
Abstract
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of the nuclear hormone receptor superfamily comprising three subtypes: PPARα, PPARγ, and PPARβ/δ. The PPAR family of nuclear receptors is centrally involved in regulating whole-body energy homeostasis and metabolic function. Endogenous ligands include free fatty acids, eicosanoids, and leukotrienes. Synthetic ligands developed to serve as full agonists aim at treating diabetes type 2, hyperlipidemia, and other metabolic-related diseases. Further, there has been a developing interest in the role of PPAR agonist’s role in neurodegenerative disease. However, many of these clinically practical therapeutics are associated with harmful effects on human health. Therefore, new approaches have led to a new class of selective PPAR modulators (SPARMs), or partial agonists meet this challenge. In addition, these partial agonists have been observed to show a favorable impact on insulin sensitivity, blood glucose levels, and dyslipidemia with significantly reduced side effects on human health. Partial agonists have been found to display differences in transcriptional and cellular outcomes by acting through distinct structural and dynamic mechanisms within the ligand-binding region when compared to full agonists. Recently, a new focus on PPAR agonists’ class has intensified for neurodegenerative diseases, as new ligands and novel biological roles have emerged particularly for its therapeutic potential in Alzheimer’s disease (AD). The present chapter critically analyzes current PPAR ligands using in silico modeling and the implication of promising new therapeutics in neurodegenerative disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
10.1 Introduction
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily that are ligand-activated transcription factors [1]. These receptors have been linked to many systemic and cellular functions including insulin sensitivity and whole-body energy regulation. PPARs exhibit isotype-specific tissue expression patterns based upon energy demand in the regulation of cellular process. PPARα is abundantly expressed in tissues that utilize fatty acid catabolism such as the heart, liver, brown adipose tissue, and kidney [2, 3]. PPARγ exists in two isoforms, γ1 and γ2, and is principally expressed in white and brown adipose tissues, where they regulate adipocyte differentiation and lipid storage [4, 5]. Although expression of PPARγ2 is mostly observed in adipose tissue, PPARγ1 is ubiquitously expressed in tissues including the gut and immune cells, where they promote anti-inflammatory processes [6]. PPARδ/β has broad expression patterns and prominent roles in the skeletal muscle, adipose tissue, skin, gut, and brain [7, 8].
10.2 Overview of PPARs and Their Structures
PPARs act as transcription factors by binding and functionally responding to endogenous small molecule ligands [9]. These ligands, either endogenous or synthetic, bind to an orthosteric pocket existing in the core of the nuclear receptor ligand-binding domain. PPARs exist as a conserved domain association, including a central DNA-binding domain (DBD) flanked by two regulatory regions, a distinct amine (N)-terminal activation function-1 (AF-1) domain. In addition there is also a carboxyl (C)-terminal ligand-binding domain (LBD) containing the activation function-2 (AF-2) domain that has a coregulator interaction surface [10, 11] (Fig. 10.1). The N-terminal regulatory domain consists of A and B domains and the AF-1 domain, which is involved in ligand-independent coregulator binding [12, 13]. This region is not conserved because it varies greatly between members of this class of nuclear receptors. Two highly conserved zinc fingers are central in the recognition of specific DNA half-sites termed peroxisome proliferator response elements (PPREs) [14]. These sites are represented as either direct or indirect repeats and are separated by a spacer of base pairs. Each zinc finger contains several cysteine residues allowing for the interactions to a zinc ion. These specific zinc finger motifs permit for the discernment of nuclear receptors from other DBDs (Fig. 10.1).

(a) Linear illustration of the PPARγ structure, where the ligand binds to the ligand-binding domain (LBD ) in the AF-2 domain. (b) Full agonist rosiglitazone bound to PPARγ in complex with the heterodimeric partner RXR. PPARγ LBD lies within the RXR-LBD and DNA (DBD) to stabilize interactions with the PPRE. Additional recruitment of coactivator peptides to the PPARγ AF-2 LBD and RXR-LBD allows for additional gene transcription through ligand-stabilized conformations. Ligand stabilization of the β-sheet region and the H2’ and H3 helices allow for enhanced stabilization of the RXR-DBD and additional binding of the PPARγ DBD to the PPRE. Specific ligand-induced modifications may allow for enhanced coactivator recruitment and enhanced gene transcription. Crystal structure PDB-(3DZY) was used for constructing the image [10]
Mechanistically, DNA binding allows for either the activation and recruitment of DNA transcription machinery or the repression of transcription. All members of the PPAR nuclear receptor superfamily bind to DNA as a heterodimer, where the DNA binding is in association with the retinoic acid receptor (RXR) [14]. Each DBD subunit binds to a separate DNA half-site. The most poorly conserved PPAR region is the flexible hinge domain, which allows for rotation between the DBD and the LBD, and contains a nuclear localization signal. The LBD is the largest domain in PPAR molecule and is highly conserved across all PPARs. Ligand binding stabilizes the AF-2 domain and facilitates the interaction with coregulator molecules to remodel chromatin resulting in the induction of gene expression [15]. Although the LBD is highly conserved, specific differences exist in the amino acid make-up of the active site and thus influences the ligand specificity. Ligand binding influences the conformation of the ligand-binding surface of the AF-2 domain, resulting in adapting the binding affinity for chromatin remodeling and transcriptional coregulator proteins. Together these modifications to the ligand-binding domain result in the activation or repression of selective gene transcription [16, 17]. Further knowledge for understanding the conformational changes associated with ligand receptor interactions have been identified by crystal structures that help define the inactive or repressive and active conformations, which result in binding of transcriptional corepressor or coactivator proteins, respectively, by stabilizing specific conformations of the AF-2 region [18].
Recent findings describe how ligands can potentially form interactions with the AF-2 ligand-binding domain via an induced fit or conformational selection mechanism [19]. In the conformational selection scenario, ligands selectively bind to the receptor resulting in selecting a specific conformation that is occupied within the ligand-binding conformational group. In the induced fit model, ligand binding occurs through an encounter complex which results in promoting the ligand-binding conformational group to transform into the final ligand-bound complex. Once this complex becomes stabilized in the active, ligand-bound position, the AF-2 site acts as a binding site for coregulator proteins [19].
