
Abstract
The mitochondrial genome is the second genome for eukaryotes. It has essential functions in many cellular processes, including producing energy, signaling, aging, and programmed cell death. Up to now, ten mitochondrial genomes from seven Ganoderma species had been released on the public database. The mitochondrial genomes of each species were described and compared here, and some intra-specific differences were also issued. The mitochondrial phylogenomic tree of Polyporales, which Ganoderma is belonged to in taxonomy, was constructed, and the potential DNA barcodes from the mitochondrial genome for this genus were discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 General Introduction
4.1.1 Concept
Mitochondria: the mitochondrion (plural mitochondria) is a highly specialized organelle with double membranes, existing in the cytoplasm of almost all eukaryotic cells (cells with clearly defined nuclei). As the cell's powerhouse, it produces large quantities of energy stored in adenosine triphosphate (ATP) through oxidative phosphorylation (Chinnery and Hudson 2013; van der Giezen and Tovar 2005). The number of mitochondria in a cell can vary significantly by organism and tissue, and each mitochondrion contains multiple mitochondrial DNA molecules (Basse 2010).
Mitochondrial genome (Mitogenome): mitochondria have their DNA, known as mitochondrial DNA or mtDNA. mtDNA was first discovered in animal cells (Nass and Nass 1963). mtDNA consists of a heavy (H) and a light (L) strand. The heavy strand/light strand terminology dates back to the 1970s and refers to the relative CsCl banding of the two single strands of the mitochondrial DNA. These strands have a G/C asymmetry leading to the sense strand being “light” and the reverse complement being “heavy” in their relative molecular weights. The sense strand (L) encodes 13 of the more than 90 subunits of the electron-transfer chain and 22 tRNAs and 2 rRNAs in humans (Homo sapiens). Humans’ mitochondrial genome is a closed circular duplex DNA of estimated 16,500 nucleotide pairs (Brown et al. 1979). Then, the first complete sequence of a mitochondrial genome was reported from humans, and its exact size of 16.569 base pairs (bp) was presented (Anderson et al. 1981). In fungi, the first complete mitochondrial genome was sequenced from the yeast Saccharomyces cerevisiae in 1998, with the size of 85,779 bp (Foury et al. 1998).
4.1.2 Principle
4.1.2.1 Mitochondrial Origin
The current theory as to the origin of eukaryotic cells is endosymbiosis. Based on the endosymbiotic theory, mitochondria are the descendants of once free-living α-proteobacteria. The proteobacteria became endosymbiotic organelles of eukaryotes two billion years ago (Andersson et al. 1998; Gray et al. 1999). It is believed that mitochondria and chloroplasts began as prokaryotic organisms engulfed by the larger cells, either as food or parasites. That is why they are enclosed in double membranes. At some point, the relationship became mutually beneficial, and the mitochondria and chloroplasts evolved into permanent members in the cells as cellular machinery.
4.1.2.2 Mitochondrial Structure
Mitochondria are typically oval, spherical, or rod-shaped. Its size ranges from 0.5 to 1.0 µm in diameter. Double membranes enclose them. The membranes are lipid bilayers with proteins embedded within the layers. The region between the two membranes is the intermembrane space. The inner membrane is folded to form cristae. This folding increases the surface area of the membrane and maximizes cellular respiration output. Inside the inner membrane is the mitochondrial matrix, and within the matrix, there are ribosomes, enzymes, and mitochondrial DNA. The mitochondrion can reproduce and synthesize proteins independently. It contains the enzymes necessary for transcription and the transfer of RNAs and ribosomes required for translation and protein formation. Recent research revealed that, instead of small, oblong, and independent entities in the eukaryotic cytoplasm, mitochondria form dynamic networks of long tubular structures undergoing fusions and separations during the cell cycle or after changes of growth conditions (Fig. 4.1; Dujon 2020; Lackner 2014).

Networks of mitochondria. a Mitochondria were forming connected, tubular networks. b Mitochondria were creating well-distributed tubular networks. Reprinted from Fig. 1 of Lackner (2014) with permissions
4.1.2.3 Mitochondrial Genetics
Mitochondrial DNA (mtDNA) is typically a small circular double-stranded DNA molecule that encodes many proteins and RNA involved primarily in cellular respiration. In some fungi and protists, mtDNA can be linear (van de Vossenberg et al. 2018). Mitochondrial DNA is well conserved within taxa.
