
Abstract
Skeletal muscle (SM) metabolism is strictly controlled by both thyroid hormones and insulin action.
SM plasticity gives the muscle the ability to regenerate after a damage thanks to proliferation, fusion, and differentiation of resident, undifferentiated, mononuclear satellite cells (SCs).
The transcription factor paired box 7 (Pax7) represents a marker of quiescent satellite stem cells (Pax7+) in skeletal muscle, while skeletal muscle-specific myogenic regulatory factors (MRFs: MyoD, myogenin, MYF-5, MRF4) drive muscle differentiation.
Based on the molecular marker expression, myoblasts fuse with damaged myofibers to repair them or fuse together to generate new multinucleated myofibers, and new self-renewed satellite cells to maintain their own population.
Thyroid plays an important role in muscle tissue contraction, formation, and repair regulation, mediated by both blood and local tissue concentration of thyroid hormones (TH: thyroxine or T4 and triiodothyronine or T3) modulated by TH transporters/receptors efficiency and by enzyme deiodinase (DIO2, DIO3) activity.
DIO3 supports the proliferation of myoblasts and is essential for the stem cell activation program, and DIO2 plays a critical role in differentiation and fusion process to form the muscle fibers.
Duration and extent of thyroid hormone deficiency are often correlated with severity of muscle pain. In this contest, hypothyroid myopathy represents the most important clinical problem consequent to mitochondrial dysfunction associated with fiber switch, fast muscle fibers loss, and muscle insulin resistance.
Muscle weakness, exacerbation of muscle fatigue, and exercise intolerance characterize both hypothyroidism and hyperthyroidism. Hyperthyroidism is also associated with vision loss and thyrotoxic periodic paralysis with serious cardiopulmonary complications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Skeletal muscle (SM) represents the largest component tissue (40–50%) of the total body mass. SM wide distribution makes it one of the tissues most involved in body energy consumption, glucose and lipid homeostasis [1].
SM metabolism is strictly controlled by both thyroid hormones and insulin action; accordingly, muscle metabolism, differentiation, repair, and contractile activity are dramatically impaired by insulin resistance condition and thyroid dysfunction [2].
Therefore, before considering the consequence of hypothyroidism and hyperthyroidism on skeletal muscle, it is useful briefly to highlight some points of skeletal muscle physiology.
1 Skeletal Muscle Physiology
Skeletal muscle is a very heterogeneous tissue consisting of different post-mitotic polynucleated fibers that develop following the fusion of many embryonic muscle cells (myoblasts) and group together to generate body movement.
Based on distinct contractile and metabolic properties, muscle fibers are divided into type I (also known as slow) having oxidative metabolism, IIA (also known as fast) having intermediate metabolic properties, and IIx (also known as fastest) having glycolytic metabolism. Their different properties are a result of an adaptive mechanism which allows the muscle to respond to different metabolic and mechanical demands [3].
Skeletal muscle shows significant plasticity in response to a multiplicity of stimuli.
Fiber type switching is a common response to an increase of exercise training or physical inactivity [4], and skeletal muscle mass changes through hypertrophic or atrophic processes [5]. In particular, skeletal muscle has a remarkable ability to regenerate after a damage [6].
The nuclei present in the muscle fibers are unable to replicate since they have irreversibly escaped the cell cycle and are in a permanent postmitotic state. It follows that these terminally differentiated cells are not able to repair any loss of tissue, which may occur due to trauma or degenerative diseases, not being able to restore the mitotic activity of its nuclei.
On the other hand, the tissue repair takes place thanks to the presence of a small population (1–6% of total muscle nuclei) of resident, undifferentiated cells that retain the ability to self-renew and differentiate, referred to as satellite cells (SCs) [7].
