
Abstract
Bronchiectasis is a disorder characterized by a persistent airway inflammation and recurrent infections, defined by a common pathological end point: irreversible bronchial dilatation due to diverse etiologies. Several factors have been related to a vicious cycle that perpetuates airway inflammation and increases the risk of bronchial infections, including various immunological disorders. Increased recruitment and migration of neutrophils to airways, in contrast to reduced phagocytic activity of neutrophils in the airways, have been recently described. In addition, proteolytic enzymes degrading extracellular matrix components of the lung, such as metalloproteinases, or airway proteins with antimicrobial effects, such as mucins, have also recently been said to play an important role in airway defense in bronchiectasis. Furthermore, the development of culture-independent techniques of microbiological analysis has contributed to better understanding of the role of microbiome in the pathogenesis and evolution of the disease. In this chapter, we review and discuss evidence and new insights into the pathophysiology, immunology, and histopathology of bronchiectasis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


5.1 Introduction
The main characteristic of bronchiectasis (BE) is permanent bronchial dilatation with chronic airway inflammation and frequent infections. There is no simple cause or mechanism to explain its pathophysiology, because bronchiectasis represents an endpoint of various causes and of a complex interplay among inflammation, immune response, and microorganisms that play a part in this chronic respiratory disease.
There are several limitations for identifying mechanisms of pathophysiology in BE: (1) the lack of animal models for replicate bronchiectasis; (2) the numerous and various pathological conditions that are implicated in its development; and (3) the limited studies investigating the pathophysiology of this condition until very recently [1].
5.1.1 Vicious Cycle
Cole in 1986 [2] proposed the first model to explain the pathogenesis of BE, calling it a “vicious cycle” hypothesis. This hypothesis suggested that after an initial infectious event that compromised mucociliary clearance, microorganisms will reproduce in the airway, provoking inflammation of and damage to the epithelium. The persistence of microorganisms that chronically infect airways would attract more inflammatory cells that release factors capable of injuring the airway and maintaining inflammation. The chronic inflammation would make microbial clearance difficult by promoting more colonization closing—hence, the vicious cycle. Although recent investigations proved that airways are not as sterile as previously believed, the Cole hypothesis has been found to be applicable as a basis for research and for clinical investigations. In fact, some mechanisms involved in the pathogenesis of BE related to inflammation, infection persistence, and tissue damage, have now been clarified, using the "vicious cycle" as a framework. Today, it is recognized that in the pathogenesis there is an inappropriate interplay between airway host and microorganisms required to perpetuate the disease, leading to inefficient resolution of inflammation and infection, structural damage, and progression of the disease. New insights provide interesting information about the role of inflammation cells and the new concept of microbiome (Fig. 5.1).

Mechanisms involved in the vicious cycle
5.2 Neutrophils
BE is considered to be a neutrophil-driven disease because these cells are crucial to its development and evolution. In the physiological host response against microorganisms, neutrophils are rapidly recruited to airways, where they degranulate their cytotoxic and immune molecules. The presence of a prominent number of neutrophils in the airway is one of the hallmarks of BE and it has been confirmed in sputum, bronchoalveolar lavage, and in bronchial biopsies in association with concomitant high levels of chemotactic molecules, such as CXCL-8 and leukotrienes LTB4 [3].
The neutrophilic airway infiltration was found also in the stable phase of the disease, and in the absence of any conventional microbiological isolate (negative culture). Nevertheless, in patients chronically infected with pathogens, the burden of the neutrophils encountered was higher [4] than in patients with negative sputum/BAL culture. Dente et al. [5], in a cross-sectional study, examined inflammatory cells in the sputum and exhaled breath condensate of stable patients. They confirmed an increase of neutrophils in sputum that was higher in those with chronic Pseudomonas aeruginosa. Moreover, they correlated inflammation with severity scores (Bronchiectasis Severity Index), respiratory functional data, and the Leicester Cough Questionnaire score. In this study, they also evaluated oxidative stress determining malondialdehyde in breath condensate that was found to correlate with the number of prior exacerbations in the previous year.