10.3 PPAR-Gamma Activation Site
PPARγ relies on cytosolic ligand binding to activate the complex and consequences in the translocation to the nucleus. For gene transcription to occur, PPARγ forms a heterodimeric complex with RXR and binds to the PPAR recognition element (PPRE ). Interestingly, the PPARγ LBD intersects the DBD and LBD of RXR, whereby stability of gene transcription is dependent upon the stabilization of this interaction. Stability is greater with the intact nuclear receptor versus the DBD alone. PHE 347 was shown to greatly impact binding to the PPRE indicating that stabilization of this residue is important for gene transcription. Other heterodimeric protein interactions along with coactivator recruitment are also possible leading to increased gene transcription through cooperation [20]. Fatty acids and cyclooxygenase-derived eicosanoids are endogenous activators of PPARγ owing to its specific role in lipid storage, adipogenesis, and glucose metabolism [21, 22]. PPARγ ligands have distinct pharmacophore properties including a carboxylic acid head followed by an aromatic ring with a hydrophobic tail. In a study done with clofibric acid analogs, extension of the hydrophobic tail showed enhanced activity for both the PPARγ and alpha subtypes [23, 24]. This indicates that stabilization of residues outside of the LBD greatly influences the transcriptional potential of novel PPAR ligands. This interaction most likely arises from the ability to influence DBD stabilization to the PPRE outside of initial LBD activation. Specificity for the different PPAR isoforms becomes evident as the length of the hydrophobic tail is increased, highlighting unique gene transcription profiles between them based on the substrate available [25].
10.4 Structure of the Ligand-Binding Domain
The understanding of the PPARγ structure was deciphered by X-ray crystallography and determined that the ligand-binding domain consists of 13 α-helices that are labeled H1–H12 and H2′, as well as one β-sheet region, as shown in Figs. 10.1 and 10.2 [11, 26, 27]. Further, the ligand-binding pocket is located in the core of the ligand-binding domain [11, 26]. The ligand-binding domain is composed of 270 amino acids and resembles a large Y-shaped cavity and thus three branches, each branch having different properties based upon binding preferences [11, 28]. For example, arm 1, which displays a hydrophobic character, includes H3, H5, H11, and H12 and is the binding site for the acidic head group of ligands such as rosiglitazone [29, 30]. In comparison, arm 2, which is surrounded by helices H2′, H3, H6, and H7, includes the β-sheet region and is hydrophobic in nature, while arm III, which is surrounded by the β-sheet and helices H2, H3, and H5, and has both hydrophobic and hydrophilic regions [29]. The large ligand-binding pocket in PPARγ allows for the promiscuous binding of many ligands (endogenous and synthesized) with lower affinity, thus allowing for targeting of variable ligand interactions. The AF-2 surface includes stands H12, H3, H4, and H5 and forms a hydrophobic binding fork on the surface of PPARγ to which the coactivators bind. Ribbon diagrams of the full-length PPARγ-RXR heterodimer on DNA can be viewed in Fig. 10.1 and partially in Figs. 10.2, 10.3, and 10.4. These structures show that the PPAR/RXR ligand-binding domains dimerize with a view of a PPARγ DNA-binding domain [27]. The hinge region is composed of coils that allow for movement of the ligand-binding domain and DNA-binding domain around each other. Surprisingly, minimal surface contact is observed between the RXR and PPARγ DNA-binding domains. The region of the PPARγ ligand-binding domain that is near the β-sheet, proximal loops, and small helices (H2 and H2′) thus contacts the RXR DNA-binding domain (rather than the ligand-binding domain surface near the AF-2). This interaction allows for understanding as to how signals are conducted from the ligand-binding domain to the DNA-binding domain and vice versa. The LBD of PPARγ consisting of the transcriptional AF-2 motif associated with helix 12 mediates most of the pharmacological actions of PPARγ agonists [31]. The importance of AF-2 domain for regulating PPARγ-targeted gene expression is based upon the mechanism of ligand-induced transcriptional activation by PPARγ [31, 32]. In close inspection (Fig. 10.2), the AF-2 domain exists in an equilibrium state between closed (active) and open (inactive) conformations in the absence of the ligand [31]. Therefore, the binding of a full agonist induces the AF-2 domain conforming into the closed (active) state, thereby allowing the recruitment of coactivators for transcriptional activation [31]. Thus, a rational mechanism for developing novel PPARγ ligands would be to stabilize AF-2 domain in distinct states between closed and open conformations. Several studies have reported that locking the AF-2 domain in its closed conformation is responsible for the anti-diabetic effects as well as unwanted adverse effects from PPARγ agonists like thiazolidinediones [31, 33]. More recently, selective PPAR agonists and dual agonists are proving to be more efficacious in eliciting therapeutic efficacy and avoiding unwanted physiological effects associated with full agonism. More research is needed to understand how specific binding motifs can promote gene transcription to better utilize this drug target and remove the stigma surrounding this class of nuclear receptors. Therefore further evaluation of the stability and conformation and cofactor recruitment if the closed conformation will yield possibilities for the design of better therapeutic agents with increased tolerability.

