
Abstract
Mitochondria are cellular organelles of outstanding importance because they are responsible for energy production and the control of many processes related to cell metabolism, survival and apoptosis. They are the only cytoplasmic organelles in the animal cells to contain their own DNA, a private genetic material, the mitochondrial DNA (mtDNA), which is frequently referred as “the mitochondrial genome”. In this article, the most important features of mtDNA are reviewed, regarding its structure, functioning, transmission and relation to diseases and aging. Moreover, mtDNA has peculiar properties such as maternal transmission and elevated rates of mutation, which make it an interesting source of information about human origins, dispersion and evolution.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Mitochondria
- mtDNA
- Mitochondrial DNA
- Mitochondrial genome
- Mitochondrial diseases
- Mitochondrial inheritance
- Heteroplasmy
- Mitochondrial mutation
- Mitochondrial variation
10.1 The Origin and the Structure of the Human Mitochondrial DNA (mtDNA)
Mitochondria are cell organelles present in almost all eukaryotic cells and are covered by a double layer of membranes. Structurally, they have four compartments: the outer membrane, the inner membrane, the intermembrane space and the matrix, the region inside the inner membrane. They are the only structures of the animal cells, besides the nucleus, that contain DNA, the mtDNA. In addition, they have their own machinery for the synthesis of RNA and proteins. In plant cells and algae cells, chloroplasts are organelles that also have their own DNA molecules [1]. Mitochondria are dynamic structures, since they are frequently observed as changing their size and shape, or undergoing processes of fusion or fission. The number of mitochondria is also variable among different cell types and tissues, and this number can vary as a response to certain stimuli, for example, frequent muscle contractions [2, 3].
Mitochondria perform many tasks such as pyruvate oxidation , the Krebs cycle and the metabolism of amino acids. Mitochondria also harbor the fatty acid (FA) oxidation machinery, producing acetyl-coenzyme A (acetyl-CoA). They are also key components in calcium signaling, steroid synthesis and apoptosis (programmed cell death), but their outstanding role is certainly the generation of energy as adenosine triphosphate (ATP), by means of the oxidative phosphorylation system (OXPHOS) , which occurs via the electron transport chain (ETC) .This process requires five protein complexes, four of which make up the mitochondrial respiratory chain (complexes I, II, III and IV) and they are involved in the transport of electrons through the complexes until their final acceptor, molecular oxygen. The four complexes are organized within the inner mitochondrial membrane. The transfer of electrons generates a proton gradient across the inner mitochondrial membrane, where the complex V is also embedded. Complex V is also known as ATP synthase , since it synthesizes ATP via chemiosmotic coupling with the ETC [reviews in [2, 4].
One of the most outstanding ideas about eukaryotic cell evolution was the proposal that mitochondria originated from endosymbiotic bacteria that were incorporated into the eukaryotic cells. Bacteria colonized primordial eukaryotic cells that lacked the ability to use oxygen in energy production. A symbiotic relationship must have become permanent and engulfed bacteria evolved into the present mitochondria. Although the idea was firstly proposed near 1890, the hypothesis was reintroduced and strongly diffused by Lynn Margulis in 1967 [5]. This proposal is referred in the literature as the “endosymbiotic theory for the origin of mitochondria” or “endosymbiotic hypothesis ”. In accordance, all present genomic evidence points to all mitochondrial genomes, present in all living organisms, as originating from one single endosymbiotic event, probably involving an aerobic alpha-proteobacterium, and they share only one common ancestor, a circular bacterial genome [6,7,8]. This cell fusion event was estimated to have occurred around 1.5–2 billion years ago. Many aspects of the structure and functioning of mtDNA reinforce the theory of its bacterial origin, such as its circular organization, its presence in multiple copies within the cell, and gene expression mediated by the transcription of large polycistronic RNAs. The same hypothesis of origin from endosymbiotic organisms was applied to explain the origin of chloroplasts in photosynthesizing organisms, organelles that also have their own DNA molecules.
Presently, mtDNA molecules in different species have different sizes and coding capacities , since all of them lost substantial amounts of genetic information, when compared to the coding capacity of the genomes of presently existing bacteria. Most of the genetic information within the ancestral mtDNA was transferred to nuclear chromosomes at different rates and amounts in different species, reducing the genetic independence of mitochondria [9].
In addition to such ancient transfer of sequences from mtDNA to nuclear DNA, there has also been documented the evolutionarily recent transfer of mitochondrial sequences to the nuclear genome. Analysis of the human reference genome sequence shows hundreds of nuclear sequences that are imperfect copies of mtDNA sequences, with varied sizes and locations. These transferred mtDNA sequences usually show inactivating mutations, which pose restrictions to their genetic expression, and are described as nuclear mtDNA sequences, or NUMTs. Some NUMT sequences are present in some individuals, but not in others, thus constituting insertion/deletion polymorphisms in human populations [9].
The first human complete “genome” sequenced was that of the mitochondria in 1981 [10], by Fred Sanger and colleagues at Cambridge, many years before the Human Genome Project began. It was subsequently referred as the Cambridge Reference Sequence (CRS) . The nucleotide numbering of mtDNA sequence presently in use is based on a revised and corrected version of this reference, the rCRS [10, 11]. The human mtDNA comprises 16,569 bp . It is a double-stranded DNA molecule, that resembles bacterial genomes because it is densely packed with genes, and circular. One of the strands is called the heavy strand (HS) because it is guanine-rich compared to the light strand (LS) , which is cytosine-rich.
MtDNA has a small non-coding region, the 1.1 kb displacement loop (D loop), also named as control region , which includes elements that regulate transcription and replication : the two major transcription initiation sites that are required to generate polycistronic transcripts (HSP1 and HSP2) and one of the origins of mtDNA replication, the one of the heavy strand (OH). Parts of the control region are variable in sequence and are referred as hypervariable segments (HVS) I, II and III . The second origin of replication, on the light strand (OL), is outside the control region and is located near 11 kb away from OH. The structure of human mtDNA is schematically represented in Fig. 10.1.

The mtDNA, containing 37 genes (Figure adapted from Picard et al. [3]). OH = Origin of replication of the heavy strain; OL = Origin of replication of light chain; PL = promoter of the light strain; PH1 = Promoter 1 of heavy strain; PH2 = Promoter 2 of the heavy strain; List of genes and their products: Cyt b Mitochondrially encoded cytochrome b (MT-CYB), ND6 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 6 (MT-ND6), ND5 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 5 (MT-ND5), ND4 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 4 (MT-ND4), ND4L Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 4 L (MT-ND4L), ND3 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 3 (MT-ND3), ND2 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 2 (MT-ND2), ND1 Mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 1 (MT-ND1), COIII Mitochondrially encoded cytochrome c oxidase III (MT-CO3), ATP6 Mitochondrially encoded ATP synthase membrane subunit 6 (MT-ATP6), ATP-8 Mitochondrially encoded ATP synthase membrane subunit 8 (MT-ATP8), COII Mitochondrially encoded cytochrome c oxidase II (MT-CO2), COI Mitochondrially encoded cytochrome c oxidase I (MT-CO1), 16S Mitochondrially encoded 16S rRNA (MT-RNR2), 12S Mitochondrially encoded 12S rRNA (MT-RNR1), P Mitochondrially encoded tRNA-Pro (CCN) (MT-TP), T Mitochondrially encoded tRNA-Thr (ACN) (MT-TT), E Mitochondrially encoded tRNA-Glu (GAA/G) (MT-TE), L (CUM): Mitochondrially encoded tRNA-Leu (CUN) 2 (MT-TL2), S (AGY): Mitochondrially encoded tRNA-Ser (AGU/C) 2 (MT-TS2), H Mitochondrially encoded tRNA-His (CAU/C) (MT-TH), R Mitochondrially encoded tRNA-Arg (CGN) (MT-TR), G Mitochondrially encoded tRNA-Gly (GGN) (MT-TG), K Mitochondrially encoded tRNA-Lys (AAA/G) (MT-TK), D Mitochondrially encoded tRNA-Asp (GAU/C) (MT-TD), S (UCN): Mitochondrially encoded tRNA-Ser (UCN)1 (MT-TS1), Y Mitochondrially encoded tRNA-Tyr (UAU/C) (MT-TSY), C Mitochondrially encoded tRNA-Cys (UGU/C) (MT-TC), N Mitochondrially encoded tRNA-Asn (AAU/C) (MT-TN), A Mitochondrially encoded tRNA-Ala (GCN) (MT-TA), W Mitochondrially encoded tRNA-Trp (UGA/G) (MT-TW), M Mitochondrially encoded tRNA-Met (AUA/G) (MT-TM), Q Mitochondrially encoded tRNA-Gln (CAA/G) (MT-TQ), I Mitochondrially encoded tRNA-Ile (AUU/C) (MT-TI), L (UUR) Mitochondrially encoded tRNA-Leu (UUA/G) 1 (MT-TL1), V Mitochondrially encoded tRNA-Val (GUN) (MT-TV), F Mitochondrially encoded tRNA-Phe (UUU/C) (MT-TF)
The circular molecular also comprises a larger coding region containing 37 genes. Thirteen genes encode 13 different proteins synthesized by mitochondrial ribosomes, and related to the ETC. All 13 proteins act as subunits of the mitochondrial enzyme complexes involved in oxidative phosphorylation (OXPHOS). Twenty-two genes are templates for the transcription of 22 transfer RNAs (tRNA) which act exclusively in the translation of mitochondrial peptides. Finally, two genes are for two ribosomal RNA (rRNA) molecules, 12S and 16S, components of mitochondrial ribosomes.