5.2.1 Inflammatory Cells Recruitment and Migration to Airways (Fig. 5.2)
Neutrophils are recruited to distal airways due to the presence of high concentrations of chemoattractants—mainly IL-1b, TNF-α, IL 8 and leukotriene b4 [4]—that are contained in the airways. During migration to airways, neutrophils are activated, and there is a shedding of L-selectin; express integrins CD11/CD18 bind to ICAM-1, VCAM-1 and selectins in the endothelial cells. The role of adhesion molecules expressed on the surface of endothelial cells and leukocytes is important in BE patients because these molecules are responsible for mediating the migration of intravascular leukocytes into inflamed tissue [3]. In BE patients, the expression of CD11b/CD18 in the neutrophil surface, and L-selectin shedding, were reported to be normal, whereas in cystic fibrosis (CF), CD11b/CD18 was up-regulated and L-selectin was decreased [6]. Zheng et al. [7] have found increased serum levels of E-selectin, ICAM-1, and VCAM-1 in stable bronchiectasis patients. ICAM-1 increases, due to the raised levels of inflammatory cytokines—mainly TNFα and IL-1b—and VCAM-1 are also expressed in the presence of LPS. The increase of these adhesion molecules implies that they actively participate in transporting neutrophils to inflamed sites. Interestingly, both E-selectin and ICAM-1 levels were inversely related to forced expiratory volume in 1 s (FEV1) and positively with the number of affected lobes. The authors suggest that the source of this up-regulation of neutrophil migration could take place in the endothelium of dilated airways.

Neutrophil recruitment to airways. From [3]
All these findings together seem to suggest that the recruitment process is rather normal, although highly activated, due to the raised levels of chemoattractants [8] capable of initiating and maintaining the process. It is worth pointing out that an increased bacterial load (≥1 × 107 cfu/ml) has been associated with higher serum intercellular adhesion molecule-1, E-selectin, and vascular cell adhesion molecule-1 [9].
5.2.2 Neutrophil Activity and Phagocytosis
The ability of neutrophils to eliminate microorganisms is based on three mechanisms: (1) phagocytosis, through pattern recognition receptors; (2) degranulation of its granules: defensins (human neutrophil peptides), proteases—mainly elastases—mieloperoxidase, and lactoferrin; and (3) extruding DNA with the development of neutrophil extracellular traps (NETs) which, acting in combination with other antimicrobial proteins, enable the killing of microorganisms [10].
Although neutrophils in airways exhibited an abnormal function, it was conserved before getting airways [11]. In blood, no differences in phagocytic capacity or superoxide generation were found in idiopathic bronchiectasis when compared with controls; however, in contrast, Ruchaud-Saparagnano et al. described an enhancement of neutrophil phagocytosis and superoxide generation induced by granulocyte-macrophage colonystimulating factor (GM-CSF) [12].
Neutrophils isolated in the sputum of both CF and BE patients exhibited defective phagocytosis [13]. That deficiency was related to a higher concentration of HNP in the lung. HNP -1, −2, −3 αHNP are proteins stored in the neutrophilic granules with an antimicrobial activity; high levels of HNP could exert an inhibitory phagocytic function. Although the exact domain of HNP that determines the disturbance in phagocytosis is not known, Voglis et al. [13] have reported depressed surface Fcγ RIII, actin-filament remodeling, enhanced intracellular Ca(2+), and degranulation. These researchers suggested that HNP could be considered a potential target for novel treatments. Despite the high number of neutrophils recruited in BE airways, its diminished phagocytosis favors a scenario that leads to inefficient bacteria killing, along with increased damage by the release of its potent proteases.
5.2.3 Neutrophil Elastase
The release of proteases, especially elastase, plays a key role in the pathogenesis. Elastase that can digest phagocytized bacteria is also capable of destroying structural proteins such as elastin, fibronectin, collagen, α1-antitrypsin, and tissue inhibitors of matrix metalloproteinase [14]. Moreover, elastase is a potent secretagogue of IL-8, IL-6 release; it stimulates muc5A gene expression and granulocyte colony-stimulating factor (G-CSF). Finally, it also has cilliotoxic and cytotoxic properties that contribute to airway damage; therefore, neutrophil elastase depresses many innate defenses, facilitating P. aeruginosa infection [14]. There is a positive correlation between elastase levels, inflammatory markers and total gelatinolytic activity in sputum [15] with spirometric alterations and radiographic findings. In fact, elastase concentration was positively correlated with a percentage of neutrophils in a 24 h sputum volume, levels of IL-8 and TNF-α [16], and even sputum purulence.
Elastase has a considerably negative effect on phagocytosis and on the process of inflammation resolution [17]. In fact, it causes a cleavage of phosphatidylserine receptors to phagocytes, thereby disrupting the phagocytosis of apoptotic cells (Fig. 5.3). The result is a delay in apoptosis clearance. Moreover, it has been reported that a higher secondary cell necrosis and reduced number of macrophages was probably due to the concomitant proinflammatory cytokines [4], contributing to the persistence of inflammation.

