
Abstract
High-throughput sequencing (HTS) technologies have enabled rapid sequencing of genomes and large-scale genome analytics with massive data sets. Traditionally, genetic variation analyses have been based on the human reference genome assembled from a relatively small human population. However, genetic variation could be discovered more comprehensively by using a collection of genomes i.e., pan-genome as a reference. The pan-genomic references can be assembled from larger populations or a specific population under study. Moreover, exploiting the pan-genomic references with current bioinformatics tools requires efficient compression and indexing methods. To be able to leverage the accumulating genomic data, the power of distributed and parallel computing has to be harnessed for the new genome analysis pipelines. We propose a scalable distributed pipeline, PanGenSpark, for compressing and indexing pan-genomes and assembling a reference genome from the pan-genomic index. We experimentally show the scalability of the PanGenSpark with human pan-genomes in a distributed Spark cluster comprising 448 cores distributed to 26 computing nodes. Assembling a consensus genome of a pan-genome including 50 human individuals was performed in 215 min and with 500 human individuals in 1468 min. The index of 1.41 TB pan-genome was compressed into a size of 164.5 GB in our experiments.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
High-throughput sequencing (HTS) technologies have enabled rapid DNA sequencing of multiple samples collected from any organism and environment including human tissues, bacteria, fungi, plants, soil, water, and air. Next-generation sequencing (NGS) technology provides relatively cheap and rapid whole-genome sequencing enabling large-scale and profound genome analytics. As a result of advanced HTS technology, the sequencing data volumes are growing quickly and the number of assembled genomes is increasing rapidly as well.
Computational pan-genomics [1] is one of the efforts to exploit the huge amount of information from multiple genomes in comparative analysis bringing new opportunities for population genetics. Marcshall et al. pointed out that a pan-genome can present: (i) the genome of a single selected individual, (ii) a consensus drawn from an entire population, (iii) a functional genome (without disabling mutations in any gene), or (iv) a maximal genome that captures all sequences ever detected and generalized where the pan-genome can refer to any collection of genomic sequences that are analyzed jointly or is used as a reference [1].
The first human reference genome draft was published in 2004 by the Human Genome Project and it has been complemented from time to time [4]. Nowadays it is used as a comparative reference in the majority of scientific contributions in human genetic studies. Pan-genomes can represent more diverse populations without disabling any population- or individual-specific genomic regions, and thus, improve the genetic variation analysis by considering the genetic recombination and emphasizing the diversity of individuals [3, 8, 17,18,19,20]. Sherman and Salzberg [2] underline the importance and advantages of using pan-genomes in genetic variation studies instead of just a single reference genome. Yet, constructing a reusable reference genome from a human pan-genome requires assembling, indexing, and aligning of multiple genomes in a population, which is computationally demanding and time-consuming. Computational limits are hit in many of the processing steps: construction of pan-genome from multiple genomes, compressing and indexing the pan-genome, alignment of donor sequences to pan-genomic index, and finally assembling a pan-genomic consensus reference.
Our goal is to enable the assembling of compressed and reusable pan-genomic reference indexes that can be used directly for sequence alignment in genetic variation analyses efficiently. We focus on the scalable assembling of a reference genome from a human pan-genome as well as indexing and compressing large pan-genomes to reduce the computation time and to improve space-efficiency. PanGenSpark is a continuation for PanVC, a sequential pan-genomic variation calling pipeline with hybrid indexing presented by Valenzuela et al. in [16, 30]. Here, we proceed with parallelizing the most compute-intensive phases in PanVC such as compressing and indexing of large pan-genomes and assembling the consensus genome from a pan-genome with distributed methods.
Analyzing a huge amount of genomic data is computationally intensive, and extremely so in the pan-genomic context. We propose a scalable distributed pan-genome analysis pipeline, PanGenSpark, to assemble a new consensus reference genome from a pan-genome for downstream analysis such as sequence alignment and variant calling. The pipeline implements distributed compressed indexing of the pan-genome, the read alignment, the consensus genome assembly method, and the support for legacy variant calling tools. The prototype pan-genome analysis pipeline is designed for the Apache Spark [22] framework. We demonstrate the pipeline with 500 human genomes in the Apache Spark cluster. The source code of PanGenSpark is publicly available in GitHubFootnote 1.