RXR in the (a) open conformation illustrating the “mouse trap” model in the heterodimerization with PPARγ. As dimerization occurs, the RXR-LBD and DBD are stabilized through the ligand-induced activation of PPARγ’s LBD. PDB(3DZY). (b) PPARγ in the closed conformation. As dimerization occurs PPARγ’s DBD is able to bind to the major and minor grooves in the DNA allowing for gene transcription to occur. PDB(3DZY) [10]

Rosiglitazone bound within the PPARγ LBD active site. Rosiglitazone’s polar head forms a tight hydrogen bond network with the AF-2 TYR 473 residue, and additional hydrogen bonds with HIS 323 allows for PPARγ activation and subsequent nuclear translocation. Additional stabilization of the H3 alpha helix in ARG 288 and SER 289 along with hydrophobic interactions to the beta-sheet region provides stabilization upon heterodimerization with RXR allowing for gene transcription to occur. PDB(3DZY)

PPARγ antagonist (BADGE) bound within the PPARγ LBD. Strong hydrogen bonding to the AF-2 leads to PPARγ activation. However, conformational changes in the H3-ARG288 result in destabilization of this region opening ARG288 to post-translational modification and gene silencing. PDB(3DZY) [10]
10.5 Structural Dynamics of PPAR Gamma
Initial predictions postulated that mechanism of action of full agonists stabilizes the AF-2 surface through H12, thus allowing less of a physical price for coactivator binding and full transcriptional yield. Likewise, it was thought that partial agonists can only partially stabilize the AF-2 domain through the H12 helix by generating more of an physical burden for coactivator binding resulting in less transcriptional output. Thus the activating ligands, particularly full agonists, induced the reposition of helix H12 according to the “mousetrap model,” and the movement of H12, following ligand binding, traps the ligand within the ligand-binding pocket (Fig. 10.2) [33, 34]. Despite these models involving helix H12, the significance toward stabilization of helix H12 for coregulator binding and transactivation may not be completely understood. In particular, partial agonists are observed to preferentially stabilize other regions of the ligand-binding domain, specifically those associated with the β-sheet region. The connection between the impact of upon stabilization by partial agonists, coactivator recruitment, and PPARγ activity, including insulin-sensitizing effects, is an important question for investigation. The mechanism of action of PPARγ is initiated by ligand binding, resulting in a conformational change of the receptor and the dissociation of any corepressor complexes, including those associated with histone deacetylase activity and the resulting recruitment of coactivators [34]. When the PPAR-RXR receptor heterodimer is unbound to a ligand (natural or synthetic), it becomes associated with corepressor proteins, including NCoR (nuclear receptor corepressor 1) and SMRT (silencing mediator of retinoic acid and thyroid hormone receptor). These inactive complex functions to prevent PPAR-activated transcription and keep homeostatic PPAR-mediated transcription minimal. Upon ligand binding (full or partial), the corepressors dissociate from the receptor (PPAR-RXR) complex, permitting for the recruitment of coactivators. These coactivators then implement diverse functions to promote transcription, including altering chromatin structure (acetylation) and recruiting transcriptional machinery to the target gene promoter. Members of the PPAR coactivator family include CBP (CREB-binding protein), MED1 (Mediator 1, also known as PBP/TRAP220/DRIP205), SRC1 (steroid receptor coactivator 1), SRC2, SRC3, and PGC1α (peroxisome proliferator-activated receptor gamma coactivator 1 α) [27].
10.6 Selective PPAR Modulators (SPPARMs)
The popularity of full PPARγ agonist as insulin-sensitizing agents has been constrained because of their association with several adverse effects, including increased plasma volume and edema that is associated with inducing or exacerbating congestive heart failure, osteoporosis, as well as weight gain. Thus, there exists a critical need to develop newer PPARγ-targeted that display robust efficacy with improved tolerability. Consequently we and others have identified and characterized promising selective PPAR modulators (SPPARMs). SPPARMs are PPARγ ligands that serve as partial agonists of the receptor in cell-based transcriptional activity and adipogenesis assays [35, 36]. They have also been shown to generate attenuated and selective gene expression patterns in adipocytes in vitro. The greater therapeutic window of several SPPARγMs has been established in preclinical species. These findings would include improved insulin-sensitizing activity with attenuated adverse effects on weight gain, adiposity, and cardiac hypertrophy relative to a potent PPARγ full agonist [37,38,39,40]. Previous reports indicate that the unique properties of the SPPARMs may be due to their selective physical interaction with the distinct amino acids in the PPARγ receptor, resulting in selective conformational stability of the receptor when compared to full agonists. Findings from X-ray co-crystallographic studies of the PPARγ ligand-binding domain (LBD ) associated with full agonist rosiglitazone indicated that the nitrogen of the thiazolidinedione (TZD) ring of rosiglitazone formed hydrogen-bonding interactions with the tyrosine (Y) 473 side-chain hydroxyl group in helix 12 of the human PPARγ LBD [11]. The tyrosine-473 is known to exist deep in the AF-2 ligand-binding domain as demonstrated by in silico analysis [30]. In contrast, X-ray co-crystallography and molecular modeling studies with PPARγ LBD and SPPARγMs indicate that the carboxylic acid moiety of such ligands avoids forming hydrogen-bonding interactions with Tyr473 [41, 42]. The result of these partial interactions demonstrated by biochemical and NMR studies demonstrates that SPPARγMs induce a unique and less stable receptor conformation of the ligand-binding domain than PPARγ full agonists [37]. This instability of the AF-2 ligand-binding domain by SPPARγMs results in the compromised interactions with the transcriptional coactivator-binding pocket of the ligand-binding domain [11, 43] and thus serves as the physical basis for the altered receptor-coactivator interactions, reduced transcriptional activity, and resulting improved tolerability observed in preclinical studies with these ligands [37,38,39,40, 44, 45]. In summation, these findings suggest that Tyr473 is a critical site of interaction between the PPARγ LBD and full agonists but not SPPARγMs.
10.7 PPAR Delta Active Site Description
PPARs are approximately 70% conserved in homology, which allows promiscuity of ligand binding between the three PPAR isotypes. Furthermore the key features of the PPARδ active site can be distinguished between PPAR gamma and alpha. Crystallography structures of PPARδ that are bound with the selective agonist GW-0742, a full PPARδ agonist, display a preference for arm 2 occupation with its hydrophobic tail. This occupation of arm 2 of PPAR delta is attributed to the unique interactions involving Valine-312 residue. The VAL312 allows for a slightly larger volume and greater flexibility to accommodate PPAR-delta ligands in the arm 2 occupation, when compared to arm 2 in PPARγ. These observations were verified by mutating the VAL-312 to MET resulting in a 2.5-fold reduction in the EC50 of GW-0742 [46]. Crystallographic data showing PPARδ bound with EPA illustrate the dynamic interactions that are crucial for determining ligand-specific binding. EPA’s hydrophobic tail can assume a tail that extends up arm 2 or a tail down that extends into the entrance conformation. These interactions induce a conformational stability that can regulate the binding of selective coactivators to PPARδ. Rational drug design for PPAR selectivity must consider not just the AF-2 interaction but more importantly the stabilizing ability of the hydrophobic tail to increase interaction in PPRE interactions [47].