Neutrophil phagocytic impairment. From [3]
Recently, elastase has also been shown to trigger the expression of senescence markers on bronchial epithelial cells [14]. In CF patients, three senescence markers –p16, gH2A.X, and phospho-Chk2—were found to be highly expressed in airway sections [18]. Elastase increased in vitro p16 expression and decreased CKD4 activity in CF bronchial epithelial cells [18]. In a small pilot study performed in eight BE patients, telomere-induced senescence was investigated. A significantly increased proportion of short telomeres was found without an increase in p16 expression, but with an increase in other senescent pathways, such as p21 and TAF, and with a decrease in SIRT1 [19].
All these elastase effects are decisive for disease progression; in fact, elastase quantification has a better predictive value for lung function decline in comparison with other biomarkers (AUROC 0.68) [20]. This points to elastase as a potential target to contain disease, with some ongoing studies using oral inhibitors [21].
5.2.4 Metalloproteases
Matrix metalloproteinases (MMPs) are activated by neutrophil elastase and are able to degrade airway matrices, therefore playing a crucial role in extracellular matrix modelling. Their levels (except for tissue inhibitors of metalloproteinases, TIMP-1) correlated positively with sputum IL-8 and TNF-α, suggesting their relationship with neutrophil airway inflammation. Sputum MMP-8, MMP-9 and MMP-9/TIMP-1 ratio were found to be significantly increased in BE patients and positively correlated with clinical measures, including high-resolution computed tomography (HRCT) scores, spirometry, P. aeruginosa isolation, and the Bronchiectasis Severity Index [22].
The environment of neutrophils, elastases, cytokines, MMP, and chemoattractants leads to ongoing inflammation, along with the destruction of airways of bronchial walls. This scenario hampers bacterial elimination and contributes to maintaining airway damage. The result is persistent infection that is enhanced by the fact that microorganisms also develop mechanisms directed to evading host response, such as biofilm or hypermutation [23].
5.3 Other Cells
5.3.1 Macrophages
An increase in macrophages has been reported [4], although their specific role is not well defined. They promote neutrophil chemotaxis [24], coordinate inflammatory response by synthesis of TNF-α, IL-8, LTB4 and elastolytic enzymes, and finally eliminate apoptotic cells. The higher number of apoptotic neutrophils reported in BE patients is probably secondary to their impaired phagocytosis by macrophages due to the presence of excessive elastase [17]. Wat et al. have also found an abundance of secondary necrotic cells macrophages in sputum, and lower numbers of macrophages capable of amplifying inflammation compared levels of neutrophil apoptosis during an exacerbation [4].
5.3.2 Natural Killer (NK)
These cells accumulate in the lung parenchyma during inflammation and recruit neutrophils and T lymphocytes as part of the host response against microorganisms. Boyton and Altmann [25] have reported a functional impairment of NK that may favor the development of bronchiectasis, with increased risk of chronic bacterial infection. These events, together with excessive NK cell activation, create a highly inflammatory lung environment which, in turn, lead to the perpetuation of chronic infection.
5.4 Mucins
Mucins are the major macromolecular component of the mucus gel in health [26]. Mucus is a protective airway coating secreted in the healthy airways, composed of water, salt, and proteins. The correct balance of these components is essential for the protective function of the mucus layer. Experimental studies and clinical studies in other chronic lung diseases have suggested the crucial role that mucins play in airway defense against bacterial infections [27, 28]. In bronchiectasis, one study has evaluated the relationship among secreted mucins (MUC2, MUC 5 AC, and MUCB) and the presence of bacterial airway colonization [29]. In this study, authors included 50 stable bronchiectasis patients, showing that chronically colonized patients had higher MUC2 sputum levels compared with those without airway colonization. In addition, those patients colonized by P. aeruginosa showed the highest levels, and there is a correlation of MUC2 and MUC5AC levels with disease severity and neutrophil elastase activity, suggesting a role of mucins in airway defense in bronchiectasis.
5.5 Microbiome
In recent years, understanding of human lung microbiome has increased. The new technology of bacterial ribosomal RNA sequencing and related techniques have transformed our understanding of the relationship between microbial ecology and human health. Healthy airways are not sterile, and the diverse bacterial communities that exist in the oral cavity and upper airways constantly enter the lungs through micro-aspiration and are eliminated via mucociliary clearance and immune response [30,31,32].
Bacterial infection is central to our understanding of the pathophysiology of bronchiectasis. Traditional culture-based microbiology techniques have revealed the importance of such well-characterized pathogens as Haemophilus influenzae and P. aeruginosa [3]. However, microbiome studies have been causing an evolution in our understanding of these diseases. Previously unrecognized organisms are found in the microbiome of patients with bronchiectasis both when clinically stable and during exacerbation. Tunney et al. reported that complex polymicrobial communities were present in the lungs of patients with bronchiectasis, including high numbers of anaerobic bacteria such as Prevotella, Veillonella. and Actinomyces [33]. In this study, the authors showed that microbial load and community composition—both before and after antibiotic treatment of patients with acute pulmonary exacerbations—were stable, suggesting that changes in lung microbiota composition do not account for pulmonary exacerbations.