Mononuclear satellite cells, residing between the basal lamina and the sarcolemma of the mature muscle fibers, are normally quiescent. Following the loss or degeneration of muscle fibers, the satellite cells are stimulated to replicate by forming a progeny of cells destined to fuse together, repeating a myogenic process similar to that of the embryonic muscle myoblasts (Fig. 12.1). The syncytial structures originated by the program of proliferation, fusion, and differentiation restore both the integrity of the muscle tissue and the pool of muscle stem cells.

Muscle stem cells in mature muscle fibers are in a quiescence state until homeostatic stimuli, i.e., hormones, cytokines, or injury, induce their self-activation, re-enter into the cell cycle and consequently their proliferation in order to promote differentiation process
During postnatal life, the activation of quiescent SCs promotes muscle fiber growth and maturation. Indeed, SC depletion produces an abnormal skeletal muscle development: myofiber size is significantly reduced at the same manner as muscle mass.
The crucial role of SCs in muscle regeneration has been extensively demonstrated [8]. Skeletal muscle injury induces the activation of a multi-step process that coordinates the SC cell cycle re-enter, proliferation and differentiation, aimed at complete tissue repair.
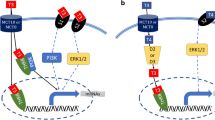
Muscle differentiation is orchestrated by four skeletal muscle-specific myogenic regulatory factors (MRFs): MyoD, myogenin, MYF-5, and MRF4.
Myogenic regulators represent nuclear phosphoproteins that bind to DNA at similar sites, activating the transcription of muscle-specific genes [9].
Several studies have proven that the transcription factor paired box 7 (Pax7) is an absolute requirement for the normal function [10] of SCs and represents a marker of quiescent satellite stem cells (Pax7+) in skeletal muscle. Pax7 is essential for the survival of the satellite cells and for the transcriptional activation of the myogenic gene MyoD; MyoD plays a critical role in satellite cell biogenesis, survival, and self-renewal and represents the earliest and crucial expression marker of SC activation [10].
Posttrauma, a marked increase in Pax-7 expression near the injury zone, has been described. The event would indicate an increase in the recruitment of satellite stem cells and the activation of the skeletal muscle regeneration cascade near the lesion area [11].
MYF5 protein levels are high during quiescence, whereas MyoD levels are negligible [12].
When satellite cells are activated rapidly induce expression of MyoD.
In proliferating SCs, MYF5 and MyoD drive transcription of genes that facilitate cell cycle progression, and regulate timely myogenic progression during regeneration. MyoD induces myogenin expression and simultaneously downregulates Myf5 expression.
MyoD and Myogenin cooperate in enhancing the expression of these genes which rapidly drive cell cycle output [13].
Moreover, MyoD and myogenin enhance MRF4 expression and other specific genes of late muscle differentiation, leading to the formation of mature myofibers. In myofibers, MRF4 together with heavy myosin chain protein (MyHC) are highly expressed and represent the main markers of mature differentiated muscle, whereas MyoD and myogenin are downregulated [13] (Fig. 12.2).

Adult activated satellite cells initially express MyoD or Myf5 or both myogenic factors. Subsequently, the proliferating myoblasts express MyoD and Myf5. MyoD induces the expression of myogenin, the downregulation of Myf5 and the production of MRF4, the main marker, together with MRF4, of mature differentiated myofibers
Based on the molecular markers, two proliferation models have been developed providing myoblasts that fuse with damaged myofibers to repair them or that fuse together to generate new myofibers, and new self-renewed satellite cells to maintain their own population.
-
1.
Following muscle damage, the quiescent Pax7+ satellite cells coexpress the MyoD factor. After proliferating as myoblasts derived from Pax7+ and MyoD+ cells, most of the cells continue to express MyoD while inhibiting Pax7 in order to differentiate for myonucleus replacement.
The other progeny, on the other hand, loses the expression of MyoD but retains that of Pax7 and contributes to the renewal of the satellite cells themselves.
-
2.
