
Abstract
In the current era of medicine where clinicians and researchers alike are seeking to personalize treatment plans to individuals, the integration of clinical data with microarray data is surprisingly absent. With this in mind, clinical covariate data was used to pre-select previously classified breast cancer tissue, and employ these classifications to new test cases. The pool of retrieved cases was then reduced further by investigating similar DNA methylation patterns. We first compared breast cancer tissue to normal tissue samples. This work was then extended to differentiating triple-negative breast cancer samples from ER-positive samples followed by investigating these subtypes at a genomic region level. In order to use the clinical covariate data, categorical distance measures were used to locate similar cases before being narrowed down with numeric DNA methylation data. Classification was then carried out using a novel, confidence-based procedure that automatically retrieves solved cases for each test sample until a threshold is met. We find that integrating clinical covariates increases the accuracy within our constructed two-stage system as opposed to using microarray data alone. Further, we outperformed random forest, naive bayes and kNN after refining the cases to a genomic region level.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
In this current era of personalized medicine, clinicians have sought after methods which specifically target the patient through carefully tailored treatment plans. Throughout this movement, clinical and molecular profiles are constructed and managed in unison for advanced treatment. While this is becoming more prevalent on the frontlines of healthcare, the integration is surprisingly absent in ’omics research. The term ’omics collectively refers to genomics, proteomics, epigenomics and similar fields. Here, analysts are typically focused on a specific subtype of ’omics data while paying little attention to the clinical information that define the research sample. Even in studies that span across ’omics, these primary variables are neglected. As these clinical variables are more descriptive, they increase focus and lend to a more explainable outcome. Therefore, it is the intent of this research study to couple a stable epigenetic biomarker, DNA methylation, with clinical data through a case-based reasoning structure built for classifying breast cancer samples. The clinical variables that were selected are the well-distinguished covariates age group, method of therapy, and race. Several studies have found age [4, 9], therapy [7, 14], and race [12] to have a significant effect on DNA methylation levels, making these common confounding variables. With this in mind, we hypothesize that drawing similarities to cases in the case base using these clinical covariates will aid in finding similar samples. Further, we assert that adding clinical covariates will improve diagnostic accuracy.
Specifically, this paper uses CBRMiC (Case-Based Reasoning for Microarray Classification), an R package designed by the authors to use clinical and microarray data for classification tasks. CBRMiC makes use of two iterative feature selection algorithms, computes distance matrices for categorical and numeric data, and classifies using a novel procedure that finds the optimal number of cases for each sample based on a confidence metric.
This paper outlines the methods used to be able to apply case-based reasoning (CBR) and instance-based learning to methylation data, most often analyzed through statistical methods. With four primary processes, retrieve, reuse, revise and retain, CBR is a powerful tool with a transparent problem-solving process. When a new case is presented to a CBR system, the similarity between the new case and previously solved cases (called the case base) is used to retrieve the most similar historic cases (the retrieve step). Then, the problem resolution of the prior cases can either be used to solve the new case (reuse step), or modified to fit any differences (revise step). Finally, the new case is stored in memory to be used in the future (retain step). The measure of similarity is often the most crucial, especially when there are different types of data as is the case within this paper. The most widely used similarity measures are often distance-based functions that compute the distance between cases using some or all of the attributes that define the case. A popular distance measure for numerical data is Euclidean distance (used within this study) though the addition of clinical data requires a categorical distance metric (discussed in Sect. 3.3). Processed DNA methylation is typically in the form of \(\beta \) values. \(\beta \) values are an estimation of the methylation levels between 0 and 1 with 0 being completely non-methylated and 1 being completely methylated. As such, these values are numerical.