1.1 Related Work
The pan-genome as a concept was first presented by Tettelin et al. in [9] where a pan-genome was used for studying genetic variation in bacteria. Since then, pan-genomes have been used successfully in various studies for identifying microbial pathogens [5,6,7]. Sherman et al. [10] assembled a human pan-genome from NGS data of 910 African descendants and revealed 10% novel genetic material that was not found from the standard human reference genome. Mallick et al. [11] studied 300 human individuals from 142 diverse populations and found 5.8 million base pairs not presented in the human reference genome. Duan et al. analyzed 275 Chinese individuals with HUPAN [12] pipeline from NGS data where they found 29.5 million base pairs of novel sequences and 188 novel genes. Zhiqiang et al. demonstrated EUPAN [13] toolkit by analyzing the pan-genome consisting of 453 rice genomes [14]. In [15] we developed ViraPipe, a scalable pipeline for mining viral sequences from a large amount of human metagenomic samples on distributed Apache Spark cluster. ViraPipe has been used in an experiment with 768 whole-genome sequenced human samples. Most of these studies are based on the De-novo method that assemblies longer sequences, contigs, from short reads sequenced from donor DNA that does not map to reference genome. The contigs are used to form the pan-genome which is eventually used for analyzing novel sequences. This work instead is based on the whole-genome re-sequencing which differs from the De-novo based approaches in that individual genomes are assembled using a reference genome. We construct a complete pan-genome from previously assembled whole-genomes where a new consensus reference genome is assembled considering all variation between the individual genomes in the pan-genome. The assembled consensus genome enables then read alignment and variant calling with a complete pan-genome.
Hadoop-BAM [26] library has been originally developed for processing genomic data formats in parallel with Apache HadoopFootnote 2 and Spark [22], and developed further under the DisqFootnote 3 project for even better Spark integration. It includes Input/Output interface for distributing genomics file formats into HDFS and tools e.g.., sorting, merging, and filtering of read alignments. Currently, supported genomics file formats are BAM, SAM, CRAM, FASTQ, FASTA, QSEQ, BCF, and VCF. Hadoop-BAM is already used in genome analytics frameworks and libraries such as GATK4Footnote 4, AdamFootnote 5, Halvade [27], SealFootnote 6 and SeqPigFootnote 7. GATK is a software package for HTS data analysis developed by Broad Institute offering best practices variant discovering pipeline for human genomes. The current GATK4 version has been developed partly on Apache Spark for enabling distributed parallelization for rapid exploratory genomic studies. They have also established an open-source FireCloud platform for managing, sharing, and analyzing genomics data. ADAM is an Apache Spark-based genome analysis toolkit developed at UC Berkeley. ADAM includes basic tools for genomics file transformations, k-mer counting, and allele frequency computation on Apache Spark cluster. Halvade is a distributed read alignment pipeline based on the Hadoop MapReduce framework [21] for enabling more efficient variant calling with GATK. Halvade uses MapReduce for distributing BWA read alignment on read chunks against the reference genome. Seal is a software suite developed in CRS4 for processing sequencing data based on the Hadoop framework and it is written in Python. It provides basic tools for parallel and distributed read demultiplexing, read alignment, identifying duplicate reads, sorting the reads, and read quality control.
2 Methods
2.1 Distributed and Parallel Data Processing in Genomics
Traditional computational genome analysis algorithms and pipelines have been developed for sequential data processing in centralized computers, whereas the current evolution of high performance computing moves towards parallel algorithms and distributed data stores for efficient computation and analysis of massive data volumes. Moreover, current genome analysis tools and pipelines are typically developed on demand by the researchers relying on existing sequential algorithms. This has led to that pipelines are utilizing a mixture of command-line tools making them often poorly scalable, computationally inefficient, inflexible, and not easily parallelizable, especially in distributed computing clusters. Distributed and parallel computing frameworks enable scalable, reliable, efficient, and relatively low-cost computing in computing clusters. Cloud services provide infrastructures for deploying computing clusters easily and cheaper. Parallel data analysis with multiple distributed computing nodes brings huge performance advantages compared to a single computer. Computing takes place in the distributed working memory over distributed data sets by minimizing the intercommunication between nodes with optimal algorithms. This is achieved by dividing each computing task to the local parts, in which each node executes computing with local data. Apache Spark [22] is an open-source framework developed for efficient in-memory distributed large-scale computing in computing clusters. Spark accelerates data analysis with in-memory processing where working sets of data can be reused and pipelined from one pipeline stage to another in-memory instead of using temporary files. Computing in Spark is based on Resilient distributed datasets (RDDs) [23], which are distributed and cached to the working memory of multiple computing nodes in a cluster. Each node assigns an executor for local tasks that are run in parallel on the multiple cores inside a node. Spark itself does not provide a data store, but it can retrieve and write data to Hadoop Distributed File System (HDFS). In addition, HDFS provides fault tolerance through data replication. Spark supports Scala, Java, Python, and R programming languages. Figure 1 shows a typical data reduction process of a genomics data analysis pipeline on Spark.

