
Abstract
Modern ideas regarding the three-dimensional organization of the genome and its role in controlling gene expression are largely based on the results of research performed using the proximity ligation protocol. It has been demonstrated that genome folding is much less regular than was previously assumed. On the other hand, the genome was found partitioned into semi-independent structural-functional units commonly referred to as topologically associating domains (TADs). TAD borders restrict the areas of enhancer action via interfering with establishment of long-distance enhancer-promoter contacts. Within TADs, spatial juxtaposing of promoters to various enhancers or silencers results in the assembly of activating or repressing chromatin hubs that constitute an important part of epigenetic mechanisms regulating gene expression in higher eukaryotes. Within the cell nucleus, the spatial organization of the genome is tightly connected with functional compartmentalization of the nucleus. Recent evidence suggests that liquid phase separation plays an important role in establishing both the 3D genome organization and nuclear compartmentalization. In this chapter, we review the present state and outline the most important trends for future research in the area of 3D genomics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Chromatin
- Topologically associating domains (TADs)
- Structural-functional domains of eukaryotic genome
- DNA loop extrusion
- Nuclear compartments
Introduction
Studies of the 3D genome organization have become a trend in modern genomics. One may say that modern genomics has acquired a third dimension. As is often the case in science, a new stage in the study of genome organization and functioning was predetermined by the development of appropriate research tools. One biochemical protocol that had a major impact on the development of 3D genomics is the chromosome conformation capture protocol (Dekker et al. 2002). The main steps of this protocol are presented in Fig. 2.1. The key step of this procedure is introduction of breaks into DNA within a fixed nucleus, followed by cross-ligation of closely located ends of broken DNA. Joining of DNA fragments located far from each other on the DNA chain but close in physical space creates chimeric DNA sequences containing information about the spatial proximity of the corresponding segments of genomic DNA. Analysis of the pools of chimeric fragments allows reconstructing the spatial organization of the genome based on the sets of captured pairwise interactions. This procedure was first successfully used to demonstrate that all remote enhancers of mouse beta-globin genes along with the promoters of genes, which are actually expressed, are organized into a common active chromatin hub (Tolhuis et al. 2002; de Laat and Grosveld 2003). This work highlighted the importance of 3D genome organization for the regulation of transcription. It has long been assumed that, to activate a gene, an enhancer should be in direct contact with this gene ((Bondarenko et al. 2003; West and Fraser 2005; Vernimmen and Bickmore 2015) and references herein). Taking into account that most enhancers are located far from the target gene, the ideal solution is to loop out the intervening segment of DNA, and 3C analysis has demonstrated that such situations are indeed quite common (Tolhuis et al. 2002; de Laat and Grosveld 2003; Gavrilov and Razin 2008; Philonenko et al. 2009; Vernimmen et al. 2007; Vernimmen et al. 2009). The number of enhancers in mammalian and Drosophila cells exceeds at least ten times the number of genes (Arnold et al. 2013; Consortium et al. 2012). The possibility of gene activation by different combinations of enhancers likely increases the regulatory capacity of the eukaryotic cell transcription control system. Disclosure of the functionally dependent mouse beta-globin gene domain 3D organization (Tolhuis et al. 2002; de Laat and Grosveld 2003) demonstrated for the first time how one gene or group of genes can be simultaneously activated by one or several enhancers.

Main steps of the chromosome conformation capture protocol. Restriction enzymes are used to cut chromatin in intact nuclei isolated from formaldehyde-fixed cells. DNA fragments located in close proximity to each other are ligated with the T4 DNA ligase. qPCR or next-generation sequencing are used for the analysis of DNA chimeras obtained
The original 3C procedure allowed studying interactions between various regions within individual genomic loci. Eventually, various derivative procedures were developed collectively known as C-methods (reviewed in de Wit and de Laat (2012)). Most of these procedures, such as 4C (van de Werken et al. 2012), Hi-C (Lieberman-Aiden et al. 2009), and ChIA-PET (Fullwood et al. 2009), allowed performing genome-wide analysis. Application of these experimental protocols has provided deep insights into the role of 3D genome organization in transcription control (Denker and de Laat 2016; Dekker and Mirny 2016; Valton and Dekker 2016; Krijger and de Laat 2016). Of special importance, the genome was found to be partitioned into semi-independent self-interacting domains termed topologically associating domains or TADs (Nora et al. 2012; Dixon et al. 2012; Sexton et al. 2012). TADs appear to restrict the areas of enhancer action and thus can be considered as structural-functional units of the eukaryotic genome (Symmons et al. 2014, 2016). Disruption of TAD borders results in development of various genetic diseases (Lupianez et al. 2015, 2016; Krumm and Duan 2018; Franke et al. 2016). In normal situations, the patterns of enhancer-promoter spatial interactions change in the course of cell differentiation accordingly to activation and/or repression of particular genes. However, most of these changes occur within TADs while the TAD borders remain relatively stable (Dixon et al. 2016; Fraser et al. 2015). Nevertheless, a certain fraction of TAD boundaries is changed in the course of cell differentiation (Bonev and Cavalli 2016). To obtain further insights into mechanisms of eukaryotic genome functioning, it is highly important to disclose the nature of both TADs and TAD borders. This task is complicated by the fact that in virtually all eukaryotic cells studied, the contact chromatin domains are hierarchical (i.e., within larger domains, it is possible to annotate several levels of smaller and more dense nested domains) (Phillips-Cremins et al. 2013; Luzhin et al. 2019; Weinreb and Raphael 2016). It is not always obvious the domain of which level should be considered as TADs. Some authors claim that TADs can be discriminated only based on their functionality (i.e., as functional units of the genome rather than the units of a particular level of hierarchical genome folding) (Zhan et al. 2017). In this review, we shall discuss mechanisms of TAD formation and the impact of TADs on genome functioning.
Hierarchical Model of DNA Packaging in Chromatin
In most textbooks, it is possible to read that, in eukaryotic cells, genomic DNA is sequentially folded into 10 nm chromatin fiber (nucleosomal chain), into 30 nm chromatin fiber (which is frequently represented as a solenoid or zigzag), and then into loops of 30 nm fiber or several levels of “supersolenoid” structures (Fig. 2.2). Remarkably, this model of chromatin folding into regular structures was proposed approximately 30 years ago (Getzenberg et al. 1991) and is poorly supported by recent data. On the contrary, it is becoming increasingly evident that the only regular level of genomic DNA folding is wrapping of DNA around the octamers of nucleosome histones, resulting in formation of 10 nm fibers (Fussner et al. 2012). The latter then aggregate to form more or less compact chromatin masses (Maeshima et al. 2014a, b, 2016). Aggregation of chromatin fibers is promoted under conditions of macromolecular crowding (Hancock 2008) typical for nucleoplasm. Although at a medium scale thus formed chromatin masses appear irregular, at larger scales, they are subdivided into self-interacting domains that are commonly interpreted as chromatin globules. Such chromatin globules were observed in a high-resolution microscopic study of cell nuclei hybridized to chromosome- or locus-specific probes (Markaki et al. 2012; Smeets et al. 2014). Furthermore, the same structures (1 Mb chromatin clusters) appear to correspond to early replicating chromatin domains (Markaki et al. 2010). In a recent study by the Cavalli laboratory, it was directly shown that TADs correspond to chromatin globules that can be visualized using FISH with TAD- and locus-specific probes (Szabo et al. 2018). Within the entire chromatin domain, TADs containing mostly active and mostly repressed chromatin are spatially segregated into the so-called A and B chromatin compartments, which likely correspond to euchromatin and heterochromatin (Lieberman-Aiden et al. 2009; Gibcus and Dekker 2013; Eagen 2018).

A classical view of hierarchical folding of DNA in the nucleus. 10 nm nucleosome fiber folds into 30 nm fiber of variable architecture, which then forms hierarchical loops and supersolenoid structures
Most of the current knowledge about higher levels of DNA packaging in chromatin is based on the results of Hi-C analysis. The contact chromatin domains were observed in different taxa including mammals (Dixon et al. 2012; Nora et al. 2012), insects (Sexton et al. 2012), and birds (Ulianov et al. 2017). Of note, in Drosophila, TADs have a size in the range of 100 Kb (Sexton et al. 2012; Hou et al. 2012), while mammalian TADs are ten times larger (Dixon et al. 2012, 2016). Some contact domains can also be revealed in the genomes of plants and lower eukaryotes (Wang et al. 2015; Hsieh et al. 2015; Eser et al. 2017; Nikolaou 2017). However, they are substantially different from the TADs of mammals and Drosophila both in size and in the levels of insulation and genome coverage.
