
Abstract
Scientific advances in technology have helped in digitizing genetic information, which resulted in the generation of the humongous amount of genetic sequences, and analysis of such large-scale sequencing data is the primary concern. This chapter introduces a scalable genome sequence analysis system, which makes use of parallel computing features of Apache Spark and its relational processing module called Spark Structured Query Language (Spark SQL). The Spark framework provides an efficient data reuse feature by holding the data in memory, increasing performance substantially. The introduced system also provides a web-based interface, by which users can specify the search criteria, and Spark SQL performs search operations on the data stored in memory. Experiments detailed in this chapter make use of publicly available 1000 genome Variant Calling Format (VCF) data (Size 1.2TB) as input. The input data are analyzed using Spark and the end results are evaluated to measure the scalability and performance of the system.
Access provided by CONRICYT-eBooks. Download conference paper PDF
Similar content being viewed by others


1 Introduction
Big data computing technologies have been proliferating fast with unprecedented volumes of data generation in several domains. Since the completion of the Human Genome Project in 2003 [1], the cost of sequencing human genomes is reduced by more than 100,000× [2]. Reduced cost and recent scientific advances in technology have helped digitize human genes and generate huge amounts of human genomic sequences that help researchers in understanding human genes. A major challenge encountered with such vast gene data generation is the analysis of the required sequence structures.
Many data processing techniques are available, but the ability to process the increasing data needs to address the performance and scalability challenges. For instance, processing of increasing data by minimizing the query response time is key to achieving scalability. One of the tools available for such large-scale analysis is Apache Hadoop [3], which is an open source framework that gives users powerful features to store and process huge datasets. While widely used, we observed it requires around 20 min for every single query to be completed against a data size of 1.2 terabytes (for the 1000 genome data set) using 16 Hadoop computing resources. This motivates us to explore other tools for scalable gene sequence analysis. Apache Spark [4] can provide enhanced performance for certain applications compared to the existing MapReduce framework using its in-memory processing feature. In detail, Spark provides Resilient Distributed Datasets (RDDs) that can be stored in memory between query executions that facilitates the reuse of intermediate outputs.
In this chapter, we examine the feasibility of Spark for the application of gene sequence variation analysis with respect to performance and scalability. In addition, optimization is an important part of system performance and we will evaluate the impact of caching in Spark with extensive experiments on performance evaluation. Another important element for such analysis systems is the degree of user friendliness to access the analysis tool since the majority of the users of this application would be scientific researchers with limited knowledge on computing systems. This chapter also introduces a graphical user interface for intuitive analysis and develops a prototype system that equips a variety of features to improve user friendliness including an automatic query generation engine.
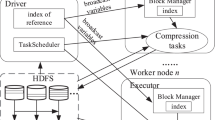
Figure 1 shows an overview of the introduced system for large-scale gene sequence analysis with low latency for processing. The system uses the Spark framework with Spark SQL and YARN resource management that provides effective configuration of the cluster computing system. The graphical user interface of the system promotes user interaction as part of data analysis. As shown in Fig. 1, researchers can simply access the system interface on existing web browsers for composing a query. Based on the users’ selection criteria, application system generates a query script automatically and executes the query script on Spark. Finally, the processing results are delivered to the end users.