Data reduction process.
2.2 Distributed and Parallel Characteristics of Genomic Data
Distributed and parallel computing has not been in major focus when widely used algorithms and data models for genomics were originally designed. Data parallelism is one promising choice for parallelizing genomics pipelines without fully rewriting all of the existing algorithms. Data locality can be achieved in the nodes of the computing cluster and data processing can be done in parallel without reloading or moving any data. Raw sequencing read data can be distributed for read alignment when reference assembly methods such as BWA [24] and Bowtie [25] are used. Assembled genomes and variant data are usually parallelizable by the chromosomes and chromosomal regions, giving an opportunity to distribute input data for parallel processing stages. Existing general genomics file formats are not designed for distributed file systems and especially binary formats BAM, BCF, BED are not distributable without external tools. However, Hadoop-BAM [26] can already handle distributed BAM and BCF files on HDFS in parallel and also in-memory with Spark.
2.3 Reference Genome Assembly
Reference genome-based assembly is preceded by a read alignment process where billions of Next-generation sequencing (NGS) reads are sequenced from a donor DNA sample and aligned to a reference genome. That is, the human genome can not be sequenced as a whole with current technology. Instead, the genome is reconstructed from short fragments, called reads, which are sequenced from a donor DNA sample and aligned to a reference genome. In the pan-genomic context, the reference is a multiple sequence alignment of N sequences. The uncompressed size of a human pan-genomic reference is approximately N \(\times \) 3 billion bases where N is the number of haploid genomes included (human genome is diploid, having two haploids of length 3 billion bases approximately). The amount of NGS data coming from a sequencing machine depends on the size of the donor genome, the used sequencing method, and the parameters given to a sequencer. Typically the size of whole-genome sequencing read data varies from 10 to 100 GB per human genome. NGS reads have to be aligned to a reference genome for assembling consensus genome of a donor which is then used to call the variants against. The assembly process requires that every short read (tens to hundreds of base pairs long) is aligned to every position in the reference genome. This work focuses on whole-genome data where pan-genomic reference is assembled from N whole-genomes.
2.4 Compressed Indexing of Pan-Genomes
The pan-genome has to be indexed in order to perform read alignment and eventually reference genome assembly. In a pan-genomic context, the index can contain thousands of individual genome sequences. The size of the pan-genome index can be reduced hugely with compression methods such as Lempel-Ziv [28] by utilizing the characteristics of identical genome sequences between the individuals, that is, a human genome includes a large proportion of repetitive sequences which can be found from every individual. There are a few relatively fast legacy read alignment tools such as Burrows-Wheeler transformation [29] based BWA [24] and Bowtie [25]. However, the BWT based aligners use suffix array-based indexes where BWT has to permute over the whole index search space for scoring the alignment. Valenzuela et al. [30] propose a CHICO indexer based on hybrid index implementation of LZ77 variant of Lempel-Ziv algorithms for compressing the pan-genome. Hereinafter, LZ77 shall be referred to as LZ. The hybrid index separates the compression part from the indexing, where the identical parts are compressed with the LZ and the LZ compressed sequence is then indexed with the legacy Bowtie2 or BWA indexer. CHIC [31] provides also read aligner tool for aligning reads against hybrid indexFootnote 8 with BWA and Bowtie2 support. The CHIC indexing with Bowtie2 was evaluated [31] on a single high-performance machine with 48 cores and 1.5 TB of main memory where they reported 35 h indexing time for 200 human genomes compressed to 180 GB index from 540 GB input data (compression ratio 3:1). Sequential PanVCFootnote 9 pipeline integrates CHIC aligner, CHICO index, and external variant calling tools such as GATKFootnote 10.
2.5 Variant Calling
Variant calling is a routine process for identifying genetic variations e.g.., Single Nucleotide Variations (SNVs) between a donor and some reference genome. Variant calling begins by aligning the NGS read sequences to a reference genome and filtering out the unaligned reads. Next, alignments are typically filtered by quality. Finally, reads are piled up over aligned reference genome base positions, and reference aligned bases are counted. This way the most probable bases in a donor genome covering a genomic position can be detected. Instead of using one reference genome, variant calling against a pan-genomic reference can provide more accurate information about genetic variation by aligning donor sequences to genomic positions in multiple genomes. Variant calling with a pan-genomic reference assembly can be done directly with legacy variant calling tools such as GATK’s best practice pipeline.