Interpretation of Hi-C maps strongly depends on resolution of the analysis. At 1 Mb resolution, only segregation of active and inactive chromatin can be registered (Lieberman-Aiden et al. 2009). 20–100 Kb resolution revealed TADs (Dixon et al. 2012, 2016; Gibcus and Dekker 2013). Finally, 1 Kb resolution maps demonstrated that TADs comprise two types of self-interacting domains, namely, looped domains and ordinary domains (Rao et al. 2014). The distinctive feature of looped domains in Hi-C maps is a spot at the top of a triangle reflecting a spatial proximity of loop bases (Fig. 2.3). In mammalian cells, chromatin loops originate due to enhancer-promoter interaction (Jin et al. 2013; Sahlen et al. 2015; Ghavi-Helm et al. 2014) or because of interactions between CTCF-binding sites (Sanborn et al. 2015). The nature of ordinary chromatin domains is less clear. It has been proposed that these domains originate due to clustering and spatial segregation of active and inactive genomic regions. Accordingly, it was proposed to call them “compartmental domains” (Rowley and Corces 2018). The mechanisms underlying the spatial segregation of chromatin compartments (or compartmental domains) are still unclear. A current model postulates that proteins enriched in different chromatin types trigger phase separation, resulting in their spatial segregation (Nuebler et al. 2018; Rada-Iglesias et al. 2018).

Potential role of CTCF in defining chromatin spatial organization and epigenetic state. (a) Chromatin loop is manifested as a filled triangle in the Hi-C heat map only if numerous interactions between loop internal regions occur. (b) In a “traffic jam” model, DNA-bound CTCF restricts the spreading of histone posttranslation modifications along the chromatin fiber, preventing binding of chromatin-modifying complexes to nucleosomes located downstream of the CTCF-binding site. (c) Point-to-point interactions between CTCF-binding sites are unable to insulate extended loops from each other in the 3D nuclear space
Functional Domains of the Eukaryotic Genome
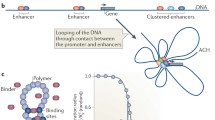
The eukaryotic genome has long been proposed to be a mosaic of semi-independent structural-functional domains (Bodnar 1988; Goldman 1988). The original model was inspired by the results of analysis of DNaseI sensitivity of individual genes and genomic segments (Weintraub and Groudine 1976; Weintraub et al. 1981; Lawson et al. 1982; Jantzen et al. 1986). It was proposed that the entire genome is built from similarly organized structural-functional units (domains) that may be either active or repressed. The transcriptional status of the domain was thought to be controlled at the level of chromatin packaging. The model stimulated research aimed to identify regulatory elements controlling the chromatin status of genomic domains. These studies resulted in identification of domain bordering elements (insulators) (Kellum and Schedl 1991, 1992; Udvardy et al. 1986), nuclear matrix attachment regions (MARs) (Cockerill and Garrard 1986), and locus control regions (LCRs) (Forrester et al. 1987, 1990; Grosveld et al. 1987; Li et al. 1990). Although in its initial form the domain model of eukaryotic genome organization cannot account for a number of recent observations, it can be upgraded taking into account the 3D genome organization (Razin and Vassetzky 2017). Considering the necessity of juxtaposition of enhancers and promoters, one may conclude that any self-interacting chromatin domain would impose certain restrictions on enhancer action. Indeed, it has been demonstrated that, in most cases, the areas of enhancers’ action are restricted to the so-called insulating neighborhoods (Sun et al. 2019), regulatory archipelagos (Montavon et al. 2011), regulatory landscapes (Spitz et al. 2003; Zuniga et al. 2004), or regulatory domains (Symmons et al. 2014). These functional genomic blocks are large (100 Kb to 1 Mb) segments of the genome within which non-related genes demonstrate similar tissue specificity of expression. Being integrated in such a domain, a reporter gene under control of a minimal promoter demonstrates a tissue-specific expression profile typical for the domain as a whole (Ruf et al. 2011; Symmons et al. 2014). Although there is still some discrepancy in the results of different authors, they all agree that insulated areas colocalize with self-interacting chromatin domains identified by Hi-C analysis, either with TADs (Montavon et al. 2011; Symmons et al. 2014) or looped domains (sub-TADs) (Sun et al. 2019). Interestingly, TADs harboring superenhancers are preferentially insulated by boundaries possessing a particularly high insulation score (Gong et al. 2018).
Partitioning of the genome into semi-independent structural-functional domains appears important for two reasons. First, it minimizes the possibility of an off-target activity of any given enhancer. To this end, it is of note that genomic rearrangements affecting TAD boundaries frequently result in compromising gene regulation networks and development of diseases (Lupianez et al. 2015; Franke et al. 2016; Valton and Dekker 2016; Ibn-Salem et al. 2014; Vicente-Garcia et al. 2017). Second, partitioning of the genome into TADs restricts the area the enhancer should explode to find a target promoter. Correspondingly, the time necessary to establish enhancer-promoter communication is reduced (Symmons et al. 2016). Lack of rigidity in the TAD structure is of importance in this context. Alternative configurations of the chromatin fiber continuously interchange within a TAD (Tiana et al. 2016). This interchange is likely to provide additional possibilities for cell adaptation to a changing environment (Razin et al. 2013). The functional relevance of genome partitioning into TADs is likely to explain the apparent conservation of this organization in the genomes of related species (Dixon et al. 2012) as well as the fact that TADs are stable against rearrangements during evolution (Krefting et al. 2018; Lazar et al. 2018). Interestingly, paralog gene pairs are enriched for colocalization in the same TAD and frequently share common enhancer elements (Ibn-Salem et al. 2017).
Besides constituting the insulation neighborhoods for transcription regulation, the TADs also contribute to the control of replication because they correspond to units of replication timing (replication domains) (Pope et al. 2014). Interestingly, after being disrupted in mitosis (Naumova et al. 2013), TADs are re-established in G1 phase of the cell cycle at about the same time with the establishment of the replication-timing program (Dileep et al. 2015a, b). It may be that exactly at the level of chromatin packaging, the link between active transcription and early replication is established.
TAD Assembly and Insulation
Taking into consideration the fact that TADs restrict the areas of enhancer action, it is particularly important to understand how they are assembled and why they are insulated. Comparison of Hi-C maps with genome-wide distribution of various epigenetic marks demonstrated that, in mammals, TAD boundaries are enriched in CTCF-binding sites and active genes (Dixon et al. 2012). Also, cohesin was found enriched at TAD boundaries (Hansen et al. 2017). Deletion of CTCF-binding sites at TAD boundaries resulted in a full or partial loss of TAD insulation (Narendra et al. 2015, 2016; Lupianez et al. 2015; Sanborn et al. 2015). The same effect was observed upon targeted degradation of CTCF in living cells (Nora et al. 2017). CTCF has long been implicated in mediation of enhancer-blocking activity of insulators (Chung et al. 1997). In addition, it mediates formation of DNA/chromatin loops (Vietri Rudan and Hadjur 2015; Holwerda and de Laat 2012). It should be mentioned, however, that by itself, formation of a chromatin loop is not sufficient for TAD assembly. Within a loop, only the bases are permanently located in a spatial proximity. On a Hi-C heat map, a DNA loop can be recognized as a high interaction signal between bases that looks like a spot at the top of a triangle. However, to “fill” the triangle, it is necessary to ensure mutual interaction of internal parts of the loop (Fig. 2.3a). It is also not clear how deposition of CTCF at TAD boundaries can prevent spatial interactions between internal regions of different TADs. Although CTCF is a large protein (~130 kDa), the octamer of histones constituting the nucleosomal core has approximately the same summary weight, and the 1 Mb mammalian TAD is composed of ~5000 nucleosomes. It is easy to speculate about a mechanism by which deposition of CTCF can interfere with spreading if signals travel along a linear chromatin fiber. Here, a traffic jam model fits perfectly (Fig. 2.3b). However, it is difficult to see how spatial interactions between internal regions of large TADs can be prevented by CTCF (Fig. 2.3c). In fact, it is easier to consider a possibility that TAD is held together by some internal links (see below). However, preferential deposition of CTCF as well as cohesin at mammalian TAD boundaries is an established fact (Sofueva et al. 2013; Nora et al. 2012; Dixon et al. 2012; Zuin et al. 2014; Wutz et al. 2017), and there should be a reason for this deposition.
The model explaining the roles of CTCF and cohesion in TAD formation was suggested by two research teams (Fudenberg et al. 2016; Sanborn et al. 2015). According to the model, cohesin mediates DNA loop extrusion. The process of extrusion may start anywhere in the genome but cannot pass CTCF-binding sites present in a certain orientation. The last supposition was based on the observation that CTCF-binding motive has a direction and that CTCF-binding motives present at TAD boundaries (and bases of sub-TAD loops) usually have convergent orientation (Sanborn et al. 2015; Vietri Rudan et al. 2015; de Wit et al. 2015). Of note, the model considers TAD as a population phenomenon. In each individual cell, only a loop or a set of loops exist within the area that is considered as a TAD. However, all Hi-C maps that have been discussed so far were obtained when cell populations were studied. That is typical for a normal biochemical experiment. In a typical Hi-C protocol, one starts with 1–10 millions of cells. The loop extrusion model assumes that filled triangles (TADs) seen on population Hi-C maps represent superimposition of signals reflecting mainly interaction of bases of a variety of loops extruded in individual cells. This model has been supported by in silico modeling (Fudenberg et al. 2016). Also, it has been demonstrated that depletion or degrading of cohesin results in partial or full disruption of TADs (Sofueva et al. 2013; Rao et al. 2017), whereas depletion of cohesin unloading factor WAPL results in generation of longer chromatin loops (Wutz et al. 2017; Haarhuis et al. 2017) as predicted by the DNA loop extrusion model. The main challenge of the model is that the ability of cohesin to extrude DNA loops was not directly demonstrated. At the same time, it is known that cohesin possesses ATPase activity (Hirano 2005) and is able to move along DNA both in vitro (Stigler et al. 2016; Kanke et al. 2016) and in vivo (Busslinger et al. 2017). Of note, this movement is restricted by CTCF (Davidson et al. 2016; Busslinger et al. 2017). Recently published results of Casellas’s lab demonstrated that loop domains are formed by a process that requires cohesin ATPases (Vian et al. 2018). Finally, a condensin complex that is closely related to cohesin was found able to extrude DNA loops (Ganji et al. 2018). Taken together, these observations strongly support a supposition that cohesin may act as a DNA loop extrusion motor in the interphase nucleus.
