
Abstract
In real-world environment, the term big data is referred to portray the huge volume of complex structured and unstructured data, which is growing exponentially very fast in time. It is mainly applicable for overgrowth of biological data which is processed by cloud source. Cloud computing refers to processing and storing the massive volume of data over the Internet instead of single computer’s hard drive. Various types of services offer to process the data. Cloud provides most of the intelligent services like security, performance, productivity, reliability, scalability, speed, and accurate access. The data is distributed among various places and various sizes. Centralize all the data into single site is to increase the processing speed and memory. A distributed approach employed in the present study is to replace the centralized data environment. The distributed refers to the collection of independent components. To access the data by distributed way, that is, processing the data based on feature selection in data source and getting the data representative based on this, the informative data will be collected into single site. The hybrid machine learning and deep learning models are used to detect the diseases in biological data to improve the computational efficiency and reduce the memory. The hybrid distributed models show the excellent performance in biological research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Big data is an emergent field which increases more number of data in the fields like marketing, medical, biological research, transaction of data, and so on. Due to growing size of data, data retrieval is more complex. Big data is classified into three V’s, that is, Volume, Velocity, and Variety of data [1]. Big data, which is a huge volume of data, is not only collected from computers but also from mobile phones, sensors in various filed, social media posts, and many other resources. Data retrieval, data analysis, quality and quantity measures of algorithm and data, and outlier detection are considered various issues in Big data [2].
Biological data is a collection of life science information, computational study, information of living organism, and high quantity of research knowledge. The progress of biological data information’s collected from DNA, RNA, protein discovered [3, 4]. The types of biological data are incorporated from genomics, proteomics, microarray, metabolomics, gene expression, and ontology, and so on. The biological data is distinguished in different data format like image, sequence, structure, patterns, graph, text, geometric, and expression [5, 6].
The cell is the basic structure of every living organism. The nucleus is the heart of the cell with chromosomes which have a part called DNA. The four bases of DNA are Adenine (A), Cytosine(C), Guanine (G), and Thymine (T). DNA is transcribed into RNA which has the base pair of Adenine (A), Cytosine(C), Guanine (G), and Uracil (U) [7]. The base pair of RNA is similar to DNA except for Thymine. RNA use Uracil instead of Thymine. RNA is translated to protein. Proteins are formed by linking different amino acid or peptide bonds [8]. A protein is normally denoted as a sequence or string on an alphabet of 20 characters, except B, J, O, U, X, and Z.
Cloud computing is the main part of the research in bioinformatics for huge volume of biological data [9]. Distributed cloud computing is one of the main roles in cloud computing that simplifies the cloud location, progress, distribution of data, and application from various sites to achieve the necessities, hence improving the performance and reducing the idleness.
Machine learning denotes to design and assess the algorithms to enable the data mining models from raw data. Generally, machine learning facilitates the two learning mechanization, that is, supervised learning and unsupervised learning [10]. Supervised learning represents the classification and prediction of the members with known features based on class label of data. Unsupervised learning, otherwise called as clustering and outliers, collects similar data into one group and dissimilar data into another. Both learning mechanisms work well in biological research for biological data. The combination of machine learning and deep learning is quite complex for biological data. Machine learning hybrid with deep learning and cloud computing enhances the performance of the algorithm.
Distributed clustering is used to solve computational issues in distributed data. Generally, the data is classified into two forms: homogeneous and heterogeneous. Homogeneous data has similar dataset attributes, and heterogeneous has different dataset attributes. In Fig. 2.1, the distributed clustering is done in two levels such as local and global [11, 12].

