
Abstract
Porphyrias are metabolic disorders of the heme biosynthesis. Clinically, they can be differentiated into acute and non-acute porphyrias. The symptomatic phase of acute hepatic porphyrias is characterized by overproduction of neurotoxic porphyrin precursors and porphyrins. Acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and Doss porphyria (ALSDP) belong to this group of metabolic disorders. The clinical presentation of the acute hepatic porphyria syndrome includes abdominal, psychiatric, neurological, and cardiovascular symptoms. The diagnosis is based on an at least tenfold increased urinary excretion of porphobilinogen (apart from Doss porphyria and lead intoxication). Besides symptomatic therapy with non-porphyrinogenic drugs, electrolyte compensation, and intensive monitoring, intravenous administration of glucose and heme arginate is established for treatment. Among the non-acute types like porphyria cutanea tarda, erythropoietic protoporphyria, and congenital erythropoietic porphyria, the accumulated porphyrins cause photosensitivity of the skin and in some cases severe liver damage. X-linked protoporphyria (XLPP) represents a new type of protoporphyria, with 5-aminolevulinic acid synthase 2 gain of function leading to high concentrations of free protoporphyrin IX. The location of the deficient enzyme within the heme biosynthetic pathway determines the pattern of the accumulated porphyrins. The cDNA of all enzymes of heme biosynthesis have been characterized, and mutations responsible for any of the porphyrias have been described. Besides light protection, there are different therapies depending on the type of non-acute porphyria. Ultimately, liver transplantation may be considered in therapy-resistant cases of acute hepatic porphyrias and bone marrow transplantation in severe cases of erythropoietic porphyrias.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Porphyria Cutanea Tarda
- Molecular Adsorbent Recirculation System
- Acute Intermittent Porphyria
- Acute Intermittent Porphyria
- Acute Porphyria
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara SummaryPorphyrias are metabolic disorders of the heme biosynthesis. Clinically, they can be differentiated into acute and non-acute porphyrias. The symptomatic phase of acute hepatic porphyrias is characterized by overproduction of neurotoxic porphyrin precursors and porphyrins. Acute intermittent porphyria (AIP), variegate porphyria (VP), hereditary coproporphyria (HCP), and Doss porphyria (ALADP) belong to this group of metabolic disorders. The clinical presentation of the acute hepatic porphyria syndrome includes abdominal, psychiatric, neurological, and cardiovascular symptoms. The diagnosis is based on an at least tenfold increased urinary excretion of porphobilinogen (apart from Doss porphyria and lead intoxication). Besides symptomatic therapy with non-porphyrinogenic drugs, electrolyte correction, and intensive monitoring, intravenous administration of glucose and heme arginate is established for treatment. Among the non-acute types like porphyria cutanea tarda, erythropoietic protoporphyria, and congenital erythropoietic porphyria, the accumulated porphyrins cause photosensitivity of the skin and in some cases severe liver damage. X-linked protoporphyria (XLPP) represents a new type of protoporphyria, with 5-aminolevulinic acid synthase 2 gain of function leading to high concentrations of free protoporphyrin IX. The location of the deficient enzyme within the heme biosynthetic pathway determines the pattern of the accumulated porphyrins. The cDNA of all enzymes of heme biosynthesis have been characterized, and mutations responsible for any of the porphyrias have been described. Besides light protection, there are different therapies depending on the type of non-acute porphyria. Ultimately, liver transplantation may be considered in therapy-resistant cases of acute hepatic porphyrias and bone marrow transplantation in severe cases of erythropoietic porphyrias.
1 Introduction
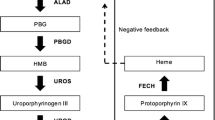
Porphyrias are a heterogeneous group of metabolic disorders, which are based on genetic deficiencies along the heme synthesis (Bonkovsky et al. 2013; Puy et al. 2010) (Fig. 33.1).
Clinically it can be differentiated between acute and non-acute porphyrias, pathogenetically between hepatic and erythropoietic porphyrias. Apart from the recessive congenital erythropoietic porphyria (Morbus Günther) and the two types of protoporphyria (EPP,XLPP) all other porphyrias are classified as hepatic.
