
Summary
Porphyrias are metabolic disorders of the heme biosynthesis. The location of the deficient enzyme within the heme biosynthetic pathway determines the pattern of the accumulated porphyrin precursors and/or porphyrins. Clinically, they can be differentiated into acute and non-acute porphyrias. Acute hepatic porphyrias (AHP) are characterized by overproduction of probably neurotoxic porphyrin precursors and porphyrins. Acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and Doss porphyria (ADP) belong to this group of metabolic disorders. The clinical presentation of acute hepatic porphyrias includes abdominal, psychiatric, neurological, and cardiovascular symptoms. They are diagnosed by an at least fourfold elevated urinary excretion of 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) (except for ADP and lead poisoning (LP)). Besides symptomatic therapy with non-porphyrinogenic drugs, the combination of electrolyte correction, intensive monitoring, intravenous caloric supply (mainly glucose), and heme is established for treatment. Recently, in the Phase 3 ENVISION study in patients with AHP, givosiran, an RNAi therapeutic which targets liver mRNA encoding ALAS1 to reduce 5-ALA/PBG, ameliorates attacks and clinical manifestations.
Non-acute types are porphyria cutanea tarda (PCT), hepatoerythropoietic porphyria (HEP), erythropoietic protoporphyria (EPP), X-linked protoporphyria (XLP), and congenital erythropoietic porphyria (CEP). Accumulated porphyrins cause photosensitivity of the skin and in some cases severe liver damage. X-linked protoporphyria (XLP) represents a new type of protoporphyria, with 5-aminolevulinic acid synthase 2 gain of function leading to high concentrations of free protoporphyrin IX (PPIX). Treatment of PCT is based on iron depletion, the use of hydroxychloroquine (HCQ), and, in case of chronic hepatitis C virus (HCV) infection, antiviral therapy. Patients with EPP or XLP profit from treatment with an analogue of α-melanocyte-stimulating hormone that reduces sunlight sensitivity and inflammation. Further therapeutic developments directly address dysfunctional steps of the heme biosynthetic pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
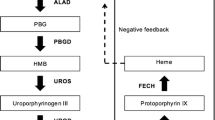
Porphyrias are a heterogeneous group of metabolic disorders, which are caused by a range enzyme deficiencies or defects due to mutations in genes along the heme synthesis pathway (Puy et al. 2010; Bonkovsky et al. 2013; Bissell et al. 2017; Stölzel et al. 2019) (Fig. 57.1). The majority of patients are carriers of heterozygous mutations. Some patients carry homozygous or compound heterozygous mutations resulting in a reduced enzyme activity. As customary, autosomal dominant disorders porphyrias can vary in penetrance and phenotype; moreover, there are considerable interactions between genetic and environmental factors. Moreover, they can result from combinations of genetic alterations (Lenglet et al. 2018) . In addition, there are considerable interactions between genetic and environmental factors.
PCT is the most common porphyria. Hepatic uroporphyrinogen decarboxylase (UROD) is reduced in all cases with PCT (acquired type 1 or familial type 2). Thus, a cross-sectional registry study comprising 4653 patients showed prevalence ratios of 91/26/9/4/2/1 for PCT/AIP/EPP/VP/HCP/CEP (Stölzel et al. 2019). Among AHPs, AIP is the most frequent type, followed by VP, HCP, and a rare autosomal recessive acute hepatic porphyria ADP (synonym: porphobilinogen synthase defect porphyria, Doss porphyria) which is biochemically very similar to lead poisoning (LP) (Doss et al. 1979) .
Clinically, acute (AIP, VP, HCP, ADP) and non-acute porphyrias (PCT, HEP, EPP, XLP, CEP) can be differentiated, while pathogenetically hepatic (PCT, HEP, AIP, VP, HCP, ALADP) and erythropoietic (EPP, XLP, CEP) porphyrias form two major groups. Dual porphyrias (biochemical findings of two porphyrias) are rare and have been confirmed by mutation analyses. Increased porphyrin precursors and porphyrins are also found in LP.
The clinical presentation of AHP and LP comprises abdominal, neuropsychiatric, and cardiovascular symptoms and hyponatremia, whereas chronic hepatic and erythropoietic porphyrias present with photodermatosis and occasionally severe liver damage. AHP is not just an “acute” disease as its name implies, but also has chronic manifestations that impact patients’ lives (Gouya et al. 2020).