Unlike nuclear DNA passed on from both parents, mitochondrial DNA is generally uniparentally inherited (with some notable exceptions). In animals, mtDNA is passed on maternally through the egg. In plants, mtDNA may be passed on maternally, paternally, or biparentally. There is also evidence for paternal leakage of mtDNA, where the offspring inherits most of their mtDNA from their mother and receives a small amount from their father. Uniparental inheritance of mitochondria is a common phenomenon in sexual eukaryotes. It leads to little opportunity for genetic recombination between different lineages of mitochondria.
4.1.2.4 Mitochondrial Evolution
Researches on mitochondrion evolution began with animal mitochondrial DNA. There is a widely accepted generalization concerning molecular evolution rates: the more important the function of a gene or protein, the more slowly it undergoes an evolutionary change in the primary structure. As mitochondria have essential cellular functions, the life of animals is crucially dependent on these functions. Mitochondrial evolution would be expected to be highly constrained. Besides, animal mitochondria genome is small in size and relatively uniform in structure among vertebrate and invertebrate animals. The implication is strong that this genome was reduced at an early stage of animal evolution to the minimum size compatible with function.
However, evidence from restriction endonuclease cleavage maps implies that mtDNA evolves 5–10 times faster than single-copy nuclear DNA (Brown et al. 1979). The mean substitution rate in the three studied mitochondrial tRNA genes is at least 100 times higher than that in nuclear tRNA genes (Brown et al. 1982). The base substitution rate of mammalian mtDNA has been estimated to be 0.5–1.0% per lineage per 106 years (Brown 1980). For this reason, mitochondrial DNA is commonly used to study evolutionary relationships and population genetics in animals. Plant mtDNA evolves reasonably slowly and is less commonly used in evolutionary studies.
4.1.2.5 Mitochondrial Function
Mitochondria host the tricarboxylic acid (TCA) cycle and oxidative phosphorylation, breaking down sugars and fats into energy through aerobic respiration (cellular respiration). This metabolic process creates ATP, the energy source of a cell, through a series of steps that require oxygen. Beyond their well-known function as energy-producing entities, mitochondria are involved in a multitude of cellular processes, including the production of iron-sulfur clusters, calcium homeostasis, calcium signaling, haem synthesis, steroid synthesis, cellular differentiation, aging, programmed cell death (apoptosis), as well as maintaining the control of the cell cycle and cell growth (Malina et al. 2018).
The role of mitochondria in disease has been expanded beyond the respiratory chain, as defects in additional mitochondrial functions and behaviors have been linked to cancer, metabolic disorders, and neurodegenerative diseases, such as Alzheimer's, Parkinson's, and Huntington's disease. Mutations in mitochondrial DNA can result in many human genetic disorders. Examples include diabetes, heart disease, myoclonic epilepsy, and Kearns-Sayre neuromuscular syndrome (Friedman and Nunnari 2014).
4.1.3 Research Methods For Mitochondrial Genome Analysis
4.1.3.1 Molecular Experiments
4.1.3.1.1 DNA Extraction
Genomic DNA could be extracted from fruiting bodies using Tiangen DNA Extraction Kit (DP305) or a fungal DNA kit (Cat. #D3390–00, Omega Bio-Tek, Norcross, GA, USA) according to the manufacturer’s instructions.
4.1.3.1.2 Next-Generation Sequencing
Whole genomic sequencing was conducted on an Illumina HiSeq 2500 Platform (Illumina, San Diego, CA, USA). Two 300 bp genomic DNA libraries were constructed for two Ganoderma species. For G. meredithiae, the produced paired-end reads were 100 bp long, while the paired-end reads of G. applanatum were 125 bp long.