The pipeline stages denoted with letters a to g are explained in the Sect. 2.7. The green stages are distributed to multiple nodes and multiple cores in parallel (with Spark). The blue stages are distributed to multiple nodes and run in parallel per chromosome (multiple cores utilized in CHIC and Bowtie). The yellow stages are run on a single node in parallel on multiple cores. (Color figure online)
2.6 Designing the Distributed Pipeline
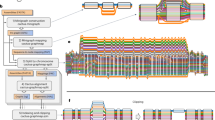
Reusability of existing genomics tools is a natural starting point for designing the workflow for the pipeline as those are widely used and well known within the bioinformatics and genomics communities, and quite efficient sequential algorithms have been already developed for the most general processing phases such as read alignment. Distributed genomic data can be processed in parallel partitions at different levels (separated chromosomes, chromosomal regions, NGS read partitions) in the pipeline. We have selected to use Spark with the Hadoop Distributed File System (HDFS) in our solution as it provides a flexible framework for scalable and efficient distributed data processing, and data management. Key challenges for implementing a distributed genome analysis pipeline are; decomposing the tasks and data to partitions for parallel execution with existing genomic data formats, processing the distributed tasks in parallel with existing tools and algorithms, and piping of multiple tools and algorithms together with minimal I/O operations while maintaining load balance. Moreover, latencies for reading data from HDFS to in-memory RDDs and writing it back to HDFS have to be taken into account as bioinformatics pipelines typically process thousands of separate files as well as big files together. This sets high requirements for computing cluster’s disk I/O, memory access, data warehousing, and networking performance. Figure 2 describes the architecture of the parallel pan-genomics pipeline at a high level.
2.7 Overall Pipeline Description.
The pipeline depicted in Fig. 2 consists of the following stages:
a) Preparing the pan-genome. The pan-genome itself is composed of multiple genomes that are aligned to a standard reference genome. Each genome in a pan-genome is assembled by applying variants from VCF files to a standard reference genome with vcf2multialignFootnote 11 tool and loading assemblies into HDFS under the same folder. When diploid genomes (e.g., humans) are used, this step generates two sequences, both haploids, per genome. The standard genome itself is applied on top of the pan-genome. If genome assemblies are already provided, the pan-genome can be constructed by simply loading all the individual genomes into HDFS under the same folder.
b) Indexing the pan-genome. Typically, the reference index data remains a magnitudes smaller than the read data sequenced from a donor genome. In pan-genomic context, the index data grows in proportion to the number of underlying individual sequences in a pan-genome where each individual sequence has to be indexed.

Distributed Relative Lempel-Ziv compression.
For reducing the compression and indexing time, we modify the CHICO indexer [30] to exploit distributed computing. CHICO supports BWA and Bowtie2 legacy indexes with the Relative Lempel-Ziv (RLZ) algorithm [32]. CHICO compresses the original pan-genome with RLZ that is eventually indexed for the read alignment purposes. The reference pan-genome is compressed to the kernel representation which reduces the repetition of similar sequences in the pan-genome. The repetitive sequences are compressed using a dictionary that is constructed from a partial pan-genome. Building the dictionary of the whole pan-genome would be too time-consuming and is not necessary due to repetitiveness, although, it can improve the compression ratio slightly. The size of the compressed kernel index depends on the number of individual genomes in the pan-genome and the similarity between the genomes.
We implement Distributed Relative Lempel-Ziv (DRLZ) compression (Fig. 3) with Spark for reducing the compression time through parallelization. The pan-genome is partitioned by chromosomes of individual reference genomes and distributed to HDFS. The chromosomal chunks are RLZ compressed in parallel with Spark. RLZ uses a suffix array which is calculated from the dictionary and broadcast to Spark for distributed RLZ compression. RadixSA library is used to construct the suffix array [33]. The RLZ compressed chromosomal partitions are eventually downloaded from the HDFS to different nodes where the kernel representation is composed and indexed with CHICO in parallel per chromosome. RAM disks can be configured here for storing and accessing data in local filesystem more rapidly.
c) Read alignment against the compressed pan-genome index. As each chromosome is indexed in parallel, read alignment is also done in parallel per chromosome. NGS reads are loaded from the HDFS to the local filesystem of the corresponding node and aligned with CHIC aligner using Bowtie2 against a compressed index of a chromosome. Multiple sequence alignment generated gaps are stored in this step into gap position files for fixing the mapping score in the next step. Then, the mapped reads are grouped by the reference sequence where they are mapped to. After the alignment process, grouped SAM files are put to HDFS for the next step. Eventually, duplicate mapped reads are removed.