All porphyrias are diagnosed and differentiated by specific biochemical patterns of elevated porphyrins and porphyrin precursors in urine, feces, and blood (Bonkovsky et al. 2013; Bissell et al. 2017; Stölzel et al. 2019). AHPs are characterized by excessive accumulation and excretion of the porphyrin precursors 5-ALA (ADP, LP) or 5-ALA and PBG (AIP, VP, HCP) as well as porphyrins. In patients with PCT, but also in patients with HEP, levels of porphyrins are greatly increased in urine and plasma, with uro- and heptacarboxyporphyrins predominating. Increased levels of metal-free protoporphyrin (>4.500 nmol/l, controls <89 nmol/l) in hemolyzed anticoagulated whole blood confirm the diagnosis of EPP or XLP. Here, patients with XLP display a significantly higher proportion of zinc- to metal-free protoporphyrin (>25%) than patients with EPP (<15%).
Erythropoietic and hepatic porphyrias are genetically determined primary porphyrias, whereas clinically asymptomatic secondary porphyrinurias and porphyrinemias are not due to functional mutations of the heme synthetic pathway but caused by a number of different (e.g., metabolic, hepatic, or hematologic) diseases and dysfunctions.
In tyrosinemia type I, the nonfunctional fumarylacetoacetate hydrolase leads to succinylacetone that inhibits the enzyme ALA dehydratase.
Nomenclature
No. | Disorder | Alt name | Abbreviation | Gene symbol | Chromosomal localization | Affected protein | OMIM no. | Inheritance |
|---|---|---|---|---|---|---|---|---|
X-linked sideroblastic anemia | Erythroid 5-aminolevulinate synthase deficiency | XLSA | ALAS2 | Xp11.21 | 5-Aminolevulinate synthase 2 | 300751 | XL | |
X-linked protoporphyria | Erythroid 5-aminolevulinate synthase gain of function | XLP | ALAS2 | Xp11.21 | 5-Aminolevulinate synthase 2 | 300752 | XL | |
5-Aminolevulinate dehydratase deficiency | Doss porphyria | ADP | ALAD | 9q32 | Delta-aminolevulinate dehydratase | 612740 | AR | |
Acute intermittent porphyria | Porphobilinogen deaminase deficiency | AIP | HMBS | 11q23.3 | Hydroxymethylbilane synthase | 176000 | AD | |
Congenital erythropoietic porphyria | Uroporphyrinogen III synthase deficiency | CEP | UROS | 10q25.2–26.3 | Uroporphyrinogen III synthase | 263700 | AR | |
Porphyria cutanea tarda types I, II and hepatoerythropoietic porphyria | Hepatic uroporphyrinogen decarboxylase deficiency | PCT/HEP | UROD | 1p34 | Uroporphyrinogen decarboxylase | 176100 | AD, AR | |
Hereditary coproporphyria | Coproporphyrinogen oxidase deficiency | HCP | CPOX | 3q12 | Coproporphyrinogen oxidase | 121300 | AD | |
Porphyria variegata | Protoporphyrinogen oxidase deficiency | VP | PPOX | 1q22-q23 | Protoporphyrinogen oxidase | 176200 | AD | |
Erythropoietic protoporphyria | Ferrochelatase deficiency | EPP | FECH | 18q21.3 | Ferrochelatase | 177000 | AD |
Metabolic Pathways

Localization of characteristic enzyme defects of heme biosynthesis and in porphyrias, lead poisoning, and type I tyrosinemia. The porphyrinogens are excreted after their oxidation to porphyrins. (a) In the liver, the first heme synthetic enzyme ALAS1 is regulated via a negative feedback loop by the end product heme. In contrast, the rate-limiting bone marrow enzyme ALAS2 is regulated by iron and erythropoietin and not by heme. (b) Three enzymes involved in heme biosynthesis are compromised in lead poisoning. (c) The two isomers uroporphyrinogen I and III that are synthesized from hydroxymethylbilane are converted to coproporphyrinogen I and III, respectively. Only isomer III is utilized for heme synthesis. The nonfunctional isomer I is excreted via the hepatobiliary and renal routes in feces and urine, respectively (Modified from Ref. Stölzel et al. 2019)
Signs and Symptoms
Overview on Symptomatology