4.1.3.2 Data Analyses
4.1.3.2.1 Mitochondrial Genome Assembly and Annotation
De novo assembly of the mitogenomes was performed using ABySS-pe version 1.5.2 (Simpson et al. 2009) for G. meredithiae, CLC Genomics Workbench Version 7.5.1 (CLC Bio, Aarhus, Denmark) for G. applanatum, and SPAdes 3.9.0 (Bankevich et al. 2012) for the five ones published in 2019. The assembled scaffolds were compared to the reference sequence using BLASTN version 2.2.23 (Altschul et al. 1990). The screened scaffolds were compared with the reference using Gepard version 1.30 (Krumsiek et al. 2007) and then assembled into a circular mitogenome sequence by manual editing. The coverage was examined using Tablet version 1.14.10.20 (Milne et al. 2013).
The mitogenome was annotated using MFannot (Lang et al. 2014) or MITOS (Bernt et al. 2013). The Sequence Manipulation Suite Version 2 (SMS2, http://www.bioinformatics.org/sms2/) was used to identify the hypothetical proteins. tRNA genes were initially identified using tRNAscan-SE (Lowe and Chan 2016). DNASTAR Lasergene (http://www.dnastar.com/) was used to analyze the base composition of the mitogenomes. The synonymous substitution rate (Ks) and the nonsynonymous substitution rate (Ka) for all PCGs in each of the mitogenomes were calculated using DnaSP (Rozas et al. 2017). Mitochondrial genome maps were generated by OGDRAW (Lohse et al. 2007).
4.1.3.2.2 Phylogenomic Analysis
The concatenated amino acid sequences were extracted from the GenBank files of the mitogenomes of Polyporales. They were then aligned using MAFFT v7.221 (Katoh et al. 2002) or MUSCLE version 3.6 (Edgar 2004). The neighbor-Joining analysis was conducted using MEGA version 6.06 (Tamura et al. 2013). Bootstrap values were calculated from 1,000 replicates.
4.2 Progress of Lingzhi Mitochondrial Genome
Up to date (October 2020), ten mitochondrial genomes of Ganoderma species were released from the NCBI database, seven of them recognized by the RefSeq database. Their information is summarized in Table 4.1. All of them were submitted by Chinese researchers.
4.2.1 Structure of Lingzhi Mitochondrial Genome
As listed in Table 4.1, three mitochondrial genomes of ‘Ganoderma lucidum (Curtis) P. Karst.’ was released online. The identity of this species is G. sichuanense J.D. Zhao and XQ. Zhang, as detailed elucidated in the previous taxonomic researches (Wang et al. 2012; Thawthong et al. 2017; Yao et al. 2020, 2013).
Strain CGMCC 5.26 was isolated from the fruiting body collected in China and then deposited to China General Microbiological Culture Collection Center (CGMCC, Beijing, China) in 1963 for medicinal usage. Its mitogenome (HF570115) was first reported in this genus Ganoderma (Li et al. 2013). This mitogenome is a typical circular DNA molecule of 60,630 bp with 26.67% GC content. About 62.69% of the mitochondrial genome contains 50 genes encoding two ribosomal RNAs, 26 transfer RNAs, one ribosomal protein gene rps3, 14 genes involved in respiratory chain complexes, four ORFs in the intron of other genes (ip1–4), and two ORFs in the intergenic regions (orf1 and orf2). These genes were in the same orientation, except those genes encoding for trnW-CCA and three ORFs. In total, thirteen introns were found in the protein-coding genes cox1, cox2, cox3, nad4, nad5, and rnl and rns genes. All introns were the group I introns, except for the single type II intron cox1 i6.
The other released mitogenome (KC763799 = NC_021750) of the same strain was submitted by Qian, but it was not published. It has only five base pairs more than the one (HF570115) reported by Li et al. (Li et al. 2013), while the GC content is almost the same. The gene order is the same between them, as listed in Table 4.2. But the introns reported in rnl and rns of HF570115 were not identified, so only 11 introns exist in the mitogenome KC763799. An ideogram of the mitogenome organization and gene classification is shown in Fig. 4.2. The two mitogenomes were from the same strain and assembled from the same raw data. Thus, the differences do not reflect the genetic variation of the same species. These differences need to be further validated using Sanger sequencing.