Two types of Pax7+ satellite cells exist, depending on whether or not they express the MYF5 marker (Pax7+/MYF5+ and Pax7+/MYF5−).
In relationship with aging, there is a functional decline of the SCs: their capacity of activation and differentiation decreases, leading to a decline in the regenerative function [14]. Aged muscle SCs respond to injury with limited expansion and differentiation capacity, contributing to the decline in muscle regenerative potential.
Although the intervention of SCs in muscle regeneration is recognized, their role in the processes of atrophy (loss of muscle mass) and hypertrophy (increase in muscle mass) in response to external stimuli, intrinsic factors, or physical activity is not yet clear [15, 16].
Muscle tissue plasticity implies a series of mass fluctuations: muscle subjected to resistance exercise becomes hypertrophic, mass and strength increase, but drastically decrease following atrophy due to immobilization, sepsis, cachexia, etc.
The consequences of atrophy have obvious health implications. Muscle weakness is an important factor for both mortality and morbidity and is associated with an increased risk of all causes of death [17]. Indeed, reducing muscle atrophy in cancer cachexia can significantly prolong life [18]. Additionally, many older individuals suffer from sarcopenia, a prolonged muscle wasting disorder that typically begins after the age of 50 years and results in a loss of approximately 1% of muscle mass per year [19]. This means that by the age of 80 years, sarcopenic individuals have lost about 40% of their muscle mass, a key factor in falls, frailty, and nursing home admissions. Consequently, understanding the mechanisms and potential therapeutic responses to atrophy is of broad clinical and basic interest.
The syncytial nature of the muscle cells and their large number of nuclei represent a unique feature that allows the muscle fibers to reach enormous lengths (up to 600 mm) and the nuclei to produce adequate amounts of mRNA to generate and maintain muscle mass.
In the past, the plastic nature of muscle and its syncytial organization have led to formulate the “myonuclear domain hypothesis” which identifies SCs as fundamental in the processes of hypertrophy and atrophy [20]. “Myonuclear domain hypothesis” was developed on the basis of “sphere of influence” concept postulated at the end of the nineteenth century by Strassburger [21] that each nucleus is able to support a limited volume of cytoplasm, thus defining the size limit of the cell. The syncytial nature allows the muscle fiber to greatly increase its size, but during hypertrophy or atrophy, new nuclei are added or lost to preserve the right ratio of nuclei/cytoplasm.
Although some controversy remains, substantial data has shown that during the hypertrophy process, the number of nuclei increases [22, 23]. The muscle fiber inherits the supernumerary nuclei from the satellite cells which, stimulated by anabolic steroids or focal lesions following resistance exercises, proliferate and finally merge with the muscle fiber, facilitating both repair and growth.
However, an interesting study, performed using an animal model characterized by a conditional depletion of SCs, has shown that SCs are not necessary to induce hypertrophic response to overload at short time [24]. However, the same authors have shown that long-term SC ablation affects muscle hypertrophy [24].
Following the myonuclear domain hypothesis, if hypertrophy involves the addition of new nuclei, atrophy implies their loss. Although numerous apoptotic cells appear within the atrophic tissue, several authors have shown that the atrophic process involves a reduction in the volume of muscle fibers, but no loss of myonuclei [25, 26]. Contrasting the myonuclear domain hypothesis, it appears that the myonuclei acquired by the fibers persist even when the muscle becomes atrophic.
2 Thyroid Hormones and Muscle Tissue
Thyroid plays an important regulatory function of metabolism, contractile function, process of formation, and repair of muscle tissue.
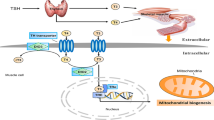
The influence of the thyroid gland on muscle is mediated not only by the blood concentration of thyroid hormones (TH: thyroxine or T4 and triiodothyronine or T3) but also by the local tissue levels of TH, consequent to the efficiency of TH transporters, TH receptors, and the activity of the enzyme deiodinase (DIO).