Others studies have detected more than 140 bacterial species in the sputum and bronchoalveolar lavage of patients with bronchiectasis [34,35,36]. H. influenzae, P. aeruginosa, Streptococcus pneumoniae, Veilonella dispar and Neiserria subflava were reported to be the most common ones. Bacterial community composition was related to lung function and neutrophils count [35], suggesting that characteristics of lower airways microbiota in bronchiectasis was correlated significantly with clinical markers of disease. In addition, stratification of patients on the basis of predominant bacterial taxa (P. aeruginosa, H. influenzae and other taxa) was more clinically informative than conventional culture [34, 36]. The predominance of P. aeruginosa, followed by the Veilonella species, was the best predictor of future exacerbations frequency, while H. influenzae's predominance communities had significantly fewer episodes. Furthermore, the presence of P. aeruginosa and H. influenzae was related to increased inflammatory disease in terms of C-reactive protein (CRP), IL-8, and IL-1β.
Other studies have characterized the airway microbiome following antibiotic treatment. Patients included in the BLESS trial [37] who had received long-term erythromycin treatment changed the composition of respiratory microbiota more than those who received the placebo [34, 36]. These changes were most substantial in patients with airway infections dominated by organisms other than P. aeruginosa, and primarily reflected reductions in the relative abundance of H. influenzae and increases in intrinsically macrolide-tolerant organisms. These findings suggested potentially deleterious consequences of maintenance macrolide treatment on the composition of airway microbial community, and were not detected using traditional cultures.
In some studies, researchers have begun to evaluate the role of "host response" and their relationship to the lung microbiome in various chronic lung diseases [38, 39]. The key to understanding the pathogenesis of these diseases may reside in deciphering the complex interactions between the host, pathogen, and resident microbiota during stable disease and exacerbations. In bronchiectasis, a recent study evaluated the relationships among lung microbiome and MMPs [40]. In this study, the authors evaluated the concentrations of nine MMPs and four tissue inhibitors of metalloproteinases in induced sputum from 86 bronchiectasis patients and eight healthy controls, and related their levels to airway microbiota classified as P. aeruginosa-dominated, H. influenzae-dominated, and dominated by other species. The main results were that increased MMP levels (particularly MMP-8 and MMP-1) and MMP/TIMP rations, were found in patients with bronchiectasis, compared with healthy controls. Regarding microbiomes, MMP profiles differed according to the dominant pathogen. Patients in whom P. aeruginosa was dominant had increased MMP-9 activity, while patients with H. influenzae dominance had increased MMP-1, MMP-2, and MMP-8 activity (Fig. 5.4). These findings suggested a possibility of differential airway remodelling according to airway microbiology.

Composition of airway microbiota and various matrix metalloproteinases. From [40]
In summary, although all of these finding may be clinically relevant, the role of lung microbiomes in the pathophysiology of bronchiectasis is not yet completely understood. Further studies concentrating on better understanding of the relationship with the host immune response are crucial for increasing our knowledge in this promising field.
5.5.1 Microbiome and Immunology
New evidence shows the role of dysbiosis and specific bacteria in modulating T-cell differentiation. Moreover, it is also possible that intestinal microbiome drives changes in lung microbiota and lung-immune differentiation. The term "immune dysregulation" has been considered in the pathogenesis of BE, since in that disease an immune deficiency of various causes, or hyperimmune activation, may be found [41]. Several immune deficiency conditions have been associated with BE as a deficit of IgG subclasses, common variable immunodeficiency, low mannose-binding lectin levels, hyper IgE syndrome, and a defect in the transporter associated with antigen presentation [42]. On the other hand, in chronic granulomatous disease, inflammatory activation may coexist along with a component of immune deficiency.
The current state-of-the-art of lung immunology in BE is still uncertain, due to the few studies carried out on airway cells. Boyton and Altmann [25, 41] proposed a pathway for TH17 immunity. According to them, diverse microbiota species may interact with innate receptors on antigen-presenting cells favoring induction and differentiation of TH 17- CD4 cells. These cells secrete IL 17 in response to bacteria, and local IL 17 leads to neutrophilia and mucous secretions. Although the main effect of the Th 17 pathway is defending against microorganisms, it may also cause damage to the airways. A persistent TH17 activation could drive to produce ectopic lymphoid follicles with CD4 T-cells and B-cells. The activation of a TH17 pathway was studied in endobronchial biopsies and in broncoalveolar lavage. Chen et al. [43] found that Th17 cytokines—IL 17A and IL 23—were significantly higher in bronchiectasis than in control subjects, and had a higher gene expression of IL-17A, IL-1β, IL-8, and IL-23 in biopsies.