Disorder | Leading clinical symptoms | Onset of symptoms | Prevalence |
|---|---|---|---|
57.1 X-linked sideroblastic anemia | Anemia | ||
57.2 X-linked protoporphyria | Photosensitivity/liver disease/anemia | Childhood | Rare |
57.3 ALA-dehydratase deficiency Lead poisoning | Abdominal and neurological symptoms Abdominal and neurological symptoms/anemia/lead blue line (Burton’s line) | Childhood Childhood/adolescence | Very rare Rare |
21.1 Tyrosinemia type 1 | Failure to thrive, abdominal and neurological symptoms, liver dysfunction | Early childhood | Rare |
57.4 Acute intermittent porphyria | Abdominal and neurological symptoms/hyponatremia | After puberty (female>male) | 1: 100,000 |
57.5 Congenital erythropoietic porphyria | Photosensitivity: Blisters, erosions, mutilations/anemia/hemolysis/hepatosplenomegaly/erythrodontia | Neonatal/childhood | Very rare |
57.6 Porphyria cutanea tarda I, II, Hepatoerythropoietic porphyria | Liver disease/skin fragility/photosensitivity/hypertrichosis | Late adolescence | 20: 100,000 Very rare |
57.7 Hereditary coproporphyria | Abdominal and neurological symptoms, photosensitivity | After puberty (adolescence And senescence) | 0.1: 100,000 |
57.8 Porphyria variegate | Abdominal and neurological symptoms, photosensitivity | After puberty (adolescence And senescence) | 0.3: 100,000 |
57.9 Erythropoietic protoporphyria | Photosensitivity, some patients develop liver disease | Infancy/childhood | 1: 100,000 |
Enzyme Defects along the Heme Biosynthesis in Porphyrias, Regulated Induction of ALAS1, and Major Clinical Manifestation
Disorder in heme synthesis | Enzyme | Induction of ALAS1 | Major clinical manifestation | |||
|---|---|---|---|---|---|---|
Neurovisceral | Cutaneous | Anemia | Liver | |||
X-linked sideroblastic anemia (57.1) | ALAS2 | – | ++ | |||
XLP (57.2) | ALAS2 | – | – | ++ | −/+ | −/+ |
ADP (Doss) (57.3) | ALAD | + | + | – | −/+ | – |
Lead poisoning (57.3) | ALAD | + | + | – | +a | +b |
Tyrosinemia type 1c (21.1) | ALAD | + | + | |||
AIP (57.4) | PBGD | + | + | – | – | −/+ |
CEP (Günther) (57.5) | UROS | – | – | + | + | −/+ |
PCT / HEP (57.6) | UROD | – | – | + | – | −/+ |
HCP (57.7) | CPOX | + | + | −/+ | – | −/+ |
VP (57.8) | PPOX | + | + | −/+ | – | −/+ |
EPP (57.9) | FECH | – | – | + | −/+ | −/+ |
Reference Ranges
Urinary Excretion of Porphyrin Precursors and Porphyrins
24-h urine collection | Urine spot sample | |
|---|---|---|
Analyte | Referred to the total excretion volume | Referred to excreted creatinine |
ALA | <49 μmol/day | <2.6 mmol/mol, BZ 2.6–6.8 |
PBG | <7.5 μmol/day | <1.0 mmol/mol, BZ 1.0–2.2 |
Uroporphyrin | <33 nmol/day | <4.5 μmol/mol |
Coproporphyrin I | <51 nmol/day | <7.0 μmol/mol |
Coproporphyrin III | <102 nmol/day | <14.0 μmol/mol |
Total porphyrins | <209 nmol/day | <26.7 μmol/mol |
Fecal Porphyrin Excretion
Analyte | Upper limit of reference |
|---|---|
Coproporphyrin I | <26 nmol/g dry weight |
Coproporphyrin III | <11 nmol/g dry weight |
Protoporphyrin | <95 nmol/g dry weight |
Erythrocytic Porphyrins
Analyte | Upper limit of reference |
|---|---|
Zinc protoporphyrin IX | <385 nmol/l or < 40μmol/mol heme |
Free protoporphyrin IX | <89 nmol/l |
Pathological Values
Biochemical Markers in Primary and Secondary Disorders of Heme Biosynthesis
Sample/metabolite | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
XLSA | XLP | ADP | LP a | TE 1 b | AIP | CEP | PCT/HEP | HCP | VP | EPP | |
Urine | |||||||||||
5-ALA | n | n | ↑↑ | ↑↑ | ↑-↑↑ | ↑↑ | n | n | ↑ | ↑ | n |
PBG | n | n | n-↑ | n-↑ | n | ↑↑ | n | n | ↑ | ↑ | n |
Porphyrin I isomers | ↑↑ | ||||||||||
Uroporphyrins, total | n-↑ | n-↑↑ | n-↑↑ | n | ↑↑ | ↑↑ | ↑↑ | n-↓ | n-↓ | n-↑ | |
Uroporphyrins I | − | − | − | ↑↑ | ↑↑ | − | − | ||||
Uroporphyrins III | ↑ | − | ↑↑ | ||||||||
Heptacarboxyporphyrin | n-↑ | n-↑ | n-↑ | n | ↑ | ↑ | ↑↑ | n-↑ | n-↑ | n-↑ | |