System overview: The system is based on the Spark framework with Spark SQL and YARN resource management. End users access the system through Web-based interfaces
This chapter is organized as follows. Section 2 provides the summary of the big data computing tools including Hadoop and Spark, with closely related studies to our work. Section 3 demonstrates the design of the system for scalable gene sequence processing on the Spark framework, and Sect. 4 presents the evaluation results conducted on a Spark computing cluster with 16 compute nodes. In Sect. 5, we present a prototyping system and we conclude our presentation in Sect. 6.
2 Background
2.1 Apache Hadoop and Spark
Hadoop is an open source software platform managed by the Apache Software Foundation. It is the most widely recognized platform for efficient and cost-effective storage and management of large volumes of data. HDFS provides a resilient and fault-tolerant storage of data by means of data duplication which is termed as data replication. Pig [5] and Hive [6] are the two high-level query languages supported by Hadoop, whose execution is based on MapReduce programming model that provides a scalable, flexible and fault-tolerant computing solution [7]. Hadoop comprises of two major components: Hadoop Distributed File System (HDFS) and the MapReduce framework. A traditional Hadoop job works by reading the data from HDFS for each run and writing back the intermediate results to HDFS which incurs the cost of data operations for each run time. Thus, Hadoop does not seem to be a good fit for iterative computations and low latency applications.
Spark is an open source framework that can process data 100× faster than Hadoop. It is a widely accepted for parallel cluster computing systems in both science and industry [8]. This framework attracts users due to the delivery speed it provides by making use of its in-memory computing capability using fault-tolerant [9] Resilient Distributed Datasets (RDDs).RDDs are created by performing operations called transformations (map, filter etc.) on the data stored in the file systems or other existing RDDs. By default, RDDs will be persisted in memory but they can even spill to the disk if there is not enough Random Access Memory (RAM). Spark also provides users with an option to specify the storage level for RDDs, such as persisting in disk only, disk by using replicating flag and both memory and disk. Spark provides Scala, Java, and Python programming interfaces to create RDDs transformations and performs actions on it [10]. Spark contains Spark SQL module that helps to achieve relational processing on the persisted data.
2.2 Gene Sequence Analysis
Genes are small pieces of a genome and they are made of DNAs. DNA contains bases that carry the genetic information which helps in understanding the behavioral pattern of organisms. Each human genome contains approximately three million DNA base pairs. Genome sequencing is about identifying the order of DNA bases that defines the characteristics of an organism. Genome sequences help researchers or scientists answer questions like why some people get infections or why some people have different behaviors that others do not have, and will the genes have any impact on others genes.
Many computational tools have been developed for digitizing the genetic information, producing genome sequences which can be further analyzed for meaningful patterns. As mentioned, one genome has up to three million base pairs and thus sequencing of multiple human genomes will quickly add up to larger data sizes and mapping, in turn, generates more. For effective analysis of such data, it is essential to store data in a fault-tolerant manner and process the data with optimal performance and scalability. To address this concern, Hadoop has been applied in the Genome Analysis Toolkit (GATK) tool [11]. In the tool, the GATK process needs to access the distributed data across the cluster, parallelize the processing, persist and share intermediate processing results. This process has drawbacks such as limited parallel processing with the significant overhead due to disk I/O between stages of walkers and expensive read while processing between stages for repeated access. The idea of caching in Spark could reduce the repeated disk access of data that eliminates the repeated I/O bottleneck and improves the performance.
Spark is used by variant calling tools for the genomic analysis. Using the advantages of Spark, the ADAM project was developed by Big Data Genomics (BDG) group at UC Berkeley. ADAM can process large-scale genome data available in various data formats such as VCF, BAM, and SAM [12]. ADAM project uses APIs that transform the input data to ADAM-defined formats and store the RDD sets generated from these formats in memory to perform sorting and other operations using Spark modules such as GraphX, MLlib (Machine Learning libraries) and Shark. Even though ADAM has overcome the traditional processing speed limitations of MapReduce, additional processing complexities such as converting VCF files to the ADAM formats and overhead for compression of inputs before processing, have led to non-optimal performance.
Another Spark based implementation for genomic information analysis is VariantSpark [13]. This system is developed in Scala using Spark and its machine learning methods (MLlib). VariantSpark reads the VCF files and transposes them into vectors based on variants, which are then transformed into key-value pairs. The key-value pairs are then zipped and saved in a distributed system. Further processing is implemented using a K-means clustering algorithm. MLlib performs complex programming logic by transforming, zipping and processing of data using machine learning algorithms.
Unlike the above Spark-based gene sequence analysis system, our system employs Spark SQL tables to process the direct genome VCF data without conversion and uses Spark in-memory data store for processing required data, in order to reduce format conversion and save processing times. The introduced system concentrates on reducing the processing complexities, maximizing the performance of the variants analysis, and increasing the ease of usability of the tool when compared to the other variant tools.
3 System Model for Scalable Gene Sequence Analysis
Utilizing the advantages of HDFS, Sparks’ relational processing module Spark SQL and YARN cluster management, a scalable model is designed as shown in Fig. 2. A series of experiments were conducted on top of this design to examine the scalability. Spark applications can run on top of existing HDFS that store data in clusters in a resilient manner. There are three possible cluster modes that control the resources of a cluster, namely, Standalone Mode, Mesos Mode, and YARN mode. Standalone Mode utilizes all the recourses available in the cluster and at least, each executor has to be running on each node. Total utilization of resources causes other applications to be in the queue until the prior process is completed, which will be a downside when processing multiple applications. To overcome this, YARN resource manager can be used as it can allocate resources dynamically within the cluster or we can choose the number of executors to be assigned to each process [14].