5.6 Histopathology
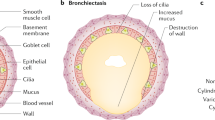
The main characteristic of BE is permanent irreversible dilatation of bronchial airways accompanied by wall thickening and the loss of distal narrowing. This general basic description comes from classic and older publications on lungs, from autopsies, or from surgery. In fact, there is a lack of recent pathology descriptions from a wide range of patients in distinct phases of these diseases. Traditionally, three morphologic types were described, from the less to the more severe: cylindrical, varicose, and cystic. Cylindrical BE presents thick-walled bronchi reaching the lung periphery (at 1 cm of lung), with peribronchial fibrosis and without normal tapering. The term "varicose BE" is due to the “varicose vein” aspect caused by the irregular bronchial wall. "Cystic BEs" are in general groups of cysts that may be filled with air or mucus, with a patchy distribution [44].
The thickening of bronchial airways is caused by inflammation, and normal mucosal and muscular layers are substituted by edema, ulceration, or fibrosis [45]. In later stages, polymorphonuclear transmural inflammation can be associated with micro-abscesses of airways. In proximal airways, the structural cartilage can be diminished, provoking a corresponding reduction of supportive structure. Proximal bronchi or distal bronchioles may be filled with mucus or necrotic debris, eventually forming plugs that obstruct airways. In the most advances phases, neovascular bronchial arterioles with thick walls have been described. In 1952, Whitwell [46] coined the term "follicular bronchiectasis," due to the presence of an excessive formation of lymphoid tissue—follicles and nodes—within the walls of dilated bronchi. That finding was more frequently accompanied by an enlargement of proximal lymph nodes. The distribution and location depends on the etiology and/or cause of bronchiectasis.
Nowadays, high-resolution computed tomography (HRCT) has become the better non-invasive method for envisaging gross pathologic features in BE: morphology, distribution, extent, and severity [47]. A bronchus is considered dilated by HRCT when the luminal airway diameter is more than 1.5 times the adjacent vessel, and mucus or plugs filling the bronchus can also be observed. When small airways are affected, peripheral, irregular, short (2-4 mm) linear branching markings are noted and the term “tree-in-bud pattern” is applicable. Cysts in the bronchial wall are a feature of more destructive bronchiectasis; in more advanced cases, the grape-like cysts appear in clusters (cystic bronchiectasis).
The most modern immune-staining techniques, and the more frequent use of bronchial biopsies, have been providing detailed information with regard to the inflammatory cell types. Zheng et al. [48] in endobronchial biopsies in stable patients, have shown higher neutrophils, macrophages, and TNFα . The higher density of MMP-8 and MMP-9 positive cells in the lamina propia of airways was correlated with neutrophils, but not with macrophages [49]. Gaga et al. [24] in research on 12 patients, described inflammation with neutrophils, CD4+ T-cells, and CD68+ macrophages, increased IL-8 expression, and mucous gland hypertrophy in up to 40% of some tissue sections. The T-cells and IL-8+ cells' infiltration was lower in patients receiving corticosteroids. The presence of higher T-cell counts—CD4+, CD8+, and IL-17+ in airways—was observed in children with bronchiectasis, whether it was from cystic fibrosis or not. Tan et al. demonstrated submucosal Th17 (CD4 + IL-17+) lymphocytes in endobronchial biopsies along with IL-17+ neutrophils, γδT cells, and natural killer cells in the BE airways [50]. Recently, Chen et al. studied the gene expression of IL-17A, IL-1β, IL-8, and IL-23 in endobronchial biopsies, and Th17 pathway cytokines in bronchoalveolar lavage fluid [7], showing no differences for IL-17A gene expression. However, gene expression of IL1β and IL- 8 was significantly higher in BAL fluid, while IL-8 and IL-1α levels showed significant relationships with clinical measures and airway microbiology.
References
Chalmers JD, Elborn JS. Reclaiming the name “bronchiectasis”. Thorax. 2015;70:399–400. https://doi.org/10.1136/thoraxjnl-2015-206956.
Cole PJ. Inflammation: a two-edged sword−the model of bronchiectasis. Eur J Respir Dis Suppl. 1986;147:6–15.
Chalmers JD, Hill AT. Mechanisms of immune dysfunction and bacterial persistence in non-cystic fibrosis bronchiectasis. Mol Immunol. 2013;55:27–34. https://doi.org/10.1016/j.molimm.2012.09.011.