Hexacarboxyporphyrin | n | n-↑ | n-↑ | n | ↑ | ↑ | ↑ | n-↑ | n-↑ | n | |
Pentacarboxyporphyrin | n | n-↑ | n-↑ | n-↑ | ↑ | ↑ | ↑ | n-↑ | n-↑ | n | |
Coproporphyrins, total | n-↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑ | ↑↑ | n-↑ | ↑↑ | ↑↑ | n-↑ | |
Coproporphyrin I > III | +++ | + | |||||||||
Coproporphyrin III > I | + | + | + | ++ | + | ||||||
Coproporphyrin III | ↑↑ | ↑↑ | ↑↑ | ↑ | ↑ | ||||||
Feces | |||||||||||
Porphyrin I isomers | ↑ | ||||||||||
Uroporphyrins | n | n | n | n-↑ | n-↑ | n | n | n | |||
Heptacarboxyporphyrin | n | n | n | n-↑ | ↑ | n | n | n | |||
Hexacarboxyporphyrin | n | n | n | ↑ | ↑ | n | n | n | |||
Pentacarboxyporphyrin | n | n | n | ↑ | ↑ | n | n | n | |||
Isocoproporphyrin | − | − | n-↑ | − | − | − | |||||
Coproporphyrins | n-↑ | n-↑ | n-↑ | n-↑ | ↑ | ↑↑ | ↑ | ||||
Coproporphyrin III > I | ++ | + | |||||||||
Coproporphyrin I/III | > > 1 | <1 | <1 | n->1 | |||||||
Protoporphyrin | n-↑ | n-↑ | n-↑ | n | n-↑ | ↑ | ↑↑ | n-↑↑ | |||
Plasma or erythrocytes, resp. | |||||||||||
Uroporphyrin | ↑ | n | |||||||||
Coproporphyrin | ↑ | ||||||||||
Zinc protoporphyrin | ↑↑ | ↑↑ | ↑↑ | ↑ | ↑c | n | n | ↑↑ | |||
Free protoporphyrin | ↑↑ | ↑ | ↑ | ↑ | ↑c | n | n | ↑↑ | |||
Protoporphyrin free/zinc | ↓ | 2: 1 | < 1 | < 1 | > > 1 | ||||||
Various | |||||||||||
PBG deaminase | n | n | n | n | n | ↓↓-n d | n-↑ | n-↑ | n | n-↓ | n |
5-ALA dehydratase | n | n | ↓↓ | ↓↓ e | n | n | n | n | n | n | |
Uroporphyrinogen-decarboxylase | n | n | n | n | n | n | n-↓↓ | n | n | n | |
Emission maximum of plasma fluorescence spectrum on excitation with 405 nm (nm) | 624–635 | 615–620 | 615–620 | 615–620 | 615–620 | 615–620 | 615–620 | 625–627 | 624–635 | ||
Diagnostic Flowcharts
Acute hepatic porphyrias | Porphyria cutanea tarda | Protoporphyrias |
|
|
|
Patients after puberty | Adult patients Age > 18 years | Children or adolescents |
|
|
|
• Unexplained gastrointestinal complaints (colic, vomiting, subileus) • Neuropsychiatric symptoms (paresthesia, seizures, paresis, depression, anxiety, hallucination) • Cardiovascular symptoms (tachycardia, hypertension) • Red-colored urine without erythrocytes or hemoglobin • Serum hyponatremia | • Blister-forming dermatosis on light-exposed skin areas • Increased skin vulnerability • Hyper- and hypopigmentation on light-exposed skin • Hypertrichosis of the cheeks, temples, and eyebrows; often associated with: – Iron overload – HCV infection – HIV infection – Alcohol consumption – Hormone (replacement) therapy – Toxic agents, e.g., hexachlorobenzene | • Burning pain • Erythema/redness on light-exposed skin areas • Angioedema-like swelling on the face, on the back of the hands, and on the forearms • Often microcytic anemia |
Key diagnostic features | ||
|
|
|
> Fourfold elevated ALA and PBG in urine | ALA and PBG in urine normal, elevated total porphyrins in urine with uroporphyrin > coproporphyrin | ALA and PBG in urine normal, metal-free erythrocyte protoporphyrin increased in blood |









Specimen Collection
Test | Material | Handling |
|---|---|---|
First-line diagnostic tests | ||
Fluorescence scan | 1 ml serum, plasma (heparin, EDTA) | Light protected and cool |
5-Aminolevulinic acid Porphobilinogen (porphyrin precursors) | 5 ml urine (spot sample or 24-h collection) | Light protected and cool |
Porphyrins | 5 ml urine (spot sample or 24-h collection), 5 g feces | Light protected and cool |
Free protoporphyrin | 3 ml EDTA blood or | |
Zinc protoporphyrin | Heparinized blood | Light protected and cool |
Second-line diagnostic tests | ||
Enzyme activity tests | 5 ml heparinized blood or ACD blood | Cool, not frozen |
Molecular genetic assays | EDTA blood or | – |
Genomic DNA | Isolated DNA | |
cDNA | EDTA blood | Rapid sample transport (<12 h) |