Case-based reasoning (CBR) within the domain of microarray analysis is mostly unexplored, especially for epigenetic data. The primary foundation for CBR is its ability to consistently update with new cases, and adapt prior solutions to fit a new problem. Within microarray analysis, however, problems exist that make updating and adaptation particularly difficult. The first problem is the high dimensionality with few samples. There are thousands of features for a small subset of samples (specifically 485,000 for the standard chipset used in DNA methylation), and these samples are often imbalanced between cases and controls. A second problem is that technical variations, called “batch effects”, often exist. Batch effects are alterations of the data that occur when different laboratories, technicians, or different equipment collects the data. Even when the same technicians operate on the same equipment within the same laboratory, subtle varying factors such as the amount of humidity can alter expression levels. These effects can be controlled to some degree through pre-processing, but need to be performed again when new cases are investigated. Lastly, clinical variables are all but ignored in any prior ventures in case-based reasoning for microarray analysis. Typically, researchers investigate similarities in genetic expression levels while disregarding whether samples are similar on a phenotypical level. [16] performed a multiple’omics study across 14 different cancer types and found that integrating clinical variables led to improved prognostic performance, while [13] found that clinical integration increased prediction performance. It is plausible that investigating samples that are similar on a clinical level as well as on a microbiological level will lend to more precise case selection, and therefore greater precision in identifying samples.
Methylation data require a preprocessing pipeline leading to improved analysis, as this article shows. First, potential confounding factors such as batch effects (discussed in Sect. 2.1) are eliminated. Following, methods which cluster the probes into possible functional regions for gene transcription are applied. Feature selection methods are also tested to further refine and select appropriate probes. Eventually, these probes are grouped into genomic regions.
This paper shows that the integration of clinical covariates improves the accuracy over microarray data alone. Further, we compare our results with four other classification algorithms. We have outperformed one of these algorithms during the first two tasks, before outperforming three after refining to a genomic region level.
Specifically, we offer the following significant contributions:
-
1.
Clinical and microarray integration: A methodology that integrates clinical and microarray data, in the form of DNA methylation values. To the best of the author’s knowledge, this is one of the first papers to take clinical covariate factors into account.
-
2.
Tailored case retrieval for each sample: A method which locates a custom-tailored number of similar cases for each sample based on an automatically defined level of confidence in each of the stored cases. Varying the number of cases upon retrieval for each test case, and the method through which it is performed are both novel contributions.
-
3.
Multi-level case elaboration and refinement which examine biological and statistical differences: Significantly different methylation levels in the DNA found at a high-order cluster of probes that serve similar functions were utilized and compared. Lastly, these probes are mapped to genetic regions to capture their precise influence upon the gene.
-
4.
One of the first applications of CBR using methylation data: While studies using gene expression data in a CBR context have been performed previously, very few applications using methylation data have been produced.
2 Background
The term epigenetics was first introduced into modern biology by Conrad Waddington as a means of defining interactions between genes and their products that result in phenotypic variations. Waddington’s landscape presents a cell becoming more differentiated as time goes on. One of the events that can cause this differentiation is methylation. Methylation is a covalent attachment of a methyl group to cytosine. Cytosine (C) is one of the four bases that construct DNA and one of only two bases that can be methylated. While adenine can be methylated as well, cytosine is typically the only base that’s methylated in mammals. Once this methyl group is added, it forms 5-methylcytosine where the 5 references the position on the 6-atom ring where the methyl group is added. Under the majority of circumstances, a methyl group is added to a cytosine followed by a guanine (G) which is known as CpG. While the methyl group is added onto the DNA, it doesn’t alter the underlying sequence but it still has profound effects on the expression of genes and the functionality of cellular and bodily functions. Methylation at these CpG sites has been known to be a fairly stable epigenetic biomarker that usually results in silencing the gene. Further, the amount of methylation can be increased (known as hypermethylation) or decreased (known as hypomethylation) and improper maintenance of epigenetic information can lead to a variety of human diseases.