It should be stressed that the DNA loop extrusion model (Fudenberg et al. 2016; Sanborn et al. 2015) considers TAD as a population phenomenon. The single-cell Hi-C studies performed so far have not provided a definitive answer to the question of whether there are TADs in individual mammalian cells due to a low resolution of Hi-C maps (Nagano et al. 2013; Flyamer et al. 2017). On the other hand, compact, and at first approximation globular, domains can be visualized in nuclei by FISH with TAD-specific probes (Bintu et al. 2018; Szabo et al. 2018). It is thus likely that there should be another mechanism that ensures compactization of entire TADs or extruded loops. It has been proposed that entropic forces primarily drive the formation of compact contact domains in a polymer confined to a limited space (Vasquez et al. 2016). This supposition made based on results of computational simulations is indirectly supported by the fact that contact domains occur in one or another form in the genomes of various organisms, including bacteria (Le et al. 2013), and special cell types, such as spermatozoa, which contain protamines in place of histones in their nuclei (Battulin et al. 2015). However, organization of nucleosomal fiber into compact domains may be also promoted by electrostatic interaction between nucleosomal particles. The ability of nucleosomal fibers to form various conglomerates is well documented. The conglomerates are stabilized by interactions between positively charged N-terminal tails of histones H3 and H4 and a negatively charged acidic patch on the surface of a nucleosomal globule (Kalashnikova et al. 2013; Pepenella et al. 2014). The same interactions facilitate the formation of 30-nm nucleosome fibers at low fiber concentrations, when between-fiber contacts are unlikely (Luger et al. 1997; Sinha and Shogren-Knaak 2010).
The main concern regarding the model of TAD assembly by condensation of nucleosomal fibers is to explain why individual TADs are separated. To this end, it should be mentioned that, in Drosophila, CTCF loops do not play a major role in 3D genome organization (Rowley et al. 2017). We and others reported that, in Drosophila cells, TAD boundaries harbor transcribed genes and are enriched in histone modifications typical for active chromatin (Ulianov et al. 2016; Sexton et al. 2012; Hou et al. 2012). Histone acetylation, which is typical of active chromatin, decreases the histone charge and prevents internucleosome interactions (Shogren-Knaak et al. 2006; Allahverdi et al. 2011). We argued that these processes may be sufficient to prevent assembly of active chromatin regions into compact domains (Ulianov et al. 2016). Thus, the distribution of active and inactive genes along a DNA molecule may determine the profile of chromosome organization in TADs. To test this idea, we performed computer modelling of self-folding of a virtual polymer that consists of alternating nucleosome blocks of two types reproducing the properties of active and inactive chromatin regions (Fig. 2.4) (Ulianov et al. 2016). The particles of inactive block (500 particles in each block) were allowed to establish a limited number of relatively unstable contacts with the particles of the same type from the same or other inactive blocks. The particles of active blocks (50 particles in each block) were not allowed to establish contacts with each other or with particles from inactive blocks. The self-folding of polymer simulated using dissipative particle dynamics algorithm resulted in formation of globular structures roughly colocalizing with inactive blocks separated by unfolded active blocks (Ulianov et al. 2016). Of course, in each individual simulation, the folding of polymer was not fully regular. In some cases, conglomerates of inactive nucleosomes fused to produce superconglomerates; in other cases, nucleosomes of one inactive block formed more than one conglomerate with less compact spacers between the conglomerates (Fig. 2.4). However, averaging of the results of 12 simulations allowed generation of a Hi-C map containing contact domains (TADs) that coincided with inactive nucleosome blocks and were separated by spacers of active nucleosomes (Ulianov et al. 2016). Other simulations have demonstrated that short patches of “active chromatin” inserted into “inactive chromatin” blocks tend to be extruded on a surface of inactive block (Gavrilov et al. 2016). Insertion of larger stretches of “active chromatin” resulted rather in splitting of inactive blocks. This observation was in agreement with experimental observations that activation of transcription of tissue-specific genes located within TADs correlates with decompacting of the corresponding region, which, in some cases, resulted in TAD splitting (Ulianov et al. 2016).

Model heteropolymer built up from long blocks of inactive particles (non-acetylated nucleosomes interacting with each other) interspersed with short blocks of active particles (acetylated nucleosomes unable to interact with other nucleosomes) recapitulates some structural properties of chromatin. Polymer simulations demonstrate that blocks of inactive particles fold into globules manifested as TADs in spatial distance maps of the polymer. The results of a typical simulation are presented
It should be mentioned that DNA loop extrusion and nucleosome condensation are not mutually exclusive. Thus, nucleosome condensation may contribute to the compaction of extruded chromatin loops in mammalian cells. There is yet another group of models postulating that TAD formation is mediated by architectural proteins that form intra-TAD links, thus pulling together remote segments of a chromatin fiber. To explain the existence of isolated TADs, the models assume a multiplicity of architectural protein groups, each ensuring the formation of a particular TAD (Barbieri et al. 2012, 2013; Pombo and Nicodemi 2014). The models are supported by computer simulations but seem implausible biologically because there are 100 times fewer architectural protein types than TADs even in Drosophila, which is known to have several architectural proteins in addition to CTCF (Zolotarev et al. 2016).
3D Organization of the Genome in the Context of Nuclear Compartmentalization
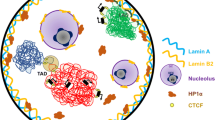
The current model of the global genome organization within the eukaryotic cell nucleus was formulated long before the development of Hi-C and other C-methods. Initially, this model was based exclusively on the results of microscopic studies. Territorial organization of interphase chromosomes and the existence of an interchromatin domain (ICD) that spans chromosomal territories are the main points of the model (Cremer and Cremer 2001, 2010, 2018; Cremer et al. 2017, 2018). The interchromatin domain is the place where various membraneless nuclear bodies such as nucleoli, splicing speckles, Cajal bodies, paraspeckles, histone locus bodies, and PML bodies are assembled (for a review, see Mao et al. (2011); Ulianov et al. (2015); Stanek and Fox (2017)). The initial version of the model placed ICD between chromosomal territories (Cremer et al. 1993; Zirbel et al. 1993). With the increase of resolution of microscopic methods, it became evident that the ICD also penetrates chromosomal territories (Cremer and Cremer 2010, 2018). Chromosome territories themselves are composed of chromatin domains and chromatin domain clusters that likely correspond to TADs and contact domains of higher order. Interestingly, internal parts of these domains appear to contain mostly inactive chromatin, whereas active genes are preferentially located at the perichromatin layer (Cremer and Cremer 2018; Cremer et al. 2018). Although individual chromosomes constitute rather separated entities within the cell nucleus, interchromosomal contacts could still be found at various reaction centers such as transcription factories, PML bodies, and splicing speckles. Such contacts were first observed using FISH to visualize various genes in combination with immunostaining to observe functional nuclear compartments (Wang et al. 2004; Sun et al. 2003; Shopland et al. 2003; Szczerbal and Bridger 2010; Moen et al. 2004) and then reanalyzed using genome-wide C-methods (Wang et al. 2016; Schoenfelder et al. 2010; Quinodoz et al. 2018).
It should be mentioned that biochemical protocols based on a proximity ligation (C-methods) allow for identification of only particularly close spatial contacts. Recruitment of several genomic regions to the same compartment is difficult, if not impossible, to detect using C-methods. Development of alternative experimental protocols based on barcoding of DNA fragments present within the same, even quite large, fixed chromatin complex (Quinodoz et al. 2018) solved the problem. Using such an experimental procedure termed “SPRITE” (split-pool recognition of interactions by tag extension) , Quinodoz et al. have identified two hubs of interchromosomal interactions that are arranged around the nucleolus (repressed hub) and nuclear speckles (active hub) (Quinodoz et al. 2018). Another genome-wide protocol that enables measuring distances between various genes and nuclear compartments is TSA-Seq (Chen et al. 2018). The procedure utilizes the tyramide amplification cascade (Wang et al. 1999) to biotinylate DNA in the vicinity of sites to which horseradish peroxidase (HRP) catalyzes the formation of tyramide-biotin free radicals recruited using an appropriate cascade of antibodies. Biotinylated DNA is then pulled down on streptavidin and sequenced. Using TSA-Seq, Belmont and coauthors confirmed clustering of active genes close to nuclear speckles. In agreement with a number of previous reports (Shevelyov and Nurminsky 2012; van Steensel and Belmont 2017), the repressed genes were found more in proximity to the nuclear lamina (Chen et al. 2018).