Mitochondrial genome map of G. lucidum CGMCC 5.26 (NC_021750)
The more recently released mitogenome (MH252532) of G. lucidum s26 was sequenced from the strain isolated from a fruiting body collected from Tai’an City, Shandong Province, China. A comparison between this one and KC763799 was described (Li et al. 2019). The nucleotide sequence similarity between the two mitogenomes was 95.18%. Furthermore, most protein-coding genes and tRNA genes were conserved between them. Ganoderma CGMCC 5.26 has longer intergenic regions than G. lucidum s26 does. In addition, single nucleotide polymorphisms were frequently observed in the intergenic region of the two mitogenomes. Besides, G. lucidum s26 had one more intron (12 introns in total, one more in rns) than G. lucidum CGMCC 5.26, indicating that the mitochondrial genomes of G. lucidum from different origin have been differentiated.
4.2.2 Evolution of Mitochondrial Genome in Genus Ganoderma
Besides the three mitogenomes of G. lucidum, there are still seven ones released in this genus (Table 4.1). A mount of inter-specific variance has been observed (Table 4.2).
4.2.2.1 Mitochondrial Genome of Ganoderma meredithiae
The strain CGMCC 5.766 equals CBS 271.88. It is the holotype culture isolated from the holotype specimen of G. meredithiae Adask. & Gilb. (JEA 345). JEA 345 is deposited in US National Fungus Collections (BPI, Beltsville, Maryland). The mitogenome of this species was reported by Wang et al. (2016b).
The 100 bp paired-end reads were assembled using ABySS-pe. The mitogenome (HF570115) of G. lucidum CGMCC 5.26 served as a reference sequence. A total of 1.35 GB clean data of 100 bp paired-end reads were produced. After assembly, 14,054 contigs were obtained. All the contigs were aligned with the reference sequence. And two contigs were identified as a mitochondrial origin. Contig 133 was 46,593 bp long, and contig 223 was 31,145 bp long. The two contigs shared a 390 bp long repeat sequence at both ends. The two contigs were assembled according to the reference sequence. Four gaps were detected in the preliminary mitochondrial genome sequence. They were then filled by comparing the assembly results produced by AbySS-pe software with parameters of k-mer 25 and k-mer 64. The authors mapped reads onto this genome sequence to validate this mitogenome's assembly and check its coverage. A total of 546,887 reads were mapped, accounting for 4.0% of all the reads produced (546,887/13,512,066). The average coverage depth was 693.5, and the maximum coverage depth was 1,552.
This mitogenome is a circular mtDNA with a total length of 78,447 bp and a GC content of 26.14%. It encodes a set of mitochondrial protein and RNA genes, including 15 conserved proteins, 29 tRNAs, large and small ribosomal RNAs, and 18 homing endonucleases. All structural genes are located on the same strand except trnW-CCA (Fig. 4.3). The gene order is listed in Table 4.2. The tRNA genes code for all 20 standard amino acids. Most amino acids are coded by only one tRNA gene; however, two trnC-GCA, two trnI (trnI-GAU and trnI-UAU), two trnL (trnL-UAA and trnL-UAG), three trnM-CAU, three trnR (one trnR-UCG and two trnR-UCU), and three trnS (one trnS-UGA and two trnS-GCU) are found in this mitogenome. Eighteen introns are detected in six genes: cob (3), cox1 (10), cox2 (1), nad4 (1), nad5 (2), and rnl (1). For each intron, there is one homing endonuclease. Three of the 18 endonucleases are GIY-YIG type (ip4, ip10, and ip17), and the others are LAGLIDADG type.

Mitochondrial genome map of G. meredithiae
Compared with the mitogenome of G. lucidum (NC_021750), G. meredithiae has larger genome content, lower GC content, three more tRNA genes (trnC-GCA, trnI-UAU, and trnR-UCG), and seven more introns in the protein-coding or rRNA genes.
4.2.2.2 Mitochondrial Genome of Ganoderma applanatum
The strain CGMCC 5.249 of G. applanatum (Pers.) Pat. was isolated from a basidiocarp collected from Changbai Mountain, Jilin Province, China. The mitogenome of this species was reported by Wang et al. (2016a).
The 125 bp pair-end reads were assembled using CLC Genomics Workbench (CLC Bio, version 7.5.1, Aarhus, Denmark). The authors used the mitogenome of G. meredithiae CGMCC 5.766 as the reference to identify the scaffolds and to determine their order. Manual comparison of the ends of the ordered scaffolds helped to assemble the scaffolds into a circular molecule.