d) Adhoc reference genome assembly. After the read alignment phase, the mapped reads of each individual are scored based on the alignment information provided in the SAM and the gap position files. That is, the pan-genome index does not include gaps, and now the read mapping is fixed to correspond the gapped positions in the pan-genome [16]. The pan-genome with gaps is read from HDFS into the scoring matrix with Spark Mllib BlockMatrix class which distributes the score matrix of size M \(\times \) N into S blocks, where M is the length of the individual sequence, N is the number of the individuals in the pan-genome, S is the number of partitions configured. The adhoc consensus genome is then assembled from the score matrix blocks by extracting the heaviest path (Fig. 2, e), that is simply, taking index of maximum scoring alignment per each column, mapping the index to the corresponding nucleotide in the reference genome and merging the blocks at the end.
e) Heaviest path. The score matrix for calculating the heaviest path is sparse; the first sequence contains most of the matches and the following genomes add only little extra information. Therefore, Apache Spark’s sparse vector representation is used to achieve the best possible performance. The score matrix is transposed to find the maximum number of matches across different genomes. After this operation, we obtain the heaviest path sequence telling the row number corresponding to individual genome which has the most matches in that position. Next, the nucleotides in the corresponding positions pointed by the heaviest path are extracted from the pan-genome. To change from this number representation to corresponding DNA reference sequence, a simple logic OR operation is applied between each individual sequence in the pan-genome and the calculated heaviest path blocks (Fig. 4). After this operation has been done for each individual, a tree-reduce RDD transformation is used to combine the sequence branches into a single adhoc reference sequence in parallel.

Transformation from calculated heaviest path to the adhoc reference with three individuals.
f) Legacy variant calling from adhoc reference genome. After the adhoc reference genome assembly, variant calling is performed with the donor genome reads. The adhoc genome is first indexed with legacy tools such as BWA or Bowtie and donor reads are aligned to that index. Any legacy variant calling pipeline that outputs VCF format can be used to call variants from the assembled adhoc reference genome. GATK4 best practices variant calling pipeline is provided with our implementation. As an alternative, basic Samtools and Bcftools variant callingFootnote 12 method is included in our pipeline.
g) Variant normalization. If a standard reference genome is used to construct the pan-genome, the variants called from an adhoc reference are normalized against the standard reference genome. That is, normalization generates a consensus genome from the standard reference genome by applying variants called from the adhoc read alignment. In practice, normalization applies SNPs, insertions, and deletions to the reference genome positions assigned in VCF files and gap files (produced already in the indexing phase). Finally, the projection between the normalized consensus and adhoc reference consensus is constructed. The projection does sequence alignment between those two consensus genomes showing indels and mismatches for comparison and further analysis. The normalization and projection are done with the tools provided in the original PanVC [16].
3 Experiments
3.1 Data Preparation
We generate a pan-genome based on the human reference genome by applying heterozygous SNPs from phased haploid VCF data to GRCh37 reference thus generating two consensus genomes per individual into pan-genome. Pan-genomes are generated from the autosomes of 1000 Genomes phase 3 VCF data including 2506 individuals totaling the pan-genome size of 13.1355 TB. Read alignment and variant calling is performed with NGS read data set sequenced from donor HG01198 genome published by 1000 genomes project. Read data contains 9.4 million paired-end reads totalling 2 GB.
3.2 Computing Environment
The experiments are run on the Apache Spark cluster in a cloud computing environment. The cluster consists of 25 Spark worker nodes having 40 GB of RAM and 16 cores (Intel(R) Xeon(R) CPU E5-2680 v3) in each and one Spark master node having 256 GB of RAM and 48 cores. The whole cluster comprises 448 CPU cores, 1.256 TB of RAM, Infiniband 40 GB/s network, 30 TB of HDD storage space in total. The Spark cluster is deployed with Apache Spark 2.3.2 and Hadoop 3.1.0 versions on virtual machines running CentOS 7 operating system. We utilize the computing resources of the Finnish IT Center for Science (CSC) in our experiments.

Scalability of PanGenSpark with increasing pan-genome size compared to single node PanVC execution.