Architecture of distributed clustering
1.1 Chapter Sections Overview
Chapter sections are organized as follows: Sect. 2.1, describes the introduction; Sect. 2.2 presents a detailed survey of previous studies, Sect. 2.3 explains about the hybrid models; Sect. 2.4, presents the results and discussion; and Sect. 2.5 presents the conclusion.
2 Literature Review
Bioinformatics is an emerging research area for storing and accessing a huge volume of data. Data access is a difficult task in the research field. The structure and function of protein based on the statistical metric based feature selection techniques, which reduced the feature vector size for evaluate the growing biological data [13]. The neural network classifiers compared with other classifiers for improving the evaluation. The accuracy of classification is to exactly identify the changes of amino acid sequence. This feature selection proves a significant upgrade in performance in terms of accuracy, sensitivity, and F-measure. This selection technique fails to manage the time complexity for accessing the data.
The distribution-based spectral clustering and cuckoo search used for cancer identification with protein sequence data reduces time complexity. Invariant sequence identified based on the similarity index, which is identified by Jaccard similarity index. Fuzzy logic used to detect the membership value of protein sequence. Based on the similarity and membership value, the sequence is detected whether cancerous or non-cancerous. This distribution-based spectral clustering improves the accuracy and reduces the time but fails to detect the features-based detection [14, 15].
TRIBE-MCL is used for the family of protein to detect the information of sequence similarity. Protein family detection is one of the main goals of functional and structural genomics. Construct a protein–protein similarity graph for proteins. Then, generate a weighted transition matrix for the constructed similarity graph by BLAST E-Values and finally transform, weight into transition probability for constructing a Markov matrix. This task is probably expensive to achieve a goal in a short period [16].
The deep learning algorithm exactly identifies the breast cancer using mammography image. Digital Database for Screening Mammography (CBIS-DDSM) test improves the sensitivity, specificity and reduces the false-positive and false-negative rates [17]. Deep learning method is highly suitable for heterogeneous mammography image, but it takes much time to produce the result of algorithm. Random forest and distributed techniques are rarely used in biological environment [18, 19].
3 Hybrid Models of Deep Learning and Machine Learning
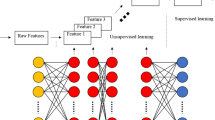
The data is distributed among various places and size. If all the data collected into single site, it takes more execution time and memory for process the data. To avoid this contingency, the distributed approach is used to cluster the data locally and form a global data based on data representative. Local cluster is done by Distributed Spectral Clustering (DSC) technique such that construct a diagonal matrix for “n” number of protein data, then find the similarity using Jaccard similarity index, then compute the Laplacian function with the help of Eigen values and Eigen vectors. Then, run the Fuzzy C-Means (FCM) to separate an object. In normal spectral clustering, K-means is used to separate a data instead of FCM. Apply the statistical metric-based feature selection in global data. This selection is done based on the scoring and length of the sequence. In this model, machine learning algorithm of spectral clustering is used to split up the data based on the similarity and the deep learning-based feature selection acts to get final informative sequence. Table 2.1. represents the Pseudo code of Distributed Spectral Clustering with Feature Selection (DSCFS).
Feature selection is done based on the length and score of amino acid. The standard 20 (A, C, D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y) amino acid is used to stipulate the protein sequence of any length for any gene. Figure 2.2 represents the architecture of Distributed Spectral Clustering with Feature Selection.

Architecture of distributed spectral clustering with feature selection
4 Experimental Results and Discussion
The clustering measures are calculated by the following values: True Positive (TP), True Negative (TN), False Positive (FP), False Negative (FN) [20, 21].
4.1 Accuracy
Accuracy is refers to defined as correctly detect the cancerous sequence by the total number of sequence. It is measured in terms of percentage (%) (Table 2.2; Fig. 2.3).

Comparison of TRIBE-MCL and DSCFS in terms of accuracy
4.2 Precision/Specificity
Precision is referred to measure the quality of accuracy and it is the ratio of correctly identified sequences and the total number of sequences. It is also measured in terms of percentage (%) (Table 2.3; Fig. 2.4).

Comparison of TRIBE-MCL and DSCFS in terms of precision
4.3 Recall/Sensitivity
Recall is referred to measure the quality of accuracy and it is defined as a fraction of correctly identified sequences and the total number of sequences. It is also measured in terms of percentage (%) (Table 2.4; Fig. 2.5).

Comparison of TRIBE-MCL and DSCFS in terms of recall
4.4 F-Measure
F-measure is referred to integrate the mean of precision and recall. It is also measured in terms of percentage (%) (Table 2.5; Fig. 2.6).

Comparison of TRIBE-MCL and DSCFS in terms of F-measure
4.5 Time
Time is referred to as starting and ending time of execution for the total number of sequence which is measured in terms of milliseconds (ms) (Table 2.6; Fig. 2.7).

Comparison of TRIBE-MCL and DSCFS in terms of time
4.6 Motif for Normal Sequence (Fig. 2.8)

Motif graph for normal sequence
4.7 Motif for Mutant Sequence (Fig. 2.9)

