patients interestingly had viruses detected in their airways, particularly during winter (92%) compared to lower rates (33%) in the summer. The main viruses detected included rhinovirus, influenza A and B and respiratory syncytial virus with greatest incidence of co-infection in the winter months [21]. Both studies clearly confirm the presence of common respiratory viruses in the stable and exacerbation states in bronchiectasis albeit without a significant association to clinical outcome. Building on their previous work, Chen et al. next identified a significantly higher frequency of viruses at exacerbation compared to the stable state in patients from the Guangzhou region further implicating viruses in bronchiectasis exacerbations where rhinovirus and influenza A and B demonstrated the strongest effects [69]. The second aspect of virology that warrants consideration in bronchiectasis is the role of viruses as mediators of acquired immune deficiency, which in turn may accelerate the disease ‘cycle’ through disrupting normal immune homeostasis. Such viruses include the Human T-cell leukaemia virus, type 1 (HTLV-1) which has been documented in association with bronchiectasis in several studies, from western and indigenous Australian populations, the latter originally describing the link [23]. More recent evidence also implicates the Epstein–Barr virus (EBV), which is associated with a shortened time to next exacerbation and more rapid decline of lung function [70]. While this latter association is less well supported, the concept of acquired immune deficiency should at least be considered and is consistent with current disease paradigms (Fig. 7.1). More recent virome research, especially in bronchiectasis relates to the role of bacteriophages and their contribution to microbiome architecture and stability. While WGS metagenomics has been performed in bronchiectasis, no study to date has directly assessed the presence and abundance of bacteriophages (Fig. 7.2) [50, 52]. Whether a distinct disease- or patient-specific bacteriophage profile exists in bronchiectasis remains to be established, and investigations should focus on characterizing the disease or specific patient groups, defined by bacteriophage pattern that associate with clinical outcomes. Emerging data from the gut in studies unrelated to bronchiectasis does suggest an individual specificity of the virome (‘phageome’), with high inter-individual differences [71]. It is probably too premature to suggest whether such variability could be correlated with clinical outcomes in bronchiectasis, but as bacteriophages possess the potential to dramatically reshape the microbiome and contribute to dysbiosis, there is certainly scope for broad assessment from diagnostic and therapeutic perspectives [72, 73]. Additionally, the relevance of bacteriophages in the context of emerging antimicrobial resistance and as facilitators of horizontal gene transfer is highly relevant in respiratory diseases including bronchiectasis and remains a key area for future investigation [74].
Microbial Networks and the ‘Multi-Biome’
While the bacteriome, mycobiome and virome have to date been considered as separate and individual entities, integrating them into a holistic ‘multi-biome’ framework appears to be the next logical step for bronchiectasis and other chronic respiratory disease states. The concept and description of the host microbiome as an integrated microbial network is emerging and has been advanced as a potential model underpinning exacerbations in CF [25]. Networks and their associated microbial interactions may better account for observed clinical differences compared to taxonomic abundance alone and therefore represents a promising platform for development of respiratory disease models of infection and exacerbation [75]. The role of fungi and their own inter-kingdom communication with bacteria remain a recognized part of a holistic ecosystem where active research is ongoing, and with clear relevance to bronchiectasis, as both kingdoms independently have been shown to be highly relevant in disease [8, 19, 41, 76]. Going beyond this, the role of viruses should also be considered, as individual common respiratory viruses, those linked to acquired immunodeficiency but also bacteriophages that can have a major influence on microbiome architecture and the mobilization of antimicrobial resistance genes. While each individual microbiome is clearly relevant, they remain to be fully investigated in the context of an integrated and holistic inter-kingdom microbial consortia, a rich avenue for future microbiome research in bronchiectasis.