The mitochondrial ribosomes have a sedimentation coefficient of 55S and are constituted by two subunits, 39S and 28S, in which are present the ribosomal RNA molecules of 16S (expressed by MT-RNR2) and 12S (expressed by MT-RNR1), respectively. There are no introns in the human mtDNA and more than 90% of the mitochondrial ‘genome’ specifies a protein or a functional RNA. The sequences of neighboring genes are continuous or separated by only a few non-coding bases, and there is extensive overlap of coding sequences between the two strands of the circle, the heavy (H) strand and the light strand (L).
It is important to highlight that there are estimates that, in human mitochondria, a proteome of 1100–1700 different proteins with varied functions is acting. Thus, the capacity of mtDNA of coding only 13 mitochondrial peptides reveals that, presently, mitochondrial functions are largely dependent on proteins encoded by nuclear genes, translated in cytoplasmic ribosomes, and imported by mitochondria. In accordance, only 13 of the 80 proteins required for oxydative phosphorylation are coded by the mitochondrial genome. Crucial proteins needed for replication, transcription and repair of mtDNA are encoded by nuclear genes. As a consequence, mitochondria are under dual genetic control , by its own DNA and the nuclear genome.
The mtDNA is comparatively protein-free , as are bacterial genomes, because it is not condensed with histones as nuclear chromosomes are. Nevertheless, it is packed with some proteins to form nucleoids, nucleoprotein structures that are associated with the inner mitochondrial membrane. The mitochondria nucleoid s contain the protein machinery required for DNA replication, transcription, repair and packaging of mtDNA, including the mtDNA polymerase, POLG (or POL gamma) and the main mtDNA transcription factor (TFAM; mitochondrial transcription factor A), as well as mtDNA helicases (TWINKLE) and other proteins, such as single strand DNA binding proteins. The major structural component of mitochondrial nucleoids is TFAM, an abundant protein involved in mtDNA transcription, acting as a mitochondrial transcription factor besides its function of packing the mtDNA into nucleoids. Some microscopy experiments suggested that each nucleoid is 100 nm in diameter and comprises one copy of mtDNA packaged with multiple TFAM molecules, but there are references to one nucleoid containing more than one copy of the mtDNA molecule [2].
The replication of both strands of the human mtDNA, heavy (H) and light (L) chains, is unidirectional and starts at specific origins, OH and OL, respectively. Although mtDNA is generally double-stranded, the repeated replication of a small segment of the H strand results in the production of a shorter third strand called 7SDNA. The 7S DNA can pair with the L-strand, displacing the H strand, which then forms a small loop, called displacement loop or D-loop. As shown in Fig. 10.1, this region also contains the major promoter regions and the origin of replication for the H-strand (OH), and this explains why it is referred as the CR/D-loop region, or the control Region. The three main factors in mtDNA replication are DNA polymerase gamma (POLG), the mitochondrial helicase (TWINKLE) and the mitochondrial single-strand DNA binding protein (mtSSB) . The POLG holoenzyme is a heterotrimer that consists of two identical subunits (PolG-B) and one catalytic subunit named PolG-A. The DNA helicase forms a hexameric structure and is required at the replication fork where it unwinds the double-stranded DNA ahead of POLG to expose the template for replication. POLG was thought to be the sole DNA polymerase in mitochondria, being involved in replication and repair, but at least four other mitochondrial polymerases were found to be related to mtDNA maintenance and repair.
The exact mechanism of mtDNA replication is unknown and there are conflicting theories to explain the process. According to one of the models of mtDNA replication, only after about 2/3 of the H strand is replicated from the OH, the replication machinery reaches the origin of replication of the L strand (OL), starting the replication of this strand in the opposite direction. This model of replication was the first to be proposed, but evidence for an alternate model of replication was also obtained, in which leading-lagging strand DNA replication is coordinated, suggesting simultaneous replication of both strands, similar to the replication strategy observed in linear nuclear chromosomes. The controversy about the two possible modes of DNA replication remains unsolved [2, 4].
MtDNA replication does not seem to be subject to a strict control of copy number. The replication rates are flexible and seem to vary. Between 100 and 10,000 mtDNA copies may be found in the inner mitochondrial compartment (matrix) in different cell types, and oocytes are estimated to have 100,000 mtDNA copies. The mitochondrial genome is frequently renewed and replication does not occur in a specific phase of cell cycle, as it happens with nuclear DNA. The number of copies of mtDNA is observed to vary with cell type and within the same cell type, and it can change in accordance to energy demands.
Mitochondria are also equipped with mechanisms to repair damaged DNA, but not all components of nuclear DNA repair have their counterparts in mitochondria. Base excision repair (BER) is active within mitochondria and repairs small lesions such as alkylated and oxidized bases. There are seven different glycosylases that initiate BER in mtDNA and there is evidence that DNA synthesis after damage removal is performed by polymerase beta. Proteins involved in recombination and translesion DNA synthesis were also identified within mitochondria, as well as some proteins related to nucleotide excision repair (NER) , although the existence of this repair pathway remains uncertain.
The expression of mtDNA is widely different from nuclear genes. The two strands of mtDNA are transcribed to give two long polycistronic transcripts that resemble bacterial polycistronic RNAs. The long transcripts are cleaved to generate individual mRNAs or functional RNAs such as tRNAs and rRNAs. Transcription initiates from three possible promoters: one located in the light-strand (LSP) and two located on the heavy strand (HSP1 and HSP2). The HSP1 enables the transcription of the 12S and 16S rRNA genes while the HSP2 enables the transcription of the entire H-strand as a polycistronic transcript. The L-strand is transcribed from the light strand promoters (LSP) . Transcription is, thus, bidirectional and requires TFAM, the mitochondrial transcription factor B2 (TFB2M) and mitochondrial RNA polymerase. The transcription factors assemble at the promoters to initiate the synthesis of the polycistronic RNAs that are later processed into smaller RNAs. TFAM appears to be regulated via post-translational modifications and these modifications may be related to epigenetic mechanisms of regulation of mitochondrial transcription. It seems that RNA transcription in mitochondria is also regulated by control of the number of copies of mtDNA. An increase in energy need is usually followed by an increase of copy number of mtDNA in tissues, resulting in enhanced expression of mitochondrial genes [2].
The mitochondrial ribosomes exclusively translate peptides coded by mtDNA genes and do not translate nuclear derived mRNAs. As a result, the mitochondrial genetic code could drift in evolution from the “Universal Genetic Code ”. The mitochondrial code is slightly different from the genetic code that is used in cytoplasmic ribosomes of almost all living organisms. According to the “Universal Genetic Code”, 61 codons specify amino acids and three are stop codons: UAA, UAG and UGA. In the human mitochondrial code, 60 codons specify amino acids and there are four stop codons: UAA, UAG (also stop codons in the nuclear code) and AGA and AGG (which would specify arginine in the nuclear code). UGA, which is a stop codon in the universal code, encodes tryptophan in mitochondria. AUA encodes methionine in mitochondria, instead of isoleucine, as in the conventional code.
The mitochondrial genome shows unique genetic characteristics, such as matrilineal inheritance, lack of recombination, and high sequence variability, which make it distinct from the nuclear genome in many genetic features detailed in the next topics of this review.
10.2 The Mutation Rates in mtDNA are Elevated When Compared to Nuclear DNA
A 10–1000 fold higher mutation rate was estimated in mtDNA when compared to the substitutional mutation rate in nuclear DNA, depending on the portion of the mtDNA evaluated. The mutational rate in the control region is the highest, leading to a large number of different mtDNA sequences in human populations. The estimated mutation rates are not uniform, ranging from 2 × 10−7 in some tRNA sequences to 5 × 10−6 in the HVS-I and HVS-II [12].