Taking together, the above observations argue that 3D organization of the genome and functional compartmentalization of the cell nucleus are mutually dependent. 3D organization is not simply a sum of enhancer-promoter and CTCF loops. It relies on a number of factors present in non-disturbed nuclei. Various fractionation procedures compromise this complex organization and drastically affect the results of analysis based on capturing pairwise interactions of remote DNA fragments (Gavrilov et al. 2013). Juxtaposition of remote genomic elements is not only ensured by interaction of proteins bound to these elements but rather represents a result of specific folding of a large genomic segment supported by numerous interactions outside the juxtaposed regions (Razin et al. 2013). These interactions include repositioning of various genomic segments to the vicinity of functional nuclear compartments. On the other hand, the folded genome as a whole provides a structural basis for nuclear compartmentalization (Misteli 2007; Schneider and Grosschedl 2007; Lanctot et al. 2007; Razin et al. 2013). The ICD where all these compartments are assembled is formed by exclusion from the areas occupied by chromatin. Segregation of interphase chromosomes resulting in the existence of chromosomal territories appears to be ensured by basic physical properties of charged polymers (Rosa and Everaers 2008; Mateos-Langerak et al. 2009; Bohn and Heermann 2010; Tark-Dame et al. 2011). It is less clear what supports the existence of channeled compartment within chromosomal territories. The simplest supposition is that repulsion between surfaces of TADs is of primary importance. The key point to be taken into account is that the surface of TADs should be more charged than the internal regions. Recent results of the Cremer team demonstrate that active chromatin is located at the surface of 1 Mb chromatin domains (TADs) (Cremer and Cremer 2018; Cremer et al. 2018) and thus lines the ICD channels. This finding is corroborated by the results of in silico modeling of TAD assembly (Gavrilov et al. 2016). High levels of histone acetylation typical for active chromatin (Shogren-Knaak et al. 2006; Allahverdi et al. 2011) should make the perichromatin layer more negatively charged compared to the internal part of chromatin domains/TADs. Thus, the perichromatin layer should stabilize and insulate inactive chromatin domains/TADs via generating electrostatic repulsion between them. This layer may prevent intermingling of TADs and ensure existence of intrachromosomal channels. The basic landscape for nuclear compartmentalization is thus directed only by physical laws (Rosa and Everaers 2008; Cook and Marenduzzo 2009; Dorier and Stasiak 2009; Kim and Szleifer 2014). Once established after mitosis, the territorial organization of interphase chromosomes becomes stabilized by interaction of certain chromosomal regions with the nuclear lamina (Guelen et al. 2008; Pickersgill et al. 2006) and nucleolus (Nemeth et al. 2010; van Koningsbruggen et al. 2010). Nucleoli are assembled at particular genomic loci harboring arrays of rRNA genes. The same is true for histone locus bodies. Transcription factories are likely to assemble stochastically by aggregation of closely located transcription complexes (Razin et al. 2011). Still, spatial positioning of the involved transcribed genes will predetermine their location. Typically for biological systems, this organization is highly dynamic. This dynamism applies to the both folding of interphase chromosomes and assembly of nuclear compartments. Live imaging studies have demonstrated that both chromosome territories and individual domains within chromosomal territories undergo constant movement (Marshall et al. 1997a, b, Marshall 2002; Levi et al. 2005; Pliss et al. 2013). The typical configuration of an interphase chromosome or shorter genomic segments represents an equilibrium of a number of possible configurations (Nagano et al. 2013; Stevens et al. 2017). The nature of functional nuclear compartments has been a matter of long-term discussions. The current model suggests that these compartments are liquid droplets formed by phase separation. They can fuse or separate into smaller droplets depending on external conditions. Although each type of compartments is rich in a particular set of proteins, the sets of proteins present in different compartments may overlap, and proteins present within compartments rapidly exchange with those proteins present in nucleoplasm. Furthermore, while speckles were reported to be positionally stable within hours (Misteli et al. 1997; Kruhlak et al. 2000), Cajal bodies and PML bodies appear to diffuse within the ICD as freely as an artificially created inert object of the same dimensions (Gorisch et al. 2004). An apparent order within the cell nucleus is thus likely to emerge out of a disorder due to a shaky equilibrium of different forces including a depletion attraction force (Cho and Kim 2012; Marenduzzo et al. 2006; Hancock 2004b; Rippe 2007). Apparently, the interplay between various functional processes that occur in the nucleus in any given moment directs both the chromosome folding and spatial compartmentalization of the nucleus (Rippe 2007; Kim and Szleifer 2014; Hancock 2004a; Razin et al. 2013; Golov et al. 2015; Sengupta 2018; Shah et al. 2018). Consequently, the cell nucleus should be considered as an integrated system, the properties of which emerge due to the interaction of numerous components and cannot be fully explained or predicted based on the properties of individual components. Further progress in understanding mechanisms of eukaryotic genome functioning will depend on reconsideration of all pull of existing data in terms of systems biology.
References
Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu CF, Nordenskiold L (2011) The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res 39(5):1680–1691. https://doi.org/10.1093/nar/gkq900
Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A (2013) Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339(6123):1074–1077. https://doi.org/10.1126/science.1232542
Barbieri M, Chotalia M, Fraser J, Lavitas LM, Dostie J, Pombo A, Nicodemi M (2012) Complexity of chromatin folding is captured by the strings and binders switch model. Proc Natl Acad Sci U S A 109(40):16173–16178. https://doi.org/10.1073/pnas.1204799109
Barbieri M, Fraser J, Lavitas LM, Chotalia M, Dostie J, Pombo A, Nicodemi M (2013) A polymer model explains the complexity of large-scale chromatin folding. Nucleus 4(4):267–273
Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL (2015) Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol 16:77. https://doi.org/10.1186/s13059-015-0642-0
Bintu B, Mateo LJ, Su JH, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X (2018) Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362(6413). https://doi.org/10.1126/science.aau1783
Bodnar JW (1988) A domain model for eukaryotic DNA organization: a molecular basis for cell differentiation and chromosome evolution. J Theor Biol 132(4):479–507
Bohn M, Heermann DW (2010) Diffusion-driven looping provides a consistent framework for chromatin organization. PLoS One 5(8):e12218. https://doi.org/10.1371/journal.pone.0012218
Bondarenko VA, Liu YV, Jiang YI, Studitsky VM (2003) Communication over a large distance: enhancers and insulators. Biochem Cell Biol 81(3):241–251. https://doi.org/10.1139/o03-051
Bonev B, Cavalli G (2016) Organization and function of the 3D genome. Nat Rev Genet 17(11):661–678. https://doi.org/10.1038/nrg.2016.112
Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, Peters JM (2017) Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544(7651):503–507. https://doi.org/10.1038/nature22063
Chen Y, Zhang Y, Wang Y, Zhang L, Brinkman EK, Adam SA, Goldman R, van Steensel B, Ma J, Belmont AS (2018) Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J Cell Biol 217(11):4025–4048. https://doi.org/10.1083/jcb.201807108
Cho EJ, Kim JS (2012) Crowding effects on the formation and maintenance of nuclear bodies: insights from molecular-dynamics simulations of simple spherical model particles. Biophys J 103(3):424–433. https://doi.org/10.1016/j.bpj.2012.07.007
Chung JH, Bell AC, Felsenfeld G (1997) Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci U S A 94(2):575–580
Cockerill PN, Garrard WT (1986) Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell 44:273–282
Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74. https://doi.org/10.1038/nature11247
Cook PR, Marenduzzo D (2009) Entropic organization of interphase chromosomes. J Cell Biol 186(6):825–834. https://doi.org/10.1083/jcb.200903083