10.8 PPAR Alpha Active Site Description
In all PPARs, 80% of the active binding site residues are conserved. PPARα is uniquely different when examining the AF-2 region, where a histidine residue is substituted for a tyrosine (TYR) residue on the H5 helix. The larger TYR can explain some of the selectivity of the polar head when designing ligands for PPAR alpha. Although the binding site volume in PPARs is conserved, PPAR alpha is narrower in nature when approaching the AF-2 due to the larger volume of the residues involved in forming the AF-2 stabilizing hydrogen bond network [48].
Compound | Structure | PPARγ | PPARβ/δ | PPARα | References |
|---|---|---|---|---|---|
Alkylphenylpropanoic acid R-7 |
| Partial activity | No apparent activity | No apparent activity | [49] |
MRL-20 |
| Partial activity | No apparent activity | No apparent activity | [50] |
GW-0742 |
| No apparent activity | Full activity | No apparent activity | [12] |
MBX-0825 |
| No apparent activity | Full activity | No apparent activity | [51] |
Rosiglitazone |
| Full activity | No apparent activity | No apparent activity | [52] |
Pioglitazone |
| Full activity | No apparent activity | No apparent activity | [52] |
Indeglitazar |
| Partial activity | Partial activity | Partial activity | [53] |
Chiglitazar |
| Full activity | Partial activity | Partial activity | [54] |
WY14643 |
| No apparent activity | No apparent activity | Full activity | [55] |
Gemfibrozil |
| No apparent activity | No apparent activity | Full activity | [56] |










10.9 PPARS and Neurodegenerative Diseases
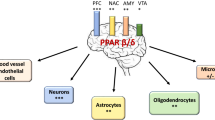
PPARγ agonists have shown efficacy in Alzheimer’s disease, Parkinson’s disease, brain and spinal injuries, and ALS. Interestingly, in the brain the relative expression of all three isoforms of PPARs are expressed; however PPARδ/β is most abundant in the brain with expression in neurons and microglial cells [57]. PPARγ is observed less in the neuron and microglia, and PPARα is observed mostly in astrocytes [57]. These findings offer PPARs as potential therapeutic targets for mitigating neurodegenerative diseases. Although PPARδ/β is the most abundant PPAR isotype in the brain, PPARγ has been the most extensively studied by using clinical applicable therapeutics in brain injury and degenerative models. The application of PPARγ agonists has been significantly investigated in rodent models of Alzheimer’s disease. Findings indicating improvement in amyloid beta burden as well as neurodegeneration have allowed PPARγ agonists to offer promise at the clinical levels. Clinical investigations using PPARγ agonists have revealed a significant reduction in amyloid beta and tau pathology measured in patient samples suffering from AD [58, 59]. More recently, PPARs have been demonstrated to modulate inflammation by inhibiting the production of pro-inflammatory molecules by peripheral immune cells as well as resident inflammatory cells. Furthermore, PPAR agonists have effectively suppressed the development of CNS inflammation in animal models of neuroinflammation and neurodegeneration [60]. In line with this oral administration of the pioglitazone (PPARγ), agonist nullified glial cell activation and the accumulation of Aβ-positive plaques in the hippocampus and cortex [61]. However, the protective signaling mechanisms mediated by central PPARγ activation resulting in improved cognition in AD have not been extensively investigated. Furthermore, PPARγ agonist has been observed to improve cognitive deficits in AD but is limited due to its poor bioavailability in the brain and off-target effects [62, 63].
Furthermore, failures at the clinical level and trials have quenched the clinical applicability of these agonists and have negated volumes of findings verifying these therapeutics for mitigating pathology and neurodegeneration associated with AD. Therefore, there is a perilous need to develop novel PPAR-targeted agents that display improved bioavailability and tolerability. To understand the significance of the chemical interactions on pharmacological consequences will help develop newer PPAR agonists for different cellular targets. Currently, no SPPARγMs have been applied to the clinical level, and mechanistically it remains unclear how to achieve selective PPARγ activation. The current review discusses the role of PPAR in modulating the pathologies of AD followed by SPPARMs under development for treating AD.
10.10 PPARS for Alzheimer’s Disease: Overview of AD
The manifestation of clinical symptoms associated with AD is due to years of pathological markers related to AD. For example, the development of AD is thought to be due to the amyloid beta cascade, which involves amyloid beta deposition and damage and leads tau hyperphosphorylation. However, alternative signaling mechanisms associated with the development and progression of pathological mechanisms promote development of clinical AD [64]. Therefore, therapeutics with multi-targets of action against AD pathology may offer potential for treatment of AD (Fig. 10.5).