Treatment
Acute Porphyrias (ALADP; 57.3, AIP; 57.4, HCP; 57.7, VP; 57.8)
Generally, patients need to be admitted to an intensive care unit, to receive pain treatment and caloric support (mainly glucose infusion) and to have their electrolyte abnormalities corrected. Porphyrinogenic medications and other triggers need to be identified and discontinued (Stölzel et al. 2019). AHP requires specific therapies (see table below).
1. Discontinuation of porphyrinogenic drugs and intensive medical monitoring | www.drugs-porphyria.org and see Fig. 57.2 |
2. Caloric support (carbohydrates, protein) And heme treatment: | Intravenous and/or oral carbohydrates as preferred source of energy; beware of dilutional hyponatremia; serum sodium, magnesium, and phosphate must be monitored daily For severe cases, neurologic manifestations and associated hyponatremia: Heme arginate (e.g., NormosangR), 3 mg/kg body weight/day in 100 ml human albumin (5%–20%), infused in 15 min, for up to four consecutive days |
3. Symptom measures: For pain: For tachycardia and hypertension: For restlessness or vomiting: For symptoms of ileus: For respiratory relief: For infections: For recurrent attacks and chronic symptoms: Physiotherapeutic measures from the very beginning | Acetylsalicylic acid, morphine derivatives, gabapentin Propranolol, metoprolol, valsartan Chlorpromazine, lorazepam, ondansetron Neostigmine Assisted or controlled ventilation (possibly tracheotomy) Penicillin, cephalosporins, imipenem, gentamicin, amikacin, vancomycin Givosiran (GivlaariR) 2.5 mg/kg body weight, s.c., monthly |
Heme therapy is clearly indicated when neurological symptoms occur (Bonkowsky et al. 1971). Usually, these and other symptoms begin to improve within 48 h of early-on intravenous administration of heme (Normosang®, Orphan Europe, Puteaux, France, Europe, and elsewhere; Panhematin®, Recordati Rare Diseases, Lebanon, NJ, United States, Mexico, and elsewhere). Heme is a feedback inhibitor of the rate-limiting hepatic enzyme ALAS1 at the transcriptional level. While being effective in most cases, lack of response can be due to insufficient dosing (<3 mg/kg/day), symptoms that are not caused by porphyria, late start of therapy, or chronified porphyria-related pathology, which includes irreversible neurological damage. Heme therapy is also effective in LP and ALADP. Prophylactic treatment with heme in fixed intervals, e.g., up to weekly in severe cases, is justified for patients with recurrent attacks (defined as more than three per year). However, regular heme infusions over prolonged periods have significant side effects, especially iron overload and venous damage and obliteration caused by heme degradation products that bind to clotting factors, platelets, and endothelial cells. These patients require an intravenous port for blood sampling and intravenous heme therapy. Administration of heme bound to albumin is an easy measure to reduce intravenous heme toxicity (see table above). We dilute the heme arginate in 100 ml of human albumin (5–20%). After infusion into a large vein or the port, physiological saline is infused for 15 min to reduce local toxicity. In rare cases that require highly frequent heme infusions, because of severe clinical manifestations, the accumulating heme can activate heme oxygenase 1, which results in accelerated heme degradation and loss of feedback inhibition of ALAS1. This can explain the more than fourfold increase of reported AIP patients that experience recurrent attacks since the introduction of heme therapy in 1985 (from 4/230 in 1985 to 40/536 in 2008), but improved survival with heme therapy may also have contributed (Schmitt et al. 2018). Taken together, if feasible, high-frequency heme therapy should be avoided.