System model, utilizing the advantages of HDFS, Sparks’ relational processing module Spark SQL and YARN cluster management
In order to process the data in a cluster, Spark application runs as an independent process in each node of the cluster and these processes are coordinated by a driver program called SparkContext. As shown in Fig. 2, the driver program interacts with Resource Manager in YARN Mode, which keeps track of the cluster resources and monitors tasks allocated to Node Mangers. Node manager acts as a worker that initiates tasks based on allocated cores for processing of data stored in its respective nodes. Each node can have multiple executors and each executor can have multiple tasks. Each node also has designated cache memory to store the data which can be accessed by all the executors in the node.
Spark provides a Thrift JDBC/ODBC server that helps to run queries from within applications or by end-users using a command-line interface called Beeline. This acts as Business Integration medium in connecting different applications to Spark. This server provides distributed engine that can run SQL queries without writing complex code for querying. Beeline command lines are used to test the JDBC server connectivity from applications to run the SQL queries. This thrift server helps in maintaining the cache as long as the server is active. Keeping the server active helps multiple users access the cache to obtain faster results. This server is used in the design of proposed prototype system for efficient and optimized gene sequence analysis.
We assume that VCF [15] format genomic data is read and processed using Spark to identify if the system is scalable to use. In addition, a web-based gene Sequence analysis system is designed to store the VCF formatted data and process the data interactively for faster and easier access as will be discussed in Sect. 5.
4 Evaluation
In this section, we present the experimental results conducted particularly to evaluate the scalability of the system.
The experiments were conducted in a computing cluster machine that consists of 16 nodes mounted in a rack. Cluster configuration used for the experiments is of 128 cores, 128 GB memory and 32TB disk space. The nodes in the cluster are interconnected through Gigabit Ethernet Switch. The Operating System used for the cluster is CentOS release 6.5 that comes with rocks application, which manages the connections between all nodes that make up a cluster machine. I have installed the Hadoop pre-built version of Apache Spark v1.4.1, Stable version of Hadoop v2.6.0 and high-level query language Pig v0.14.0, Hive v1.2.0 and MySQL Server v5.6 for the experiments.
The experimental data set used has VCF formatted F data file [16], which contains genotype data of 1,092 individuals which equal to 37,644,734 records that approximate to 1.2TB. VCF represents Variant Calling Format that stores the genome data in the form of variants or subjects arranged as columns in the data file. The data are distributed to the fault tolerant HDFS across 16 nodes in the cluster. To consider the impact of query characteristics, three types of queries that have different coverage with respect to the fraction of the records hit are defined. Table 1 presents a summary of the queries with the description and hit coverage. Query2 and Query3 are extreme cases with a very rare hit (“small hit”) and a large hit (“large hit”), respectively. SQL aggregate functions executed on data have less output display time compared to the select functions on the data. Hence, the queries are chosen to perform the count operations on data to minimize the output display time and thereby to measure the computational performance exclusively.
4.1 Spark SQL vs. Other High-Level Query Tools (Pig and Hive)
For big data analytic tools, high-level query support is essential for easy usage and reducing programming complexities. Input dataset is loaded into HDFS, and high-level query languages are often used to process the loaded data. We compared several high-level query tools such as Pig and Hive with Spark SQL.
Table 2 shows the query completion time in seconds. The results show that Spark’s iterative response time gives approximately 90–100% decreases the response, increasing the performance, when compared to Spark’s initial response time (Cache Time) and approximately 200% increase when compared to Pig and Hive’s response time.
4.2 Scale-Out and Scale-Up Performance
To check the scalability of using Spark for analysis the experiments are conducted in two folds. First, we evaluate scale-out performances of the proposed system. The scale-out check is to evaluate the impact of increasing computing capability for processing of data. Second, we evaluate scale-up performance of the proposed system. This scale-up check is to examine the impact of increasing data sizes for processing the data using the proposed system.
Figures 3 and 4 show scale-out performance in case of in-memory data access (Fig. 3) and in-disk data access (Fig. 4). The experiments are carried on the cluster using one executor and three cores on each node. The two figures show a certain degree of scalability. The benefit is the greatest from four nodes to eight nodes in both settings. As expected, Query 3 (large hit) shows the greatest improvement with an increasing number of compute nodes. When we compare the two settings, in-disk data access shows the greater scale-out performance. We presume that it is due to the basic overhead (around 5 s) to run an individual query.