DNA methylation, tested with a chip known as a microarray, has recently become more prevalent in genetic research studies in oncology. This paper proposes to apply these findings in a study of the diagnostic accuracy of DNA methylation signatures for classifying breast cancer samples when samples are first compared on a clinical level. The first study will be breast cancer samples versus normal tissue samples, while the second study is classifying two specific subtypes of breast cancer. These subtypes are ER-positive samples and Triple Negative samples. Breast cancer cells can be hormone receptor-positive, hormone receptor-negative or triple-negative. Hormone receptor-positive samples have either estrogen (ER) receptors, progesterone (PR) receptors or both. ER-positive breast cancer, specifically, is the most common type of breast cancer that’s currently diagnosed. Triple-negative breast cancer cells do not have estrogen or progesterone receptors and do not make much of the protein called HER2. These cancers tend to grow and spread faster and do not respond to hormone therapies or drugs that target HER2. Due to the common nature of ER-positive breast cancer, and the aggressive nature of Triple Negative breast cancer, we sought a method that could help distinguish the two.
2.1 Research Background
An additive nonparametric margin maximum for case-based reasoning method (ANMM4CBR) was proposed in [15]. ANMM4CBR focuses on the retrieving and reusing stages of CBR and feature selects using additive nonparametric margin maxima. The nonparametric margin maximum is defined based on the nearest between-class distance maximization and the furthest within-cluster distance minimization. They first perform pre-selection and then cluster using hierarchical clustering. Finally, they apply an additive approach where, at each iteration, a feature is selected. When a feature is selected, for each sample the nearest between-class neighbor and furthest within-cluster neighbor may change. They state that maintaining the distance between any two samples in each iteration is computationally expensive and therefore maximize instead of directly optimizing. This allows them to test each feature on a training set and select the top-ranked. In order to reduce redundancy among features, they assign weights to training samples and update the weights where a sample that has a larger margin will receive a lower weight. Surprisingly, their case base is samples in one class. Testing with 10, 20, 30, 40 and 50 iterations on 4 different datasets, ANMM4CBR performed better than LogitBoost, SVM and kNN at every iteration on colon cancer. For leukemia, small round, blue cell tumors (SRBT), and global cancer map (GCM) data, ANMM4CBR had comparable results but did not outperform the others.
[2] built a framework with the kNN classifier as its backing. Also using gene expression data, they tested on child leukemia, colon cancer and prostate cancer. First, they preprocess the training data. Beginning with feature selection, they apply a Balanced Iterative Random Forest (BIRF) algorithm to select the relevant features (discussed further in Sect. 3.5). They follow this stage with dimensionality reduction through principal component analysis and weight features either through eigenvalues or a genetic algorithm. If classes are imbalanced, they oversample using the SMOTE algorithm. During testing, samples are reduced to the selected features and kNN is used to retrieve similar cases. Revision and retention is not employed through their framework. On leukemia, the best results were with a k of 5 which resulted in an average accuracy of 73%. A balanced accuracy of 93% was achieved on the colon data, and 98% on the prostate data.
More recently, [10] proposed a CBR method that visualizes results. The CBR system was rather straight-forward, retrieving cases through a distance measure, though their specialization was in the explainability. Qualitative attributes between cases were shown using rainbow boxes, where labeled and colored rectangles extend through columns that represent the cases, clearly showing what was similar or dissimilar between cases. Quantitative attributes are provided in scatter plots that center on the query case and accurately display the similar cases.
In the domain of instance-based retrieval methods is the work of [3]. Constructed for gene expression data, [3] proposed a modified k-nearest neighbor algorithm. Their methodology consists of projecting the data through the gene expression values, computing the center of each class, and computing the distance between each class item and the center of the class. With this data, they compute each item’s weight. They then compute the distance between the center of the class and a test point and pick the smallest and largest of these distances (Ds and Dl). Neighbors are selected within the circles created with radius Ds and Dl, with the distances between the test item and these neighbors calculated to determine the strength of the neighbors. The test point is then assigned to the class with the highest summation of item strength.
3 Methods
Methylation data for breast cancer (BRCA,Footnote 1) was downloaded from The Cancer Genome Atlas (TCGA,Footnote 2) using the R package TCGAbiolinksGUI [6]. Molecular data was filtered for only the Illumina Human Methylation 450 microarray platform and prepared as an RStudio object. This data pertained to 892 samples and the 485,577 probes that exist on the Illumina Human Methylation 450 beadchip. The methylation \(\beta \) values were then extracted. \(\beta \) values are an estimation of the methylation levels between 0 and 1 with 0 being completely non-methylated and 1 being completely methylated. Tissue samples were either from the primary cancer tumor, normal breast tissue (typically from the opposite, non-cancerous breast), or the metastasized site. For the validation set, cancer tumor tissue was subsetted to only those samples having an ER positive or a Triple Negative status. Similarly, the BRCA clinical data was downloaded. Variables of interest in the clinical data were the age at initial diagnosis, race, and therapy method.