The intracellular availability of TH depends on the efficiency of the facilitated transport of TH across the plasma membrane, mediated by primary monocarboxylate transporters MCT10 and MCT8 [27].
Type 2 (DIO2) and 3 (DIO3) iodothyronine deiodases also contribute to the control of intracellular TH levels: production and inactivation of T3 are closely related to DIO2 and DIO3 activities [28].
DIO2 increases the availability and effect of T3 by converting T4 to T3, while DIO3 conversely reduces them by converting T4 to reverse T3 (rT3) and T3 to diiodothyronine (T2), unable to interact with TH receptors.
T3 is transported and concentrated in the cell nucleus where it is bound by specific free receptors α and β (TRA mainly present in SM and TRB). Each receptor has a binding site for DNA and a distinct site for binding T3. The T3-TR complex, after dimerization of the receptor and interaction with transcription factors, binds to DNA and promotes the interaction with TH response elements (TREs), activating the transcription of target genes and therefore the synthesis of proteins necessary for the physiological effect of T3 in the muscle cell occurs [2, 29].
Consequently, the intramuscular levels of T3, their link with TRs, and the consequent effects depend on the balance of activity between DIO2 and DIO3.
In this scenario, thyroid exerts a control over the myogenic progression of activated satellite cells, in a spatial- and temporal-regulated manner, showing an important impact on the proliferation and differentiation balance of muscle stem cells.
The expression of D3 and D2 is finely regulated during the different stages of myogenesis within SCs. D3 is expressed only in the proliferative and therefore early phase of myogenesis, while D2 is expressed in the late phase, playing an essential role in the differentiation process [30].
It is conceivable that the early expression of D3 is aimed at keeping the TH signal inactive to allow proliferation and prevent the differentiation of stem cells. In fact, both D3-depletion and TH treatment induce the expression of pro-apoptotic genes in muscle stem cells.
In this context, D3 supports the proliferation of myoblasts and is essential for the stem cell activation program.
At the end of the proliferation phase, the activated myoblasts undergo the differentiation process, fusing to form the muscle fibers. The production of T3 by DIO2 plays a critical role in this process. T3 production contributes to the induction of myogenic factors such as MyoD, the main regulator of the myogenic development and regeneration program [31] (Fig. 12.3).

Differential expression of DIO3 and DIO2 determines the increase of active thyroid hormone T3 destined to induce and complete the myogenesis process
3 Hypothyroidism and Muscle Tissue
Most patients with hypothyroidism mention various muscle pains, from stiffness to cramps.
Muscle symptoms are often underestimated without considering that they can represent an important manifestation of hypothyroidism. Sometimes the muscular symptoms dominate the clinical picture or even can present themselves as the only onset manifestation of hypothyroidism, so the differential diagnosis with other causes of myopathy becomes difficult.
The severity of muscle pain correlates with the duration and extent of thyroid hormone deficiency [2, 32].
Hypothyroid myopathy represents the most important clinical problem [33]. This muscle pathology includes four different subtypes: (1) myasthenic syndrome, beginning in childhood and usually causes mobility loss in aging [34], (2) Kocher–Debre–Semelaigne syndrome, which in children causes severe muscle atrophy [35], (3) Hoffmann syndrome, usually associated with primary hypothyroidism in adults that often report painful spasms, slow movements, and proximal muscle weakness [36], and (4) severe muscle atrophy. In rare cases, hypothyroid myopathy evolves into rhabdomyolysis [37].
Increased level of creatine kinase and other muscle enzymes, such as lactic dehydrogenase, are typical biochemical alterations of hypothyroid myopathy [33, 37], and recently other markers, i.e. titin and desmin, are proposed to evaluate skeletal muscle damage associated with thyroid dysfunction [38].