Scale-out performance in case of in-memory data access

Scale-out performance in case of in-disk data access
We next discuss scale-up performance with increasing data sizes. The 1.2TB Genome dataset is divided into multiple datasets of increasing sizes from 10% to 100% of the total. For the Spark initial run or Cache run, an increase in data sizes results in greater processing time. Figure 5 shows the processing time that was taken to cache the data in memory and on disk and increasing data sizes retain the linear scale-up performance of the system. Figure 6 shows scale up of the scan operation performed on the records cached in memory and on disk. After loading 10% percent genome, 16 columns of data is cached in memory or on disk for iterative runs. From both figures, we can see that initial loading time is tremendous; however after the data is loaded, we can see relatively very small scan time with a good degree of scalability.

Scale-up performance for cache loading time and initial run

Scale-up performance for cache scan
4.3 Impact of the Number of Cached Columns
We also evaluated the impact of the number of cached columns. As shown from Fig. 7, increasing the number of columns cached does not much impact the data response times. To test this, a series of query runs are implemented on the cached data by increasing the number of columns but with the same number of rows every time. Despite the increase in the amount of data loaded in cache, the processing time of queries on data remains consistent.

Impact of number of columns cached
4.4 Impact of Data Spills
Spark caches the data in the memory based on configurations provided. If the memory available to cache is less than the amount of data to be cached, then Spark spills the leftover data after caching on to disk to avoid data loss. A series of queries are executed on cache data to test the impact of data spill from memory on performance. The configuration for this experiment is set to 16 executors with three cores and 1GB of memory allocated. From this configuration, the total memory capacity available for storing of data is up to 8.2GB.
As shown in Table 3, due to limited memory capacity, if users try to load more data into cache beyond the capacity that it can accommodate, then the remaining data will be spilled onto disk across each computing node within the cluster. The data spill on each node of the cluster is based on the size of data distributed across the nodes. From the previous experiment, we observe that each query executed on cached data with an increasing number of columns cached gives consistent processing time. However, an increasing number of columns cached will increase the size of data spill from memory due to limited capacity, which in turn impacts the processing time of the cached data. Figure 8 shows that increase in data spilled to disk increases the cached data processing time, and thus, decreasing the performance.

Impact of data spills from memory
4.5 Impact of Record Hits
Accessing in memory data using different query options with varied output results will impact the data processing time. Within the cached data, increase in the number of records accessed (i.e. number of record hits) increases the processing time for each run. To demonstrate this, we loaded 16 columns of genome data in memory with 3.9 million records and performed a set of scan count queries. These queries hit the cache data with increasing percentage of output records. The results are plotted in Fig. 9, which shows that increasing access to the records from the cache data increases the processing time which ultimately leads to lower performance.

Impact of record hits
5 System Prototyping
A prototype system is implemented to analyze the genome data using Spark. Figure 10 shows the proposed system architecture for gene sequence analysis that is designed to read the input VCF genome data and analyze the data using query scripts.

Gene sequence analysis system architecture
A web-based interface system designed to store and processes the VCF data in a more user-friendly manner. Figure 11 gives the flow diagram of this interface based on which the web interface is designed. Figure 12 displays the GUI interface designed for providing the query support to users, especially scientists with minimal programming skills.

Execution flows of the prototype system

Web-based user interfaces for query support
This prototype system provides an option to execute queries in count only mode to maximize the performances. It minimizes the time that is taken to transfer the queried results between execution engine and the graphical user interface compared to transfer time without count mode. Larger the queried results the more will be transferred time of results.
Based on the above criteria editable query will be generated on the right-hand side of the interface for execution, as shown in Fig. 12. The generated query is executed and the output of executed query displays results along with record count and run time taken by Spark, as shown in Fig. 13. The query runtime does not include the data transfer time between Spark engine and the user interface module.