3.1 Data Preprocessing
Metastatic tissue samples (those pertaining to the metastasized site, not the primary cancer site) were discarded from the methylation data, as well as samples from males. Age group was determined based on which decade the sample’s diagnosed age fell into. A sample extracted from an individual who was diagnosed with breast cancer at age 45 would be in group 4, while a sample from someone who was diagnosed at age 53 would be in group 5 for example. Batch effects were then located and removed using the R package ComBat. TCGA barcodes have a plate identifier, and this identifier was used to determine the batch. After pre-processing and batch correction, 782 cancer samples, 96 normal samples and 364,464 features (DNA methylation probes) were used. These probes were then used to locate differentially methylated regions (DMR) using the TCGAbiolinks R package. Differentially methylated regions are clusters of probes that are a possible functional region for gene transcriptional regulation. Here, this process served as a feature reduction mechanism using a biological methodology. The number of features were reduced to 8,722. Once this data was constructed, it was passed into the CBRMiC system where it was tested.
3.2 System Overview
Retrieval and classification was carried out using an author-constructed R package called CBRMiC. CBRMiC is a modular system that allows a user to split data into K-folds, perform class balancing, undergo feature selection, calculate distance matrices for categorical and numeric features and finally classify.
When a dataset is loaded in, it is split into training and testing folds. Information is gleaned from the training folds so that it may serve as a case base for the testing folds. The training data can then be class-balanced and/or feature selected prior to distance calculation.
3.3 Distance Measures
Distance for the microarray features is calculated for a novel test case using Euclidean distance, while clinical (categorical) variables are calculated using either the Goodall3 or Lin measures [5]. The Goodall measure tries to normalize the similarity between two objects by the probability that the similarity value observed could be observed in a random sample of two points. A higher similarity is assigned to a match if the value is infrequent. In its third iteration, the measurement assigns a higher similarity if the matching values are infrequent regardless of the frequencies of the other values. To contrast, the Lin measure assigns a higher weight to mismatches on infrequent values and if there are few other infrequent values. A lower weight is assigned to mismatches if either of the mismatching values are frequent or if there are several values that have frequency in between the mismatching values. Operating these measures results in a distance matrix where test samples have a notated distance to each of the stored cases in the case base.

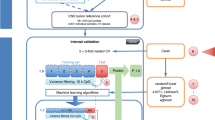
Three query cases and their retrieved cases using the two-stage process. Retrieved cases are based upon similar clinical covariate data as well as a similar microarray signature.
3.4 Retrieval Framework
During classification, similar cases are retrieved for each test sample using a confidence metric. This confidence metric is the computed average of all distances to samples of a different classification minus the average of all distances to samples from the same classification for each case in the case base (shown in Algorithm 1). To frame it in terms of finding a cancerous sample among normal samples, the best cancer sample during training would have the highest average distance to all normal samples and the lowest average distance to all cancer samples. These values are normalized, giving this prototypical case a perfect score of 1.0 to imply 100% confidence.

During the two-stage process, cases in the case base are retrieved for each test case by first finding cases with a similar age group, therapy method and race (shown in Algorithm 2). This method uses the clinical covariate distance matrix calculated with the categorical distance measures outlined above to determine the distance between the test case and the case base. Then, once a pool of these cases has been retrieved, it is further refined using a Euclidean distance matrix from the DNA methylation features. Cases are continuously retrieved based on each case’s confidence value until a threshold is reached. While this paper uses a threshold of 1.0 in its methods, a threshold greater than 1.0 can be established if the user wishes to retrieve more cases. In this manner, a distinct number of cases in the case base are retrieved for each test case. The classification label assigned to a testing case is then the majority label of the retrieved training cases. An example of this methodology in action is depicted in Fig. 1. In this figure, three randomly selected query cases are paired with their retrieved cases. Each query case has a different number of retrieved cases, though they share highly similar clinical traits which aids to the power and explainability of our system.