Watt AP, Brown V, Courtney J, Kelly M, Garske L, Elborn JS, Ennis M. Neutrophil apoptosis, proinflammatory mediators and cell counts in bronchiectasis. Thorax. 2004;59:231–6. https://doi.org/10.1136/thx.2003.008037.
Dente FL, Bilotta M, Bartoli ML, Bacci E, Cianchetti S, Latorre M, Malagrinò L, Nieri D, Roggi MA, Vagaggini B, Paggiaro P. Neutrophilic bronchial inflammation correlates with clinical and functional findings in patients with noncystic fibrosis bronchiectasis. Mediat Inflamm. 2015;2015:642503. https://doi.org/10.1155/2015/642503.
McShane PJ, Naureckas ET, Tino G, Strek ME. Non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2013;188:647–56. https://doi.org/10.1164/rccm.201303-0411CI.
Zheng L, Tipoe G, Lam WK, Leung RYH, Ho JC, Shum IH, Ooi GC, Ip MS, Tsang KW. Up-regulation of circulating adhesion molecules in bronchiectasis. Eur Respir J. 2000;16:691–6. https://doi.org/10.1034/j.1399-3003.2000.16d21.x.
Russell DW, Gaggar A, Solomon GM. Neutrophil fates in bronchiectasis and alpha-1 antitrypsin deficiency. Ann Am Thorac Soc. 2016;13(Suppl 2):S123–9. https://doi.org/10.1513/AnnalsATS.201512-805KV.
Chalmers JD, Smith MP, McHugh BJ, Doherty C, Govan JR, Hill AT. Short- and long-term antibiotic treatment reduces airway and systemic inflammation in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2012;186:657–65. https://doi.org/10.1164/rccm.201203-0487OC.
Cooper PR, Palmer LJ, Chapple ILC. Neutrophil extracellular traps as a new paradigm in innate immunity: friend or foe? Periodontol. 2013;2000(63):165–97. https://doi.org/10.1111/prd.12025.
Hardy CL, King SJ, Mifsud NA, Hedger MP, Phillips DJ, Mackay F, de Kretser DM, Wilson JW, Rolland JM, O’Hehir RE. The activin a antagonist follistatin inhibits cystic fibrosis-like lung inflammation and pathology. Immunol Cell Biol. 2015;93:567–74. https://doi.org/10.1038/icb.2015.7.
Ruchaud-Sparagano M-H, Gertig H, Hester KL, Macfarlane JG, Corris PA, Simpson AJ, de Soyza A. Effect of GM-CSF on neutrophil function in idiopathic bronchiectasis. Respirology. 2013;18(8):1230–5. https://doi.org/10.1111/resp.12138.
Voglis S, Quinn K, Tullis E, Liu M, Henriques M, Zubrinich C, Peñuelas O, Chan H, Silverman F, Cherepanov V, Orzech N, Khine AA, Cantin A, Slutsky AS, Downey GP, Zhang H. Human neutrophil peptides and phagocytic deficiency in bronchiectatic lungs. Am J Respir Crit Care Med. 2009;180:159–66. https://doi.org/10.1164/rccm.200808-1250OC.
Gifford AM, Chalmers JD. The role of neutrophils in cystic fibrosis. Curr Opin Hematol. 2014;21:16–22. https://doi.org/10.1097/MOH.0000000000000009.
Goeminne PC, Vandooren J, Moelants EA, Decraene A, Rabaey E, Pauwels A, Seys S, Opdenakker G, Proost P, Dupont LJ. The sputum colour chart as a predictor of lung inflammation, proteolysis and damage in non-cystic fibrosis bronchiectasis: a case-control analysis. Respirology. 2014;19:203–10. https://doi.org/10.1111/resp.12219.
Tsang KW, Chan KN, Ho PL, Zheng L, Ooi GC, Ho JCM, Lam WK. Sputum elastase in steady-state bronchiectasis. Chest. 2000;117:420–6. https://doi.org/10.1378/chest.117.2.420.
William Vandivier R, Fadok VA, Hoffmann PR, Bratton DL, Penvari C, Brown KK, Brain JD, Accurso FJ, Henson PM. Elastase-mediated phosphatidylserine receptor cleavage impairs apoptotic cell clearance in cystic fibrosis and bronchiectasis. J Clin Invest. 2002;109:661–70. https://doi.org/10.1172/JCI200213572.
Fischer BM, Wong JK, Degan S, Kummarapurugu AB, Zheng S, Haridass P, Voynow JA. Increased expression of senescence markers in cystic fibrosis airways. Am J Phys Lung Cell Mol Phys. 2013;258:L394–400. https://doi.org/10.1152/ajplung.00091.2012.