Query result
6 Conclusions
Big data computing and analytics open the doors for researchers to play with an unprecedentedly large set of data in a cost-effective manner with efficient parallel computing clusters. This chapter makes use of these advanced analytic tools to develop a scalable system for gene sequence variation analysis. The key summary of this work are as follows:
-
In this work, we examined the feasibility of using Spark for our own application with respect to performance and scalability, with the comparison with other analytics tools of Pig and Hive. We observed that Spark SQL significantly outperforms Pig and Hive. After loading the data in memory, it takes no longer than 15 s by Spark to complete any type of queries (execution plus response time) in our experimental setting with 16 computing nodes, while the other two tools require greater than 18 min for any individual query. The architecture used in this system with Spark and its ecosystem also demonstrated a high degree of scalability, particularly for scale-up over data size, suggesting it as a good choice for the bioinformatics application.
-
We conducted extensive experiments to analyze ways to achieve optimal performance of the system. Even though iterative operations on the cached data will give results in seconds, the maximum number of active tasks or cores allocated for performing of cached data results in further minimizing the latency of jobs. Caching of any number subjects does not impact the processing time of queries on the cached data. Extended caching of data beyond the available cache memory degrades the performance of the system.
-
We implemented a prototype system that can process VCF files of any number of subjects. In this prototype system, users will load the data into the cache, after which the system generates and executes queries for the selected criteria’s giving optimal results. The data will remain in the cache as long as user explicitly removes the cache. Hence, the holding of data in cache for longer duration helps users perform various query operations on data iteratively.
The prototype system can be further enhanced to process all formats of genome data along with VCF. In addition, providing a function for customization to individual users would be helpful for more flexible analysis. For example, users may want to configuration certain properties, such as the number of subjects to be cached, the size of the cache memory to be used, the size of the cache memory that is to be made available, and so forth. This work leaves such functions for greater flexibility for composing queries as one of the future studies.
Since the genome data contains sensitive information of an individual, it is important to protect the data from unauthorized access. To ensure the privacy of the data, the system can be enhanced to implement encryption or privacy preserved algorithms and evaluate if there is any performance trade-off between processing data before encryption and after encryption. The presented system in this chapter does not contain a technical module for privacy and it will be an interesting piece of the future research.
References
Human Genome Project: [Online]. Available: https://www.genome.gov/10001772. Accessed 22 Nov 2015 (2003)
DNA Sequencing Costs: [Online]. Available: http://www.genome.gov/sequencingcosts/. Accessed 31 Jan 2016 (2016)
Lewis, S., Csordas, A., Killcoyne, S., Hermjakob, H., Hoopmann, M., Moritz, R., Deutsch, E., Boyle, J.: Hydra: a scalable proteomic search engine which utilizes the Hadoop distributed computing framework. BMC Bioinf., 13, 6 (2012)
Apache Spark Project: [Online]. Available: http://spark.apache.org. Accessed 2015 (2015)
Apache Pig: [Online]. Available: http://pig.apache.org/ (2015)
Apache Hive: [Online]. Available: https://cwiki.apache.org/confluence/display/Hive/Home (2016)
Weil, S.A., Brandt, S.A., Miller, E.L., Long, D.D.E., Maltzahn, C.: Ceph: a scalable, high-performance distributed file system. In: Proceedings of the 7th Symposium on Operating Systems Design and Implementation (2006)
Zaharia, M., Chowdhury, M., Franklin, M.J., Shenker, S., Stoica, I.: Spark: cluster computing with working sets. In: Proceedings of the 2nd short Title USENIX Conference on Hot Topics in Cloud Computing, HotCloud’10, Berkeley, CA, USA (2010)
Zaharia, M., Chowdhury, M., Das, T., Dave, A., Ma, J., McCauley, M., Franklin, M.J., Shenker, S., Stoica, I.: Resilient distributed datasets: a fault-tolerant abstraction for in-memory cluster computing. UCB/EECS (2011)
Apache Spark Documentation: [Online]. Available: http://spark.apache.org (2016)
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., DePristo, M.A.: The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010)
Massie, M., Nothaft, F.A., Hartl, C., Kozanitis, C., Schumacher, A., Joseph, A.D., Patterson, D.: ADAM: Genomics Formats and Processing Patterns for Cloud Scale Computing. DEECS Department, University of California, Berkeley (2013)
O’Brien, A.R., Saunders, N.F.W., Guo, Y., Buske, F.A., Scott, R.J., Bauer, D.C.: VariantSpark: population scale clustering of genotype information. BMC Genomics. 16(1), 1052 (2015)
White, T.: Hadoop: The Definitive Guide. O’Reilly Media, Newton, MA (2015)
VCF File processing: [Online]. Available: http://vcftools.sourceforge.net (2014)
1000 Genome Data: [Online]. Available: http://www.1000genomes.org/data (2014)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this paper

Cite this paper
Syed, M., Hwang, T., Kim, J. (2017). Scalable Gene Sequence Analysis on Spark. In: Suh, S., Anthony, T. (eds) Big Data and Visual Analytics. Springer, Cham. https://doi.org/10.1007/978-3-319-63917-8_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-63917-8_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-63915-4
Online ISBN: 978-3-319-63917-8
eBook Packages: Computer ScienceComputer Science (R0)