At cellular level, mitochondrial dysfunction is the main feature of hypothyroid-induced muscle impairment. Hypothyroidism induces a metabolic myopathy, with a reduction of the energetic production and mitochondrial metabolism, due to an inhibition of the main oxidative pathways and the respiratory chain. T3 influences mitochondrial activity by modulating the expression of proteins encoded by both the nuclear and mitochondrial genome.
This metabolic modification is associated with fiber switch: hypothyroidism causes the loss of fast muscle fibers. Skeletal muscles of hypothyroid patients are primarily constituted by slow fibers having numerous fibrotic depots that aggravate inflammation condition [33, 39]. As known, T3 plays a crucial role in the metabolism of the connective tissue, and its deficiency is associated with an altered increase in the synthesis of glycosaminoglycans as demonstrated by an increased urinary excretion of glycosaminoglycans in hypothyroid patients [33, 40]. Moreover, slow muscle fibers are characterized by an internal area called “core-like areas,” not having enzymatic activity and intermyofibrillar material [41]. The loss of muscle morphology and activity is corroborated by high abnormal expression of vimentin, desmin, fetal, and neonatal MyHC isoforms, the latter marker of muscle regeneration process [33].
In addition, hypertrophy, typical compensatory response observed in hypothyroid muscle, is insufficient [33], and hypothyroid patients have a reduced capacity to perform exercise training [34] due to mitochondrial dysfunction that causes abnormal accumulation of protons and ions, in particular Ca2+ the main regulator of actin–myosin interaction, membrane excitability, and glucose metabolism [35]. Moreover, thyroid hormone loss promotes glycogen accumulation in muscle, but mitochondrial oxidative pathway impairment causes defective utilization of glycogen and simultaneously activates anaerobic metabolism that exacerbates the risk of cramps and fatigue [42]. Considering the peculiar interconnection between hypothyroidism and obesity, the reduced ability to exercise creates a vicious circle that further exacerbates the metabolic condition of patients with hypothyroidism.
In addition, different works have demonstrated how in hypothyroid rats skeletal muscle show a less response to insulin, and in general, insulin impairment action in hypothyroid skeletal muscle causes leads to insulin resistance in hypothyroid patients [43].
Finally, it is important to note that hypothyroidism causes not only muscle damage but also peripheral nervous system impairment, and then muscle damage induced by hypothyroidism should be seen from a neuromuscular perspective.
4 Hyperthyroidism and Muscle Tissue
The only muscle clinical manifestation in common between patients with hypothyroidism and hyperthyroidism is represented by muscle weakness, comprising both the upper and lower extremities, and exacerbation of muscle fatigue and exercise intolerance [44].
Different authors have proposed that these muscle alterations are caused by weight loss because rarely hyperthyroid patients refer only muscle weakness as only symptom of their pathology. In contrast to hypothyroid myopathy, biochemical markers, such as creatine kinase, have normal levels in hyperthyroid patients [45].
Hyperthyroidism is associated with Grave’s ophthalmopathy disease, characterized by severe damage of muscles that control movement of the eyelids and eye leading to vision loss, and with thyrotoxic periodic paralysis, characterized by overproduction of TH and a simultaneous hypokalemia, not due to altered intake of potassium but due to a sudden intracellular intake.
Thyrotoxic periodic paralysis is a rare complication of hyperthyroidism which causes complete momentaneous immobility that involves not only proximal and distal limb muscles but also respiratory musculature. Moreover, cardiovascular alterations are important and typical manifestations of thyrotoxic periodic paralysis, and to avoid serious cardiopulmonary complications, it is fundamental to restore immediately optimal potassium concentration [46, 47].
References
Salvatore D, Simonides WS, Dentice M, Zavacki AM, Larsen PR. Thyroid hormones and skeletal muscle—new insights and potential implications. Nat Rev Endocrinol. 2014;10:206–14. https://doi.org/10.1038/nrendo.2013.238.