3.3 Results
The scalability in terms of pan-genome size is in the main focus of our experiments. Figure 5 shows the execution time in different pipeline phases from Relative Lempel-Ziv (RLZ) compression to reference genome assembly with accumulating pan-genome size. For a baseline, a pan-genome of size 50 haploid genomes is assembled with the original PanVC on a single node. The total runtime with 50 genomes is 1624 min consisting of following execution times: 150 min for RLZ, 555 min for Bowtie2 indexing, and 919 min for adhoc reference genome assembly. Multithreaded Bowtie2 indexer is executed using 16 threads. The same multithreading configurations have been used in the distributed pipeline as well. Variant calling is not included in the results as it is not part of the distributed implementation. To note, the variant calling takes the equal amount of time with all pan-genome sizes as the adhoc reference genome is always the same size.
With the distributed pipeline and 50 haploid genomes (141 GB input data), the RLZ compression time is 68 min with a compression ratio of 5.71:1 shown in Table 1. Indexing time with Bowtie2 indexer is 58 min while the largest indexed kernel is 3.2 GB for chromosome 2 (chromosomes are indexed on distributed nodes in parallel using 16 threads per node). Adhoc reference assembly from 50 genomes takes 89 min.
With 100 haploid genomes (282 GB input data) the distributed RLZ compression time is 95 min with a compression ratio of 7.74:1. Distributed indexing time with Bowtie2 is 84 min while the largest indexed kernel is 3.8 GB for chromosome 2. Assembling the adhoc reference genome with 100 genomes takes 125 min. With 500 genomes (1.41 TB GB input data) the distributed RLZ compression time is 165 min with a compression ratio of 8.57:1. Bowtie2 indexing becomes a bottleneck with 500 genomes kernel (largest chromosomal kernel 16 GB) and indexing time increases to 1140 min. Adhoc reference genome assembly from 500 genomes takes only 163 min.
4 Discussion
Preliminary results are promising, but also show some limitations of the pipeline. Figure 5 shows how the different parts of the pipeline perform. When the pan-genome size increases, the distributed Relative Lempel-Ziv (DRLZ) compression scales well. DRLZ execution takes relatively long with small pan-genomes as suffix array files are the same size (30 GB) for all pan-genome sizes and broadcasting the data took almost 40 min. The speedup with 50 genomes compared to PanVC is 7.6x with 25 worker nodes. Indexing step with Bowtie2, that is distributed by chromosomes and executed with 16 cores in parallel per node, scales the worst and takes most of the computation when the pan-genome size grows up to 500. The adhoc genome assembly scales the best from all of the distributed parts. We were able to compress the index of 500 haploid genomes to the size of 58 genomes with a compression ratio of 8.57:1. For one genome the compression ratio is 3.53:1 (Table 1). The index compression ratio with sequential PanVC is equal to the distributed version as is expected. RLZ dictionary size affects the compression ratio the most (the longer it is the better the compression), and secondly the chosen maximum read sequence length parameter (the shorter it is the better the compression). We used the whole chromosomes as a dictionary for DRLZ and the maximum read length of 80 bases. Compression ratio increases in proportion to the number of genomes which is obvious due to sequence repetition.
Apache Spark is extremely good at scaling up to an unlimited number of rows, but the limiting issue here is that whole genome sequences can not be processed as one piece as the length of the string is restricted by Java Integer.MAX_VALUE constant which is roughly 2 billion. To improve the compression ratio we would need to use the whole genome as a dictionary part for the RLZ compression which is currently not possible due to Integer.MAX_VALUE issue with Spark and Hadoop frameworks. Therefore, we read the pan-genome by chromosome, compress by chromosome, and index the chromosomal kernels. BWA indexer could not handle longer than 4 GB kernel text, thus we chose to use Bowtie2. However, Bowtie2 does not scale linearly and as it can be seen from the Fig. 5: with 50 haploid genomes indexing time is 58 min whereas with 500 haploid genomes indexing time increases 20 times (the longest kernel text with 50 genomes is 3.2 GB and with 500 genomes 16 GB). However, the whole pipeline runtime increases only 6.8 times with 10 times larger pangenome showing still good scalability. Bowtie2 uses a “large” index method with 64-bit numbers when the input data size grows larger than 4 GB which seems to slow down the indexing. Bowtie2 legacy indexing time can be still decreased by harnessing more cores and memory for the indexing nodes. Tuning the cluster with all Hadoop and Spark configuration parameters turns out to be a complex task as there are plenty of those and optimal configurations vary in different processing steps. Minimizing the I/O operations is crucial when processing large data sets in the complex pipeline. Moreover, this poses challenges to the pipeline development as input data structure varies in different steps making data partitioning for parallel processing more complex. In the near future, we focus on improving the DRLZ compression as the better compression would also reduce the Bowtie2 indexing time due to shortened kernel text.