nTZDpa a partial agonist bound within the PPARγ LBD. Partial agonism of the PPARγ LBD progresses in the absence of interactions with the full AF-2 domain. Therefore full stabilization of the AF-2 domain stabilization by the ligand is not required for PPARγ activation. Binding to the ARG288 and SER342 permits stabilization to occur between the RXR-LBD and DBD, and dimerization can proceed. However the recruitment of coactivators may be a factor that arise in regulating gene transcription because of partial agonism PDB(3DZY) [10]
10.11 PPARs and Neuroinflammation
Neuroinflammation is now considered one of the hallmark’s early mediators for developing the pathological process of AD [65]. The inflammatory process is associated with abeta plaque formation as well as microglial activation. Over time, the increase in inflammatory signals associated wit microglia or infiltrating monocytes and macrophages induces cytokine expression and reactive oxygen species in neurons, resulting in spine loss and reduced neural plasticity (Fig. 10.6). The development of memory dysfunction in the later stages of AD is correlated with the levels of synaptic destruction and severity of tau pathology [67]. Therefore, an ideal AD drug would target multiple facets of the disease including Aβ formation and/or clearance, provide anti-inflammatory properties , and reduce tau-related pathologies. However, failures at the clinical level in late-stage AD clinical trials have encouraged researchers to focus on prophylactically administration of PPARγ agonists in the pre-symptomatic phase where Aβ and inflammation play a critical role in the neurodegenerative process and progression. One of the many potential characteristics of PPARγ is to suppress inflammatory signaling pathways in immune cells [68, 69]. For example, PPARγ activation reduces the Aβ burden by inducing microglial phagocytosis of Aβ and consequences in reduced cytokine levels as well [70]. In addition, the activation of PPARγ suppresses transcription factors associated with neuroinflammation including nuclear factor-kB, Stat-1, and transcription factor activator protein-1 [71]. Additionally, PPARγ also downregulates cyclooxygenase-2 (COX-2), metalloproteinase-9 (MMP-9), inducible nitric oxide synthase (iNOS), pro-inflammatory cytokines, chemokines, and interleukins [72, 73]. Similarly, the anti-inflammatory effects of full PPARγ agonists including rosiglitazone and pioglitazone were observed to be efficacious in several rodent models [74]. Specifically, pioglitazone reduced Aβ levels as well as astrocyte and microglial activation in the cortex and hippocampus in APP695SWE mice that are associated with over-expression of Aβ and TGF-β1 [75]. Mechanistically, PPARγ reduces macrophage polarization from M1 to M2, in neurodegenerative diseases. M1 microglia are pro-inflammatory and neurotoxic via secretion of pro-inflammatory cytokines including interleukin IL-1α, IL-1β, tumor necrosis factors (TNF), and nitric oxide (NO) (Fig. 10.6). The alteration of microglia polarization axis from M1 to M2 when treated with PPARγ agonist results in increased anti-inflammatory gene expression profile and increased neurotrophin expression [76, 77]. Surprisingly 12-month-old APP/PS1 mice treated with pioglitazone showed a significant alteration of M1 to M2 microglial in the area surrounding Aβ deposits and a reduction in GFAP-immunopositive astrocytes adjacent to the amyloid plaques [78]. These data validate PPARγ agonist inducing an anti-inflammatory phenotype in microglia and astrocytes and in the process also facilitate the removal of Aβ pathology. However, further research in understanding the mechanisms how TZDs and PPARs confer their anti-inflammatory properties for AD will fill the gaps in knowledge for how newer PPAR agonists can be developed. For example, PPARδ is the most prominent form of PPAR expressed in the brain from the PPAR family. To this, GW501516, a potent PPARδ agonist, demonstrated anti-inflammatory activity [79]. Alternatively, a study by Malm et al. applied a short-term treatment of a PPARd agonist GW0742 to 5XFAD mice and observed a reduction in the parenchymal Aβ load. This was associated with a decrease in overall microglial activation (M1 levels) and reduction in pro-inflammatory cytokines. Instead, microglial immunoreactivity around Aβ deposits was increased [80]. Importantly, the reduction in the pro-inflammatory condition induced by GW0742 resulted in reduction of neuronal loss in the 5XFAD mice.

Schematic illustration demonstrates that (1) microglia polarization/activation from M2 to M1 results in secretion of cytokines. Increased cytokines and neuroinflammation are associated with the progression of Alzheimer’s disease which is associated with the resulting synaptic deficits and loss in dendritic spines. (2) Further advancement in spine loss potentiates neuronal degeneration. (3) PPARγ/δ agonists are known to reduce neuroinflammation and induce an increase in neurotrophins, including BDNF [66]. (4) The neurotrophins then enhance spine formation resulting in increased spine density
10.12 PPARs and Microglia and Neurotrophins
Microglia are associated with synaptic pruning by regulating spine formation and reduction as a normal process associated with alterations in memory and aging. However advancing AD from moderate to severe stages is highlighted by the development of synaptic deficits and memory impairment. Moreover, neuronal plasticity is associated with synaptic dysfunction, which is due to the loss of dendritic spines. However, neurotrophins mediated by microglia are significantly involved in regulating spine formation and dendritic spine density (Fig. 10.6). Brain-derived neurotrophic factor (BDNF), a major neurotrophin, is known to progressively decrease in expression as AD progresses from moderate to severe stages of AD [81]. Microglia are central mediators associated with inducing neurotrophins including BDNF. Rosiglitazone has been shown to prevent dendritic spine loss and improve synaptic function in hippocampal neurons treated with Aβ oligomers [82]. These findings can be explained mechanistically by the findings that demonstrated ligand activation of PPARγ induced the BDNF promoter in a dose-dependent manner [66]. Supportive findings from alternative studies in Aβ-injected rats treated with pioglitazone demonstrated reduced levels of active caspase-3 and enhanced BDNF levels yielding improved synaptic plasticity [83]. These observations suggest that PPARγ agonists prevent the development of synaptic deficits by improving BDNF expression and the resulting dendrite spine density.
Alternative protective signaling mechanisms are observed in studies that confirm that the full PPARγ agonist, rosiglitazone, increases the expression of neurotrophic factor-α1 (NF-α1), a neuroprotective protein, which results in the increase of the pro-survival protein BCL 2 expression in the hippocampus [84]. These observations are important because the use of PPARγ agonists improves mitochondrial function and synaptic plasticity and mitigates memory loss. In summation, PPARγ agonism can promote mitochondrial viability while also improving metabolic and energy regulation, modulate neuroinflammation, stimulate spine growth, and clear toxic Aβ from the brain [85]. Findings from our lab has observed that direct PPARg activation induces an increase in BDNF and the ensuing post-synaptic density marker 95 (PSD95), thus representing an increase in spine formation [66].