Several facts account for the observation of elevated rates of mutations. First, mtDNA is constantly attacked by reactive oxygen species (ROS) generated by oxidative phosphorylation , because of its close proximity to the components of the respiratory chain. ROS are potent genotoxic agents and they are responsible for higher nucleotide instability. In spite of being packed in mitochondrial nucleoids , with some DNA-associated proteins such as TFAM and polymerases, mtDNA is not as tightly packed with proteins as nuclear chromosomes. Nuclear DNA exists in a complex chromatin structure mainly organized by histones (see Chap. 2), and histones are not present in mitochondria. Thus, it is assumed that mtDNA is less protected than nuclear DNA from genotoxic agents. Some DNA repair pathways that can partly cope with oxidative damage are present in mitochondria, but they are not as complex as those acting in nuclear DNA. Besides, given that the number of copies of mtDNA in each cell is usually much higher than sequences in the nucleus (in which a specific sequence is replicated only once every cell cycle), the replication history of any mtDNA molecule will be longer than a nuclear sequence, since there are more replication events per unit of time. This increases the probability of replication errors. It has also been pointed that mtDNA, because of its unusual mode of replication, spends more of its time in single-stranded form and more exposed to damage, and this is especially true for the D-loop region [2, 12].
10.3 Rare mtDNA Variants Lead to Hereditary Diseases with Maternal Transmission
Mitochondrial diseases is a term used to describe a clinically heterogeneous group of genetic disorders characterized by defective oxidative phosphorylation. They result from dysfunction of mitochondria and may lead to a variety of symptoms that can be detected in neonatal phase, childhood or adulthood. The dysfunction of mitochondria results in a chronic loss of cellular energy or incapacity to meet cellular energy demands. As a consequence, the resulting symptoms can be present in isolated organs, but they often cause multiple system impairment. They are either a consequence of pathogenic variants in nuclear genes encoding protein products that are relevant to mitochondria, or result from pathogenic alterations in mitochondrial genes that code mitochondrial proteins or RNA molecules. Given an estimated proteome of 1100–1700 different proteins, in fact, most proteins involved in mitochondrial metabolism are nuclearly encoded. Nevertheless, a number of human genetic diseases is due to pathogenic alterations in mtDNA and these are exclusively maternally inherited [14]. A woman carrying a mutated mtDNA sequence passes it to all children, but men will not transmit mtDNA to their progeny. We will only review in this chapter diseases that result from pathogenic variants in the mtDNA.
Mutations in mtDNA may affect specific proteins of the respiratory chain, when they occur in protein coding genes, or they may affect the synthesis of mitochondrial proteins as a whole, if they occur in tRNA or rRNA genes. These variants in mtDNA may result in many clinical features and syndromes with overlapping clinical symptoms, variable expressivity and penetrance, thus representing a real challenge for their clinical recognition and accurate classification. Mitochondria are ubiquitous and every tissue can be affected by a mtDNA pathogenic variant, and this is why mitochondrial diseases are usually multisystemic. However, there are some clinical features that are recurrent in many of the mitochondria-related syndromes, since the basic biological defect underlying many of these diseases is impairment of energy production. Thus, mtDNA alterations usually impair more severely organs or tissues with high energy needs, such as the brain, skeletal muscles and heart, and those that have to respond quickly to abrupt changes in the environment, at the expenses of consuming more ATP [4]. Impairment of neurologic functions and of sensorial systems as hearing and vision, muscle weakness, diabetes and other endocrine diseases are frequent features of mitochondrially inherited diseases.
The first report of inherited disease resulting from mutation in the mtDNA was that of Wallace et al. in [13], who reported that mtDNA point mutations and deletions caused MERFF (Myoclonus, Epilepsy and Ragged-red Fibers mitochondrial encephalomyopathy) and this finding was considered a breakthrough for molecular medicine. Since then, it has been established that many inherited and acquired mtDNA defects are at the roots of many pediatric and adult diseases. Many different disorders were described as resulting from mtDNA alterations, which range from single nucleotide substitutions to large mtDNA deletions. Genetic mitochondrial disorders have initially remained in the domain of neurology, but the discovery of a broader range of diseases, with clinically complex phenotypes, have placed mitochondrial diseases in many different medical specialties, such as cardiology, endocrinology, immunology, oncology, and others.
Large-scale mtDNA deletions are usually associated to three main phenotypes: chronic progressive, external opthalmoplegia (PEO), Kearns-Sayre syndrome (KSS) and Pearson syndrome . Pearson syndrome shows the most severe clinical presentation: patients present sideroblastic anemia and pancreatic dysfunction early in life, and the condition may be fatal. KSS patients present with ptosis, PEO and pigmentary retinopathy and may have multisystem impairment including myopathy, ataxia and cardiac conduction defects. Deletions were also described in cases of MELAS, with severe neurological symptoms including encephalopathy and stroke-like episodes. Although large deletions often arise sporadically, they result in devastating syndromes [14, 15].
In contrast to many mitochondrial diseases that are multisystemic , it is puzzling that some mitochondrial variants result only in tissue-specific effects. This is the case of m.1555A > G in the 12S rRNA gene (MT-RN1), causative to non-syndromic hearing loss. This and some other hearing loss-associated mtDNA variants located mainly in the MT-TRN1 and MT-TS1 genes are rarely accompanied by other clinical features. Many variants in the MT-TRN1 gene are related to hearing loss, which is anticipated or intensified after administration of aminoglycoside antibiotics, because they increase susceptibility to aminoglycoside-induced hearing loss. This example is iconic in the demonstration of the interaction of mitochondrial functions with environment, and how this interaction affects age of onset, severity and progression of the disease phenotypes. It is widely recognized that the most frequent mtDNA variant associated with non-syndromic hearing loss is m.1555A > G and it was also the first to be described, in 1993 [16]. The penetrance of hearing loss is estimated near 40–50%, in pedigrees that show exclusively maternal inheritance. The age at onset and severity of hearing loss are variable within pedigrees, and besides being correlated to aminoglycoside treatment, it is possibly influenced by other factors, such as nuclear modifier genes [17]. It was speculated by some authors that the m.1555A > G substitution makes the mitochondrial ribosome RNA more similar to the bacterial counterpart, increasing the affinity of the human mitochondrial ribosome to aminoglycosides, and mitochondrial translation is consequently compromised.
A summary of the clinical characteristics of the most frequent mitochondrially inherited diseases, with the corresponding mtDNA variants associated, is presented in Table 10.1. There are no straightforward correlations between genotypes and phenotypes regarding mtDNA variants [14, 18]. In other words, no clear correlations are seen between the site of the mutation and the clinical phenotype, even within the same gene, except for some variants. For instance, variants in the tRNALeu gene MT-TL1 (UUA/G) may be associated with mitochondrial encephalopathy, lactic acidosis and stroke like episodes (MELAS) syndrome , but they can be causative to other syndromes. On the other hand, mutations in different genes can cause the same syndrome, and MELAS is one of the examples (Table 10.1). Moreover, it is striking that some small size variants (substitutions) relate to different clinical findings. An estimate based on a cohort study in England pointed to a prevalence of diseases caused by mtDNA pathogenic alterations near 9.6 cases per 100,000.
Given the difficulty in clinical classification of mitochondrial inherited diseases and the lack of clear correlation between specific variants and clinics, the molecular diagnosis of mitochondrial diseases has always been troublesome and expensive. Sanger sequencing of many mitochondrial genes was needed until the causative variant was found [4]. Besides, heteroplasmy, as explained in the next section, has always been a challenge to molecular analysis since, in many patients, the mutated mtDNA lineage is present in very low frequencies in circulating blood or it is detected only in the mostly affected tissues, such as skeletal muscle.
The introduction of next generation sequencing (NGS or massive parallel sequencing ) to the study of mtDNA and its rapid transfer to clinical practice speeded up and increased the precision in the molecular diagnosis of mitochondrial diseases (see below). Besides allowing detection of heteroplasmic mtDNA sequences in low frequencies considerably better than previously possible with conventional techniques (e.g. Sanger sequencing ), NGS allowed simultaneous sequencing of all mitochondrial genes and many nuclear genes related to mitochondrial diseases in only one experiment, for instance, after their capture and selection for NGS, constituting a panel including mitochondrial genes and nuclear mitochondrial-disease related genes [19]. Nevertheless, challenges still remain in the clinical recognition of mitochondrial diseases.
10.4 Heteroplasmy Is a Generalized Phenomenon in mtDNA Inheritance and Relates to the Clinical Expression of Diseases
Each cell, depending on the type, may contain hundreds of mitochondria, and each mitochondrion harbors some copies of its genome. Thus, thousands of mtDNA molecules may be present in each cell and pathogenic mtDNA variants may be present in some, but not all of these molecules.