Motif graph for mutant sequence
5 Conclusion
The Distributed Spectral Clustering with Feature Selection techniques is done in two models such as local and global models to reduce the time complexity, and feature selection is used to enhance the accuracy, precision, recall, and F-measures. Local model acts as a clustering and global model acts as Cloud, which provide most of the intelligent services like security, performance, productivity, reliability, scalability, speed, and accurate access. This method is mainly applicable for huge volume of distributed data. The results achieved are based on similarity, length, and score of the sequence. This novel technique is compared with TRIBE-MCL to show better performance to get mutant protein sequence. Every measure in this technique shows better performance than literature TRIBE-MCL method.
References
Driscoll, A., Daugelaite, J., & Sleator, R. D. (2013). Big data- hadoop and cloud computing in genomics. Journal of Biomedical Informatics, 46(5), 774–781. https://doi.org/10.1016/j.jbi.2013.07.001
Thenmozhi, K., Visalakshi, N. K., & Shanthi, S. (2017). Optimized data retrieval in big data environment using PPFC approach. Asian Journal of Research in Social Sciences and Humanities, 7(3), 683–690. https://doi.org/10.5958/2249-7315.2017.00198.8
Pan, T., & Uhlenbeck, O. C. (1993). Circularly permuted DNA, RNA and proteins - A review. Gene, 125(2), 111–114. https://doi.org/10.1016/0378-1119(93)90317-v
Konathala, G. K., Mandarapu, R., & Godi, S. (2017). Oncogenic mutations of PIK3CA and HRAS in carcinoma of cervix in south Indian women. Journal of Oncological Sciences, 3(3), 112–116. https://doi.org/10.1016/j.jons.2017.10.004
Diniz, W. J. S., & Canduri, F. (2017). Bioinformatics: An overview and its applications. Genetics and Molecular Research, 16(1), 1–21. https://doi.org/10.4238/gmr16019645
Zou, D., Ma, L., Yu, J., & Zhang, Z. (2015). Biological databases for human research. Genomics, Proteomics & Bioinformatics, 13(1), 55–63. https://doi.org/10.1016/j.gpb.2015.01.006
Rodrigues, M. J. F., & Mering, C. V. (2014). HPC-CLUST: Distributed hierarchical clustering for large sets of nucleotide sequences. Bioinformatics, 30(2), 287–288. https://doi.org/10.1093/bioinformatics/btt657
Abdul, M., Safdar, A., Mubashar, I., & Nabeela, K. (2014). Prediction of human breast and colon cancers from imbalanced data using nearest neighbor and support vector machines. Computer Methods and Programs in Biomedicine, 113(3), 792–808. https://doi.org/10.1016/j.cmpb.2014.01.001
Shakil, K, & Alam, M. (2018). Cloud computing in bioinformatics and big data analytics: Current status and future research. Advances in Intelligent Systems and Computing (pp. 629–654). https://doi.org/10.1007/978-981-10-6620-7.
Xu, C., & Jackson, S. A. (2019). Machine learning and complex biological data. Genome Biology, 20, 76. https://doi.org/10.1186/s13059-019-1689-0
Januzaj E, Kriegel H-P, & Pfeifle M (2003). Towards effective and efficient distributed clustering. Workshop on clustering large data sets (ICDM2003), Melbourne (Vol. 1, pp. 1–10).
Bendechache, M., & Kechadi, T. (2015). Distributed clustering algorithm for spatial data mining. Second IEEE international conference on spatial data mining and geographical knowledge services, China (Vol. 15, pp. 60–65). https://doi.org/10.1109/ICSDM.2015.7298026.
Iqbal, M. J., Faye, I., Samir, B. B., & Said, A. M. (2014). Efficient feature selection and classification of protein sequence data in bioinformatics. Hindawi Publishing Corporation, Scientific World Journal (pp. 1–12). https://doi.org/10.1155/2014/173869.
Thenmozhi, K., Visalakshi, N. K., & Shanthi, S. (2018). Distribution based fuzzy estimate spectral clustering for Cancer detection with protein sequence and structural motifs. Asian Pacific Journal of Cancer Prevention, 19(7), 1935–1940. https://doi.org/10.22034/APJCP.2018.19.7.1935
Thenmozhi, K., Visalakshi, N. K., & Shanthi, S. (2018). Distributed ICSA clustering approach for large scale protein sequences and Cancer diagnosis. Asian Pacific Journal of Cancer Prevention, 19(11), 3105–3109. https://doi.org/10.31557/APJCP.2018.19.11.3105
Enright, A. J. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research, 30(7), 1575–1584. https://doi.org/10.1093/nar/30.7.1575
Shen, L., Margolies, L. R., Rothstein, J. H., Fluder, E., McBride, R., & Sieh, W. (2019). Deep learning to improve breast Cancer detection on screening. Scientific Reports, 9(1), 12495. https://doi.org/10.1038/s41598-019-48995-4
Ramesh, K., Vinitha, A., Dhamodharan, M., & Shanmuga, V. M. (2020). An improved random Forest algorithm for effective stock market prediction trending towards machine learning. International Journal of Grid and Distributed Computing, 13(1), 873–881.
Thenmozhi, K., Pyingkodi, M., & Kumaravel, S. (2018). Tapered assessment on distributed clustering vital in protein sequence environment. International Journal of Recent Trends in Engineering & Research (IJRTER), 4(7) https://doi.org/10.23883/IJRTER.2018.4350.D8R7D.
Sokolova, M., Japkowicz, N., & Szpakowicz, S. (2006). Beyond accuracy, F-score and ROC: A family of discriminant measures for performance evaluation. Advances in Artificial Intelligence (pp. 1015–1021). https://doi.org/10.1007/11941439_114.
Huda, S., Yearwood, J., Jelinek, H. F., Hassan, M. M., Fortino, G., & Buckland, M. (2016). A hybrid feature selection with ensemble classification for imbalanced healthcare data: A case study for brain tumor diagnosis. IEEE Access, 4, 9145–9154. https://doi.org/10.1109/ACCESS.2016.2647238
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Thenmozhi, K., Pyingkodi, M., Ramesh, K. (2022). Hybrid Machine Learning Models for Distributed Biological Data in Multi-Cloud Environment. In: Nagarajan, R., Raj, P., Thirunavukarasu, R. (eds) Operationalizing Multi-Cloud Environments. EAI/Springer Innovations in Communication and Computing. Springer, Cham. https://doi.org/10.1007/978-3-030-74402-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-74402-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-74401-4
Online ISBN: 978-3-030-74402-1
eBook Packages: EngineeringEngineering (R0)