Clinical Applications
The increasingly recognized heterogeneity of clinical endophenotypes in bronchiectasis helps to account for the failure of most clinical trials in this disease [36]. Given the associations between the microbiome and disease outcomes, it seems plausible to consider an integration of microbiome data into clinical trial design. This may prove beneficial, allowing for adequate adjustment of confounding microbiome-associated variables through appropriate patient stratification (Table 7.1) [41]. In addition to targeting microbial endophenotypes, the microbiome also offers potential as a secondary outcome measure and possible prognostic marker in assessing treatment effects directed at it. Surveillance of potentially undesirable changes in the microbial community such as the emergence of potentially pathogenic taxa or antimicrobial resistance genes is also of value [50, 52]. Host genetics is an additional key factor that, at least partially, predicts microbiome composition and should be integrated into patient stratification modalities where available [77]. Expanding the assessed microbial kingdoms in bronchiectasis through multi-biome analysis has already uncovered new perspectives, for example, the identification of fungal sensitization, a feature that may be to endophenotype-targeting precision medicine approaches [64]. Furthermore, the early application of metagenomics in bronchiectasis has already demonstrated a clear potential in terms of charting the emergence of antimicrobial resistance and the environmental exposome. The role of the bronchiectasis virome remains to be established; however, emerging large-scale studies do suggest an involvement with exacerbation risk. While an untargeted appraisal of the virome or ‘phageome’ associated with the bronchiectasis microbiome remains to be examined, the clear individualized and stable phage profiles recently determined in gut microbiomes provide a clear framework for future lung investigations and in particular the examination of bacteriophages in bronchiectasis. From a diagnostic perspective, in-depth analysis of the microbiome (integrating trans-kingdom analysis with temporally and anatomically distinct samplings) may provide the ultimate stratification system by which specific bronchiectasis subtypes or overlap syndromes may be robustly defined and in turn provide a more focused management and precision medicine approach in this disease state (Table 7.1).
Future Directions
Against the backdrop of ageing global demographics and an increased awareness of the disease, there is a growing appreciation of the clinical burden of bronchiectasis [36]. This has led to renewed focus on the disease and increased research including the increasing number of microbiome studies described in this chapter (Fig. 7.2). Analysis of the microbiome has uncovered the complexity of the microbial consortia in bronchiectasis and its potential for patient stratification according to profile rather that the simple presence or absence of individual taxa [40]. A major challenge, however, in the translation of these findings will be the standardization of methodologies across studies, including the integration of data from distinct microbiome profiles: i.e. profiles obtained using different platforms, i.e. bacterial vs fungal vs viral, or profiles from distinct anatomical sites, i.e. gut vs lung in a given patient [41, 78]. Integrative approaches for patient stratification based on multiple ‘omic’ datasets have shown early promise, and the further development and application of these approaches to ‘multi-biome’ datasets may provide even deeper resolution of patient microbiome subtypes in bronchiectasis [79]. Larger studies with greater numbers of patients (approaching those already achieved in culture-based studies – Fig. 7.2) will be required, and the inclusion of longitudinal sampling and comparing geographically distinct cohorts will help further in attaining the necessary resolution required for clinical use. We must ensure to continue to improve the methodologies used for microbiome analysis, making them more accessible, refined and scalable. While targeted amplicon sequencing has now been applied in several studies, metagenomic analyses of bronchiectasis remain limited, and meta-transcriptomic analyses are yet to be described. The insights that may be derived from a functional appraisal of the metagenome remains an important area for future work. Likewise, the virome and ‘phageome’ remain to be fully characterized in bronchiectasis. Recent work from our group demonstrates the potential of metagenomics to allow sampling of the environment (patient inhaler devices and both indoor and outdoor air) in addition to the airway microbiome for identifying important exposures such as resistome-harbouring microbes and airborne fungi to which exposed patients can demonstrate sensitization responses that associate with negative clinical outcomes [52, 80]. The air microbiome and that of the built environment are therefore emerging as important factors in respiratory health and are likely to be relevant to bronchiectasis [80,81,82]. Air pollution is a well-established risk associated with adverse outcomes in chronic respiratory disease including bronchiectasis including exacerbations and hospital admission [83,84,85,86]. In this context, the microbial composition of both indoor and outdoor air appear an important consideration, yet air remains an under-sampled and under-studied planetary ecosystem with potential relevance to respiratory disease [82, 83]. Metagenomics has yielded our first insight into the dynamic nature of the air microbiome revealing its composition and diel fluctuation in microbial content [82]. Furthermore, microbes have the propensity to persist in air, on surfaces and within water systems supplying the built environment, and the indoor microbiome is undoubtedly influenced by factors inherent to building design including the use of modern materials [81, 87]. It therefore seems logical that this could impact lung health as demonstrated in our recent work focused on fungal sensitization in COPD [80]. As sensitization also represents an important clinical correlate in bronchiectasis, comparable host-environment interplay is likely involved and amenable to metagenomic study [64]. This early work highlights great potential for environmental-related intervention studies in chronic respiratory disease states including bronchiectasis where modifying environmental factors could potentially provide a cost-effective and non-invasive alternative to pharmacological therapy. The application of such an approach to much larger bronchiectasis patient populations is desirable, where host and environmental metagenomes are characterized together, coupled to an assessment of the host response to better evaluate bronchiectasis endophenotypes in relation to their surrounding environment (Table 7.1).