To summarize, our method is distinguished from the traditional k-Nearest neighbor through its usage of two distance matrices (one computed with a categorical distance measure and one computed with a numeric distance measure), and a confidence threshold that tells the algorithm when to cease retrieving cases.

3.5 Feature Selection
Feature selection algorithms used were Balanced Iterative Random Forest (BIRF), and random KNN (rKNN). BIRF was introduced in [1] and begins with the entire set of features and reduces features with zero importance value at each iteration. It continues to do so while the classification error rate of the training set is less than the classification error of the validation set. At this point, it considers the training set as being overfitted and concludes. rKNN is discussed in [11] and is an ensemble of k-Nearest Neighbor models that are constructed from a random subset of the input variables. A support criterion is used to rank features until the most relevant features can be used for classification. Additionally, it was tested with only the microarray data being used to retrieve cases until the confidence threshold was achieved and again with the two-stage clinical and microarray process.
4 Results
4.1 Test for Confounding Clinical Variables
To test whether the clinical variables had a significant effect on DNA methylation when comparing cancer and normal tissue samples, we first computed the average methylation for the samples using the differentially methylated regions. Then, each sample’s average was separated into its respective age group, therapy group or racial group with samples having an NA status being excluded. A series of single-factor ANOVAs was used to determine if this status had a significant effect on the sample’s average methylation level. For the samples used to differentiate breast cancer tissue from normal tissue, age group was found to have a significant effect on the average methylation level (F(7,855) = 2.29, p = 0.025). Therapy group was not found to have a significant effect on the average methylation level (F(3, 573) = 1.79, p = 0.147). Racial group also did not have a significant effect on the average methylation level (F(4, 874) = 1.93, p = 0.102).
A similar test was performed for the validation set of Triple Negative and ER Positive samples.The average methylation per sample was calculated using the differentially methylated regions. Each sample’s average was separated into its respective age group, therapy group or racial group depending on the test. Again, null values were removed. A series of single-factor ANOVAs was used to determine if these groups had a significant effect on the sample’s average methylation levels. For the samples used to differentiate ER positive from Triple Negative tissue, age group was again found to have a significant effect on the average methylation level (F(6,125) = 2.40, p = 0.03). Therapy group (F(2,82) = 2.30, p = 0.10) and racial group (F(3,128) = 1.53, p = 0.20) were again found to have non-significant effects.
Despite the non-significant effects in therapy and racial group for our datasets, we decided to continue with their usage since there were important differences between the groups, and also due to the support from the medical literature.
4.2 Classification Tasks
Two different stages were conducted, with the first being a subjectively easier classification task while the second was a more arduous task to validate the constructed system by trying to identify specific subtypes. The first task was accurately identifying whether a new sample (query case) was cancerous breast tissue or normal breast tissue. For this test, 782 cancer samples and 96 normal tissue samples were used. After identifying the differentially methylated regions, 8,722 features remained corresponding to probes on the DNA methylation chip. Once this data was constructed, it was passed into the CBRMiC system where it was tested with and without the two feature selection algorithms and with each of the categorical distance measures. These tests were performed using tenfold cross validation.
The second task was to try and differentiate two specific subtypes of breast cancer: ER Positive breast cancer and Triple Negative breast cancer. 48 ER positive samples and 84 Triple Negative samples were found and used in the TCGA BRCA dataset that met our selection criteria. After preprocessing and differentially methylated region analysis, 1,123 features remained. It is important to note that these samples were also used in the previous series of analyses. The tests within the CBRMiC followed the same methodology as the first task.
4.3 First Task: Cancer Classification
For these tests, a confidence threshold of 1.0 was set. Balanced accuracy (computed using the average of per-class accuracy), the F statistic and the Kappa statistic were used for performance metrics. Table 1 has the results for CBRMiC.