10.13 PPARS, TREM2, and Amyloid Beta
Dysfunction of microglial appears implicit to the etiology of late-onset Alzheimer’s disease (LOAD), as explained in findings from genetic studies that discovered variants in LOAD risk-associated genes that are highly expressed in microglia [86, 87]. One gene in particular from the study, the triggering receptor expressed on myeloid cells 2 (TREM2), a single-pass transmembrane immune receptor was observed to be expressed selectively in microglia within the CNS. TREM2 is a phagocytic receptor and has been demonstrated to be involved in the phagocytosis of apoptotic neurons (Fig. 10.7). However, recent findings demonstrate that TREM2 over-expressing macrophages accumulate on Aβ plaques where they exhibit an inflammatory phenotype yet paradoxically and phagocytically ineffective, as verified by the progressive increase in plaque burden through the course of the disease. Conversely, work by Zhao et al. has observed that TREM2 directly binds to Aβ oligomers with high affinity [88]. Further that TREM2 deficiency results in preventing Aβ degradation in primary microglial culture and in TREM 2 knock-out mice. It is well known that TREM2 suppresses inflammatory gene expression, based on findings from knockdown or genetic knock-out models of TREM2 that show higher levels of pro-inflammatory cytokines and increased levels of Aβ accumulation [89,90,91]. Thus, neurobiological functions of TREM2 and its pathophysiological ligands remain controversial and need further investigation to understand the role of TREM2 in AD. Clinically, TREM2 levels are regulated by proteolytic cleavage by ADAM10 and ADAM17 at the amino acids His157–Ser158 peptide bonds, resulting in the release of the soluble TREM2 (sTREM2) into cerebrospinal fluid [92, 93]. This soluble form of TREM2 is considered a new biomarker for AD because it is abundantly detected in human cerebrospinal fluid (CSF) and its levels are elevated in the CSF of patients with sporadic AD [94, 95]. Recent work by Savage et al. [95] provides evidence that nuclear receptors (PPARγ and PPARδ) act to stimulate the Aβ plaques from the brain, by inducing the expression of the phagocytic receptors Axl and MerTK on macrophages. The observation that these cells are from circulating monocytes and express TREM2 suggests that the actions of TREM2 and myeloid cells are involved in ameliorating AD pathogenesis. Additionally, Wang et al. observed that brain-residing TREM2 over-expressing microglia sense lipids that accumulate during Aβ deposition and thus more efficiently clear the Aβ plaques [96]. Further work on understanding the neuroprotective signaling mechanism of TREM 2 is required, including understanding the mechanism and consequence of PPAR-mediated increase in expression of TREM2 on reducing Aβ and tau phosphorylated levels in AD (Fig. 10.8).

Schematic illustration demonstrating that PPARγ ligands promote amyloid beta clearance by microglia. Mechanistically, PPARγ agonists induce an increase in TREM2 expression by transcriptional regulation. The increase in TREM2 via Syk-PI3K signaling results in diffuse and reduced levels of amyloid beta plaques in the brain by microglia activity

Schematic illustration shows that (1) glutamate regulation is compromised in moderate to late stages of AD. (2) PPARγ ligands can induce an increase in expression of GLT1/EAAT2 glutamate uptake receptor in astrocytes. (3) The increase in surface levels of GLT1/EAAT2 receptors results in improved glutamate handling in the synaptic cleft
PPARs and Astrocytes
Astrocytes play a crucial role in brain homeostasis. Among other functions, they provide metabolic support for neurons, uptake neurotransmitters such as glutamate, and blood-brain barrier maintenance [97]. Similar to microglia, astrocytes rapidly react to a wide array of insults or damaging events. Reactive astrocytes, which are characterized by increased expression of glial fibrillary acidic protein (GFAP), a constituent of the intermediate filaments, are typical of most brain pathologies. Thus, astrocytes represent an important target for anti-inflammatory and neuroprotective therapeutic strategies. In addition, rosiglitazone and the non-TZD agonist L-796,449 induced a concentration-dependent increase in glutamate transporter EAAT2/GLT-1 expression and glutamate uptake in primary rat astrocytes , which may help in improving the glutamate dysregulation associated with the progression of AD from mild cognitive impairment to full dementia (Fig. 10.2). In addition, the authors identified six putative PPREs in the promoter region of GLT1/EAAT2 gene, suggesting GLT1/EAAT2 glutamate transporter is a novel PPARγ target gene [98]. These findings suggest that PPAR ligands can reduce glutamate-mediated neurotoxicity; however critical intervention with the ligands will offer protection against the progression of glutamate-mediated cytotoxicity.
10.14 Conclusion
PPARs offer a unique advantage in treating disease states brought on by chronic inflammation and metabolic dysregulation. Current clinical PPAR therapeutics offer a molecular tool to investigate PPAR roles in neurodegenerative pathologies, yet barriers remain with these compounds as CNS activity is poor at best. High dosages required to achieve CNS activity only compound the unwanted systemic side effects seen at normal therapeutic levels, further complicating in vivo results. Advancements in the design and development of novel selective PPAR agonists may allow researchers clinically relevant compounds that are capable of achieving more efficacious CNS activity without excessive dosing. Achieving selective PPAR agonism may also be beneficial as specific gene transcription can avoid systemic side effects and elicit cellular-specific gene profiles in targeting various stages of disease pathology. When designing novel selective PPAR agonists, a greater emphasis on understanding the specific roles each arm in the PPAR LBD plays in the ability to recruit coactivators and promotes gene transcription is imperative. PPARs have long been stigmatized as clinically irrelevant therapeutics, but lack of understanding on how to regulate such a powerful molecular tool should not rule out further investigation. Furthermore, understanding the dynamic roles of PPARs in immune cell regulation is crucial to mitigate the pro-inflammatory signaling cascade and improve tissue regeneration. Energy dysregulation and metabolism are at the heart of all disease pathologies, and PPARs offer a unique tool to modulate reorganization of metabolic pathways in various tissue types.
References
Feige JN, Gelman L, Michalik L, Desvergne B, Wahli W. From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res. 2006;45:120–59.
Kersten S, Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie. 2017;136:75–84.
Bougarne N, et al. Molecular actions of PPARalpha in lipid metabolism and inflammation. Endocr Rev. 2018;39:760–802.
Festuccia WT, Blanchard PG, Richard D, Deshaies Y. Basal adrenergic tone is required for maximal stimulation of rat brown adipose tissue UCP1 expression by chronic PPAR-gamma activation. Am J Physiol Regul Integr Comp Physiol. 2010;299:R159–67.
Escher P, Wahli W. Peroxisome proliferator-activated receptors: insight into multiple cellular functions. Mutat Res. 2000;448:121–38.
Angeli V, et al. Peroxisome proliferator-activated receptor gamma inhibits the migration of dendritic cells: consequences for the immune response. J Immunol. 2003;170:5295–301.
Aleshin S, Reiser G. Role of the peroxisome proliferator-activated receptors (PPAR)-alpha, beta/delta and gamma triad in regulation of reactive oxygen species signaling in brain. Biol Chem. 2013;394:1553–70.
Hall MG, Quignodon L, Desvergne B. Peroxisome proliferator-activated receptor beta/delta in the brain: facts and hypothesis. PPAR Res. 2008;2008:780452.