Bloise FF, Cordeiro A, Ortiga-Carvalho TM. Role of thyroid hormone in skeletal muscle physiology. J Endocrinol. 2018;236(1):R57–68. https://doi.org/10.1530/JOE-16-0611.
Dave HD, Shook M, Varacallo M. Anatomy, skeletal muscle. In: StatPearls [Internet]. Treasure Island, FL: StatPearls; 2020.
Widmann M, Nieß AM, Munz B. Physical exercise and epigenetic modifications in skeletal muscle. Sports Med. 2019;49(4):509–23. https://doi.org/10.1007/s40279-019-01070-4.
Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun. 2021;12(1):330. https://doi.org/10.1038/s41467-020-20123-1.
Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr Physiol. 2015;5(3):1027–59. https://doi.org/10.1002/cphy.c140068.
Morgan JE, Partridge TA. Muscle satellite cells. Int J Biochem Cell Biol. 2003;35(8):1151–6.
Chen B, Shan T. The role of satellite and other functional cell types in muscle repair and regeneration. J Muscle Res Cell Motil. 2019;40(1):1–8. Epub 2019 Apr 9. https://doi.org/10.1007/s10974-019-09511-3.
Asfour HA, Allouh MZ, Said RS. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp Biol Med (Maywood). 2018;243(2):118–28. Epub 2018 Jan 7. PMID: 29307280; PMCID: PMC5788151. https://doi.org/10.1177/1535370217749494.
Olguín HC, Pisconti A. Marking the tempo for myogenesis: Pax7 and the regulation of muscle stem cell fate decisions. J Cell Mol Med. 2012;16(5):1013–25. https://doi.org/10.1111/j.1582-4934.2011.01348.x.
Kansal R, Kanojia RK, Kumar V, Vaiphei K, Dhillon MS. Role of PAX-7 as a tissue marker in mangled extremity: a pilot study. Eur J Orthop Surg Traumatol. 2019;29(5):1131–40. Epub 2019 Mar 9. https://doi.org/10.1007/s00590-019-02410-w.
Yoshida N, Yoshida S, Koishi K, et al. Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J Cell Sci. 1998;111(Pt 6):769–79.
Zammit PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. 2017;72:19–32. https://doi.org/10.1016/j.semcdb.2017.11.011.
Hwang AB, Brack AS. Muscle stem cells and aging. Curr Top Dev Biol. 2018;126:299–322. Epub 2017 Nov 16. https://doi.org/10.1016/bs.ctdb.2017.08.008.
Fukada SI, Akimoto T, Sotiropoulos A. Role of damage and management in muscle hypertrophy: different behaviors of muscle stem cells in regeneration and hypertrophy. Biochim Biophys Acta Mol Cell Res. 2020;1867(9):118742. https://doi.org/10.1016/j.bbamcr.2020.118742.
Schwartz LM. Skeletal muscles do not undergo apoptosis during either atrophy or programmed cell death-revisiting the Myonuclear domain hypothesis. Front Physiol. 2019;9:1887. https://doi.org/10.3389/fphys.2018.01887.
Metter EJ, Talbot LA, Schrager M, Conwit R. Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J Gerontol A Biol Sci Med Sci. 2002;57(10):B359–65. https://doi.org/10.1093/gerona/57.10.b359.
Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142(4):531–43. https://doi.org/10.1016/j.cell.2010.07.011.
Woo J. Sarcopenia. Clin Geriatr Med. 2017;33(3):305–14. Epub 2017 May 13. https://doi.org/10.1016/j.cger.2017.02.003.
Qaisar R, Larsson L. What determines myonuclear domain size? Indian J Physiol Pharmacol. 2014;58(1):1–12.
Strassburger E. Ûber die wirkungssphäre der kerne und die zellgrösse. Histol Beitr. 1893;5:97–124.