The clinical presentation of acute hepatic porphyrias includes an abdominal – neuropsychiatric – cardiovascular syndrome, whereas chronic hepatic and erythropoietic porphyrias present with dermatological symptoms due to photodermatosis (Stölzel et al. 1987; Stölzel and Doss 2009).
The symptomatic phase of acute hepatic porphyria is characterized by excessive accumulation and excretion of the porphyrin precursors 5-aminolevulinic acid (ALA) and porphobilinogen (PBG) as well as porphyrins (Bonkovsky et al. 2013; Puy et al. 2010).
Acute porphyrias are biochemically distinguished according to the localization of the affected enzyme. Among acute porphyrias, the acute intermittent porphyria (AIP) is the most common, followed by the variegate porphyria (VP), the hereditary coproporphyria (HCP), and a rare recessive acute hepatic porphyria with 5-aminolevulinic acid-dehydratase deficiency (ALADP, synonym: porphobilinogen synthase defect porphyria, Doss porphyria), which is biochemically very similar to lead poisoning (Doss et al. 1979). Therefore, lead poisoning as toxic and toxo-genetic acute porphyria can be included into the acute types of porphyrias.
In contrast to the acute type, chronic porphyrias such as porphyria cutanea tarda (PCT) as a chronic hepatic porphyria (CHP) and the erythropoietic porphyria (EPP) display characteristics of chronic diseases (Stölzel and Doss 2009).
The diagnosis of clinical symptomatic porphyrias is based on the biochemical analysis, in which each porphyria expresses its own specific diagnostic (excretory) pattern.
Porphyrias are characterized by a huge molecular genetic heterogeneity. They can be caused by a multitude of different mutations. Genetic analysis of clinically “homozygous” porphyrias show that there is quite often a compound heterozygosity. Rare homozygous forms can also be found among dominant autosomal hereditary porphyrias. DNA analysis can be helpful to screen asymptomatic carriers. However, since asymptomatic gene carriers of porphyric disorder are common in the normal population, genetic testing is not recommended for diagnosing acute porphyrias. Recently, modifying genes have been identified that contribute to clinically overt disease and moreover disease severity. In addition there is a considerable interaction between gene defect and environmental factors.
Dual porphyrias were diagnosed in rare cases with clinical and biochemical findings of two porphyrias. For example, coexistent AIP and PCT in the same individual was described.
Apart from the genetic predisposed erythropoietic and hepatic porphyrias, clinical asymptomatic secondary porphyrinurias and porphyrinemias can be detected among a number of different diseases and dysfunctions.
2 Nomenclature
No. | Disorder | Alternative name | Abbreviation | Gene symbol | Chromosomal localization | Affected protein | OMIM no. | Subtype |
|---|---|---|---|---|---|---|---|---|
33.1 | X-linked sideroblastic anemia | Erythroid 5-aminolevulinate synthase deficiency | XLSA | ALAS2 | Xp11.21 | 5-aminolevulinate synthase 2 | 300751 | All forms |
33.2 | X-linked protoporphyria | Erythroid 5-aminolevulinate synthase gain of function | XLPP | ALAS2 | Xp11.21 | 5-aminolevulinate synthase 2 | 300752 | All forms |
33.3 | 5-aminolevulinate dehydratase porphyria | Doss porphyria | ALADP | ALAD | 9q34 | Delta-aminolevulinate dehydratase | 125270 | Autosomal recessive |
33.4 | Acute intermittent porphyria | Porphobilinogen deaminase deficiency | AIP | HMBS | 11q23-11qter | Porphobilinogen deaminase | 176000 | Autosomal dominant |
33.5 | Congenital erythropoietic porphyria | Uroporphyrinogen III synthase deficiency Morbus Günther | CEP | UROS | 10q25.2-26.3 | Uroporphyrinogen III synthase | 263700 | Classic form |
33.6 | Porphyria cutanea tarda types I, II, and III and hepatoerythropoietic porphyria | Hepatic uroporphyrinogen decarboxylase deficiency chronic hepatic porphyria | PCT/HEP | UROD | 1p34 | Uroporphyrinogen decarboxylase | 176100 | All forms |
33.7 | Hereditary coproporphyria | Coproporphyrinogen oxidase deficiency | HC | CPOX | 3q12 | Coproporphyrinogen oxidase | 121300 | Autosomal dominant |