Birch J, Victorelli V, Rahmatika D, Anderson R, Jiwa K, Moisey E, Ward C, Fisher A, De Soyza A. Telomere dysfunction and senescence-associated pathways in bronchiectasis. Am J Respir Crit Care Med. 2016;193:929–31.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med. 2012;186:857–65. https://doi.org/10.1164/rccm.201203-0507OC.
Stockley R, De Soyza A, Gunawardena K, Perrett J, Forsman-Semb K, Entwistle N, Snell N. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. 2013;107:524–33. https://doi.org/10.1016/j.rmed.2012.12.009.
Guan WJ, Gao YH, Xu G, Lin ZY, Tang Y, Gu YY, Liu GH, Li HM, Chen RC, Zhong NS. Sputum matrix metalloproteinase-8 and -9 and tissue inhibitor of metalloproteinase-1 in bronchiectasis: clinical correlates and prognostic implications. Respirology. 2015;20:1073–81. https://doi.org/10.1111/resp.12582.
Whitters D, Stockley R. Immunity and bacterial colonisation in bronchiectasis. Thorax. 2012;67:1006–13. https://doi.org/10.1136/thoraxjnl-2011-200206.
Gaga M, Bentley AM, Humbert M, Barkans J, O’Brien F, Wathen CG, Kay AB, Durham SR. Increases in CD4+ T lymphocytes, macrophages, neutrophils and interleukin 8 positive cells in the airways of patients with bronchiectasis. Thorax. 1998;53:685–91. https://doi.org/10.1136/thx.53.8.685.
Boyton RJ, Altmann DM. Immune regulation in idiopathic bronchiectasis. Ann N Y Acad Sci. 2012;1272:68–72. https://doi.org/10.1111/j.1749-6632.2012.06756.x.
Rose MC. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86:245–78. https://doi.org/10.1152/physrev.00010.2005.
Henke MO, Renner A, Huber RM, Seeds MC, Rubin BK. MUC5AC and MUC5B mucins are decreased in cystic fibrosis airway secretions. Am J Respir Cell Mol Biol. 2004;31:86–91. https://doi.org/10.1165/rcmb.2003-0345OC.
Roy MG, Livraghi-butrico A, Fletcher AA, Melissa M, Evans SE, Boerner RM, Alexander SN, Lindsey K, Song AS, Petrova YM, Tuvim MJ, Adachi R, Romo I, Bordt AS, Bowden MG, Sisson JH, Prescott G, Thornton DJ, Rousseau K, Garza MMD, Seyed J, Karmouty-Quintana H, Blackburn MR, Drouin SM, Davis W, Terrell KA, Grubb BR, Neal WKO, Flores SC, Cota-gomez A, Lozupone CA, Donnelly JM, Watson AM, Hennessy CE, Keith RC, Yang IV, Barthel L, Peter M, Janssen WJ, Schwartz DA, Boucher RC, Burton F. Muc5b is required for airway defence. Nature. 2014;505:412–6. https://doi.org/10.1038/nature12807.Muc5b.
Sibila O, Suarez-Cuartin G, Rodrigo-Troyano A, Fardon TC, Finch S, Mateus EF, Garcia-Bellmunt L, Castillo D, Vidal S, Sanchez-Reus F, Restrepo MI, Chalmers JD. Secreted mucins and airway bacterial colonization in non-CF bronchiectasis. Respirology. 2015;20:1082–8. https://doi.org/10.1111/resp.12595.
Dickson RP, Erb-Downward JR, Huffnagle GB. Homeostasis and its disruption in the lung microbiome. Am J Phys Lung Cell Mol Phys. 2015;309:L1047–55. https://doi.org/10.1152/ajplung.00279.2015.
Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir Med. 2014;2(3):238–46. https://doi.org/10.1016/S2213-2600(14)70028-1.
Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The microbiome and the respiratory tract. Annu Rev. Physiol. 2015;78:1–24. https://doi.org/10.1146/annurev-physiol-021115-105238.
Tunney MM, Einarsson GG, Wei L, Drain M, Klem ER, Cardwell C, Ennis M, Boucher RC, Wolfgang MC, Elborn JS. Lung microbiota and bacterial abundance in patients with bronchiectasis when clinically stable and during exacerbation. Am J Respir Crit Care Med. 2013;187:1118–26. https://doi.org/10.1164/rccm.201210-1937OC.
Rogers GB, Bruce KD, Martin ML, Burr LD, Serisier DJ. The effect of long-term macrolide treatment on respiratory microbiota composition in non-cystic fibrosis bronchiectasis: an analysis from the randomised, double-blind, placebo-controlled BLESS trial. Lancet Respir Med. 2014;2:988–96. https://doi.org/10.1016/S2213-2600(14)70213-9.