Holzer G, Markov GV, Laudet V. Evolution of nuclear receptors and ligand signaling: toward a soft key-lock model? Curr Top Dev Biol. 2017;125:1–38.
Chandra V, et al. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008;456:350–6.
Nolte RT, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–43.
Batista FA, et al. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-delta) selective ligand binding. PLoS One. 2012;7:e33643.
Wu CC, et al. Structural basis for specific ligation of the peroxisome proliferator-activated receptor delta. Proc Natl Acad Sci U S A. 2017;114:E2563–70.
IJpenberg A, Jeannin E, Wahli W, Desvergne B. Polarity and specific sequence requirements of peroxisome proliferator-activated receptor (PPAR)/retinoid X receptor heterodimer binding to DNA. A functional analysis of the malic enzyme gene PPAR response element. J Biol Chem. 1997;272:20108–17.
Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes. 2003;52:434–41.
Santos GM, Fairall L, Schwabe JW. Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol Metab. 2011;22:87–93.
Kojetin DJ, Burris TP. Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery. Mol Pharmacol. 2013;83:1–8.
Weikum ER, Liu X, Ortlund EA. The nuclear receptor superfamily: a structural perspective. Protein Sci. 2018;27:1876–92.
Zhou HX. From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys J. 2010;98:L15–7.
Chandra V, et al. Structure of the intact PPAR-γ–RXR-α nuclear receptor complex on DNA. Nature. 2008;456:350–6.
Kliewer SA, et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–23.
Desvergne B, Ijpenberg A, Devchand PR, Wahli W. The peroxisome proliferator-activated receptors at the cross-road of diet and hormonal signalling. J Steroid Biochem Mol Biol. 1998;65:65–74.
Lewis DF, Jacobs MN, Dickins M, Lake BG. Molecular modelling of the peroxisome proliferator-activated receptor alpha (PPAR alpha) from human, rat and mouse, based on homology with the human PPAR gamma crystal structure. Toxicol In Vitro. 2002;16:275–80.
dos Santos JC, et al. Different binding and recognition modes of GL479, a dual agonist of peroxisome proliferator-activated receptor alpha/gamma. J Struct Biol. 2015;191:332–40.
Giampietro L, et al. Synthesis and structure–activity relationships of fibrate-based analogues inside PPARs. Bioorg Med Chem Lett. 2012;22:7662–6.
Jang JY, et al. Structural basis for the enhanced anti-diabetic efficacy of lobeglitazone on PPARgamma. Sci Rep. 2018;8:31.
Kaupang A, Hansen TV, The PPAR. Omega pocket: renewed opportunities for drug development. PPAR Res. 2020;2020:9657380.
Cronet P, et al. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure. 2001;9:699–706.
Huang P, Chandra V, Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu Rev Physiol. 2010;72:247–72.
Lewis SN, Bassaganya-Riera J, Bevan DR. Virtual screening as a technique for PPAR modulator discovery. PPAR Res. 2010;2010:861238.
Hughes TS, et al. Ligand and receptor dynamics contribute to the mechanism of graded PPARgamma agonism. Structure. 2012;20:139–50.
Frkic RL, et al. PPARgamma in complex with an antagonist and inverse agonist: a tumble and trap mechanism of the activation helix. iScience. 2018;5:69–79.
Shang J, et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARgamma. Elife. 2018;7:e43320.
de Vera IMS, et al. Synergistic regulation of coregulator/nuclear receptor interaction by ligand and DNA. Structure. 2017;25:1506–18, e1504.
Einstein M, et al. The differential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is a physical basis for their unique biological activities. Mol Pharmacol. 2008;73:62–74.
Gathiaka S, et al. Design, development and evaluation of novel dual PPARdelta/PPARgamma agonists. Bioorg Med Chem Lett. 2013;23:873–9.
Berger JP, et al. Distinct properties and advantages of a novel peroxisome proliferator-activated protein [gamma] selective modulator. Mol Endocrinol. 2003;17:662–76.
Acton JJ 3rd, et al. Benzoyl 2-methyl indoles as selective PPARgamma modulators. Bioorg Med Chem Lett. 2005;15:357–62.
Dropinski JF, et al. Synthesis and biological activities of novel aryl indole-2-carboxylic acid analogs as PPARgamma partial agonists. Bioorg Med Chem Lett. 2005;15:5035–8.
Minoura H, et al. Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer, FK614. Eur J Pharmacol. 2004;494:273–81.
Oberfield JL, et al. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte differentiation. Proc Natl Acad Sci U S A. 1999;96:6102–6.
Burgermeister E, et al. A novel partial agonist of peroxisome proliferator-activated receptor-gamma (PPARgamma) recruits PPARgamma-coactivator-1alpha, prevents triglyceride accumulation, and potentiates insulin signaling in vitro. Mol Endocrinol. 2006;20:809–30.
Sheu SH, Kaya T, Waxman DJ, Vajda S. Exploring the binding site structure of the PPAR gamma ligand-binding domain by computational solvent mapping. Biochemistry. 2005;44:1193–209.
Kintscher U, et al. Peroxisome proliferator-activated receptor and retinoid X receptor ligands inhibit monocyte chemotactic protein-1-directed migration of monocytes. Eur J Pharmacol. 2000;401:259–70.
Schupp M, et al. Molecular characterization of new selective peroxisome proliferator-activated receptor gamma modulators with angiotensin receptor blocking activity. Diabetes. 2005;54:3442–52.
Batista FAH, et al. Structural insights into human peroxisome proliferator activated receptor delta (PPAR-Delta) selective ligand binding. PLoS One. 2012;7:e33643.
Wu C-C, et al. Structural basis for specific ligation of the peroxisome proliferator-activated receptor δ. Proc Natl Acad Sci. 2017;114:E2563–70.
Zoete V, Grosdidier A, Michielin O. Peroxisome proliferator-activated receptor structures: ligand specificity, molecular switch and interactions with regulators. Biochim Biophys Acta. 2007;1771:915–25.
Ohashi M, et al. Design, synthesis, and structural analysis of phenylpropanoic acid-type PPARgamma-selective agonists: discovery of reversed stereochemistry-activity relationship. J Med Chem. 2011;54:331–41.