A total of 1.54 GB clean data of 125 bp paired-end reads were produced. After assembly, 21,718 contigs were obtained. All the contigs were aligned with the reference sequence, and three contigs were identified as a mitochondrial origin. Contigs 21, 22, and 47 were 35,819 bp, 83,529 bp, and 304 bp long, respectively. The contigs were assembled in the order: 22-47-21-47. A 287 bp long repeat sequence was shared by the tail of contig 22 and the head of contig 47; a 21 bp long repeat sequence was shared by the tail of contig 47 and the heads of contigs 21 and 22; another 21 bp long repeat sequence was shared by the tail of contig 21 and the head of contig 47. No gap was detected in the assembled mitochondrial genome sequence. To validate the mitogenome assembly, the authors mapped reads onto this genome sequence. A total of 2,000,647 reads were mapped, accounting for 16.2% of all the reads produced. The average coverage depth was 2,086.9, and the maximum coverage depth was 3,458.
The mitogenome of G. applanatum is 119,803 bp long, with the GC content being 26.66%. It encodes 15 conserved proteins, 25 tRNAs, and the large and small ribosomal RNAs. All structural genes are located on the same strand except trnW-CCA (Fig. 4.4). The gene order is listed in Table 4.2. The tRNA genes contain codons for all 20 standard amino acids except Cysteine. Most amino acids are represented by only one tRNA gene; however, two trnL (trnL-UAA and trnL-UAG), two trnS (trnS-GCU and trnS-UGA), three trnM-CAU, and three trnR (one trnR-UCG and two trnR-UCU) genes are found in this mitogenome. Thirty-five introns are detected in seven genes, i.e., cob (6), cox1 (14), cox2 (1), cox3 (1), nad4 (2), nad5 (5), and rnl (6).

Mitochondrial genome map of G. applanatum
Compared with the mitogenome of G. lucidum (NC_021750), G. applanatum has larger genome content, almost the same GC content, one less tRNA gene, and 24 more introns in the protein-coding or rRNA genes.
4.2.2.3 Mitochondrial Genome of Ganoderma sinense
The strain CGMCC 5.69 of G. sinense J.D. Zhao, L.W. Hsu, and X.Q. Zhang was isolated from a basidiocarp collected from Hainan Province, China. This mitogenome was submitted by J. Qian, Institute of Medicinal Plant Development, Beijing, China.
The mitogenome of G. sinense is 86,451 bp long, with the GC content being 26.78%. It encodes 15 conserved proteins, 27 tRNAs, and the large and small ribosomal RNAs. All of them are located on the same strand except trnW-CCA. The gene order is listed in Table 4.2. A total of 30 introns exist in this mitogenome. Compared with the mitogenome of G. lucidum (NC_021750), G. sinense has larger genome content, higher GC content, one more tRNA gene, and 19 more introns in the protein-coding or rRNA genes. In detail, trnS-GCU is missing; two trnR-UCG and one trnR-UCU are added; three more introns in rnl, six more in cox1, five more in cob, two more in cox2 and rns, respectively, and one more in cox3.
4.2.2.4 Mitochondrial Genome of Ganoderma tsugae
The strain s90 of G. tsugae Murrill was isolated from a basidiocarp collected from Wangqing County, Jilin Province, China. This mitogenome was published by Li et al. (2019).
The mitogenome of G. tsugae is 92,511 bp long, with the GC content being 26.67%. It encodes 15 conserved proteins, 25 tRNAs, and the large and small ribosomal RNAs. All structural genes are on the same strand except atp6, trnS-GCU, and trnW-CCA. The gene order is listed in Table 4.2. A total of 24 introns exist in this mitogenome. Compared with the mitogenome of G. lucidum (NC_021750), G. tsugae has larger genome content, the same GC content, one less tRNA gene, and 13 more introns in the protein-coding or rRNA genes. That is, trnC-GCA is missing; the locations of trnS-GCU and trnR-UCG are changed; one more intron in rnl, five more in cox1, four more in cob, two more in nad1 and nad5, respectively.