While it is natural to focus on appraisal of the lung microbiome in chronic respiratory diseases, microbiome composition at other anatomical sites is also an important consideration. The oral microbiome – which forms a continuum with the upper and lower respiratory tract along an ecological gradient – is of relevance, as its composition may influence or predict immunological status in the lower airway or even the presence of other respiratory conditions [56, 88]. The composition of the gastrointestinal microbiome is also important both because of the potential for sub-clinical micro-aspiration of gut microbes and their accompanying inflammatory consequences. In addition, the interplay between the gut microbiome and immune homeostasis may further influence the pathogenic response to microbial encounters in the lung through the lung-gut axis [38, 89]. The role of gastrointestinal disorders such as gastroesophageal reflux disease (GERD) and irritable bowel syndrome (IBS), both reported as bronchiectasis comorbidities, may signal the presence of a dysbiotic gut microbiome [90, 91]. The development of integrative methodologies for the sequential analysis of multiple biomes are now advancing, paving the way for analysis of multiple microbiome samples from individual patients that will likely provide even more granularity for patient stratification across patient cohorts and even anatomical sites [75, 92]. The era of microbiome medicine is arriving and learning from the lessons of the past; bronchiectasis must not be left behind.
References
Barker AF. Bronchiectasis. N Engl J Med. 2002;346(18):1383–93.
Chalmers JD, Chang AB, Chotirmall SH, Dhar R, McShane PJ. Bronchiectasis. Nat Rev Dis Primers. 2018;4(1):45.
Juliusson G, Gudmundsson G. Diagnostic imaging in adult non-cystic fibrosis bronchiectasis. Breathe (Sheff). 2019;15(3):190–7.
Cole PJ. Inflammation: a two-edged sword--the model of bronchiectasis. Eur J Respir Dis Suppl. 1986;147:6–15.
King PT. The pathophysiology of bronchiectasis. Int J Chron Obstruct Pulmon Dis. 2009;4:411–9.
Flume PA, Chalmers JD, Olivier KN. Advances in bronchiectasis: endotyping, genetics, microbiome, and disease heterogeneity. Lancet. 2018;392(10150):880–90.
Mac Aogáin M, Chotirmall SH. Bronchiectasis and cough: an old relationship in need of renewed attention. Pulm Pharmacol Ther. 2019;57:101812.
Chandrasekaran R, Mac Aogain M, Chalmers JD, Elborn SJ, Chotirmall SH. Geographic variation in the aetiology, epidemiology and microbiology of bronchiectasis. BMC Pulm Med. 2018;18(1):83.
Wurzel DF, Marchant JM, Yerkovich ST, Upham JW, Petsky HL, Smith-Vaughan H, et al. Protracted bacterial bronchitis in children: natural history and risk factors for bronchiectasis. Chest. 2016;150(5):1101–8.
Tamburini S, Shen N, Wu HC, Clemente JC. The microbiome in early life: implications for health outcomes. Nat Med. 2016;22(7):713–22.
Chotirmall SH, Burke CM. Aging and the microbiome: implications for asthma in the elderly? Expert Rev Respir Med. 2015;9(2):125–8.
King PT, Holdsworth SR, Freezer NJ, Villanueva E, Holmes PW. Microbiologic follow-up study in adult bronchiectasis. Respir Med. 2007;101(8):1633–8.
Foweraker J, Wat D. Chapter 6. Microbiology of non-CF bronchiectasis. Eur Respir monogr. 2011;52:68.
Dhar R, Singh S, Talwar D, Mohan M, Tripathi SK, Swarnakar R, et al. Bronchiectasis in India: results from the European Multicentre Bronchiectasis Audit and Research Collaboration (EMBARC) and Respiratory Research Network of India Registry. Lancet Glob Health. 2019;7(9):e1269–e79.
Kunst H, Wickremasinghe M, Wells A, Wilson R. Nontuberculous mycobacterial disease and Aspergillus-related lung disease in bronchiectasis. Eur Respir J. 2006;28(2):352–7.