33.8 | Porphyria variegata | Protoporphyrinogen oxidase deficiency | PV | PPOX | 1q22-q23 | Protoporphyrinogen oxidase | 176200 | Autosomal dominant |
33.9 | Erythropoietic protoporphyria | Ferrochelatase deficiency | EPP | FECH | 18q21.3 | Ferrochelatase | 177000 | Autosomal dominant |
3 Metabolic Pathways

The enzymes of the heme biosynthesis, their subcellular localizations, and associated diseases. There are two differently regulated isoforms of 5-aminolevulinic acid synthase (ALA synthase). ALAS-1 is a ubiquitous enzyme. ALAS-2 is an erythroid-specific enzyme. X-linked protoporphyria is caused by ALAS-2 gain of function mutations
4 Signs and Symptoms
5 Reference Values
6 Pathological Deviations and Values
7 Diagnostic Flowcharts
8 Specimen Collection
Test | Material | Handling |
|---|---|---|
First-line diagnostic tests: | ||
Fluorescence scan | 1 ml serum, plasma (heparin, EDTA) | Light protected and cool |
5-Aminolevulinic acid Porphobilinogen (Porphyrin precursors) | 5 ml urine (spot sample or 24 h collection) | Light protected and cool |
Porphyrins | 5 ml urine (spot sample or 24 h collection, 5 g feces | Light protected and cool |
Free protoporphyrin Zinc protoporphyrin | 3 ml EDTA blood or heparinized blood | Light protected and cool |
Second-line diagnostic tests: | ||
Enzyme activity tests | 5 ml heparinized blood or ACD blood | Cool, not frozen |
Molecular genetic assays | ||
Genomic DNA | EDTA blood or isolated DNA | – |
cDNA | EDTA blood | Rapid sample transport (<12 h) |
9 Treatment
Acute Porphyrias (ALADP; 33.3, AIP; 33.4, HCP; 33.7, VP; 33.8)
General treatment strategies include admitting the patient in to an intensive care unit, analgesic therapy, glucose infusion, correction of electrolyte abnormalities and stopping porphyrinogenic medication (Anderson et al. 2005). The most important therapeutic measures are summarized in the table below.
Therapy of Acute Porphyrias
1. Withdraw unsafe medication and intensive care unit observation |
2. Treatment with glucose or together with heme |
Intravenous glucose (300–500 g/day) should be given for mild symptoms (moderate pain, no paresis or hyponatremia). Glucose combined with insulin is considered to be more effective than glucose alone |
CAVE: Large volumes of 10 % glucose may increase risk for hyponatremia |
Severe symptoms and neurological complications should be treated as soon as possible with intravenously given human heme arginate (Normosang®, Orphan Europe) (3 mg/kg body weight for four consecutive days). After infusion, preferably into large vein normal saline should be infused for 15 minutes to reduce local toxicity |
We suggested to dilute the human heme arginate in 100 ml of human albumin (4–20 % depending on country availability) instead of normal saline solution to avoid damage of the veins. Heme as a lyophilized powder (Panhematin®; RECORDATI Rare Disease) is available in the United States |
3. Monitoring and correction of electrolytes including sodium and magnesium |
Analgesic therapy: aspirin, gabapentin, morphine derivatives |
In case of tachycardia and hypertension: metoprolol, propranolol, valsartan |
In case of unrest and vomiting: lorazepam, phenothiazines, ondansetron |
In case of ileus: neostigmine |
In case of respiratory failure: mechanical ventilation |
In case of infection: penicillin and derivatives, cephalosporins, imipenem, aminoglycosides |
In case of paresis: physiotherapy |
Monitoring: urinary excretion of ALA, PBG, and porphyrins in AIP and additionally fecal excreted porphyrins in HCP and VP |