Alterations in the mtDNA sequence occur very frequently, and they can exist, at least transiently, as two or more different molecules with distinct nucleotide sequences within a single mitochondrion, cell, tissue or organism. The multicopy nature of mtDNA thus allows the phenomenon of heteroplasmy, a unique aspect of mitochondrial inheritance . Heteroplasmy is defined as the co-existence, in cells, tissues, organs or individuals, of mtDNA molecules with different nucleotide sequences. Heteroplasmy was firstly recognized as frequent, and clinically relevant, in patients from pedigrees in which mitochondrially inherited diseases were segregating (review in [4]).
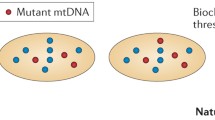
The existence of multiple mtDNA copies within a cell greatly affects the impact of pathogenic variants of mtDNA. Wild-type copies of mtDNA encode normal copies of mitochondrial proteins or RNA molecules, while mutated mtDNA encode abnormal products. Besides, mitochondria are continuously involved in fusion and fission, which allow exchange of proteins, RNA and other components between mitochondria located within the same cell. Variability in clinical expression and age at onset, or lack of penetrance, are features of many diseases with classical Mendelian transmission. However, these features are enormously enhanced in hereditary diseases that result from mtDNA mutations, partly because of heteroplasmy. The clinical expression of a pathogenic variant in mtDNA correlates with the proportion of wild-type and mutant copies. In many of the diseases, a minimum amount of mutated mtDNA must be present before any kind of cellular dysfunction occurs and clinical signs of disease become apparent, in a threshold effect. The threshold seems to be lower in tissues that are largely dependent on oxidative metabolism, such as brain, heart, muscle, retina, endocrine glands and kidney, thus explaining why these are frequently compromised in mitochondrial diseases (review in [18]).
The random distribution of mitochondria at the time of cell divisions can lead to fluctuations in the proportion of mutated mtDNA that is received by daughter cells. Whenever a pathogenic threshold is surpassed, the cell phenotype can change. This explains some age-related and some tissue-related variability of clinical symptoms in individuals with mtDNA disorders. The investigation of families in which mtDNA pathogenic variants are segregating has shown that onset of clinical manifestation and severity of disease may be correlated to the frequency of heteroplasmy. For instance, different mutation proportions explain the different degrees of severity of neuropathy, ataxia and retinitis pigmentosa in Leigh’s syndrome. Heteroplasmy is probably one of the major explanations for the wide variation of phenotypes between maternally related individuals sharing a mtDNA variant that leads to inherited disease, and it has been investigated for decades in the context of mitochondrial diseases.
On theoretical grounds, heteroplasmy should be expected to be a very common event, since each oocyte contains many mitochondria, each one with many mtDNA copies, and mutation rates are elevated in mitochondria. Indeed, all presently existing inherited mtDNA variants must have existed transiently in heteroplasmic states when they first rose after mutation, before their fixation in the germ cells. However, conventional strategies of molecular assessment of heteroplasmy, for instance, Sanger sequencing, had shown severe sensitivity limitations to detect alternative sequences present in low number of copies.
More recently, the NGS technology provided excellent opportunities to reassess the matter of heteroplasmy, allowing more precise and quantitative approaches of investigation of mutated mtDNA in healthy and abnormal tissues, because of its deep coverage. This has largely confirmed that heteroplasmy is a generalized phenomenon, much more frequent than initially suspected [20,21,22].
The observation that individuals from the same sibship or pedigree may have different proportions of heteroplasmy always puzzled geneticists. Furthermore, large shifts in the frequency of heteroplasmy can be observed in only one transmission, from mother to child. This also contributes to explain why individuals in the same pedigree show different clinical presentations of the mitochondrial disease, with striking differences in severity, and some hypothesis were proposed to explain the findings. It is now widely recognized that heteroplasmy frequency shifts can be explained by a so-called mitochondrial “bottleneck ” [2, 4]. In oogenesis, the population of mitochondria that is present in a mammalian oocyte (near 100,000) results from the amplification of a reduced initial number of mitochondria, containing a small number of mtDNA copies. During female embryogenesis, the primordial germ cells (oogonia) develop and early in this process there is a bottleneck of a few hundred mtDNA copies and, by chance or selection, some mtDNA lineages containing variants may be eliminated. Thus, the rapid amplification of mtDNA from a reduced pool of molecules may result in different oocytes bearing widely different proportions of mutated and wild-type mtDNA, leading to abrupt changes of the frequency of heteroplasmy in one generation. It has also been pointed that, in mice, a subsequent bottleneck occurs in the early postnatal period. Fertilized oocytes develop into blastocysts by day 4.5 after conception and this step is known as preimplantation development. The mtDNA copy number remains constant during the preimplantation period while cells divide. It was proposed that, in this period, there is a reduced number of mtDNA molecules per cell and unequal segregation mtDNA molecules in the following cell divisions, widening differences in the frequency of heteroplasmic variants.
Nevertheless, one must always bear in mind thaty is far from being the sole explanation for differences in expressivity of mitochondrial disorders. Some mtDNA variants, for instance, m.1555A > G that is causative of hearing loss, has been detected in in most of the pedigrees in which it was found. Age of onset and severity of hearing loss are extremely variable within these pedigrees and penetrance of hearing loss hardly exceeds 50% [17]. In other diseases, clinical and phenotypic variability may exist among patients with similar levels of heteroplasmy. Many other factors may affect clinical variability and progression of mitochondrial diseases and these are probably environmental factors. As previously mentioned in the case of m.1555A > G, the penetrance of hearing loss is known to be strongly influenced by the administration of aminoglycosides. Lifestyle, exercise, smoking, exposure to oxidant molecules and aging are the most likely environmental modifiers of the progression of mitochondrial disease. Besides, since the majority of mitochondria proteome is coded by nuclear genes, variability in genotypes in nuclear genes that code mitochondrial components must have a role in phenotypic expression of mitochondrial dysfunction. In fact, attempts were made to map nuclear modifiers of expression of mtDNA related diseases. It also remains plausibl e that single nucleotide polymorphisms (SNPs) in the mitochondrial genome, for instance, polymorphic sites associated to the definition of haplogroups, may have a subtle effect on manifestation of mitochondrial diseases.
Although molecular diagnosis has recently improved its accuracy, genetic counseling of families with mtDNA pathogenic variants remains a complex task, since it is almost impossible to predict precisely the recurrence risk or severity of a disease in the following generation, given the many uncertainties regarding the prediction of heteroplasmy levels and other factors that affect the clinical outcome [18, 19].
10.5 Human mtDNA Is Maternally Transmitted , But Striking Exceptions Have Been Described
The transmission of human mtDNA is strictly maternal. All mtDNA in the zygote derive from the ovum. Therefore, a mother carrying a mtDNA variant in homoplasmy passes it to all her children, but only their daughters will transmit it to progeny. In other words, no one is expected to inherit mtDNA from the father. Although maternal inheritance was long ago assumed as a dogma in Biology, the cellular mechanisms underlying this assumption came to light more recently.
Although some basic facts about the biology of human and mammalian fertilization are known since the decade of 1930, unfortunately, many biology textbooks still replicate a wrong concept to explain the exclusively maternal inheritance of mtDNA: that sperm midpiece and tail do not enter oocytes in mammalian fertilization, thus excluding paternal mitochondria from the zygote. Although this happens in some exceptional cases, such as in the Chinese hamster, this was equivocally extrapolated to other mammals and humans. This is clearly a misconception and it is surprising that it has survived so long, since it has been long demonstrated that sperm fully enters the oocytes in fertilization, and sperm mitochondria are indeed observed within the oocytes after fertilization. In mammalian fertilization, the mitochondria-rich midpiece of the sperm tail enters the oocyte and it is a fact that sperm contributes to the fertilized oocyte’s pool of mitochondria. Tail and midpiece can be traced within the zygote for several division cycles after fertilization [23].
The number of mtDNA molecules within a single spermatozoon is certainly much lower than the copy number in oocytes. It was estimated that a human spermatozoon may contain something in between 100 and 1200 mtDNA copies, while a human oocyte contains between 100,000 and 250,000 molecules. This effect of “dilution” of paternal mtDNA allows to predict a reduced contribution of paternal mtDNA in offspring [25]. However, even under such scenario, one occasionally would see transmission of paternal mtDNA, but these exceptions are extremely rare. Thus, additional mechanisms that halt paternal mtDNA transmission were expected to be revealed.