Sufficient caloric support (especially with carbohydrates and protein) is a central basic therapy for AHP. Attacks are often induced by low caloric intake and worsened by nausea and vomiting (Doss et al. 1985). The infused glucose inhibits the peroxisome proliferator-activated receptor-γ (PPAR-γ) coactivator-1α that otherwise upregulates transcription of ALAS1 (Handschin et al. 2005). Glucose combined with insulin may be more effective than glucose alone, but controlled studies are lacking. A too rapid glucose infusion may lead to a dangerous refeeding syndrome, promoting, e.g., hyponatremia. Therefore, patients with hyponatremia should first receive heme therapy followed by careful glucose infusion.
Pain, another component of the vicious cycle, needs to be treated immediately and rigorously prevented. Well-tolerated drugs used by us are opiates and gabapentin. They do not induce hepatic ALAS1 and are excreted via the kidneys. Mild acute hepatic porphyria cases should be treated with pain medication and caloric support alone. Figure 57.2 lists safe drugs, based on numerous experimental studies, pharmacological data, and clinical reports (Stölzel et al. 2009). We recommend to consult the International Guidelines (www.drugs-porphyria.org, www.porphyria-europe.com).

Non-porphyrinogenic drugs
In women with acute porphyria, pregnancy is in general not at risk, although progesterone potently induces liver heme production. However, pregnancy-associated vomiting and subsequent caloric deficiency should be normalized promptly by caloric supply with parenteral nutrition. For those women suffering from frequent attacks related to menstrual cycle, gonadotropin-releasing hormone analogues, combined subsequently with low-dose estrogen patch to suppress menopausal symptoms, can be helpful.
Ultimately, in severe and complicated disease, liver transplantation was shown to cure the disease (Seth et al. 2007). Complete normalization of porphyrin metabolism after liver transplantation proves that acute porphyrias are diseases of the liver.
Small interfering RNA (givosiran, Givlaari®, Alnylam, USA) silences hepatic ALAS1-mRNA, normalizes ALA und PBG overproduction (Tschudy et al. 1965), and significantly reduces the annualized rate of porphyria attacks (Balwani et al. 2020). After 12 months of follow-up, 62% of patients on givosiran were attack-free. Safety profile was acceptable. However, 10 (11%) and 16 (17%) of the treated patients had renal and/or hepatic adverse events, respectively.
Overall, the rigorous elimination of precipitating factors in daily life remains the mainstay of prevention and therapy. Avoiding porphyrinogenic medication, including alcohol, smoking, and physical stress, is of major importance, as well as a balanced diet with a high percentage of carbohydrates. Patients with acute porphyria should take special care to avoid infections and other diseases, and the porphyrin precursors ALA and PBG should be monitored. We recommend liberal vaccination.
Porphyria Cutanea Tarda/Hepatoerythropoietic Porphyria (PCT I, II/HEP; 57.6)
Vitamin D supplementation and adequate sun protection is indispensable. The skin should not be exposed to intensive artificial light sources. Patients are advised to avoid known precipitating factors, especially alcohol and smoking, that upregulate CyP450 enzymes and thus the heme synthetic machinery. Alcohol further contributes by downregulating hepcidin, which increases iron resorption and enhances oxidative stress. Women must discontinue hormonal contraception or replacement therapy. Photoprotection, phlebotomy, and treatment with hydroxychloroquine (HCQ) (100 mg twice per week, respectively) are effective first-line therapies (Kordac and Semrádová 1973). Phlebotomy is employed to remove excess iron. Initially, a biweekly phlebotomy up to 500 ml is performed and monitored by serum ferritin concentrations (target value near the lower limit of normal) to avoid iron deficiency. When phlebotomy is not possible, such as in severe anemia, oral iron chelators or low-dose HCQ can be given. Phlebotomy and low-dose HCQ are also effective baseline therapies for the majority of patients with PCT, with comparable efficacy. HCQ can mobilize cellular porphyrin aggregates with subsequent elimination mainly via the urine. With HCQ, urinary porphyrin excretion usually increases at least twofold, and skin photosensitivity can worsen during the first 3 months, but then starts to decrease, followed by clinical remission which is accompanied by normalization of elevated liver enzyme activities in 95% of patients. Long-term HCQ therapy can lead to retinopathy, requiring (baseline and annual) regular ophthalmologic monitoring. Patients with PCT and iron overload related to HFE mutations should preferentially undergo phlebotomy (Stölzel et al. 2003). In general, in patients with increased serum ferritin, the combination of phlebotomy with low-dose HCQ shortens the time of remission. Even advanced liver damage and siderosis, such as in patients that are homozygous for the HFE C282Y mutation, can regress after combined phlebotomy and HCQ therapy.