Cremer T, Cremer C (2001) Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat Rev Genet 2(4):292–301
Cremer T, Cremer M (2010) Chromosome territories. Cold Spring Harb Perspect Biol 2(3):a003889. https://doi.org/10.1101/cshperspect.a003889
Cremer M, Cremer T (2018) Nuclear compartmentalization, dynamics, and function of regulatory DNA sequences. Genes Chromosomes Cancer. https://doi.org/10.1002/gcc.22714
Cremer T, Kurz A, Zirbel R, Dietzel S, Rinke B, Schrock E, Speicher MR, Mathieu U, Jauch A, Emmerich P, Scherthan H, Ried T, Cremer C, Lichter P (1993) Role of chromosome territories in the functional compartmentalization of the cell nucleus. Cold Spring Harb Symp Quant Biol 58:777–792
Cremer M, Schmid VJ, Kraus F, Markaki Y, Hellmann I, Maiser A, Leonhardt H, John S, Stamatoyannopoulos J, Cremer T (2017) Initial high-resolution microscopic mapping of active and inactive regulatory sequences proves non-random 3D arrangements in chromatin domain clusters. Epigenetics Chromatin 10(1):39. https://doi.org/10.1186/s13072-017-0146-0
Cremer T, Cremer M, Cremer C (2018) The 4D nucleome: genome compartmentalization in an evolutionary context. Biochemistry (Mosc) 83(4):313–325. https://doi.org/10.1134/S000629791804003X
Davidson IF, Goetz D, Zaczek MP, Molodtsov MI, Huis In’t Veld PJ, Weissmann F, Litos G, Cisneros DA, Ocampo-Hafalla M, Ladurner R, Uhlmann F, Vaziri A, Peters JM (2016) Rapid movement and transcriptional re-localization of human cohesin on DNA. EMBO J 35(24):2671–2685. https://doi.org/10.15252/embj.201695402
de Laat W, Grosveld F (2003) Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11:447–459
de Wit E, de Laat W (2012) A decade of 3C technologies: insights into nuclear organization. Genes Dev 26(1):11–24
de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W (2015) CTCF binding polarity determines chromatin looping. Mol Cell 60(4):676–684. https://doi.org/10.1016/j.molcel.2015.09.023
Dekker J, Mirny L (2016) The 3D genome as moderator of chromosomal communication. Cell 164(6):1110–1121. https://doi.org/10.1016/j.cell.2016.02.007
Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295(5558):1306–1311
Denker A, de Laat W (2016) The second decade of 3C technologies: detailed insights into nuclear organization. Genes Dev 30(12):1357–1382. https://doi.org/10.1101/gad.281964.116
Dileep V, Ay F, Sima J, Vera DL, Noble WS, Gilbert DM (2015a) Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res 25(8):1104–1113. https://doi.org/10.1101/gr.183699.114
Dileep V, Rivera-Mulia JC, Sima J, Gilbert DM (2015b) Large-scale chromatin structure-function relationships during the cell cycle and development: insights from replication timing. Cold Spring Harb Symp Quant Biol. https://doi.org/10.1101/sqb.2015.80.027284
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485(7398):376–380. https://doi.org/10.1038/nature11082
Dixon JR, Gorkin DU, Ren B (2016) Chromatin domains: the unit of chromosome organization. Mol Cell 62(5):668–680. https://doi.org/10.1016/j.molcel.2016.05.018
Dorier J, Stasiak A (2009) Topological origins of chromosomal territories. Nucleic Acids Res 37(19):6316–6322. https://doi.org/10.1093/nar/gkp702
Eagen KP (2018) Principles of chromosome architecture revealed by Hi-C. Trends Biochem Sci 43(6):469–478. https://doi.org/10.1016/j.tibs.2018.03.006
Eser U, Chandler-Brown D, Ay F, Straight AF, Duan Z, Noble WS, Skotheim JM (2017) Form and function of topologically associating genomic domains in budding yeast. Proc Natl Acad Sci U S A 114(15):E3061–E3070. https://doi.org/10.1073/pnas.1612256114
Flyamer IM, Gassler J, Imakaev M, Brandao HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, Tachibana-Konwalski K (2017) Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544(7648):110–114. https://doi.org/10.1038/nature21711
Forrester WC, Takegawa S, Papayannopoulou T, Stamatoyannopoulos G, Groudine M (1987) Evidence for a locus activating region: the formation of developmentally stable hypersensitive sites in globin expressing hybrids. Nucleic Acids Res 15:10159–10175
Forrester WC, Epner E, Driscoll MC, Enver T, Brice M, Papayannopoulou T, Groudine M (1990) A deletion of the human b-globin locus activation region causes a major alteration in chromatin structure and replication across the entire b-globin locus. Gene Dev 4:1637–1649
Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schopflin R, Kraft K, Kempfer R, Jerkovic I, Chan WL, Spielmann M, Timmermann B, Wittler L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538(7624):265–269. https://doi.org/10.1038/nature19800
Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, Barbieri M, Moore BL, Kraemer DC, Aitken S, Xie SQ, Morris KJ, Itoh M, Kawaji H, Jaeger I, Hayashizaki Y, Carninci P, Forrest AR, Consortium F, Semple CA, Dostie J, Pombo A, Nicodemi M (2015) Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol 11(12):852. https://doi.org/10.15252/msb.20156492
Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA (2016) Formation of chromosomal domains by loop extrusion. Cell Rep 15(9):2038–2049. https://doi.org/10.1016/j.celrep.2016.04.085
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, Chew EG, Huang PY, Welboren WJ, Han Y, Ooi HS, Ariyaratne PN, Vega VB, Luo Y, Tan PY, Choy PY, Wansa KD, Zhao B, Lim KS, Leow SC, Yow JS, Joseph R, Li H, Desai KV, Thomsen JS, Lee YK, Karuturi RK, Herve T, Bourque G, Stunnenberg HG, Ruan X, Cacheux-Rataboul V, Sung WK, Liu ET, Wei CL, Cheung E, Ruan Y (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462(7269):58–64
Fussner E, Strauss M, Djuric U, Li R, Ahmed K, Hart M, Ellis J, Bazett-Jones DP (2012) Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep 13(11):992–996. https://doi.org/10.1038/embor.2012.139
Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, Dekker C (2018) Real-time imaging of DNA loop extrusion by condensin. Science 360(6384):102–105. https://doi.org/10.1126/science.aar7831
Gavrilov AA, Razin SV (2008) Spatial configuration of the chicken {alpha}-globin gene domain: immature and active chromatin hubs. Nucleic Acids Res 36:4629–4640
Gavrilov AA, Gushchanskaya ES, Strelkova O, Zhironkina O, Kireev II, Iarovaia OV, Razin SV (2013) Disclosure of a structural milieu for the proximity ligation reveals the elusive nature of an active chromatin hub. Nucleic Acids Res 41:3563–3575. https://doi.org/10.1093/nar/gkt067
Gavrilov AA, Shevelyov YY, Ulianov SV, Khrameeva EE, Kos P, Chertovich A, Razin SV (2016) Unraveling the mechanisms of chromatin fibril packaging. Nucleus 7(3):319–324. https://doi.org/10.1080/19491034.2016.1190896
Getzenberg RH, Pienta KJ, Ward WS, Coffey DS (1991) Nuclear structure and the three-dimensional organization of DNA. J Cell Biochem 47(4):289–299. https://doi.org/10.1002/jcb.240470402
Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EE (2014) Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512(7512):96–100. https://doi.org/10.1038/nature13417
Gibcus JH, Dekker J (2013) The hierarchy of the 3D genome. Mol Cell 49(5):773–782. https://doi.org/10.1016/j.molcel.2013.02.011
Goldman MA (1988) The chromatin domain as a unit of gene regulation. BioEssays 9:50–55
Golov AK, Gavrilov AA, Razin SV (2015) The role of crowding forces in juxtaposing beta-globin gene domain remote regulatory elements in mouse erythroid cells. PLoS One 10(10):e0139855. https://doi.org/10.1371/journal.pone.0139855
Gong Y, Lazaris C, Sakellaropoulos T, Lozano A, Kambadur P, Ntziachristos P, Aifantis I, Tsirigos A (2018) Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries. Nat Commun 9(1):542. https://doi.org/10.1038/s41467-018-03017-1
Gorisch SM, Wachsmuth M, Ittrich C, Bacher CP, Rippe K, Lichter P (2004) Nuclear body movement is determined by chromatin accessibility and dynamics. Proc Natl Acad Sci U S A 101(36):13221–13226. https://doi.org/10.1073/pnas.0402958101
Grosveld F, van Assandelt GB, Greaves DR, Kollias B (1987) Position-independent, high-level expression of the human b-globin gene in transgenic mice. Cell 51:975–985
Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453(7197):948–951. https://doi.org/10.1038/nature06947
Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, Brummelkamp TR, de Wit E, Rowland BD (2017) The cohesin release factor WAPL restricts chromatin loop extension. Cell 169 (4):693-707. e614. doi:https://doi.org/10.1016/j.cell.2017.04.013
Hancock R (2004a) Internal organisation of the nucleus: assembly of compartments by macromolecular crowding and the nuclear matrix model. Biol Cell 96(8):595–601
Hancock R (2004b) A role for macromolecular crowding effects in the assembly and function of compartments in the nucleus. J Struct Biol 146(3):281–290. https://doi.org/10.1016/j.jsb.2003.12.008