Glucose and more importantly intravenously applied heme downregulate the metabolic dysregulation (Doss and Verspohl 1981). Mild cases can be treated with glucose alone (Anderson et al. 2005). Neurological symptoms should be immediately treated with heme in order to reverse and avoid progress or persistence. The so-called glucose effect was recently confirmed by elucidating regulatory links between hepatic heme biosynthesis – and glucose metabolism via nuclear receptors including PGC-1α (Handschin et al. 2005). Glucose combined with insulin is considered to be more effective than glucose alone. On one hand, fasting worsens; on the other hand, glucose can help to treat mild clinical manifestations of acute porphyria. If tolerated, diet rich in carbohydrates or oral glucose can be added to or replace intravenous application. Large volumes of 10 % glucose may increase risk for hyponatremia. Intravenously given human heme (3 mg/kg body weight for four consecutive days, Normosang®, Orphan Europe) increases the hepatic heme storage, improves the function of hepatic hemoproteins, represses ALA synthase, and decreases the overproduction and consequently urinary excretion of ALA and PBG within days (Bonkowsky et al. 1971; Bonkovsky 1993).
After infusion, preferably into a large vein to reduce local toxicity, normal saline should be infused for 15 minutes to reduce local toxicity. We suggest to dilute the human heme arginate in 100 ml of human albumin (4–20 % depending on country specific availability) instead of normal saline solution in order to avoid damage of the veins. Heme as a lyophilized powder (Panhematin®; RECORDATI Rare Disease) is available in the United States where human hemin (Normosang®) lacks FDA approval. Prophylactic heme application within periods ranging from weekly to monthly is used to prevent repeated clinical manifestations. Frequent treatment with heme leads to iron overload since 100 mg of heme contains 8 mg iron. Moreover, heme induces the hemoxygenase 1. Recently heme arginate-associated inflammatory reactions such as elevation of interleukin 6 and metabolic stress were observed. Taken together these effects may explain tachyphylaxis with subsequent need to shorten the interval between therapeutic applications (Bonkovsky et al. 2013). Long-term application of heme arginate should be scrutinized critically.
Heme therapy was found to be helpful to relieve symptoms in lead intoxication and ALADP.
Intravenously applied recombinant PBD reduced PBG levels but did not show any benefit on clinical endpoints.
Ultimately, in severe and complicated disease, liver transplantation can cure the disease (Seth et al. 2007). Complete normalization of porphyrin metabolism after liver transplantation proves the fact that acute porphyrias are diseases of the liver. Using adenoviral vectors the hepatic enzymes defects were reversed. So far seven patients have been included, and patients as well as physicians wait for hopefully encouraging short- and long-term results. Transplantation of hepatocytes may be an option for the future. Using small interfering RNA in order to silence hepatic ALAS 1 may be helpful to inhibit ALA und PBG overproduction.
In women with acute porphyria, pregnancy is in general not at risk, although progesterone potently induces liver heme production (Kühnel et al. 2002). However, pregnancy-associated vomiting and subsequent caloric deficiency should be normalized promptly by glucose infusion.
For those women suffering from frequent attacks related to menstrual cycle, gonadotropin-releasing hormone analogues, combined with low-dose estrogen patch to suppress menopausal symptoms, can be helpful. Before treatment with LH-RH- agonist is considered, low-dose hormonal oral contraceptives should be used under strict control of the urinary excretion of porphyrin precursors and porphyrins.
The H2 receptor antagonist cimetidine was successfully used in a few patients. The reports on LH-RH agonists and even on cimetidine are non consistent, and more data from placebo controlled double blinded studies are not available so far. The H2 blocker cimetidine can obviously be used safely on patients with acute porphyria. Parenteral injected magnesium was reported to be helpful in case of epileptic seizures.
Avoiding porphyrinogenic medication, alcohol and physical stress is of major importance, as well as a balanced diet with a high percentage of carbohydrates. The patients should be advised not to smoke. Patients with acute porphyria should take special care to avoid infections and other diseases, and the porphyrin precursors ALA and PBG should be monitored in order to detect and to effectively treat a renewed porphyria manifestation as soon as possible.