Dalbo VJ, Roberts MD, Mobley CB, Ballmann C, Kephart WC, Fox CD, Santucci VA, Conover CF, Beggs LA, Balaez A, Hoerr FJ, Yarrow JF, Borst SE, Beck DT. Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia. 2017;49(3). https://doi.org/10.1111/and.12622.
Masschelein E, D’Hulst G, Zvick J, Hinte L, Soro-Arnaiz I, Gorski T, von Meyenn F, Bar-Nur O, De Bock K. Exercise promotes satellite cell contribution to myofibers in a load-dependent manner. Skelet Muscle. 2020;10(1):21. https://doi.org/10.1186/s13395-020-00237-2.
Englund DA, Murach KA, Dungan CM, Figueiredo VC, Vechetti IJ Jr, Dupont-Versteegden EE, McCarthy JJ, Peterson CA. Depletion of resident muscle stem cells negatively impacts running volume, physical function, and muscle fiber hypertrophy in response to lifelong physical activity. Am J Physiol Cell Physiol. 2020;318(6):C1178–88. https://doi.org/10.1152/ajpcell.00090.2020.
Bruusgaard JC, Johansen IB, Egner IM, Rana ZA, Gundersen K. Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc Natl Acad Sci U S A. 2010;107:15111–6. https://doi.org/10.1073/pnas.0913935107.
Duddy WJ, Cohen T, Duguez S, Partridge TA. The isolated muscle fibre as a model of disuse atrophy: characterization using PhAct, a method to quantify f-actin. Exp Cell Res. 2011;317:1979–93. https://doi.org/10.1016/j.yexcr.2011.05.013.
Van Mullem AA, van Gucht ALM, Visser WE, Meima ME, Peeters RP, Visser TJ. Effects of thyroid hormone transporters MCT8 and MCT10 on nuclear activity of T3. Mol Cell Endocrinol. 2016;437:252–60. https://doi.org/10.1016/j.mce.2016.07.037.
Louzada RA, Carvalho DP. Similarities and differences in the peripheral actions of thyroid hormones and their metabolites. Front Endocrinol (Lausanne). 2018;9:394. PMID: 30072951. https://doi.org/10.3389/fendo.2018.00394.
Rurale G, Di Cicco E, Dentice M, Salvatore D, Persani L, Marelli F, Luongo C. Thyroid hormone hyposensitivity: from genotype to phenotype and back. Front Endocrinol (Lausanne). 2020;10:912. https://doi.org/10.3389/fendo.2019.00912.
Dentice M, Ambrosio R, Damiano V, Sibilio A, Luongo C, Guardiola O, Yennek S, Zordan P, Minchiotti G, Colao A, Marsili A, Brunelli S, Del Vecchio L, Larsen PR, Tajbakhsh S, Salvatore D. Intracellular inactivation of thyroid hormone is a survival mechanism for muscle stem cell proliferation and lineage progression. Cell Metab. 2014;20(6):1038–48.
Dentice M, Marsili A, Ambrosio R, Guardiola O, Sibilio A, Paik JH, Minchiotti G, DePinho RA, Fenzi G, Larsen PR, Salvatore D. The FoxO3/type 2 deiodinase pathway is required for normal mouse myogenesis and muscle regeneration. J Clin Invest. 2010;120(11):4021–30. Epub 2010 Oct 11. PMID: 20978344; PMCID: PMC2964991. https://doi.org/10.1172/JCI43670.
Rodolico C, Bonanno C, Pugliese A, Nicocia G, Benvenga S, Toscano A. Endocrine myopathies: clinical and histopathological features of the major forms. Acta Myol. 2020;39(3):130–5. https://doi.org/10.36185/2532-1900-017.
Sindoni A, Rodolico C, Pappalardo MA, Portaro S, Benvenga S. Hypothyroid myopathy: a peculiar clinical presentation of thyroid failure. Review of the literature. Rev Endocr Metab Disord. 2016;17(4):499–519. https://doi.org/10.1007/s11154-016-9357-0.