Near the 1960s, it was already known that in rat, all sperm structure s penetrate the oocyte, but in the first cell divisions after fertilization, paternal mitochondria swell, lose their cristae and disintegrate, being completely eliminated in the preimplantation embryos. In a remarkable contribution, Sutovsky and co-workers [24] provided the first evidence of the molecular mechanisms acting on the elimination of paternal mitochondria after fertilization in mammals. They demonstrated that ubiquitination of sperm mitochondria during mammalian spermatogenesis is a key factor that leads to elimination of sperm mitochondria by means of proteolysis in the oocyte cytoplasm. Poly-ubiquitination is one of the cellular processes in which a protein is tagged to proteolysis. This is achieved by a covalent binding of the ubiquitin peptide to lysine residues of the targeted proteins. Mammalian mitochondria are ubiquitinated probably in the male reproductive tract and ubiquitin tagging of the sperm mitochondrial membranes culminates in their recognition by the ubiquitin-proteasome-dependent proteolytic machinery. According to Sutovsky and co-workers [25], the mitochondria inner membrane protein prohibitin would be the best candidate as an ubiquitin substrate. This serves as a death sentence for paternal mitochondria after fertilization, since it triggers their elimination. During mammalian spermatogenesis, mitochondrial ubiquitination is already detected at the secondary spermatocyte phase. It is not clear whether, besides degradation by the ubiquitin proteasome system, autophagy also plays a role in the elimination of sperm mitochondria [26,27,28]. Sutovsky and collaborators also showed that the elimination of ubiquinated sperm mitochondria could be prevented by injection of anti-ubiquitin antibodies. In parallel to mitochondria elimination, paternal mtDNA degradation probably involves a mitochondrial endonuclease that degrades mtDNA within paternal mitochondria after fertilization. However, there are reports suggesting that elimination or reduction of number of copies of paternal mtDNA molecules in mammalian sperm may have happened before fertilization [29]. As a consequence, it is nowadays largely recognized that reduction or elimination of mtDNA copies in sperm mitochondria, followed by elimination of sperm mitochondria after fertilization is the most plausible biological explanation for the strictly maternal inheritance of mammalian mtDNA [27].
Nevertheless, some striking exceptions to maternal inheritance in humans were reported. In 2002, a patient with mitochondrial myopathy, due to a deletion of 2 bp in the MT-ND2 gene was described. The patient, born from an unaffected couple, showed mutated mtDNA in high frequency of heteroplasmy in muscle. Molecular analysis revealed that, besides heteroplasmy regarding the 2 bp deletion, the patient also showed other heteroplasmic sites, in nucleotide positions known for harboring SNPs that allowed to define mtDNA haplogroups. This indicated that he had mtDNA molecules from two different haplogroups, one inherited from the mother and the second, from the father. Haplotype analysis also allowed the conclusion that the deletion was present in the paternally derived mtDNA haplogroup, although it was not detected in the father, at least in blood. It was demonstrated in that study that the paternally derived mtDNA contributed with 90% to the mtDNA pool in skeletal muscle. The deletion probably arose de novo in early embryogenesis or, more likely, in the paternal germ line [30]. After this report, many other series of patients with mitochondrial diseases were investigated and no other paternally inherited cases of mtDNA were reported [31].
The recognized capacity of NGS in detecting DNA sequences even if they are present in very low frequencies in biological samples gave opportunity of reappraising the issue of the escape of paternal mtDNA after fertilization in humans. Studies demonstrated that low frequency heteroplasmy is a generalized phenomenon in human cells, both in normal and cancer tissues [20, 21, 32]. The comparison of heteroplasmic variants present in trios (father, mother and child) allowed the conclusion that low frequency heteroplasmic variants in children do not result from inheritance of variants present in paternal mtDNA molecules, but they raise probably due to chance, from post-zygotic changes, leading to somatic mosaicism [33].
In 2018, Luo et al. [34] reported three unrelated Chinese pedigrees in which high levels of mtDNA heteroplasmy were detected in more than one generation. Investigation of mtDNA in the three pedigrees showed evidence of biparental mtDNA transmission, and the capacity of transmitting male mtDNA seemed to be inherited as an autosomal dominant character. One of the hypothesis to explain the findings was an inherited disruption of normal cell processes related to the prevention of inheritance of paternal mtDNA. Nevertheless, these findings were recently contested by other authors that highlighted that the transmission of NUMTs can lead to the false conclusion that paternal mtDNA is transmitted, and this would provide a more likely explanation for the recent reports of paternal transmission of mtDNA [35]. Meanwhile, while the controversy remains, it seems that paternal transmission of mtDNA must be considered as an extremely rare event and it does not change the fact that, for genetic counseling purposes, diseases that result from pathogenic variations in the mtDNA are maternally transmitted as a rule and the risk of transmission of paternal DNA is negligible [17]. Furthermore, no theoretical premises in studies about human evolution and dispersion must change to account for paternal transmission of mtDNA, because of its rarity.
10.6 Frequent mtDNA Variants Define Haplogroups Correlated to the Geographical Patterns of Dispersion of Modern Humans
The elevated variability of sequences within the mtDNA and the lack of recombination between paternal and maternal genomes allowed maternal lines in mtDNA, in all different human populations, to be transmitted as haplotype blocks. Diversity within mtDNA has been investigated all over the world by sequencing the most variable parts of the control region, the hypervariable segments I and II (HVS-I and HVS-II), often complemented by genotyping some informa tive SNPs from the coding region. Commonly inherited mtDNA variants have thus created stable population subgroups sharing maternal lines that can be identified according to the presence or absence of some polymorphic sequence variants. The classification of maternal lines in groups sharing some ancestral variants resulted in what we call mtDNA haplogroups [12, 36, 37].
Mitochondrial haplogroups are groups of mitochondrial sequences sharing some nucleotide variants, which indicate common ancestors. Since some variants originated in specific geographical regions and were spread with human populations’ dispersal, a mitochondrial haplogroup can be a marker of geographical or even continental ancestry, on the maternal side. Most of the mtDNA of Europeans belong to one of 10 major (top level) haplogroups: H, I, J, K, R, U, T, V, W and X. A, B, C, D, E, F, G, M, N, O, P, Q, S, Z and Y are the haplogroups found originally in Asia. All L* lineages are African: L0, L1, L2, L3, L4, L5, L6. In Native American populations , only the haplogroups A, B, C, and D, which originally rose in Asia, are usually present. Each major (top level) haplogroup can be divided into many sub-haplogroups or lineages, when detailed information about mtDNA sequence is available. This has offered valuable opportunities for investigating human origins, the dispersal of human populations in the continents, its timing, and to assess genetic diversity and admixture of human populations. In addition, when coupled to Y chromosome variation studies, mtDNA allows investigation of sex-biased admixture. In other words, genetic admixture estimates based on uniparental markers, such as the Y chromosomes and mtDNA, allow the identification of sex bias in the makeup of an admixed population. For instance, admixed populations in South America result from recent colonial admixture from European colonizers. While high frequency of Y chromosome of European ancestry indicate male-biased transfer of European DNA, mtDNA in South America comparatively reveals low frequencies of European mtDNA haplogroups. On the other hand, Native American and African mtDNA lineages are more abundant. This reveals that in South America, the European genetic contribution originated mainly from males, and that Native American and African women, and not European women, were prominent in the origin of the admixed populations [12].
MtDNA is frequently used in evolutionary studies as a genetic marker of diversity, since the rate of substitution of nucleotides is increased when compared to nuclear DNA. Remarkable variability of sequences is found mainly in the “hypervariable regions ”, HVS-1, HVS-2 and HVS-3, located in the D-loop region, where the rates of mutation were estimated to be the highest. Since mtDNA is present in cells in high number of copies, when compared to nuclear genes, it can be easily amplified by PCR (polymerase chain reaction ). In forensic science and practice, amplification of mtDNA followed by genotyping or sequencing is possible even when samples are obtained from poorly conserved biological material, in situations when very little DNA is available or it is partially degraded. These properties of mtDNA allowed the investigation of samples obtained from ancient human remains, including bones of thousands of years of age, resulting in interesting academic outcomes to archeology and to studies of human evolution.
10.7 Frequent mtDNA Variants Correlate with Increased Susceptibility to Complex Disorders
Some mtDNA variants were clearly correlated with the origin of known maternally inherited diseases, being causative of dysfunctions with profound effects on quality of life, as already reviewed in this chapter. However, some mtDNA variants, including single nucleotide substitutions with milder effects on mitochondria functioning, can confer increased susceptibility to disease. Many mtDNA SNPs have historically segregated in haplogroups , in human evolution and migrations . Although they were often treated as “neutral” from the point of view of evolution, there are examples in which they have been found to be important in adaptation of human populations to new environments and in modulating risk of developing disease [38, 39]. Many studies correlated mitochondrial haplogroups with longevity, athletic performance, adaptation to high altitude, and risks for diabetes, Alzheimer and Parkinson diseases, some psychiatric disorders and cancer. The mtDNA haplogroups may also influence the penetrance of autosomal genetic defects or even mtDNA defects [17]. The same rare mtDNA variant can cause different degrees of severity of disease, depending on the mtDNA haplogroup on which is present. Modest but relevant differences in respiratory chain and mtDNA copy number may be present in individuals with different haplogroups . In one study, different patterns of gene expression were found in stem cells harboring different haplogroups. ROS and metabolic intermediates derived from mitochondrial metabolism act as signals that convey information between mitochondria and the nucleus. These metabolites act as substrates and co-factors for chromatin remodeling complexes, resulting in epigenetic marks that may alterate nuclear gene expression. Thus, mito-nuclear crosstalk and its link to the epigenomes may provide a way to explain why common variation in mtDNA may influence susceptibility to diseases and physiological responses to environment [40].