Based on experimental work, pharmacological data, and clinical reports, safe und unsafe drugs are listed (Table 33.10) (Stölzel et al. 2009). International guidelines are useful to individually handle problems (www.drugs-porphyria.org, www.porphyria-europe.com).
Chro Porphyria cutanea trada; Hepatoerythropoietic Porphyria (PCT/HEP; 33.6)
Phlebotomy and treatment with low-dose chloroquine are therapeutically effective in PCT. The therapeutic aim is the elimination of accumulated porphyrins from the liver and other organs. The patients should be advised to avoid known precipitation factors. For patients with advanced liver cirrhosis and reduced albumin synthesis, phlebotomy is contraindicated.
In most cases, the dosage of 125 mg chloroquine twice a week leads to metabolic and clinical remission (Koestler and Wollina 2005). In case of excessive urinary porphyrin excretion of more than 10 mg/day, the treatment can be started with weekly phlebotomy and after 1 month continued with chloroquine for several months up to 1 year.
The treatment can be abandoned when the urinary porphyrin excretion stabilizes in a subclinical level (<0.3 mg/day).
A complete normalization of the porphyrinuria is rare, and in particular, it cannot be expected in case of a genetic disposed form. Since a low to a moderate increased porphyrinuria does not cause clinical symptoms, further treatment is not recommended. Steatosis of liver cells and siderosis can regress after phlebotomy as well as after a therapy with chloroquine (Stölzel et al. 2003). Chronic hepatitis C is associated with PCT (Stölzel et al. 1995). After iron depletion, treatment of chronic hepatitis C should be initiated with combination of protease inhibitor, pegylated, interferon-alpha-2a, and ribavirin (Bonkovsky et al. 2013).
Erythropoietic Protoporphyria (EPP; 33.9)
The light sensitivity is symptomatically treated with beta-carotene with a dosage of 50 up to 200 (300) mg/day. Monitoring of serum carotene levels is recommended (11–15 μmol/L). EPP is characterized by sensitivity to visible light; conventional sunscreens that protect against ultraviolet radiation (particularly UVB) are usually not effective. Reflectant sunscreens based on titanium dioxide or zinc oxide that cover both UVA, UVB, and visible light to a degree will be more effective. Afamelanotide is a new synthetic analogue of naturally occurring alpha-melanocyte-stimulating hormone (α-MSH). After binding to the melanocortin-1 receptor, it induces physiologic melanogenesis independent of the potentially harmful effects of UV light. This leads to increased skin pigmentation and photoprotection. In contrast to acute porphyrias, for patients with EPP, there is in general no restriction for porphyrinogenic substances or medication.
Colestyramine and ursodeoxycholic acid are used for the treatment of EPP-induced liver disease. Colestyramine and other absorbents such as activated charcoal may interrupt the enterohepatic circulation of protoporphyrin. Ursodeoxycholic acid should help to slow the progress of cholestasis and to enhance the biliary elimination of protoporphyrin. Clinical benefit can obviously only be achieved in the early phase of hepatobiliary damage. In order to protect the liver, otherwise damaging agents should be completely avoided. Therefore, vaccination against hepatitis A und B is generally recommended for patients with EPP. Vitamin D substitution is advisible since patients avoid sunlight. Although intravenous heme does not downregulate ALAS 2 in bone marrow, clinical improvement of liver function was described in patients with EPP treated with heme. In some cases, red cell transfusions improved EPP. Plasmapheresis and extracorporeal albumin dialysis (molecular adsorbents recirculation system (MARS), Prometheus) can be used for the elimination of protoporphyrin. Liver transplantation is the therapy of choice in EPP patients with cholestatic liver cirrhosis. Recently for the first time, bone marrow transplantation has been reported in EPP (Wahlin et al. 2007).
Iron supplementation improves protoporphyrin overload, liver damage, and anemia in a new type of X-linked protoporphyria.