In patients with chronic HCV infection, treatment with iron depletion combined with highly effective antiviral therapy induces rapid clinical and biochemical remission (Combalia et al. 2017).
Treatment should be discontinued once urinary porphyrin levels stabilize around 400 nmol/day (normal <209 nmol/day). Such mild porphyrinuria usually persists during clinical remission. Still, biochemical and clinical relapse occurred in 36% and 20% of patients in the first year after discontinuation of HCQ or phlebotomy, respectively (Salameh et al. 2018).
Erythropoietic Protoporphyrias (EPP/XLP 57.1/57.9)
EPP and XLP require effective sun protection, including protection from intensive artificial light sources. Conventional sunscreens are insufficient, since photosensitivity is mainly due to visible blue light (Soret band: near 400 nm). Appropriate skin protectants contain zinc oxide or titanium oxide. Since sunlight exposure triggers pain, patients quickly learn and adopt light protection measures. Vitamin D substitution (1000–2000 U of D3 daily) is necessary. Afamelanotide (Scenesse®, Clinuvel Pharmaceuticals, Melbourne, VIC, Australia), an α-melanocyte-stimulating hormone analogue, promotes skin pigmentation independent of sunlight via the activation of the melanocyte melanocortin-1 receptor and improves sunlight protection and tolerance (Langendonk et al. 2015).
In uncontrolled observational studies, ursodeoxycholic acid appears to increase hepatic clearance, and cholestyramine may bind excess protoporphyrin IX (PPIX) in the gut to interrupt its enterohepatic circulation. Excess metal-free PPIX has also been removed by plasma exchange in the treatment of liver failure or for prevention of hepatic decompensation. Erythrocytapheresis may be useful, since patients’ red blood cells contain high amount of toxic PPIX. Moreover, iron depletion should be beneficial, since iron stimulates ALAS2. Patients with advanced cholestasis or cirrhosis should receive liver transplants. Before liver transplantation, excess circulating PPIX must be removed. During transplantation, but also during other abdominal surgeries, the use of special yellow filters prevents light-induced damage of visceral organs. Unfortunately, and in contrast to patients with AHP, liver transplantation does not cure patients with EPP or XLP, where the excessively elevated PPIX originates from the bone marrow. Consequently, allogeneic hematopoietic stem cell transplantation has led to a PPIX reduction of up to 85% and a resolution of inflammatory liver damage, regardless of prior liver transplantation.
Patients with EPP or XLP are not sensitive to numerous drugs, as are patients with AHP. Paradoxical on a first glance, iron substitution can decrease PPIX concentrations and improve symptoms in patients with XLP, which can be explained by iron serving as secondary substrate to promote conversion of toxic PPIX to heme by FECH (Landefeld et al. 2016). In contrast, EPP is exacerbated by iron substitution, since iron induces the bone marrow enzyme ALAS2 (Barman-Aksoezen et al. 2017). In this line, mild iron deficiency may rather protect patients with EPP. Should iron substitution in patients with EPP be necessary, as in cases of severe anemia, it should be done during the darker seasons with low-intensity sunlight. There is some hope for gene therapeutic approaches to EPP.
Congenital Erythropoietic Porphyria (CEP; 57.5)
As in EPP and XLP, baseline treatment and prevention for CEP are light protection and vitamin D supplementation. Some patients with anemia have benefited from splenectomy. The indication for splenectomy must be personalized, since clinical presentation is highly variant, with different degrees of splenomegaly, anemia, and thrombocytopenia. Allogeneic hematopoietic stem cell transplantation is curative and should be performed at younger age. A single case was reported, where iron depletion with deferasirox improved photosensitivity, likely by reducing the activity of ALAS2. This mechanism is supported by another case, where an ALAS2 gain-of-function mutation increased the severity of CEP. Furthermore, proteasome inhibitors or chemical chaperones could stabilize the otherwise dysfunctional UROS variants to increase their activity, reduce porphyrin accumulation, and ameliorate skin photosensitivity in CEP patients.
References
Balwani M, Sardh E, Ventura P, et al. Phase 3 trial of RNAi therapeutic givosiran for acute intermittent porphyria. N Engl J Med. 2020;382:2289–301.