Aksamit TR, O’Donnell AE, Barker A, Olivier KN, Winthrop KL, Daniels MLA, et al. Adult patients with bronchiectasis: a first look at the US Bronchiectasis Research Registry. Chest. 2017;151(5):982–92.
Chotirmall SH, Martin-Gomez MT. Aspergillus species in bronchiectasis: challenges in the cystic fibrosis and non-cystic fibrosis airways. Mycopathologia. 2018;183(1):45–59.
Cuthbertson L, Felton I, James P, Cox MJ, Bilton D, Schelenz S, et al. The fungal airway microbiome in cystic fibrosis and non-cystic fibrosis bronchiectasis. J Cyst Fibros. 2021;20(2):295–302.
Mac Aogáin M, Chandrasekaran R, Lim AYH, Low TB, Tan GL, Hassan T, et al. Immunological corollary of the pulmonary mycobiome in bronchiectasis: the CAMEB study. Eur Respir J. 2018;52(1):1800766.
Mac Aogáin M, Tiew PY, Lim AYH, Low TB, Tan GL, Hassan T, et al. Distinct ‘immuno-allertypes’ of disease and high frequencies of sensitisation in non-cystic-fibrosis bronchiectasis. Am J Respir Crit Care Med. 2019;199(7):842–53.
Mitchell AB, Mourad B, Buddle L, Peters MJ, Oliver BGG, Morgan LC. Viruses in bronchiectasis: a pilot study to explore the presence of community acquired respiratory viruses in stable patients and during acute exacerbations. BMC Pulm Med. 2018;18(1):84.
Gao YH, Guan WJ, Xu G, Lin ZY, Tang Y, Lin ZM, et al. The role of viral infection in pulmonary exacerbations of bronchiectasis in adults: a prospective study. Chest. 2015;147(6):1635–43.
Normando VMF, Dias Á, da Silva ALSE, da Silva PD, de Souza Santos MC, Rodrigues CL, et al. HTLV-I induces lesions in the pulmonary system: a systematic review. Life Sci. 2020;256:117979.
Segal LN, Clemente JC, Tsay JC, Koralov SB, Keller BC, Wu BG, et al. Enrichment of the lung microbiome with oral taxa is associated with lung inflammation of a Th17 phenotype. Nat Microbiol. 2016;1:16031.
Layeghifard M, Li H, Wang PW, Donaldson SL, Coburn B, Clark ST, et al. Microbiome networks and change-point analysis reveal key community changes associated with cystic fibrosis pulmonary exacerbations. NPJ Biofilms Microbiomes. 2019;5:4.
Chalmers JD, Moffitt KL, Suarez-Cuartin G, Sibila O, Finch S, Furrie E, et al. Neutrophil elastase activity is associated with exacerbations and lung function decline in bronchiectasis. Am J Respir Crit Care Med. 2017;195(10):1384–93.
Angrill J, Agustí C, De Celis R, Filella X, Rañó A, Elena M, et al. Bronchial inflammation and colonization in patients with clinically stable bronchiectasis. Am J Respir Crit Care Med. 2001;164(9):1628–32.
Taylor SL, Rogers GB, Chen AC, Burr LD, McGuckin MA, Serisier DJ. Matrix metalloproteinases vary with airway microbiota composition and lung function in non-cystic fibrosis bronchiectasis. Ann Am Thorac Soc. 2015;12(5):701–7.
Fuschillo S, De Felice A, Balzano G. Mucosal inflammation in idiopathic bronchiectasis: cellular and molecular mechanisms. Eur Respir J. 2008;31(2):396–406.
Chalmers JD, Hill AT. Mechanisms of immune dysfunction and bacterial persistence in non-cystic fibrosis bronchiectasis. Mol Immunol. 2013;55(1):27–34.
Bedi P, Davidson DJ, McHugh BJ, Rossi AG, Hill AT. Blood neutrophils are reprogrammed in bronchiectasis. Am J Respir Crit Care Med. 2018;198(7):880–90.
Chotirmall SH. One small step for neutrophils, one giant leap for bronchiectasis. Am J Respir Crit Care Med. 2018;198(7):828–30.
Finch S, Shoemark A, Dicker AJ, Keir HR, Smith A, Ong S, et al. Pregnancy zone protein is associated with airway infection, neutrophil extracellular trap formation, and disease severity in bronchiectasis. Am J Respir Crit Care Med. 2019;200(8):992–1001.
Chotirmall SH. Stratifying bronchiectasis: getting to within a zone’s throw. Am J Respir Crit Care Med. 2019;200(8):952–4.