10.8 Somatic Variation Accumulated in the mtDNA is Related to Aging and Diseases
Accumulation of mutations in the mitochondrial genome seems to be a natural feature of aging . In a set of postulates, known in the literature as the “mitochondrial theory of aging”, it was proposed that the progressive accumulation of somatic mutations in the mtDNA during lifetime leads to mitochondrial abnormalities and decline in mitochondrial function. Mitochondrial abnormalities and mtDNA mutations are instigators of multisystem degeneration and energy deficits, and one of the most important factors in this process is supposed to be the production of ROS during normal functioning of the respiratory chain taking place within mitochondria. The accumulated somatic mtDNA mutations due to ROS production can impair the function of the respiratory chain and lead to increased ROS production. As a result, more mutations are accumulated, in a vicious cycle. This cycle is believed to account for increase in oxidative damage during aging. The consequence of this cycle is loss of cellular functions, increasing energy insufficiency, cell senescence and apoptosis [4, 41, 42].
Some studies have consistently shown that mitochondrial respiration decreases with age, attributed to reduced activity in each of the four OXPHOS complexes of the mitochondrial electron transport chain (ETC). The detection of human cells deficient in COX (Cytochrome C oxidase) in aging post-mitotic tissues was the first biochemical evidence of the mitochondrial theory of aging. Loss of structure in cristae, alteration of mitochondrial morphology and dysregulation of mitochondrial metabolism are considered senescence markers, placing mitochondria as a senescence gatekeeper. Reduced mitochondrial quality and content in tissues is indeed implicated in several aging conditions such as cancer, type 2 diabetes, osteoporosis, dementia, neurologic and neurometabolic syndromes. Sustained mitochondrial dysfunction leads to activation of the caspase cascade culminating in DNA fragmentation, a characteristic of apoptosis , observed in aged tissues and in many disorders. Morphological changes in mitochondria with aging correlate with increased level of oxidative mtDNA damage, for instance, 8-oxoguanine, as well as the presence of mtDNA deletions and point mutations. Hence, mtDNA replication errors accumulating during the lifespan were proposed to be the driving force of mitochondrial metabolic failure and aging.
Aging cells show some mitochondrial properties similar to those of patients with inherited mitochondrial diseases. Not surprisingly, many of the frequent characteristics present in individuals with advanced age, such as diabetes, hearing loss, cataract, neurologic alterations and muscle weakness, are present in inherited diseases due to mtDNA pathogenic variants. This reinforces the biological connection between mutated mtDNA molecules and aging. Some mutated mtDNA variants can clonally expand to high levels in individual cells and the question remains whether this happens because abnormal copies of mtDNA are selectively replicated, but it seems that drift could also explain the findings [4].
Many studies performed with aging humans and animal models confirm the connection between age and the frequency of mtDNA mutations, which is in accordance to some of the steps of the “mitochondrial theory of aging”. A remarkable advance in the field was the development, by two different groups, of mouse models expressing a defective version of the PolG mtDNA polymerase, lacking its proof-reading exonuclease activity. These mice acquired mtDNA mutations at a higher rate than controls, thus being called “mutators”. They showed remarkable marks of premature aging such as osteoporosis, hunched appearance, weight loss, reduced adipose tissue and muscle mass [43]. These studies confirmed that somatic mtDNA alterations contribute to aging phenotypes. In spite of this compelling evidence, still many questions remain unanswered. A lot is still required to confirm that mtDNA mutations per se are causal of the aging process or if they represent collateral findings of this process. Mitochondrial gene expression, in particular, efficiency in mitochondrial translation, is likely another important issue in aging. Damage to mitochondrial components other than DNA may be also key factors in this vicious cycle.
Modern chronic diseases are boosted by excessive food intake and sedentary lifestyle, and mitochondrial biology can be the conceptual link to explain many of the epidemiological observations in the field. Oversupply, which is the excess supply of energy substrates, mainly glucose and lipids, was correlated to biochemical mitochondrial overload, which leads to increased ROS production, mitochondrial fission, oxidative stress and these culminate in mtDNA damage, possibly increasing cellular aging, with shortening of telomeres (see Chap. 7) [3, 39].
It has already been shown that ROS is an important factor in telomere damage and mitochondria are the source of ROS. Aging in primary cells is known to be associated with a gradual increase in ROS production due to progressive mitochondrial failure and is concomitant to telomere shortening. The neutralization of ROS does not restore the mitochondrial function but inhibits telomere shortening thus establishing ROS as probably causative of telomere shortening. In addition, in human syndromes with excess of ROS production, such as some mitochondriopathies, a decrease in telomere length was observed. It is remarkable the recent increase in the number of studies aiming to connect mitochondrial processes related to senescence and aging to telomere biology [44].
Many epidemiological studies in humans demonstrate that exercise reduces the risk of several chronic diseases and contributes to increased life expectancy. Safdar and collaborators, in 2011 [45], demonstrated that endurance training was able to rescue mitochondrial biogenesis, increase mitochondria oxidative capacity, restore mitochondrial morphology, and reduce apoptosis in many tissues of the mtDNA mutator mice. Exercise attenuated the decline in mtDNA copy number and decreased the frequency of point mutations in the mtDNA, which appeared as related to the progeroid phenotype in the mutator mice. Endurance training also contributed to mitigate apoptosis, and the premature mortality was also prevented. They hypothesized that exercise may impose selective mitochondrial biogenesis of healthy mitochondria via modulation of mitochondrial dynamics, by promotion of fusion and fission and by destruction of mitochondria carrying high levels of mutated mtDNA. Their findings support exercise as an approach to improving systemic mitochondrial dysfunction caused by aging or diseases.
Exercise has been also shown to positively affect the brain and reverse age-related brain atrophy. Exercise increases whole-body oxygen consumption and accelerates mitochondrial energy production. Increased energy demand engages adaptive signaling pathways that increase mitochondrial content and optimize their function via mitochondrial biogenesis, inducing the expression of genes that restrict inflammation and may therefore counteract pro-aging mitochondrial signaling. There is also evidence that exercise stimulates mitochondrial biogenesis in the brain. In accordance, sedentary behavior is a known major risk for Alzheimer’s disease . This might be explained by the fact that physical inactivity promotes metabolic stress, possibly through disruption of mitochondrial dynamics and accumulation of mtDNA damage [3].
It can be predicted that advances in understanding mitochondrial biology will soon help to develop strategies to prevent the adverse effects of aging and to treat mitochondrial diseases.
10.9 mtDNA Disorders
Current treatment strategies for conditions related to mitochondrial dysfunction are limited, since they address some of the symptoms but do not mitigate the reduced mitochondrial oxidative capacity in aged tissues. The research of novel strategies to mitigate mitochondrial dysfunction is desirable to improve the quality of life of aging subjects or of the ones affected by mitochondrial disorders. Some studies have attempted to address mitochondrial diseases focusing on different strategies of treatment: promotion of increase the oxidizing capacity of mitochondria; administration of lacking substances or adding substances that increase the energy capacity of mitochondria; reduction of the quantity of mutated mtDNA and induction of mitochondrial biogenesis. For instance, vitamins and co-factors, such as vitamins C and E, coenzyme Q10 and folic acid were tried. Some of these substances provided some beneficial effects, depending on the type of disease. However, there is remarkable interest in academic research related to the fourth strategy, the stimulation of mitochondrial biogenesis as a consequence of exercise, as mentioned in the previous paragraphs.
The most effective strategy presently available is the prevention through appropriate genetic counseling . There are empiric recurrence risks available in the case of transmission of the most common , if they are in homoplasmy . However, whenever the causative variants are in heteroplasmi c state, the genetic bottleneck is a barrier to the precise prediction of disease risk in the following generations.
Pre-implantation genetic diagnosis (PGD) was shown to be of utility to some women with pathogenic variants in heteroplasmy. Embryos obtained after in vitro fertilization can be genetically analyzed, through biopsy of one or few blastomeres, and those embryos which are mutation-free or show the lowest mutation levels can be selected to be transferred to the uterus. Such strategy can minimize the probability of severe disease, but it may not be able to eliminate completely the risk of disease [46].