4.2.2.5 Mitochondrial Genome of Ganoderma calidophilum
The strain s136 of G. calidophilum J.D. Zhao, L.W. Hsu, and X.Q. Zhang was isolated from a basidiocarp collected from Qiongzhong County, Hainan Province, China. This mitogenome was published by Li et al. (2019).
The mitogenome of G. calidophilum is the largest one in this genus up to now. It is 124,588 bp long, with the GC content of 25.43%. It encodes 15 conserved proteins, 25 tRNAs, and the large and small ribosomal RNAs. All of them are on the same strand except trnW-CCA. The gene order is listed in Table 4.2. A total of 31 introns exist in this mitogenome. Compared with the mitogenome of G. lucidum (NC_021750), G. calidophilum has larger genome content, lower GC content, one less tRNA gene, and 20 more introns in the protein-coding or rRNA genes. Specifically, trnS-GCU is missing, and the locations of trnR-UCU and trnR-UCG are changed; trnS-GCU and atp8 are shifted to the region between rps3 and cox2. In particular, there are four more introns in rnl, eight more in cox1, six more in cob, one more in nad1, and two more in nad5.
4.2.2.6 Mitochondrial Genome of Ganoderma leucocontextum
The strain s116 of G. leucocontextum TH Li et al. was isolated from a basidiocarp collected from Huidong County, Sichuan Province, China. This mitogenome was published by Li et al. (2019).
The mitogenome of G. leucocontextum is 88,194 bp long, with the GC content being 27.08%. It encodes 15 conserved proteins, 25 tRNAs, and the large and small ribosomal RNAs. A total of 22 genes are on the same strand, and 20 genes are on the anti-sense strand. The gene order is listed in Table 4.2. A total of 19 introns exist in this mitogenome. Compared with the mitogenome of G. lucidum (NC_021750), the gene order of G. leucocontextum is much different, as shown in Table 4.2. The tRNA genes (gene order 4 to 13) are not located close to atp6 (gene order 3); in contrast, they are shifted to the region before cox2 (gene order 36). And there is a big reverse region including genes from order 16 to 35. This arrangement of mitochondrial genes has not been seen in other Ganoderma species. It has larger genome content, higher GC content, one less tRNA gene, and eight more introns in the protein-coding or rRNA genes. In detail, trnC-GCA is missing. There are two more introns in rnl, three more in cox1, one more in nad3, and four more in cob.
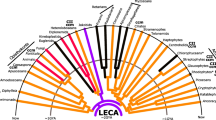
4.2.2.7 Mitochondrial Phylogenomics of Ganoderma
To investigate the mitochondrial phylogeny of Ganoderma, the accessible mitogenomes in Polyporales were all included: Phlebia radiate (NC_020148), Taiwanofungus camphoratus (NC_042771), Trametes cingulata (NC_013933), Trametes hirsute (NC_037239), and Wolfiporia cocos (NC_050681). Amino acid sequences of the 15 conserved protein-coding genes were extracted from the mitogenomes. The 15 proteins included subunits of the respiratory chain complexes (cox1, cox2, cox3, and cob), ATPase subunits (atp6, atp8, and atp9), NADH:quinone reductase subunits (nad1, nad2, nad3, nad4, nad4L, nad5, and nad6), and ribosomal protein S3 (rps3). The alignment of the concatenated 15 genes is 5837 amino acid long.
As shown in Fig. 4.5, the genus Ganoderma is monophyly and contains nine sequenced members. Ganoderma lucidum and G. sp. s8 clustered in the same branch with very short branch lengths and very strong support, hinting that they are the same species. Ganoderma meredithae has the closest relationship with G. lucidum. Trametes is the sister group of Ganoderma, consistent with the previous results (Wang et al. 2016a, b; Li et al. 2019).

Mitochondrial phylogenomic tree of Polyporales
Ganoderma calidophilum has the largest mitogenome among the Ganoderma members, while G. sp. s8 has the smallest one. The former is more than twice in mitogenome size as the latter (Table 4.1). Except for G. lucidum (including G. sp. s8), G. meredithae has the smallest mitogenome. This might suggest that the evolution of the mitochondrial genome of Ganoderma correlates with the phylogeny in some branch to some extent. For GC content, G. leucocontextum has the highest value, while G. calidophilum has the lowest one. For the number of tRNAs, G. meredithae has the most genes (29), while G. applanatum, G. calidophilum, G. leucocontextum, and G. tsugae have the least genes (25). For the number of introns, G. applanatum has the most (35); in contrast, G. lucidum has the least (11–13). The mitogenomes of Ganoderma presented highly genetic diversity in terms of genome content, gene order and structure, and so on.