5 Conclusions
To exploit massive amounts of genomic data, it is essential to compress and store genomic data sets in an efficient and reusable form for supporting bioinformatics tools. Pan-genomic indexes and reference genomes are urgent for efficient large-scale read alignment, variant calling, and sequence matching purposes. In this work, we design a prototype pipeline, PanGenSpark, for scalable compressed indexing of pan-genomes and assembling of reference genomes from pan-genomes. The PanGenSpark assembled a reference genome from a pan-genome of 50 human haploid genomes in 215 min and 500 haploid genomes in 1468 min. The index of 1.41 TB pan-genome was compressed into a size of 164.5 GB. The experiments have been run on a distributed Spark cluster consisting of 448 cores on 26 computing nodes. Altogether, our distributed pipeline allows now assembling the pan-genomic consensus reference in a tolerable time from hundreds of human genomes in practice. Moreover, the compressed pan-genomic index can be reused for efficient NGS read alignment and sequence matching purposes as well.
Notes
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
References
Marcshall, T., Marz, M., Abeel, T., et al.: Computational pan-genomics: status, promises and challenges. The Computational Pan-Genomics Consortium. Brief. Bioinform. (2016). https://doi.org/10.1093/bib/bbw089
Sherman, R.M., Salzberg, S.L.: Pan-genomics in the human genome era. Nat. Rev. Genet. 21, 243–254 (2020). https://doi.org/10.1038/s41576-020-0210-7
Dilthey, A., Cox, C., Iqbal, Z., Nelson, M., McVean, G.: Improved genome inference in the MHC using a population reference graph. Nat. Genet. 47, 682–688 (2015). https://doi.org/10.1038/ng.3257
Auton, A., Abecasis, G., Altshuler, D., et al.: A global reference for human genetic variation. Nature 526, 68–74 (2015). https://doi.org/10.1038/nature15393
Rouli, L., Merhej, V., Fournier, P.E., Raoult, D.: The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85 (2015)
Rasko, D.A., Rosovitz, M.J., Myers, G.S.A., et al.: The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190, 6881–6893 (2008)
Trost, E., Blom, J., Soares, S.C., et al.: Pangenomic study of Corynebacterium diphtheriae that provides insights into the genomic diversity of pathogenic isolates from cases of classical diphtheria, endocarditis, and pneumonia. J. Bacteriol. 194, 3199–3215 (2012). https://doi.org/10.1128/jb.00183-12
Kehr, B., Helgadottir, A., Melsted, P., et al.: Diversity in non-repetitive human sequences not found in the reference genome. Nat. Genet. 49, 588–593 (2017). https://doi.org/10.1038/ng.3801
Tettelin, H., Masignani, V., Cieslewicz, M.J., et al.: Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial ‘pan-genome’. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955 (2005). https://doi.org/10.1073/pnas.0506758102
Sherman, R.M., Forman, J., Antonescu, V., et al.: Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 51, 30–35 (2019). https://doi.org/10.1038/s41588-018-0273-y
Mallick, S., Li, H., Lipson, M., et al.: The Simons genome diversity project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). https://doi.org/10.1038/nature18964
Duan, Z., Qiao, Y., Lu, J., et al.: HUPAN: a pan-genome analysis pipeline for human genomes. Genome Biol. 20, 149 (2019). https://doi.org/10.1186/s13059-019-1751-y
Hu, Z., et al.: EUPAN enables pan-genome studies of a large number of eukaryotic genomes. Bioinformatics 33(15), 2408–2409 (2017). https://doi.org/10.1093/bioinformatics/btx170
Zhao, Q., Feng, Q., Lu, H., et al.: Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278–284 (2018). https://doi.org/10.1038/s41588-018-0041-z
Maarala, A.I., Bzhalava, Z., Dillner, J., Heljanko, K., Bzhalava, D.: ViraPipe: scalable parallel pipeline for viral metagenome analysis from next generation sequencing reads. Bioinformatics 34(6), 928–935 (2018). https://doi.org/10.1093/bioinformatics/btx702
Valenzuela, D., Norri, T., Välimäki, N., et al.: Towards pan-genome read alignment to improve variation calling. BMC Genomics 19, 87 (2018). https://doi.org/10.1186/s12864-018-4465-8