Bruning JB, et al. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure. 2007;15:1258–71.
Choi YJ, et al. Effects of the PPAR-delta agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis. 2012;220:470–6.
Toyota Y, Nomura S, Makishima M, Hashimoto Y, Ishikawa M. Structure-activity relationships of rosiglitazone for peroxisome proliferator-activated receptor gamma transrepression. Bioorg Med Chem Lett. 2017;27:2776–80.
Artis DR, et al. Scaffold-based discovery of indeglitazar, a PPAR pan-active anti-diabetic agent. Proc Natl Acad Sci U S A. 2009;106:262–7.
Sullivan HJ, Wang X, Nogle S, Liao S, Wu C. To probe full and partial activation of human peroxisome proliferator-activated receptors by pan-agonist chiglitazar using molecular dynamics simulations. PPAR Res. 2020;2020:5314187.
Veiga FMS, et al. Anti-obesogenic effects of WY14643 (PPAR-alpha agonist): hepatic mitochondrial enhancement and suppressed lipogenic pathway in diet-induced obese mice. Biochimie. 2017;140:106–16.
Almad A, Lash AT, Wei P, Lovett-Racke AE, McTigue DM. The PPAR alpha agonist gemfibrozil is an ineffective treatment for spinal cord injured mice. Exp Neurol. 2011;232:309–17.
Warden A, et al. Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep. 2016;6:27618.
Perez MJ, Quintanilla RA. Therapeutic actions of the thiazolidinediones in Alzheimer’s disease. PPAR Res. 2015;2015:957248.
Jojo GM, Kuppusamy G. Scope of new formulation approaches in the repurposing of pioglitazone for the management of Alzheimer’s disease. J Clin Pharm Ther. 2019;44:337–48.
Racke MK, Drew PD. PPARs in neuroinflammation. PPAR Res. 2008;2008:638356.
Quan Q, Qian Y, Li X, Li M. Pioglitazone reduces beta amyloid levels via inhibition of PPARgamma phosphorylation in a neuronal model of Alzheimer’s disease. Front Aging Neurosci. 2019;11:178.
Cao B, et al. Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: a systematic review and network meta-analysis. Diabetes Obes Metab. 2018;20:2467–71.
Kermani A, Garg A. Thiazolidinedione-associated congestive heart failure and pulmonary edema. Mayo Clin Proc. 2003;78:1088–91.
Kumar A, Singh A. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacol Rep. 2015;67:195–203.
Bronzuoli MR, Iacomino A, Steardo L, Scuderi C. Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res. 2016;9:199–208.
Kariharan T, et al. Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol Aging. 2015;36:1451–61.
Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4:a005777.
Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–6.
Ricote M, et al. Expression of the peroxisome proliferator-activated receptor gamma (PPARgamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1998;95:7614–9.
Yamanaka M, et al. PPARgamma/RXRalpha-induced and CD36-mediated microglial amyloid-beta phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci. 2012;32:17321–31.
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82.
Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front Biosci. 2008;13:1813–26.
Heneka MT, Klockgether T, Feinstein DL. Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci. 2000;20:6862–7.
Toledo EM, Inestrosa NC. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry. 2010;15:272–85.
Chapman PF, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–6.
Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12.
Odegaard JI, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20.
Martinez B, Peplow PV. Amelioration of Alzheimer’s disease pathology and cognitive deficits by immunomodulatory agents in animal models of Alzheimer’s disease. Neural Regen Res. 2019;14:1158–76.
Defaux A, Zurich MG, Braissant O, Honegger P, Monnet-Tschudi F. Effects of the PPAR-beta agonist GW501516 in an in vitro model of brain inflammation and antibody-induced demyelination. J Neuroinflammation. 2009;6:15.
Malm T, Mariani M, Donovan LJ, Neilson L, Landreth GE. Activation of the nuclear receptor PPARdelta is neuroprotective in a transgenic mouse model of Alzheimer’s disease through inhibition of inflammation. J Neuroinflammation. 2015;12:7.
Falkenberg T, et al. Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett. 1992;138:153–6.
Chiang MC, Cheng YC, Chen HM, Liang YJ, Yen CH. Rosiglitazone promotes neurite outgrowth and mitochondrial function in N2A cells via PPARgamma pathway. Mitochondrion. 2014;14:7–17.
Prakash A, Kumar A. Role of nuclear receptor on regulation of BDNF and neuroinflammation in hippocampus of beta-amyloid animal model of Alzheimer’s disease. Neurotox Res. 2014;25:335–47.
Thouennon E, Cheng Y, Falahatian V, Cawley NX, Loh YP. Rosiglitazone-activated PPARgamma induces neurotrophic factor-alpha1 transcription contributing to neuroprotection. J Neurochem. 2015;134:463–70.
Zolezzi JM, et al. PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol Rev Camb Philos Soc. 2017;92:2046–69.
Carmona S, Hardy J, Guerreiro R. The genetic landscape of Alzheimer disease. Handb Clin Neurol. 2018;148:395–408.
Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43.
Zhao Y, et al. TREM2 is a receptor for beta-amyloid that mediates microglial function. Neuron. 2018;97:1023–31, e1027.
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201:647–57.
Turnbull IR, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177:3520–4.
Jay TR, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–95.
Feuerbach D, et al. ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after histidine 157. Neurosci Lett. 2017;660:109–14.
Schlepckow K, et al. An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med. 2017;9:1356–65.
Suarez-Calvet M, et al. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med. 2016;8:466–76.
Suarez-Calvet M, et al. Early changes in CSF sTREM2 in dominantly inherited Alzheimer’s disease occur after amyloid deposition and neuronal injury. Sci Transl Med. 2016;8:369ra178.
Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–71.
Nedergaard M, Dirnagl U. Role of glial cells in cerebral ischemia. Glia. 2005;50:281–6.
Romera C, et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J Cereb Blood Flow Metab. 2007;27:1327–38.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Steinke, I., Amin, R. (2021). Design of Novel PPAR Agonist for Neurodegenerative Disease. In: Badr, M.Z. (eds) Nuclear Receptors. Springer, Cham. https://doi.org/10.1007/978-3-030-78315-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-78315-0_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-78314-3
Online ISBN: 978-3-030-78315-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)