4.3 Future Perspective
4.3.1 Problems
Ganoderma species have high medicinal values (Wang et al. 2012). There are three Ganoderma species recorded in the Chinese pharmacopeia, i.e., G. lucidum, G. sinense, and G. tsugae. The mitogenomes of them had been published or recovered in the previous researches and compared here. Nevertheless, most of the mitogenomes of the species in this genus Ganoderma have not been studied. More than 80 phylogenetic species of Ganoderma were recognized in a meta-analysis of internal transcribed spacer (ITS) rDNA sequences (Fryssouli et al. 2020), and many of them have been proven to be with significant medicinal or ecological values. Only seven Ganoderma species (Table 4.1 and Fig. 4.5) were investigated for their mitochondrial genomes. More intense sampling is in need for the future mitogenome researches of Ganoderma at both inter- and intra-specific levels.
4.3.2 Topics of Interests
ITS has demonstrated high efficacy in resolving relationships amongst most of the Ganoderma taxa; however, it was not equally useful at elucidating species boundaries across the entire genus (Fryssouli et al. 2020). Mitochondrial small subunit (mtSSU = rns) gene was used to infer this genus's phylogeny (Hong and Jung 2004). However, the incongruence between mitochondrial and nuclear phylogenies of the genus was detected based on three mitochondrial genes and seven nuclear genes (Wang 2012).
On the other hand, phylogenetic analyses based on different single genes of mitogenome recovered incongruent tree topologies. But the BI phylogeny based on the cox1 gene was mostly consistent with that generated using combined datasets, indicating this gene could be a potential molecular marker for phylogenetic analysis (Li et al. 2019). These findings suggest that selecting suitable molecular markers is essential for studying phylogenetic relationships in the Ganoderma genus.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290(5806):457–465
Andersson SG, Zomorodipour A, Andersson JO, Sicheritz-Ponten T, Alsmark UC, Podowski RM, Naslund AK, Eriksson AS, Winkler HH, Kurland CG (1998) The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396(6707):133–140
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19(5):455–477
Basse CW (2010) Mitochondrial inheritance in fungi. Curr Opin Microbiol 13(6):712–719
Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M, Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69(2):313–319
Brown WM (1980) Polymorphism in mitochondrial-DNA of humans as revealed by restriction endonuclease analysis. P Natl Acad Sci-Biol 77(6):3605–3609
Brown WM, George M Jr, Wilson AC (1979) Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A 76(4):1967–1971
Brown WM, Prager EM, Wang A, Wilson AC (1982) Mitochondrial DNA sequences of primates: tempo and mode of evolution. J Mol Evol 18(4):225–239
Chinnery PF, Hudson G (2013) Mitochondrial genetics. Br Med Bull 106:135–159
Dujon B (2020) Mitochondrial genetics revisited. Yeast 37(2):191–205
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797
Foury F, Roganti T, Lecrenier N, Purnelle B (1998) The complete sequence of the mitochondrial genome of Saccharomyces cerevisiae. FEBS Lett 440(3):325–331
Friedman JR, Nunnari J (2014) Mitochondrial form and function. Nature 505(7483):335–343
Fryssouli V, Zervakis GI, Polemis E, Typas MA (2020) A global meta-analysis of ITS rDNA sequences from material belonging to the genus Ganoderma (Basidiomycota, Polyporales) including new data from selected taxa. Mycokeys 75:71–143
Gray MW, Burger G, Lang BF (1999) Mitochondrial evolution. Science 283(5407):1476–1481
Hong SG, Jung HS (2004) Phylogenetic analysis of Ganoderma based on nearly complete mitochondrial small-subunit ribosomal DNA sequences. Mycologia 96(4):742–755
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30(14):3059–3066
Krumsiek J, Arnold R, Rattei T (2007) Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23(8):1026–1028