Hancock R (2008) Self-association of polynucleosome chains by macromolecular crowding. Eur Biophys J: EBJ 37(6):1059–1064. https://doi.org/10.1007/s00249-008-0276-1
Hansen AS, Pustova I, Cattoglio C, Tjian R, Darzacq X (2017) CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6. https://doi.org/10.7554/eLife.25776
Hirano T (2005) SMC proteins and chromosome mechanics: from bacteria to humans. Philos Trans R Soc Lond B Biol Sci 360(1455):507–514. https://doi.org/10.1098/rstb.2004.1606
Holwerda S, de Laat W (2012) Chromatin loops, gene positioning, and gene expression. Front Genet 3:217. https://doi.org/10.3389/fgene.2012.00217
Hou C, Li L, Qin ZS, Corces VG (2012) Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell 48(3):471–484. https://doi.org/10.1016/j.molcel.2012.08.031
Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ (2015) Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162(1):108–119. https://doi.org/10.1016/j.cell.2015.05.048
Ibn-Salem J, Kohler S, Love MI, Chung HR, Huang N, Hurles ME, Haendel M, Washington NL, Smedley D, Mungall CJ, Lewis SE, Ott CE, Bauer S, Schofield PN, Mundlos S, Spielmann M, Robinson PN (2014) Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol 15(9):423. https://doi.org/10.1186/s13059-014-0423-1
Ibn-Salem J, Muro EM, Andrade-Navarro MA (2017) Co-regulation of paralog genes in the three-dimensional chromatin architecture. Nucleic Acids Res 45(1):81–91. https://doi.org/10.1093/nar/gkw813
Jantzen K, Friton HP, Igo-Kimenes T (1986) The DNase I sensitive domain of the chicken lyzozyme gene spans 24 kb. Nucleic Acids Res 14:6085–6099
Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B (2013) A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503(7475):290–294. https://doi.org/10.1038/nature12644
Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, Hansen JC (2013) The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc, Interface/ R Soc 10(82):20121022. https://doi.org/10.1098/rsif.2012.1022
Kanke M, Tahara E, Huis In’t Veld PJ, Nishiyama T (2016) Cohesin acetylation and Wapl-Pds5 oppositely regulate translocation of cohesin along DNA. EMBO J 35(24):2686–2698. https://doi.org/10.15252/embj.201695756
Kellum R, Schedl P (1991) A position-effect assay for boundaries of higher-order chromosomal domains. Cell 64:941–950
Kellum R, Schedl P (1992) A group of scs elements function as boundaries in enhancer-blocking assay. Mol Cell Biol 12:2424–2431
Kim JS, Szleifer I (2014) Crowding-induced formation and structural alteration of nuclear compartments: insights from computer simulations. Int Rev Cell Mol Biol 307:73–108. https://doi.org/10.1016/B978-0-12-800046-5.00004-7
Krefting J, Andrade-Navarro MA, Ibn-Salem J (2018) Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol 16(1):87. https://doi.org/10.1186/s12915-018-0556-x
Krijger PH, de Laat W (2016) Regulation of disease-associated gene expression in the 3D genome. Nat Rev Mol Cell Biol 17(12):771–782. https://doi.org/10.1038/nrm.2016.138
Kruhlak MJ, Lever MA, Fischle W, Verdin E, Bazett-Jones DP, Hendzel MJ (2000) Reduced mobility of the alternate splicing factor (ASF) through the nucleoplasm and steady state speckle compartments. J Cell Biol 150(1):41–51
Krumm A, Duan Z (2018) Understanding the 3D genome: emerging impacts on human disease. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2018.07.004
Lanctot C, Cheutin T, Cremer M, Cavalli G, Cremer T (2007) Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet 8(2):104–115
Lawson GM, Knoll BJ, March CJ, Woo SLC, Tsai M-J, O’Malley BW (1982) Definition of 5′ and 3′ structural boundaries of the chromatin domain containing the ovalbumin multigene family. J Biol Chem 257:1501–1507
Lazar NH, Nevonen KA, O’Connell B, McCann C, O’Neill RJ, Green RE, Meyer TJ, Okhovat M, Carbone L (2018) Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res 28(7):983–997. https://doi.org/10.1101/gr.233874.117
Le TB, Imakaev MV, Mirny LA, Laub MT (2013) High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342(6159):731–734. https://doi.org/10.1126/science.1242059
Levi V, Ruan Q, Plutz M, Belmont AS, Gratton E (2005) Chromatin dynamics in interphase cells revealed by tracking in a two-photon excitation microscope. Biophys J 89(6):4275–4285. https://doi.org/10.1529/biophysj.105.066670
Li Q, Zhou B, Powers P, Enver T, Stamatoyannopoulos G (1990) b-globin locus activations regions: conservation of organization, structure and function. Proc Natl Acad Sci USA 87:8207–8211
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326(5950):289–293
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161(5):1012–1025. https://doi.org/10.1016/j.cell.2015.04.004
Lupianez DG, Spielmann M, Mundlos S (2016) Breaking TADs: how alterations of chromatin domains result in disease. Trends Genet 32(4):225–237. https://doi.org/10.1016/j.tig.2016.01.003
Luzhin AV, Flyamer IM, Khrameeva EE, Ulianov SV, Razin SV, Gavrilov AA (2019) Quantitative differences in TAD border strength underly the TAD hierarchy in Drosophila chromosomes. J Cell Biochem 120(3):4494–4503. https://doi.org/10.1002/jcb.27737
Maeshima K, Imai R, Hikima T, Joti Y (2014a) Chromatin structure revealed by X-ray scattering analysis and computational modeling. Methods 70(2-3):154–161. https://doi.org/10.1016/j.ymeth.2014.08.008
Maeshima K, Imai R, Tamura S, Nozaki T (2014b) Chromatin as dynamic 10-nm fibers. Chromosoma 123(3):225–237. https://doi.org/10.1007/s00412-014-0460-2
Maeshima K, Rogge R, Tamura S, Joti Y, Hikima T, Szerlong H, Krause C, Herman J, Seidel E, DeLuca J, Ishikawa T, Hansen JC (2016) Nucleosomal arrays self-assemble into supramolecular globular structures lacking 30-nm fibers. EMBO J. https://doi.org/10.15252/embj.201592660
Mao YS, Zhang B, Spector DL (2011) Biogenesis and function of nuclear bodies. Trends Genet 27(8):295–306
Marenduzzo D, Finan K, Cook PR (2006) The depletion attraction: an underappreciated force driving cellular organization. J Cell Biol 175(5):681–686. https://doi.org/10.1083/jcb.200609066
Markaki Y, Gunkel M, Schermelleh L, Beichmanis S, Neumann J, Heidemann M, Leonhardt H, Eick D, Cremer C, Cremer T (2010) Functional nuclear organization of transcription and DNA replication: a topographical marriage between chromatin domains and the interchromatin compartment. Cold Spring Harb Symp Quant Biol 75:475–492. https://doi.org/10.1101/sqb.2010.75.042
Markaki Y, Smeets D, Fiedler S, Schmid VJ, Schermelleh L, Cremer T, Cremer M (2012) The potential of 3D-FISH and super-resolution structured illumination microscopy for studies of 3D nuclear architecture: 3D structured illumination microscopy of defined chromosomal structures visualized by 3D (immuno)-FISH opens new perspectives for studies of nuclear architecture. BioEssays 34(5):412–426. https://doi.org/10.1002/bies.201100176
Marshall WF (2002) Order and disorder in the nucleus. Curr Biol 12(5):R185–R192
Marshall WF, Fung JC, Sedat JW (1997a) Deconstructing the nucleus: global architecture from local interactions. Curr Opin Genet Dev 7(2):259–263
Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, Sedat JW (1997b) Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7(12):930–939
Mateos-Langerak J, Bohn M, de Leeuw W, Giromus O, Manders EM, Verschure PJ, Indemans MH, Gierman HJ, Heermann DW, van Driel R, Goetze S (2009) Spatially confined folding of chromatin in the interphase nucleus. Proc Natl Acad Sci U S A 106(10):3812–3817. https://doi.org/10.1073/pnas.0809501106
Misteli T (2007) Beyond the sequence: cellular organization of genome function. Cell 128(4):787–800
Misteli T, Caceres JF, Spector DL (1997) The dynamics of a pre-mRNA splicing factor in living cells. Nature 387(6632):523–527. https://doi.org/10.1038/387523a0
Moen PT Jr, Johnson CV, Byron M, Shopland LS, de la Serna IL, Imbalzano AN, Lawrence JB (2004) Repositioning of muscle-specific genes relative to the periphery of SC-35 domains during skeletal myogenesis. Mol Biol Cell 15(1):197–206. https://doi.org/10.1091/mbc.E03-06-0388
Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, Splinter E, de Laat W, Spitz F, Duboule D (2011) A regulatory archipelago controls Hox genes transcription in digits. Cell 147(5):1132–1145. https://doi.org/10.1016/j.cell.2011.10.023
Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P (2013) Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502(7469):59–64. https://doi.org/10.1038/nature12593
Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D (2015) CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347(6225):1017–1021. https://doi.org/10.1126/science.1262088
Narendra V, Bulajic M, Dekker J, Mazzoni EO, Reinberg D (2016) CTCF-mediated topological boundaries during development foster appropriate gene regulation. Genes Dev 30(24):2657–2662. https://doi.org/10.1101/gad.288324.116
Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, Dekker J (2013) Organization of the mitotic chromosome. Science 342(6161):948–953. https://doi.org/10.1126/science.1236083
Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, Solovei I, Cremer T, Dopazo J, Langst G (2010) Initial genomics of the human nucleolus. PLoS Genet 6(3):e1000889. https://doi.org/10.1371/journal.pgen.1000889
Nikolaou C (2017) Invisible cities: segregated domains in the yeast genome with distinct structural and functional attributes. Curr Genet. https://doi.org/10.1007/s00294-017-0731-6
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485(7398):381–385. https://doi.org/10.1038/nature11049
Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG (2017) Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169(5):930–944. e922. https://doi.org/10.1016/j.cell.2017.05.004
Nuebler J, Fudenberg G, Imakaev M, Abdennur N, Mirny LA (2018) Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc Natl Acad Sci U S A 115(29):E6697–E6706. https://doi.org/10.1073/pnas.1717730115
Pepenella S, Murphy KJ, Hayes JJ (2014) Intra- and inter-nucleosome interactions of the core histone tail domains in higher-order chromatin structure. Chromosoma 123(1-2):3–13. https://doi.org/10.1007/s00412-013-0435-8
Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG (2013) Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153(6):1281–1295. https://doi.org/10.1016/j.cell.2013.04.053
Philonenko ES, Klochkov DB, Borunova VV, Gavrilov AA, Razin SV, Iarovaia OV (2009) TMEM8 – a non-globin gene entrapped in the globin web. Nucleic Acids Res 37(22):7394–7406
Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B (2006) Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet 38(9):1005–1014. https://doi.org/10.1038/ng1852
Pliss A, Malyavantham KS, Bhattacharya S, Berezney R (2013) Chromatin dynamics in living cells: identification of oscillatory motion. J Cell Physiol 228(3):609–616. https://doi.org/10.1002/jcp.24169
Pombo A, Nicodemi M (2014) Physical mechanisms behind the large scale features of chromatin organization. Transcription 5(2):e28447. https://doi.org/10.4161/trns.28447
Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, Thurman RE, Cheng Y, Gulsoy G, Dennis JH, Snyder MP, Stamatoyannopoulos JA, Taylor J, Hardison RC, Kahveci T, Ren B, Gilbert DM (2014) Topologically associating domains are stable units of replication-timing regulation. Nature 515(7527):402–405. https://doi.org/10.1038/nature13986
Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, Trinh V, Aznauryan E, Russell P, Cheng C, Jovanovic M, Chow A, Cai L, McDonel P, Garber M, Guttman M (2018) Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174(3):744–757. e724. https://doi.org/10.1016/j.cell.2018.05.024
Rada-Iglesias A, Grosveld FG, Papantonis A (2018) Forces driving the three-dimensional folding of eukaryotic genomes. Mol Syst Biol 14(6):e8214. https://doi.org/10.15252/msb.20188214
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7):1665–1680. https://doi.org/10.1016/j.cell.2014.11.021
Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, Huang X, Shamim MS, Shin J, Turner D, Ye Z, Omer AD, Robinson JT, Schlick T, Bernstein BE, Casellas R, Lander ES, Aiden EL (2017) Cohesin loss eliminates all loop domains. Cell 171(2):305–320. e324. https://doi.org/10.1016/j.cell.2017.09.026
Razin SV, Vassetzky YS (2017) 3D genomics imposes evolution of the domain model of eukaryotic genome organization. Chromosoma 126:59–69. https://doi.org/10.1007/s00412-016-0604-7
Razin SV, Gavrilov AA, Pichugin A, Lipinski M, Iarovaia OV, Vassetzky YS (2011) Transcription factories in the context of the nuclear and genome organization. Nucleic Acids Res 39(21):9085–9092. https://doi.org/10.1093/nar/gkr683
Razin SV, Gavrilov AA, Ioudinkova ES, Iarovaia OV (2013) Communication of genome regulatory elements in a folded chromosome. FEBS Lett 587(13):1840–1847. https://doi.org/10.1016/j.febslet.2013.04.027
Rippe K (2007) Dynamic organization of the cell nucleus. Curr Opin Genet Dev 17(5):373–380. https://doi.org/10.1016/j.gde.2007.08.007
Rosa A, Everaers R (2008) Structure and dynamics of interphase chromosomes. PLoS Comput Biol 4(8):e1000153. https://doi.org/10.1371/journal.pcbi.1000153
Rowley MJ, Corces VG (2018) Organizational principles of 3D genome architecture. Nat Rev Genet 19(12):789–800. https://doi.org/10.1038/s41576-018-0060-8
Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, Corces VG (2017) Evolutionarily conserved principles predict 3D chromatin organization. Mol Cell. https://doi.org/10.1016/j.molcel.2017.07.022
Ruf S, Symmons O, Uslu VV, Dolle D, Hot C, Ettwiller L, Spitz F (2011) Large-scale analysis of the regulatory architecture of the mouse genome with a transposon-associated sensor. Nat Genet 43(4):379–386. https://doi.org/10.1038/ng.790
Sahlen P, Abdullayev I, Ramskold D, Matskova L, Rilakovic N, Lotstedt B, Albert TJ, Lundeberg J, Sandberg R (2015) Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol 16:156. https://doi.org/10.1186/s13059-015-0727-9
Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, Geeting KP, Gnirke A, Melnikov A, McKenna D, Stamenova EK, Lander ES, Aiden EL (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112(47):E6456–E6465. https://doi.org/10.1073/pnas.1518552112
Schneider R, Grosschedl R (2007) Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 21(23):3027–3043. https://doi.org/10.1101/gad.1604607
Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, Eskiw CH, Luo Y, Wei CL, Ruan Y, Bieker JJ, Fraser P (2010) Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat Genet 42(1):53–61
Sengupta K (2018) Genome 3D-architecture: its plasticity in relation to function. J Biosci 43(2):417–419
Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G (2012) Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148(3):458–472. https://doi.org/10.1016/j.cell.2012.01.010
Shah FR, Bhat YA, Wani AH (2018) Subnuclear distribution of proteins: Links with genome architecture. Nucleus 9(1):42–55. https://doi.org/10.1080/19491034.2017.1361578
Shevelyov YY, Nurminsky DI (2012) The nuclear lamina as a gene-silencing hub. Curr Issues Mol Biol 14(1):27–38
Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL (2006) Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311(5762):844–847. https://doi.org/10.1126/science.1124000
Shopland LS, Johnson CV, Byron M, McNeil J, Lawrence JB (2003) Clustering of multiple specific genes and gene-rich R-bands around SC-35 domains: evidence for local euchromatic neighborhoods. J Cell Biol 162(6):981–990. https://doi.org/10.1083/jcb.200303131
Sinha D, Shogren-Knaak MA (2010) Role of direct interactions between the histone H4 Tail and the H2A core in long range nucleosome contacts. J Biol Chem 285(22):16572–16581. https://doi.org/10.1074/jbc.M109.091298
Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, Sterr M, Fiedler S, Demmerle J, Popken J, Leonhardt H, Brockdorff N, Cremer T, Schermelleh L, Cremer M (2014) Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenet Chromatin 7:8. https://doi.org/10.1186/1756-8935-7-8
Sofueva S, Yaffe E, Chan WC, Georgopoulou D, Vietri Rudan M, Mira-Bontenbal H, Pollard SM, Schroth GP, Tanay A, Hadjur S (2013) Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J 32(24):3119–3129. https://doi.org/10.1038/emboj.2013.237
Spitz F, Gonzalez F, Duboule D (2003) A global control region defines a chromosomal regulatory landscape containing the HoxD cluster. Cell 113(3):405–417
Stanek D, Fox AH (2017) Nuclear bodies: news insights into structure and function. Curr Opin Cell Biol 46:94–101. https://doi.org/10.1016/j.ceb.2017.05.001
Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, Cramard J, Faure AJ, Ralser M, Blanco E, Morey L, Sanso M, Palayret MGS, Lehner B, Di Croce L, Wutz A, Hendrich B, Klenerman D, Laue ED (2017) 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544(7648):59–64. https://doi.org/10.1038/nature21429
Stigler J, Camdere GO, Koshland DE, Greene EC (2016) Single-molecule imaging reveals a collapsed conformational state for DNA-bound cohesin. Cell Rep 15(5):988–998. https://doi.org/10.1016/j.celrep.2016.04.003
Sun Y, Durrin LK, Krontiris TG (2003) Specific interaction of PML bodies with the TP53 locus in Jurkat interphase nuclei. Genomics 82(2):250–252
Sun F, Chronis C, Kronenberg M, Chen XF, Su T, Lay FD, Plath K, Kurdistani SK, Carey MF (2019) Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol Cell 73(2):250–263. e255. https://doi.org/10.1016/j.molcel.2018.10.039