Chalmers JD, Haworth CS, Metersky ML, Loebinger MR, Blasi F, Sibila O, et al. Phase 2 trial of the DPP-1 inhibitor brensocatib in bronchiectasis. N Engl J Med. 2020;383(22):2127–37.
Chalmers JD, Chotirmall SH. Bronchiectasis: new therapies and new perspectives. Lancet Respir Med. 2018;6(9):715–26.
Oriano M, Gramegna A, Terranova L, Sotgiu G, Sulaiman I, Ruggiero L, et al. Sputum neutrophil elastase associates with microbiota and Pseudomonas aeruginosa in bronchiectasis. Eur Respir J. 2020;56(4):2000769.
Budden KF, Shukla SD, Rehman SF, Bowerman KL, Keely S, Hugenholtz P, et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir Med. 2019;7(10):907–20.
Boaventura R, Sibila O, Agusti A, Chalmers JD. Treatable traits in bronchiectasis. Eur Respir J. 2018;52(3):1801269.
Rogers GB, Zain NM, Bruce KD, Burr LD, Chen AC, Rivett DW, et al. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc. 2014;11(4):496–503.
Richardson H, Dicker AJ, Barclay H, Chalmers JD. The microbiome in bronchiectasis. Eur Respir Rev. 2019;28(153):190048.
Chotirmall SH, Chalmers JD. RESPIRE: breathing new life into bronchiectasis. Eur Respir J. 2018;51(1):1702444.
Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle GB, et al. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2015;12(6):821–30.
Finch S, McDonnell MJ, Abo-Leyah H, Aliberti S, Chalmers JD. A comprehensive analysis of the impact of Pseudomonas aeruginosa colonization on prognosis in adult bronchiectasis. Ann Am Thorac Soc. 2015;12(11):1602–11.
Huang HY, Chung FT, Lo CY, Lin HC, Huang YT, Yeh CH, et al. Etiology and characteristics of patients with bronchiectasis in Taiwan: a cohort study from 2002 to 2016. BMC Pulm Med. 2020;20(1):45.
Dickson RP, Erb-Downward JR, Huffnagle GB. The role of the bacterial microbiome in lung disease. Expert Rev Respir Med. 2013;7(3):245–57.
Rogers GB, Hart CA, Mason JR, Hughes M, Walshaw MJ, Bruce KD. Bacterial diversity in cases of lung infection in cystic fibrosis patients: 16S ribosomal DNA (rDNA) length heterogeneity PCR and 16S rDNA terminal restriction fragment length polymorphism profiling. J Clin Microbiol. 2003;41(8):3548–58.
Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, et al. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med. 2013;187(10):1118–26.
Rogers GB, Bruce KD, Martin ML, Burr LD, Serisier DJ. The effect of long-term macrolide treatment on respiratory microbiota composition in non-cystic fibrosis bronchiectasis: an analysis from the randomised, double-blind, placebo-controlled BLESS trial. Lancet Respir Med. 2014;2(12):988–96.
Taylor SL, Leong LEX, Mobegi FM, Choo JM, Burr LD, Wesselingh S, et al. Understanding the impact of antibiotic therapies on the respiratory tract resistome: a novel pooled-template metagenomic sequencing strategy. Multidiscip Respir Med. 2018;13(1):30.
Taylor SL, Leong LEX, Mobegi FM, Choo JM, Wesselingh S, Yang IA, et al. Long-term azithromycin reduces Haemophilus influenzae and increases antibiotic resistance in severe asthma. Am J Respir Crit Care Med. 2019;200(3):309–17.
Mac Aogáin M, Lau KJX, Cai Z, Kumar Narayana J, Purbojati RW, Drautz-Moses DI, et al. Metagenomics reveals a core macrolide resistome related to microbiota in chronic respiratory disease. Am J Respir Crit Care Med. 2020;202(3):433–47.
Cox MJ, Turek EM, Hennessy C, Mirza GK, James PL, Coleman M, et al. Longitudinal assessment of sputum microbiome by sequencing of the 16S rRNA gene in non-cystic fibrosis bronchiectasis patients. PLoS One. 2017;12(2):e0170622.
Woo TE, Lim R, Heirali AA, Acosta N, Rabin HR, Mody CH, et al. A longitudinal characterization of the non-cystic fibrosis bronchiectasis airway microbiome. Sci Rep. 2019;9(1):6871.