Lackner LL (2014) Shaping the dynamic mitochondrial network. BMC Biol 12:35
Lang BF, Jakubkova M, Hegedusova E, Daoud R, Forget L, Brejova B, Vinar T, Kosa P, Fricova D, Nebohacova M, Griac P, Tomaska L, Burger G, Nosek J (2014) Massive programmed translational jumping in mitochondria. Proc Natl Acad Sci U S A 111(16):5926–5931
Li J, Zhang J, Chen H, Chen X, Lan J, Liu C (2013) Complete mitochondrial genome of the medicinal mushroom Ganoderma lucidum. PLoS ONE 8(8):e72038
Li Q, Xiang D, Wan Y, Wu Q, Wu X, Ma C, Song Y, Zhao G, Huang W (2019) The complete mitochondrial genomes of five important medicinal Ganoderma species: features, evolution, and phylogeny. Int J Biol Macromol 139:397–408
Lohse M, Drechsel O, Bock R (2007) OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52(5–6):267–274
Lowe TM, Chan PP (2016) tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44(W1):W54–W57
Malina C, Larsson C, Nielsen J (2018) Yeast mitochondria: an overview of mitochondrial biology and the potential of mitochondrial systems biology. FEMS Yeast Res 18(5):foy040
Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D (2013) Using tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14(2):193–202
Nass MM, Nass S (1963) Intramitochondrial fibers with DNA characteristics. I. Fixation and electron staining reactions. J Cell Biol 19:593–611
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE, Sanchez-Gracia A (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34(12):3299–3302
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19(6):1117–1123
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Thawthong A, Hapuarachchi KK, Wen TC, Raspe O, Thongklang N, Kang JC, Hyde KD (2017) Ganoderma sichuanense (Ganodermataceae, Polyporales) new to Thailand. Mycokeys 22:27–43
van de Vossenberg B, Brankovics B, Nguyen HDT, van Gent-Pelzer MPE, Smith D, Dadej K, Przetakiewicz J, Kreuze JF, Boerma M, van Leeuwen GCM, Andre Levesque C, van der Lee TAJ (2018) The linear mitochondrial genome of the quarantine chytrid Synchytrium endobioticum; insights into the evolution and recent history of an obligate biotrophic plant pathogen. BMC Evol Biol 18(1):136
van der Giezen M, Tovar J (2005) Degenerate mitochondria. EMBO Rep 6(6):525–530
Wang XC (2012) Phylogenetic study on Ganodermataceae Donk. University of Chinese Academy of Sciences, Beijing
Wang XC, Xi RJ, Li Y, Wang DM, Yao YJ (2012) The species identity of the widely cultivated Ganoderma, ‘G. lucidu’ (Ling-zhi), in China. PLoS ONE 7(7):e40857
Wang XC, Shao J, Liu C (2016a) The complete mitochondrial genome of the medicinal fungus Ganoderma applanatum (Polyporales, Basidiomycota). Mitochondrial DNA A DNA Mapp Seq Anal 27(4):2813–2814
Wang XC, Wu K, Chen H, Shao J, Zhang N, Chen X, Lan J, Liu C (2016b) The complete mitochondrial genome of the white-rot fungus Ganoderma meredithiae (Polyporales, Basidiomycota). Mitochondrial DNA A DNA Mapp Seq Anal 27(6):4197–4198
Yao YJ, Wang XC, Wang B (2013) Epitypification of Ganoderma sichuanense J.D. Zhao & XQ. Zhang (Ganodermataceae). Taxon 62(5):1025–1031
Yao YJ, Li Y, Du Z, Wang K, Wang XC, Kirk PM, Spooner BM (2020) On the typification of Ganoderma sichuanense (Agaricomycetes)-the widely cultivated Lingzhi medicinal mushroom. Int J Med Mushrooms 22(1):45–54
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31700014, 31750001), Key Research Program of Frontier Science, Chinese Academy of Sciences (QYZDY-SSW-SMC029), Beijing Natural Science Foundation (7154224), and China Postdoctoral Science Foundation (2014M550659).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Wang, XC. (2021). Lingzhi Mitochondrial Genome. In: Liu, C. (eds) The Lingzhi Mushroom Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-030-75710-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-75710-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-75709-0
Online ISBN: 978-3-030-75710-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)