Rogers GB, van der Gast CJ, Cuthbertson L, Thomson SK, Bruce KD, Martin ML, Serisier DJ. Clinical measures of disease in adult non-CF bronchiectasis correlate with airway microbiota composition. Thorax. 2013;68:731–7. https://doi.org/10.1136/thoraxjnl-2012-203105.
Rogers GB, Zain NMM, Bruce KD, Burr LD, Chen AC, Rivett DW, McGuckin MA, Serisier DJ. A novel microbiota stratification system predicts future exacerbations in bronchiectasis. Ann Am Thorac Soc. 2014;11:496–503. https://doi.org/10.1513/AnnalsATS.201310-335OC.
Serisier DJ, Martin ML, McGuckin MA, Lourie R, Chen AC, Brain B, Biga S, Schlebusch S, Dash P, Bowler SD. Effect of long-term, low-dose erythromycin on pulmonary exacerbations among patients with non-cystic fibrosis bronchiectasis. JAMA. 2013;309:1260. https://doi.org/10.1001/jama.2013.2290.
Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, Erb-Downward JR, Huffnagle GB, Hayashi S, Elliott WM, Cooper J, Sin DD, Lenburg ME, Spira A, Mohn WW, Hogg JC. Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;192:438–45. https://doi.org/10.1164/rccm.201502-0223OC.
O’Dwyer DN, Dickson RP, Moore BB. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. 2016;196:4839–47. https://doi.org/10.4049/jimmunol.1600279.
Taylor SL, Rogers GB, Chen ACH, Burr LD, McGuckin MA, Serisier DJ. Matrix metalloproteinases vary with airway microbiota composition and lung function in non-cystic fibrosis bronchiectasis. Ann Am Thorac Soc. 2015;12:701–7. https://doi.org/10.1513/AnnalsATS.201411-513OC.
Boyton RJ, Altmann DM. Bronchiectasis: current concepts in pathogenesis, immunology, and microbiology. Annu Rev Pathol. 2016;11:523–54. https://doi.org/10.1146/annurev-pathol-012615-044344.
Daheshia M, Prahl JD, Carmichael JJ, Parrish JS, Seda G. The immune response and its therapeutic modulation in bronchiectasis. Pulm Med. 2012;2012:280528. https://doi.org/10.1155/2012/280528.
Chen ACH, Martin ML, Lourie R, Rogers GB, Burr LD, Hasnain SZ, Bowler SD, McGuckin MA, Serisier DJ. Adult non-cystic fibrosis bronchiectasis is characterised by airway luminal Th17 pathway activation. PLoS One. 2015;10:e0119325. https://doi.org/10.1371/journal.pone.0119325.
Dodd JD, Lavelle LP, Fabre A, Brady D. Imaging in cystic fibrosis and non-cystic fibrosis bronchiectasis. Semin Respir Crit Care Med. 2015;36:194–206. https://doi.org/10.1055/s-0035-1546749.
Moulton BC, Barker AF. Pathogenesis of bronchiectasis. Clin Chest Med. 2012;33(2):211–7. https://doi.org/10.1016/j.ccm.2012.02.004.
Whitwell F. A study of the pathology and pathogenesis of bronchiectasis. Thorax. 1952;7:213–39.
Webb WR, Müller NJ, Naidich DP. High-resolution CT of the lung. Airway diseases. 3rd ed. Philadelphia, PA: Lippincot Williams; 2009. p. 492–529.
Zheng L, Shum IH, Tipoe GL, Leung R, Lam WK, Ooi GC, Tsang KW. Macrophages, neutrophils and tumour necrosis factor-?? Expression in bronchiectatic airways in vivo. Respir Med. 2001;95:792–8. https://doi.org/10.1053/rmed.2001.1155.
Zheng L, Lam WK, Tipoe GL, Shum IH, Yan C, Leung R, Sun J, Ooi GC, Tsang KW. Overexpression of matrix metalloproteinase-8 and -9 in bronchiectatic airways in vivo. Eur Respir J. 2002;20:170–6. https://doi.org/10.1183/09031936.02.00282402.
Tan H-L, Regamey N, Brown S, Bush A, Lloyd CM, Davies JC. The Th17 pathway in cystic fibrosis lung disease. Am J Respir Crit Care Med. 2011;184:252–8. https://doi.org/10.1164/rccm.201102-0236OC.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter
Cite this chapter
Menéndez, R., Sibila, O. (2018). Pathophysiology, Immunology, and Histopathology of Bronchiectasis. In: Chalmers, J., Polverino, E., Aliberti, S. (eds) Bronchiectasis. Springer, Cham. https://doi.org/10.1007/978-3-319-61452-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-61452-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-61451-9
Online ISBN: 978-3-319-61452-6
eBook Packages: MedicineMedicine (R0)