Symmons O, Uslu VV, Tsujimura T, Ruf S, Nassari S, Schwarzer W, Ettwiller L, Spitz F (2014) Functional and topological characteristics of mammalian regulatory domains. Genome Res 24(3):390–400. https://doi.org/10.1101/gr.163519.113
Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, Spitz F (2016) The Shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Dev Cell 39(5):529–543. https://doi.org/10.1016/j.devcel.2016.10.015
Szabo Q, Jost D, Chang JM, Cattoni DI, Papadopoulos GL, Bonev B, Sexton T, Gurgo J, Jacquier C, Nollmann M, Bantignies F, Cavalli G (2018) TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci Adv 4(2):eaar8082. https://doi.org/10.1126/sciadv.aar8082
Szczerbal I, Bridger JM (2010) Association of adipogenic genes with SC-35 domains during porcine adipogenesis. Chromosome Res 18(8):887–895. https://doi.org/10.1007/s10577-010-9176-1
Tark-Dame M, van Driel R, Heermann DW (2011) Chromatin folding--from biology to polymer models and back. J Cell Sci 124(Pt 6):839–845. https://doi.org/10.1242/jcs.077628
Tiana G, Amitai A, Pollex T, Piolot T, Holcman D, Heard E, Giorgetti L (2016) Structural fluctuations of the chromatin fiber within topologically associating domains. Biophys J 110(6):1234–1245. https://doi.org/10.1016/j.bpj.2016.02.003
Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W (2002) Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 10(6):1453–1465. https://doi.org/10.1016/S1097-2765(02)00781-5
Udvardy A, Schedl P, Sander M, Hsieh T-S (1986) J Mol Biol 191:231–246
Ulianov SV, Gavrilov AA, Razin SV (2015) Nuclear compartments, genome folding, and enhancer-promoter communication. Int Rev Cell Mol Biol 315:183–244. https://doi.org/10.1016/bs.ircmb.2014.11.004
Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A, Gelfand MS, Shevelyov YY, Razin SV (2016) Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26(1):70–84. https://doi.org/10.1101/gr.196006.115
Ulianov SV, Galitsyna AA, Flyamer IM, Golov AK, Khrameeva EE, Imakaev MV, Abdennur NA, Gelfand MS, Gavrilov AA, Razin SV (2017) Activation of the alpha-globin gene expression correlates with dramatic upregulation of nearby non-globin genes and changes in local and large-scale chromatin spatial structure. Epigenet Chromatin 10(1):35. https://doi.org/10.1186/s13072-017-0142-4
Valton AL, Dekker J (2016) TAD disruption as oncogenic driver. Curr Opin Genet Dev 36:34–40. https://doi.org/10.1016/j.gde.2016.03.008
van de Werken HJ, Landan G, Holwerda SJ, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Oz Y, Bouwman BA, Verstegen MJ, de Wit E, Tanay A, de Laat W (2012) Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods 9(10):969–972. https://doi.org/10.1038/nmeth.2173
van Koningsbruggen S, Gierlinski M, Schofield P, Martin D, Barton GJ, Ariyurek Y, den Dunnen JT, Lamond AI (2010) High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol Biol Cell 21(21):3735–3748. https://doi.org/10.1091/mbc.E10-06-0508
van Steensel B, Belmont AS (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169(5):780–791. https://doi.org/10.1016/j.cell.2017.04.022
Vasquez PA, Hult C, Adalsteinsson D, Lawrimore J, Forest MG, Bloom K (2016) Entropy gives rise to topologically associating domains. Nucleic Acids Res 44(12):5540–5549. https://doi.org/10.1093/nar/gkw510
Vernimmen D, Bickmore WA (2015) The hierarchy of transcriptional activation: from enhancer to promoter. Trends Genet 31(12):696–708. https://doi.org/10.1016/j.tig.2015.10.004
Vernimmen D, De Gobbi M, Sloane-Stanley JA, Wood WG, Higgs DR (2007) Long-range chromosomal interactions regulate the timing of the transition between poised and active gene expression. EMBO J 26(8):2041–2051. https://doi.org/10.1038/sj.emboj.7601654
Vernimmen D, Marques-Kranc F, Sharpe JA, Sloane-Stanley JA, Wood WG, Wallace HA, Smith AJ, Higgs DR (2009) Chromosome looping at the human alpha-globin locus is mediated via the major upstream regulatory element (HS -40). Blood 114(19):4253–4260. https://doi.org/10.1182/blood-2009-03-213439
Vian L, Pekowska A, Rao SSP, Kieffer-Kwon KR, Jung S, Baranello L, Huang SC, El Khattabi L, Dose M, Pruett N, Sanborn AL, Canela A, Maman Y, Oksanen A, Resch W, Li X, Lee B, Kovalchuk AL, Tang Z, Nelson S, Di Pierro M, Cheng RR, Machol I, St Hilaire BG, Durand NC, Shamim MS, Stamenova EK, Onuchic JN, Ruan Y, Nussenzweig A, Levens D, Aiden EL, Casellas R (2018) The energetics and physiological impact of cohesin extrusion. Cell 173(5):1165–1178. e1120. https://doi.org/10.1016/j.cell.2018.03.072
Vicente-Garcia C, Villarejo-Balcells B, Irastorza-Azcarate I, Naranjo S, Acemel RD, Tena JJ, Rigby PWJ, Devos DP, Gomez-Skarmeta JL, Carvajal JJ (2017) Regulatory landscape fusion in rhabdomyosarcoma through interactions between the PAX3 promoter and FOXO1 regulatory elements. Genome Biol 18(1):106. https://doi.org/10.1186/s13059-017-1225-z
Vietri Rudan M, Hadjur S (2015) Genetic tailors: CTCF and cohesin shape the genome during evolution. Trends Genet 31(11):651–660. https://doi.org/10.1016/j.tig.2015.09.004
Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, Hadjur S (2015) Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 10(8):1297–1309. https://doi.org/10.1016/j.celrep.2015.02.004
Wang G, Achim CL, Hamilton RL, Wiley CA, Soontornniyomkij V (1999) Tyramide signal amplification method in multiple-label immunofluorescence confocal microscopy. Methods 18(4):459–464. https://doi.org/10.1006/meth.1999.0813
Wang J, Shiels C, Sasieni P, Wu PJ, Islam SA, Freemont PS, Sheer D (2004) Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J Cell Biol 164(4):515–526. https://doi.org/10.1083/jcb.200305142
Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D (2015) Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 25(2):246–256. https://doi.org/10.1101/gr.170332.113
Wang Q, Sawyer IA, Sung MH, Sturgill D, Shevtsov SP, Pegoraro G, Hakim O, Baek S, Hager GL, Dundr M (2016) Cajal bodies are linked to genome conformation. Nat Commun 7:10966. https://doi.org/10.1038/ncomms10966
Weinreb C, Raphael BJ (2016) Identification of hierarchical chromatin domains. Bioinformatics 32(11):1601–1609. https://doi.org/10.1093/bioinformatics/btv485
Weintraub H, Groudine M (1976) Chromosomal subunits in active genes have an altered conformation. Science 73:848–856
Weintraub H, Larsen A, Groudine M (1981) Alpha-Globin-gene switching during the development of chicken embryos: expression and chromosome structure. Cell 24(2):333–344
West AG, Fraser P (2005) Remote control of gene transcription. Hum Mol Genet 14 Spec No 1:R101–R111
Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, Walther N, Koch B, Kueblbeck M, Ellenberg J, Zuber J, Fraser P, Peters JM (2017) Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J 36(24):3573–3599. https://doi.org/10.15252/embj.201798004
Zhan Y, Mariani L, Barozzi I, Schulz EG, Bluthgen N, Stadler M, Tiana G, Giorgetti L (2017) Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res 27(3):479–490. https://doi.org/10.1101/gr.212803.116
Zirbel RM, Mathieu UR, Kurz A, Cremer T, Lichter P (1993) Evidence for a nuclear compartment of transcription and splicing located at chromosome domain boundaries. Chromosome Res 1(2):93–106
Zolotarev N, Fedotova A, Kyrchanova O, Bonchuk A, Penin AA, Lando AS, Eliseeva IA, Kulakovskiy IV, Maksimenko O, Georgiev P (2016) Architectural proteins Pita, Zw5, and ZIPIC contain homodimerization domain and support specific long-range interactions in Drosophila. Nucleic Acids Res 44(15):7228–7241. https://doi.org/10.1093/nar/gkw371
Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, Brouwer RW, van de Corput MP, van de Werken HJ, Knoch TA, van IWF, Grosveld FG, Ren B, Wendt KS (2014) Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A 111(3):996–1001. https://doi.org/10.1073/pnas.1317788111
Zuniga A, Michos O, Spitz F, Haramis AP, Panman L, Galli A, Vintersten K, Klasen C, Mansfield W, Kuc S, Duboule D, Dono R, Zeller R (2004) Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes Dev 18(13):1553–1564. https://doi.org/10.1101/gad.299904
Acknowledgments
This work was supported by the Russian Science Foundation (grant 19-14-00016) and by the Interdisciplinary Scientific and Educational School of Moscow University «Molecular Technologies of the Living Systems and Synthetic Biology».
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Razin, S.V., Gavrilov, A.A., Ulianov, S.V. (2020). Eukaryotic Genome in Three Dimensions. In: Iourov, I., Vorsanova, S., Yurov, Y. (eds) Human Interphase Chromosomes. Springer, Cham. https://doi.org/10.1007/978-3-030-62532-0_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-62532-0_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-62531-3
Online ISBN: 978-3-030-62532-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)