Some advances were also made in the field of pro-nuclear transfer with the aim of correcting mitochondrial disease. This involves the transfer of nuclear DNA from the donor zygote (obtained from a donor couple in which the mother has mtDNA disease) to an enucleated recipient zygote, by means of fusion. The zygote retains nuclear DNA from the parents, but the mtDNA, with wild-type sequence, comes from the recipient zygote. Other groups used a similar technique but they utilized spindle transfer, instead of pro-nuclear transfer, with equivalent results. These advances are far from being a routine in the clinical setting and they create debate because of potential long-term effects and ethical issues. Legalization of these procedures is not world spread and will be an ongoing debate [19, 46].
In parallel to investigations aiming at treating genetic disorders caused by variants in nuclear DNA, there is hope that, in the next years, research in gene therapy and genomic edition will bring some relief to carriers of mitochondrial dysfunctions caused by mtDNA alterations.
10.10 mtDNA is Related to Inflammation and Immunity
In mammalian cells, mitochondria contribute to immune and inflammation processes in different ways. Following mitochondrial damage due to oxidative stress, circular mtDNA can leak out into the cytoplasm. As a result, the NLRP3 (leucine-rich repeat (LRR)-containing proteins (NLR) family member 3) inflammasome is triggered by mtDNA outside the mitochondria. Mitochondria also act in immune signaling affecting anti-viral responses. Third, mtDNA can also leak into the systemic circulation where it is recognized by TLR9 (Toll-like receptor 9) and this leads to tissue lesion in heart, vascular system and may cause neurological degenerative states. Circulating cell-free mtDNA (ccf-mtDNA) and other molecules that are free in the blood are thus putative markers of early stages of diseases related to mitochondrial stress. Some studies suggested that ccf-mtDNA increases with age, contributing to the overall trend of increasing inflammation that happens with age [3]. There is an emerging notion that mitochondria is related to factors that can trigger inflammatory and pathological processes that underlie many common chronic diseases that increase with age, such as diabetes.
10.11 mtDNA and Epigenetics
Epigenetics is an important layer of information on DNA sequences and is a key factor for establishing profiles of gene expression. DNA can be epigenetically modified via methylation of citosines, a process that frequently leads to transcriptional silencing of genes if it occurs near promoter regions. Histones, key proteins in the assembly of nuclear chromatin, can also be epigenetically altered by post-translational modification such as acetylation, phosphorylation, methylation, sumoylation and ubiquitination of their N-terminal tails. Non-coding RNAs also play key roles in the definition of epigenetic landscapes, regulating gene expression. Histone modifications, DNA methylation and non-coding RNA expression are epigenetic modifications that explain differential nuclear gene expression in different cells types and in different stages of development, because they induce changes in chromatin states that affect initiation of transcription [47]. It has only recently been recognized that gene expression in mitochondria may also be regulated via epigenetic mechanisms, as it happens to nuclear gene expression. In parallel to epigenetic processes that regulate gene expression in nucleus, DNA methylation, non-coding RNAs and post-translational modification of proteins associated to the nucleoid, were identified within mitochondria and they are probably linked to the regulation of gene expression, although the matter is still the subject of ongoing debate [38].
The mechanisms of methylation and demethylation in mtDNA are not clearly understood. Several studies identified mtDNA methylation in cell lines and tissue samples, including of human origins. Methylation was reported in mtDNA many decades ago and it is a matter of controversy ever since. Some methodological issues complicate the estimates of overall methylation in mtDNA, such as contamination with NUMTs. The first studies focused on methylation of CpG sites, with heterogeneous and conflicting results. In the nucleus, cytosine methylation frequently occurs within CpG nucleotides clustered in CpG islands (see Chap. 4), but it may also occur in other sites. However, CpG islands are absent in mtDNA and methylation occurs within dispersed CpGs sites. Besides, non-CpG methylation, such as CpC, CpA and CpT, and adenine methylation were also observed. The studies altogether point that methylation indeed occurs within mtDNA. The endosymbiotic theory of mitochondrial origin combined with the findings of abundant adenine methylation in mtDNA suggest that adenine methylation in mitochondria may be more relevant than cytosine methylation, as seen in nuclear DNA. A mitochondrial localized DNA methyltransferase 1 (mtDNMT1) was identified in 2011, raising the debate on the role of methylation in gene regulation within mitochondria.
The mitochondrial D-loop is one of the most important regions to the expression of the mtDNA due to its role in controlling transcription and replication. Differential methylation within the D-loop, especially in cytosine nucleotides, has been described in many studies, but its precise function is unknown; it is tempting to speculate that different methylation profiles in the D-loop region would be related to mtDNA gene expression. Apart from D-loop, gene bodies also have regions in which methylation may have an effect on gene expression. Changes in methylation of mitochondrial genes were shown to correlate with changes in gene expression [38].
With the ongoing recognition that epigenetic modifications may play a role in mtDNA expression, the role of several factors on levels of mtDNA methylation was investigated. Several external factors such as air pollutants, smoking, diet and drugs were demonstrated as affecting mtDNA methylation. Some air pollutants and smoking were associated to D-loop and gene methylation in mtDNA, with important correlations to human diseases. In pigs, maternal diet was shown to alter mtDNA methylation levels in newborns, affecting their OXPHOS capacity. Differential mtDNA methyation was also correlated to many frequent human diseases such as Alzheimer, Parkinson, cancer and metabolic disorders, including obesity [38, 47].
In mitochondria, histone proteins are absent but modifications of nucleoid proteins were shown to play a role in the regulation of gene expression. Many proteins localized within mitochondria contain potential acetylation sites. TFAM, the main structural component of mitochondrial nucleoids, is a protein that promotes replication, transcription and general maintenance of mtDNA. TFAM can be modified by acetylation, glycosylation and phosphorylation. Acetylation and phosphorylation alter the affinity of TFAM to DNA, thus affecting mtDNA compaction; mtDNA compaction, as consequence, probably affects mtDNA replication and transcription. Furthermore, it has been shown that levels of TFAM occupancy in mtDNA affect the access of DNMTs to methylate DNA, showing that TFAM plays a role in the pattern of mtDNA methylation.
Acetylation and phosphorylation sites were identified in other nucleoid-associated proteins, including mtSSB and DNA polG, but their role in the regulation of gene expression is unknown.
Different classes of non-coding RNAs (ncRNAs ) involved in the epigenetic regulation of gene expression in mitochondria are known, but it is not clear in some cases if they are transcribed inside the mitochondria or if they are derived from NUMTs. Several long non-coding RNAs (lncRNA) encoded by the mtDNA have been identified and there is evidence that they participate in the regulation of mitochondrial gene expression. Some of these lncRNAs are transported into the nucleus and act as retrograde signaling molecules, and they are believed to act in mito-nuclear crosstalk. Moreover, significant changes in their levels were observed in cancer, suggesting they function in cell cycle progression. There are also lncRNA molecules encoded by nuclear genes and transported into the mitochondria, where they regulate mitochondrial processes of metabolism and apoptosis.
MicroRNAs were also identified within mitochondria and they were termed mitochondrial microRNAs (mitomiRs) . They are short (17–25 bp) single-stranded RNA molecules that are transcribed in nucleus from nuclear templates and are transported to mitochondria, but some of them are transcribed using mtDNA as template. They regulate expression of nuclear-encoded and mtDNA-encoded proteins. There is evidence that they can enhance or repress gene expression, at transcriptional and translational levels, modulating metabolic activities [38, 47].
There is compelling evidence that there are many pathways allowing exchange of information between mitochondria and nucleus, and this exchange may affect gene expression in the nucleus. The signals that convey information between mitochondria and nucleus are ROS and other metabolic intermediates from mitochondrial metabolism. Some of these metabolites are required substrates and co-factors in chromatin remodeling complexes, influencing post-translation histone tail modifications by histone acetylases, histone deacetylases, for instance, or leading to DNA modifications via DNA methyltransferases or demethylases. The resulting biochemical changes influence the epigenetic state of nuclear genes, resulting in changes in gene expression. It can be concluded that epigenetic communication between nuclear and mitochondrial genomes occurs at multiple levels, ensuring a coordinated gene expression between these two different genetic compartments. Metabolic changes stimulated, for example, by environment factors, such as diet or physical activity, alter the relative abundances of various metabolites, directly affecting the epigenetic machinery, both in mitochondria and nucleus [3, 47].
References
Alberts B, Hopkin K, Johnson AD, Morgan D, Raff M, Roberts K, Walter P. Essential cell Biology, 5th International Student Edition. 2018. Wiley.