Amin S, Aung M, Gandhi FR, Pena Escobar JA, Gulraiz A, Malik BH. Myasthenia gravis and its association with thyroid diseases. Cureus. 2020;12(9):e10248. https://doi.org/10.7759/cureus.10248.
Mishra D, Juneja M. Kocher-Debre-Semelaigne syndrome. J Pediatr Neurosci. 2014;9(3):289–90. https://doi.org/10.4103/1817-1745.147570.
Achappa B, Madi D. Hoffmann’s syndrome- a rare form of hypothyroid myopathy. J Clin Diagn Res. 2017;11(5):OL01–2. https://doi.org/10.7860/JCDR/2017/21234.9913.
Giampietro O, Clerico A, Buzzigoli G, Del Chicca MG, Boni C, Carpi A. Detection of hypothyroid myopathy by measurement of various serum muscle markers—myoglobin, creatine kinase, lactate dehydrogenase and their isoenzymes. Correlations with thyroid hormone levels (free and total) and clinical usefulness. Horm Res. 1984;19(4):232–42. https://doi.org/10.1159/000179893.
Zybek-Kocik A, Sawicka-Gutaj N, Domin R, Szczepanek-Parulska E, Krauze T, Guzik P, Ruchała M. Titin and dystrophin serum concentrations changes in patients affected by thyroid disorders. Endokrynol Pol. 2021;72(1):1–7. Epub ahead of print. https://doi.org/10.5603/EP.a2020.0083.
Modi G. Cores in hypothyroid myopathy: a clinical, histological and immunofluorescence study. J Neurol Sci. 2000;175(1):28–32. https://doi.org/10.1016/s0022-510x(00)00266-5.
Musielak MC, Chae JH. Hypothyroid-induced acute compartment syndrome in all extremities. J Surg Case Rep. 2016;2016(12):rjw215. https://doi.org/10.1093/jscr/rjw215.
Khaleeli AA, Gohil K, McPhail G, Round JM, Edwards RH. Muscle morphology and metabolism in hypothyroid myopathy: effects of treatment. J Clin Pathol. 1983;36(5):519–26. https://doi.org/10.1136/jcp.36.5.519.
Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94(2):355–82. https://doi.org/10.1152/physrev.00030.2013.
Czech MP, Malbon CC, Kerman K, Gitomer W, Pilch PF. Effect of thyroid status on insulin action in rat adipocytes and skeletal muscle. J Clin Invest. 1980;66(3):574–82. https://doi.org/10.1172/JCI109889.
Mathew P, Rawla P. Hyperthyroidism. In: StatPearls [Internet]. Treasure Island, FL: StatPearls; 2020.
McGrowder DA, Fraser YP, Gordon L, Crawford TV, Rawlins JM. Serum creatine kinase and lactate dehydrogenase activities in patients with thyroid disorders. Niger J Clin Pract. 2011;14(4):454–9. https://doi.org/10.4103/1119-3077.91755.
Iqbal QZ, Zia Z, Niazi M, Sattar SBA, Quyyumi S. Thyrotoxic muscle paralysis as a rare cause of reversible muscle weakness: a case report. Cureus. 2020;12(9):e10634. https://doi.org/10.7759/cureus.10634.
Lu KC, Hsu YJ, Chiu JS, Hsu YD, Lin SH. Effects of potassium supplementation on the recovery of thyrotoxic periodic paralysis. Am J Emerg Med. 2004;22(7):544–7. https://doi.org/10.1016/j.ajem.2004.09.016.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Terruzzi, I. (2021). Muscle Tissue in Hypothyroidism and Hyperthyroidism. In: Luzi, L. (eds) Thyroid, Obesity and Metabolism. Springer, Cham. https://doi.org/10.1007/978-3-030-80267-7_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-80267-7_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-80266-0
Online ISBN: 978-3-030-80267-7
eBook Packages: MedicineMedicine (R0)