Zemanick ET, Wagner BD, Robertson CE, Ahrens RC, Chmiel JF, Clancy JP, et al. Airway microbiota across age and disease spectrum in cystic fibrosis. Eur Respir J. 2017;50(5):1700832.
Huffnagle GB, Dickson RP, Lukacs NW. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 2017;10(2):299–306.
Sulaiman I, Wu BG, Li Y, Scott AS, Malecha P, Scaglione B, et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur Respir J. 2018;52(4):1800810.
Fowler SJ, French J, Screaton NJ, Foweraker J, Condliffe A, Haworth CS, et al. Nontuberculous mycobacteria in bronchiectasis: prevalence and patient characteristics. Eur Respir J. 2006;28(6):1204–10.
Bonaiti G, Pesci A, Marruchella A, Lapadula G, Gori A, Aliberti S. Nontuberculous mycobacteria in noncystic fibrosis bronchiectasis. Biomed Res Int. 2015;2015:197950.
Nguyen LD, Viscogliosi E, Delhaes L. The lung mycobiome: an emerging field of the human respiratory microbiome. Front Microbiol. 2015;6:89.
Knutsen AP, Bush RK, Demain JG, Denning DW, Dixit A, Fairs A, et al. Fungi and allergic lower respiratory tract diseases. J Allergy Clin Immunol. 2012;129(2):280–91; quiz 92–3
Mac Aogáin M, Vidaillac C, Chotirmall SH. Fungal infections and ABPA. In: Cystic fibrosis. Cham: Springer; 2020. p. 93–126.
Poh TY, Tiew PY, Lim AYH, Thng KX, Binte Mohamed Ali NA, Narayana JK, et al. Increased chitotriosidase is associated with aspergillus and frequent exacerbations in south-east Asian patients with bronchiectasis. Chest. 2020;158(2):512–22.
Mac Aogáin M, Tiew PY, Lim AYH, Low TB, Tan GL, Hassan T, et al. Distinct “immunoallertypes” of disease and high frequencies of sensitization in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2019;199(7):842–53.
Delhaes L, Touati K, Faure-Cognet O, Cornet M, Botterel F, Dannaoui E, et al. Prevalence, geographic risk factor, and development of a standardized protocol for fungal isolation in cystic fibrosis: results from the international prospective study “MFIP”. J Cyst Fibros. 2019;18(2):212–20.
Tiew PY, Mac Aogáin M, Ali NABM, Thng KX, Goh K, Lau KJX, et al. The mycobiome in health and disease: emerging concepts, methodologies and challenges. Mycopathologia. 2020;185(2):207–31.
Ali N, Mac Aogáin M, Morales RF, Tiew PY, Chotirmall SH. Optimisation and benchmarking of targeted amplicon sequencing for mycobiome analysis of respiratory specimens. Int J Mol Sci. 2019;20(20):4991.
Walsh TJ, Wissel MC, Grantham KJ, Petraitiene R, Petraitis V, Kasai M, et al. Molecular detection and species-specific identification of medically important Aspergillus species by real-time PCR in experimental invasive pulmonary aspergillosis. J Clin Microbiol. 2011;49(12):4150–7.
Chen CL, Huang Y, Yuan JJ, Li HM, Han XR, Martinez-Garcia MA, et al. The roles of bacteria and viruses in bronchiectasis exacerbation: a prospective study. Arch Bronconeumol. 2020;56(10):621–9.
Chen CL, Huang Y, Martinez-Garcia MA, Yuan JJ, Li HM, de la Rosa-Carrillo D, et al. The role of Epstein-Barr virus in adults with bronchiectasis: a prospective cohort study. Open Forum Infect Dis. 2020;7(8):ofaa235.
Shkoporov AN, Hill C. Bacteriophages of the human gut: the “known unknown” of the microbiome. Cell Host Microbe. 2019;25(2):195–209.
Mitchell AB, Oliver BG, Glanville AR. Translational aspects of the human respiratory Virome. Am J Respir Crit Care Med. 2016;194(12):1458–64.
Santiago-Rodriguez TM, Hollister EB. Human virome and disease: high-throughput sequencing for virus discovery, identification of phage-bacteria dysbiosis and development of therapeutic approaches with emphasis on the human gut. Viruses. 2019;11(7):656.
Rolain JM, Fancello L, Desnues C, Raoult D. Bacteriophages as vehicles of the resistome in cystic fibrosis. J Antimicrob Chemother. 2011;66(11):2444–7.