Chinnery PF, Hudson G. Mitochondrial genetics. Br Med Bull. 2013;106(1):135–59. https://doi.org/10.1093/bmb/ldt017. Epub 2013 May 22. PMID: 23704099; PMCID: PMC3675899.
Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105–16.
Taylor RW, Turnbull DM. MtDNA mutations in human disease. Nat Rev Genet. 2005 May;6(5):389–402.
Margulis L. The origin of plant and animal cells. Am Sci. 1971;59(2):230–5. PMID:5170543.
Gray MW. Rickettsia, typhus and the mitochondrial connection. Nature. 1998;396(6707):109–10. https://doi.org/10.1038/24030. PMID:9823885.
Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2(6):REVIEWS1018. https://doi.org/10.1186/gb-2001-2-6-reviews1018.
Thrash JC, Boyd A, Huggett MJ, et al. Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade. Sci Rep. 2011;1:13. https://doi.org/10.1038/srep00013.
Bernt M, Braband A, Schierwater B, Stadler PF. Genetic aspects of mitochondrial genome evolution. Mol Phylogenet Evol. 2013;69(2):328–38. https://doi.org/10.1016/j.ympev.2012.10.020. Epub 2012 Nov 7. PMID:23142697.
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981 Apr 9;290(5806):457–65.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mtDNA. Nat Genet. 1999 Oct;23(2):147.
Jobling M, Hollox E, Kivisild T, Tyler-Smith C. Human evolutionary genetics. 2nd ed. New York: Garland Science; 2013 June 25.
Wallace DC, Zheng XX, Lott MT, Shoffner JM, Hodge JA, Kelley RI, Epstein CM, Hopkins LC. Familial mitochondrial encephalomyopathy (MERRF): genetic, pathophysiological, and biochemical characterization of a mtDNA disease. Cell. 1988;55(4):601–10.
DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348(26):2656–68.
Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW. The genetics and pathology of mitochondrial disease. J Pathol. 2017;241(2):236–250. https://doi.org/10.1002/path.4809. Epub 2016 Nov 2. PMID: 27659608; PMCID:PMC5215404.
Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4(3):289–94. https://doi.org/10.1038/ng0793-289. PMID:7689389.
Estivill X, Govea N, Barceló E, Badenas C, Romero E, Moral L, Scozzri R, D’Urbano L, Zeviani M, Torroni A. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet. 1998;62(1):27–35. https://doi.org/10.1086/301676. PMID:9490575; PMCID:PMC1376822.
Wei W, Chinnery PF. Inheritance of mitochondrial DNA in humans: implications for rare and common diseases. J Inter Med. 2020;287(6):634–44.
Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet. 2017a Aug 31;18:257–75.
Li M, Schönberg A, Schaefer M, Schroeder R, Nasidze I, Stoneking M. Detecting heteroplasmy from high-throughput sequencing of complete human mtDNA genomes. Am J Hum Genet. 2010 Aug 13;87(2):237–49.
Sosa MX, Sivakumar IK, Maragh S, Veeramachaneni V, Hariharan R, Parulekar M, Fredrikson KM, Harkins TT, Lin J, Feldman AB, Tata P, Ehret GB, Chakravarti A. Next-generation sequencing of human mitochondrial reference genomes uncovers high heteroplasmy frequency. PLoS Comput Biol. 2012;8(10):e1002737.
Ankel-Simons F, Cummins JM. Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution. Proc Natl Acad Sci U S A. 1996 Nov 26;93(24):13859–63.
Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature. 1999;402(6760):371–2.
Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334(6059):1144–7. https://doi.org/10.1126/science.1211878. Epub 2011 Oct 27. PMID:22033522.
Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitinated sperm mitochondria, selective proteolysis, and the regulation of mitochondrial inheritance in mammalian embryos. Biol Reprod. 2000 Aug;63(2):582–90.
Luo SM, Schatten H, Sun QY. Sperm mitochondria in reproduction: good or bad and where do they go? J Genet Genomics. 2013 Nov 20;40(11):549–56.
Song WH, Ballard JW, Yi YJ, Sutovsky P. Regulation of mitochondrial genome inheritance by autophagy and ubiquitin-proteasome system: implications for health, fitness, and fertility. Biomed Res Int. 2014;2014:981867.
Zhou Q, Li H, Li H, Nakagawa A, Lin JL, Lee ES, Harry BL, Skeen-Gaar RR, Suehiro Y, William D, Mitani S, Yuan HS, Kang BH, Xue D. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science. 2016;353(6297):394–9. https://doi.org/10.1126/science.aaf4777. Epub 2016 Jun 23. PMID:27338704; PMCID:PMC5469823.
Schwartz M, Vissing J. Paternal inheritance of mtDNA. N Engl J Med. 2002 Aug 22;347(8):576–80.
Schwartz M, Vissing J. New patterns of inheritance in mitochondrial disease. Biochem Biophys Res Commun. 2003 Oct 17;310(2):247–51.
He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. Heteroplasmic mtDNA mutations in normal and tumour cells. Nature. 2010 Mar 25;464(7288):610–4.
Pyle A, Hudson G, Wilson IJ, Coxhead J, Smertenko T, Herbert M, Santibanez-Koref M, Chinnery PF. Extreme-depth re-sequencing of MtDNA finds no evidence of paternal transmission in humans. PLoS Genet. 2015 May 14;11(5):e1005040.
Luo S, Valencia CA, Zhang J, Lee NC, Slone J, Gui B, Wang X, Li Z, Dell S, Brown J, Chen SM, Chien YH, Hwu WL, Fan PC, Wong LJ, Atwal PS, Huang T. Biparental inheritance of MtDNA in humans. Proc Natl Acad Sci U S A. 2018 Dec 18;115(51):13039–44.
Wei W, Pagnamenta AT, Gleadall N, Sanchis-Juan A, Stephens J, Broxholme J, Tuna S, Odhams CA, Genomics England Research Consortium, NIHR BioResource, Fratter C, Turro E, Caulfield MJ, Taylor JC, Rahman S, Chinnery PF. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat Commun. 2020;11(1):1740. https://doi.org/10.1038/s41467-020-15336-3. Erratum in:Nat Commun. 2020;11(1):3741. PMID: 32269217; PMCID: PMC7142097.
MITOMAP https://www.mitomap.org/MITOMAP
Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010 Jun;51(5):440-50. https://doi.org/10.1002/em.20586. PMID: 20544884.
Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P, Wallace DC. MITOMAP: a human mitochondrial genome database--2004 update. Nucleic Acids Res. 2005 Jan 1;33(Database issue):D611–3.
Mposhi A, Van der Wijst MG, Faber KN, Rots MG. Regulation of mitochondrial gene expression, the epigenetic enigma. Front Biosci (Landmark Ed). 2017 Mar 1;22:1099–113.
Wallace DC. Genetics: Mitochondrial DNA in evolution and disease. Nature. 2016;535(7613):498–500. https://doi.org/10.1038/nature18902.
Bonawitz ND, Shadel GS. Rethinking the mitochondrial theory of aging: the role of mitochondrial gene expression in lifespan determination. Cell Cycle. 2007;6(13):1574–8. https://doi.org/10.4161/cc.6.13.4457. Epub 2007. PMID:17603300.
Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. https://doi.org/10.1146/annurev-biochem-060408-093701. PMID:20350166.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mtDNA polymerase. Nature. 2004 May 27;429(6990):417–23.
Sahin E, Colla S, Liesa M, Moslehi J, Müller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–65. https://doi.org/10.1038/nature09787. Epub 2011 Feb 9. Erratum in:Nature. 2011 Jul 14;475(7355):254. PMID:21307849; PMCID:PMC3741661.
Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011 Mar 8;108(10):4135–40.
Craven L, Tang MX, Gorman GS, De Sutter P, Heindryckx B. Novel reproductive technologies to prevent mitochondrial disease. Hum Reprod Update. 2017b Sep 1;23(5):501–19.
Vaiserman AM, Koliada AK, Jirtle RL. Non-genomic transmission of longevity between generations: potential mechanisms and evidence across species. Epigenetics Chromatin. 2017;10(1):38. https://doi.org/10.1186/s13072-017-0145-1. PMID:28750655; PMCID:PMC5531095.
Sharma N, Pasala MS, Prakash A. MtDNA: epigenetics and environment. Environ Mol Mutagen. 2019 Oct;60(8):668–82.
Acknowledgements
I am greatly indebted to Dr. Lilian Kimura and Ms. Vinícius Magalhães Borges for formatting and checking references and figure.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mingroni-Netto, R.C. (2021). The Human Mitochondrial DNA. In: Haddad, L.A. (eds) Human Genome Structure, Function and Clinical Considerations. Springer, Cham. https://doi.org/10.1007/978-3-030-73151-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-73151-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-73150-2
Online ISBN: 978-3-030-73151-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)