Congenital Erythropoietic Porphyria (CEP; 33.5)
Therapeutic measures have been dissatisfactory up to now. Light protection, blood transfusion in case of a severe anemia, and splenectomy can be considered.
Bone marrow transplantation was performed in some cases. Two children were reported to be disease-free for 3 and 2 years posttransplantation. In a mouse model, it has recently been demonstrated to repair mutations of UROS in bone marrow cells with the help of viral vectors. Advance in gene therapy could be a real hope for affected patients in the future.
References
Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ (2005) Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med 142:439–450
Bonkovsky HL (1993) Advances in understanding and treating ‘the little imitator’, acute porphyria. Gastroenterology 105:590–594
Bonkovsky HL, Guo JT, Hou W, Li T, Narang T, Thapar M (2013) Porphyrin and heme metabolism and the porphyrias. Compr Physiol 3:365–401
Bonkowsky HL, Tschudy DP, Collins A, Doherty J, Bossenmaier I, Cardinal R, Watson CJ (1971) Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Proc Natl Acad Sci U S A 68:2725–2729
Doss M, Verspohl F (1981) The “glucose effect” in acute hepatic porphyrias and in experimental porphyria. Klin Wochenschr 59:727–735
Doss M, von Tiepermann R, Schneider J, Schmid H (1979) New type of hepatic porphyria with porphobilinogen synthase defect and intermittent acute clinical manifestation. Klin Wochenschr 57:1123–1127
Handschin C, Lin J, Rhee J, Peyer AK, Chin S, Wu PH, Meyer UA, Spiegelman BM (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 122:505–515
Kostler E, Wollina U (2005) Therapy of porphyria cutanea tarda. Expert Opin Pharmacother 6:377–383
Kühnel A, Groß U, Doss MO (2002) Porphyrien. In: Schmailzl KJG, Hachelöer BJ (eds) Schwangerschaft und Krankheit. Blackwell Verlag, Berlin/Wien, pp S440–S453
Puy H, Gouya L, Deybach JC (2010) Porphyrias. Lancet 375:924–937
Seth AK, Badminton MN, Mirza D, Russell S, Elias E (2007) Liver transplantation for porphyria: who, when, and how? Liver Transpl 13:1219–1227
Stölzel U, Doss M (2009) Porphyrias. In: Dancygier H (ed) Clinical hepatology, principles and practice of hepatobiliary diseases, vol 2. Springer, Berlin, pp 1077–1092
Stölzel U, Doss MO, Dissmann T, Cervós-Navarro J, Riecken EO (1987) Gastroenterologic and neurologic manifestations in acute intermittent porphyria. Med Klin (Munich) 82:520–525
Stölzel U, Köstler E, Koszka C, Stöffler-Meilicke M, Schuppan D, Somasundaram R, Doss MO, Habermehl KO, Riecken EO (1995) Low prevalence of hepatitis C virus infection in porphyria cutanea tarda in Germany. Hepatology 21:1500–1503
Stölzel U, Köstler E, Schuppan D, Richter M, Wollina U, Doss MO, Wittekind C, Tannapfel A (2003) Hemochromatosis (HFE) gene mutations and response to chloroquine in porphyria cutanea tarda. Arch Dermatol 139:309–313
Stölzel U, Brosche C, Koszka C, Stauch T, Teubner A, Doss MO (2009) Safe and probably safe drugs in acute porphyria. Cell Mol Biol 55:147–151
Wahlin S, Aschan J, Björnstedt M, Broomé U, Harper P (2007) Curative bone marrow transplantation in erythropoietic protoporphyria after reversal of severe cholestasis. J Hepatol 46:174–179
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Stölzel, U., Stauch, T., Doss, M.O. (2014). Heme Synthesis Defects and Porphyrias. In: Blau, N., Duran, M., Gibson, K., Dionisi Vici, C. (eds) Physician's Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-40337-8_33
Download citation
DOI: https://doi.org/10.1007/978-3-642-40337-8_33
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-40336-1
Online ISBN: 978-3-642-40337-8
eBook Packages: MedicineMedicine (R0)