Layeghifard M, Hwang DM, Guttman DS. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 2017;25(3):217–28.
Tipton L, Muller CL, Kurtz ZD, Huang L, Kleerup E, Morris A, et al. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome. 2018;6(1):12.
Taylor SL, Woodman RJ, Chen AC, Burr LD, Gordon DL, McGuckin MA, et al. FUT2 genotype influences lung function, exacerbation frequency and airway microbiota in non-CF bronchiectasis. Thorax. 2017;72(4):304–10.
Faner R, Sibila O, Agustí A, Bernasconi E, Chalmers JD, Huffnagle GB, et al. The microbiome in respiratory medicine: current challenges and future perspectives. Eur Respir J. 2017;49(4):1602086.
Li CX, Wheelock CE, Skold CM, Wheelock AM. Integration of multi-omics datasets enables molecular classification of COPD. Eur Respir J. 2018;51(5):1701930.
Tiew PY, Ko FWS, Pang SL, Matta SA, Sio YY, Poh ME, et al. Environmental fungal sensitisation associates with poorer clinical outcomes in COPD. Eur Respir J. 2020;56(2):2000418.
Horve PF, Lloyd S, Mhuireach GA, Dietz L, Fretz M, MacCrone G, et al. Building upon current knowledge and techniques of indoor microbiology to construct the next era of theory into microorganisms, health, and the built environment. J Expo Sci Environ Epidemiol. 2020;30(2):219–35.
Gusareva ES, Acerbi E, Lau KJX, Luhung I, BNV P, Kolundzija S, et al. Microbial communities in the tropical air ecosystem follow a precise diel cycle. Proc Natl Acad Sci U S A. 2019;116(46):23299–308.
Landrigan PJ, Fuller R, Acosta NJR, Adeyi O, Arnold R, Basu NN, et al. The Lancet Commission on pollution and health. Lancet. 2018;391(10119):462–512.
Garcia-Olivé I, Stojanovic Z, Radua J, Rodriguez-Pons L, Martinez-Rivera C, Ruiz MJ. Effect of air pollution on exacerbations of bronchiectasis in Badalona, Spain, 2008–2016. Respiration. 2018;96(2):111–6.
Raji H, Riahi A, Borsi SH, Masoumi K, Khanjani N, AhmadiAngali K, et al. Acute effects of air pollution on hospital admissions for asthma, COPD, and bronchiectasis in Ahvaz, Iran. Int J Chron Obstruct Pulmon Dis. 2020;15:501–14.
Goeminne PC, Cox B, Finch S, Loebinger MR, Bedi P, Hill AT, et al. The impact of acute air pollution fluctuations on bronchiectasis pulmonary exacerbation: a case-crossover analysis. Eur Respir J. 2018;52(1):1702557.
Poh TY, Ali NABM, Mac Aogáin M, Kathawala MH, Setyawati MI, Ng KW, et al. Inhaled nanomaterials and the respiratory microbiome: clinical, immunological and toxicological perspectives. Part Fibre Toxicol. 2018;15(1):46.
Yuan BC, Yeh YT, Lin CC, Huang CH, Liu HC, Chiang CP. Clinical detection of chronic rhinosinusitis through next-generation sequencing of the oral microbiota. Microorganisms. 2020;8(6):959.
Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell. 2019;176(6):1340–55.e15
Deshpande NP, Riordan SM, Castaño-Rodríguez N, Wilkins MR, Kaakoush NO. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome. 2018;6(1):227.
Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569(7758):655–62.
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–50.
Shkoporov AN, Clooney AG, Sutton TDS, Ryan FJ, Daly KM, Nolan JA, et al. The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe. 2019;26(4):527–41.e5
Soret P, Vandenborght LE, Francis F, Coron N, Enaud R, Avalos M, et al. Respiratory mycobiome and suggestion of inter-kingdom network during acute pulmonary exacerbation in cystic fibrosis. Sci Rep. 2020;10(1):3589.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mac Aogáin, M., Chalmers, J.D., Chotirmall, S.H. (2022). Bronchiectasis. In: Huang, Y.J., Garantziotis, S. (eds) The Microbiome in Respiratory Disease. Respiratory Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-87104-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-87104-8_7
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-87103-1
Online ISBN: 978-3-030-87104-8
eBook Packages: MedicineMedicine (R0)