Barman-Aksoezen J, Girelli D, Aurizi C, Schneider-Yin X, Campostrini N, Barbieri L, Minder EI, Biolcati G. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J Inherit Metab Dis. 2017;40:433–41.
Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017;377:2101.
Bonkovsky HL, Guo J-T, Hou W, Li T, Narang T, Thapar M. Porphyrin and heme metabolism and the porphyrias. In: Compr. Physiol: American Cancer Society; 2013. p. 365–401.
Bonkowsky HL, Tschudy DP, Collins A, Doherty J, Bossenmaier I, Cardinal R, Watson CJ. Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Proc Natl Acad Sci U S A. 1971;68:2725–9.
Combalia A, To-Figueras J, Laguno M, Martínez-Rebollar M, Aguilera P. Direct-acting antivirals for hepatitis C virus induce a rapid clinical and biochemical remission of porphyria cutanea tarda. Br J Dermatol. 2017;177:e183–4.
Doss M, Sixel-Dietrich F, Verspohl F. “Glucose effect” and rate limiting function of uroporphyrinogen synthase on porphyrin metabolism in hepatocyte culture: relationship with human acute hepatic porphyrias. J Clin Chem Clin Biochem. 1985;23:505–13.
Doss M, von Tiepermann R, Schneider J, Schmid H. New type of hepatic porphyria with porphobilinogen synthase defect and intermittent acute clinical manifestation. Klin Wochenschr. 1979;57:1123–7.
Gouya L, Ventura P, Balwani M, et al. EXPLORE: a prospective, multinational, natural history study of patients with acute hepatic porphyria with recurrent attacks. Hepatology. 2020;71:1546–58.
Handschin C, Lin J, Rhee J, Peyer A-K, Chin S, Wu P-H, Meyer UA, Spiegelman BM. Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell. 2005;122:505–15.
Kordac V, Semrádová M. Therapy of patients with porphyria cutanea tarda with antimalarials. Cas Lek Cesk. 1973;112:762–5.
Landefeld C, Kentouche K, Gruhn B, Stauch T, Rößler S, Schuppan D, Whatley SD, Beck JF, Stölzel U. X-linked protoporphyria: iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br J Haematol. 2016;173:482–4.
Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015;373:48–59.
Lenglet H, Schmitt C, Grange T, et al. From a dominant to an oligogenic model of inheritance with environmental modifiers in acute intermittent porphyria. Hum Mol Genet. 2018;27:1164–73.
Puy H, Gouya L, Deybach J-C. Porphyrias. Lancet. 2010;375:924–37.
Salameh H, Sarairah H, Rizwan M, Kuo Y-F, Anderson KE, Singal AK. Relapse of porphyria cutanea tarda after treatment with phlebotomy or 4-aminoquinoline antimalarials: a meta-analysis. Br J Dermatol. 2018;179:1351–7.
Schmitt C, Lenglet H, Yu A, et al. Recurrent attacks of acute hepatic porphyria: major role of the chronic inflammatory response in the liver. J Intern Med. 2018;284:78–91.
Seth AK, Badminton MN, Mirza D, Russell S, Elias E. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:1219–27.
Stölzel U, Brosche C, Koszka C, Stauch T, Teubner A, Doss MO. Safe and probably safe drugs in acute hepatic porphyria. Cell Mol Biol (Noisy-le-Grand). 2009;55:147–51.
Stölzel U, Doss MO, Schuppan D. Clinical guide and update on Porphyrias. Gastroenterology. 2019;157:365–81.e4
Stölzel U, Köstler E, Schuppan D, Richter M, Wollina U, Doss MO, Wittekind C, Tannapfel A. Hemochromatosis (HFE) gene mutations and response to chloroquine in porphyria cutanea tarda. Arch Dermatol. 2003;139:309–13.
Tschudy DP, Perlroth MG, Marver HS, Collins A, Hunter G, Rechcigl M. Acute intermittent porphyria: the first “overproduction disease” localized to a specific enzyme. Proc Natl Acad Sci U S A. 1965;53:841–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Stölzel, U., Kubisch, I., Stauch, T., Schuppan, D. (2022). Disorders of Heme Metabolism. In: Blau, N., Dionisi Vici, C., Ferreira, C.R., Vianey-Saban, C., van Karnebeek, C.D.M. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-67727-5_57
Download citation
DOI: https://doi.org/10.1007/978-3-030-67727-5_57
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-67726-8
Online ISBN: 978-3-030-67727-5
eBook Packages: MedicineMedicine (R0)