Siren, J., Välimäki, N., Mäkinen, V.: Indexing graphs for path queries with applications in genome research. IEEE/ACM Trans. Comput. Biol. Bioinform. 11(2), 375–388 (2014). https://doi.org/10.1109/TCBB.2013.2297101
Huang, L., Popic, V., Batzoglou, S.: Short read alignment with populations of genomes. Bioinformatics 29, 361–370 (2013). https://doi.org/10.1093/bioinformatics/btt215
Schneeberger, K., Hagmann, J., Ossowski, S., et al.: Simultaneous alignment of short reads against multiple genomes. Genome Biol. 10, R98 (2009)
Paten, B., Novak, A., Haussler, D.: Mapping to a reference genome structure. ArXiv http://arxiv.org/abs/1404.5010 (2014)
Jeffrey, D., Sanjay, G.: MapReduce: simplified data processing on large clusters. Commun. ACM 51, 107–113 (2008). https://doi.org/10.1145/1327452.1327492
Zaharia, M., Chowdhury, M., Franklin, M.J., Shenker, S., Stoica, I.: Spark: cluster computing with working sets. In Proceedings of the 2nd USENIX conference on Hot topics in cloud computing (HotCloud 2010), p. 10. USENIX Association, USA (2010)
Zaharia, M., Chowdhury, M., Das, T., et al.: Resilient distributed datasets: a fault-tolerant abstraction for in-memory cluster computing. In: Proceedings of the 9th USENIX conference on Networked Systems Design and Implementation (NSDI 2012), Berkeley, CA, USA, p. 2 (2012)
Li, H., Durbin, R.: Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14), 1754–1760 (2009). https://doi.org/10.1093/bioinformatics/btp324
Langmead, B., Trapnell, C., Pop, M., Salzberg, S.L.: Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). https://doi.org/10.1186/gb-2009-10-3-r25
Niemenmaa, M., Kallio, A., Schumacher, A., Klemelä, P., Korpelainen, E., Heljanko, K.: Hadoop-BAM: directly manipulating next generation sequencing data in the cloud. Bioinformatics 28(6), 876–877 (2012). https://doi.org/10.1093/bioinformatics/bts054
Decap, D., Reumers, J., Herzeel, C., Costanza, P., Fostier, J.: Halvade: scalable sequence analysis with MapReduce. Bioinformatics 31(15), 2482–2488 (2015)
Ziv, J., Lempel, A.: A universal algorithm for sequential data compression. IEEE Trans. Inf. Theory 23(3), 337–343 (1977). https://doi.org/10.1109/TIT.1977.1055714
Burrows, M., Wheeler, D.J.: A block-sorting lossless data compression algorithm. Technical report 124, Palo Alto, CA, Digital Equipment Corporation (1994)
Valenzuela, D.: CHICO: a compressed hybrid index for repetitive collections. In: Goldberg, A.V., Kulikov, A.S. (eds.) SEA 2016. LNCS, vol. 9685, pp. 326–338. Springer, Cham (2016). https://doi.org/10.1007/978-3-319-38851-9_22
Valenzuela, D., Mäkinen, V.: CHIC: a short read aligner for pan-genomic references. bioRxiv 178129 (2017). https://doi.org/10.1101/178129
Hoobin, C., Puglisi, S.J., Zobel, J.: Relative Lempel-Ziv factorization for efficient storage and retrieval of web collections. Proc. VLDB Endow. 5(3), 265–273 (2011). https://doi.org/10.14778/2078331.2078341
Rajasekaran, S., Nicolae, M.: An elegant algorithm for the construction of suffix arrays. J. Discrete Algorithms 27, 21–28 (2014). https://doi.org/10.1016/j.jda.2014.03.001
Acknowledgements
The financial support of Academy of Finland project #336092 and #309048 are gratefully acknowledged. The computing capacity from CSC - IT Center for Science and the FGCI2 Academy of Finland Infrastructure project are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Maarala, A.I., Arasalo, O., Valenzuela, D., Heljanko, K., Mäkinen, V. (2020). Scalable Reference Genome Assembly from Compressed Pan-Genome Index with Spark. In: Nepal, S., Cao, W., Nasridinov, A., Bhuiyan, M.Z.A., Guo, X., Zhang, LJ. (eds) Big Data – BigData 2020. BIGDATA 2020. Lecture Notes in Computer Science(), vol 12402. Springer, Cham. https://doi.org/10.1007/978-3-030-59612-5_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-59612-5_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-59611-8
Online ISBN: 978-3-030-59612-5
eBook Packages: Computer ScienceComputer Science (R0)