As a means of comparison, four other classification algorithms were tested. A kNN algorithm that uses a distance weight of 1/d where d corresponds to the distance and k is found through cross-validation was tested. During this test, the optimal k was found to be 10. Random Forest with 500 iterations, a Support Vector Machine (SVM) and Naive Bayes were also tested. These tests were conducted using the Waikato Environment for Knowledge Analysis (WEKA) [8]. Additionally, we tested CBRMiC using microarray data alone. Table 2 displays the results of these tests with our algorithm for comparison (the microarray stage uses the rKNN and Goodall algorithms).
4.4 Second Task: Cancer Subtype
As aforementioned, the second series of tests was to try and differentiate two specific subtypes of breast cancer: ER Positive breast cancer and Triple Negative breast cancer. This stage follows the same pipeline as in the first task. Table 3 displays the results with our two-stage algorithm.
Similarly, four comparison methods were tested following the same paradigm as with the cancer versus normal tissue dataset. The results of these tests are available in Table 4 (the microarray stage uses the rKNN and Goodall algorithms).
4.5 Initial Findings
As shown, our proposed algorithm outperformed random forest during the first task and SVM during the second task. Further, it can outperform using microarray data alone. While our two-stage algorithm performed better than one traditional algorithm at each of these tasks, we believed that it would produce stronger results when refining the cases to a genomic region level. As the high dimensionality was reduced through integrating probes to a gene level, we believed the additional dimension of the clinical covariate data would aid in differentiating samples.
4.6 Effect of Clinical Covariate Integration
First, we looked to see if integrating the clinical covariates had a significant effect. A series of paired-samples two-tailed t-tests with an alpha of 0.05 were conducted to determine if integrating the clinical covariate variables had a significant effect on the balanced accuracy, F statistic, and Kappa statistic. We utilized the scores obtained when using only the microarray data, and when using the clinical variables in our two-stage process. While we did not observe a significant increase when differentiating cancer from normal samples, we did find a significant increase for ER Positive and Triple Negative samples. Specifically, we found a significant effect on the balanced accuracy (t(5) = −3.238, p = 0.022), a significant effect on the F statistic (t(5) = −3.415, p = 0.018) and a significant effect on the Kappa statistic (t(5) = −3.627, p = 0.015). The effect on balanced accuracy is shown in Table 5. A possible explanation of why we did not observe the significant effect for our first dataset is that the cancer versus normal dataset holds a greater number of samples and a greater number of features. Without the larger sample and feature size, the algorithm may require more data through which to differentiate the samples and draw upon the clinical covariate differences to a greater degree. More tests will need to be conducted to determine whether this theory is correct.
4.7 Case Refinement for Subtype Classification
After these initial tests, we wished to see if refining the cases further would increase our ability to differentiate cancer subtypes. Towards this end, we mapped each DNA methylation probe after DMR analysis to its associated gene and genomic region. Probes within each genomic region had their mean \(\beta \) value calculated. After mapping and calculating the means for each genomic region, 133 regions for the ER positive and Triple Negative data existed. Tests were performed with the two-stage process, as well as with the traditional algorithms as a means of comparison. A k of 15 was chosen when testing the kNN algorithm. As displayed in Table 6, our proposed two-stage algorithm outperformed Naive Bayes, kNN, and SVM. Additionally, we re-display results prior to case refinement to show that associating probes to genomic regions increased classification accuracy for our proposed method. This indicates that initial hypotheses were correct that integrating methylated probes to a genomic level would not only reduce dimensionality, but lead to better performance from our constructed system.
5 Discussion
Results indicate that integrating clinical covariates performs better than microarray data alone within our proposed system during all tasks, and holds strong results after refining the cases to a genomic region level. During the first task, our two-stage system CBRMiC outperformed random forest, outperformed SVM during the second task, and outperformed Naive Bayes, kNN and SVM after case refinement. We believe that this current iteration of CBRMiC performs stronger on datasets with reduced dimensionality and will seek to strengthen the system to more appropriately handle the high dimensionality of DNA methylation data. We believe that a further increase in performance may occur with the addition of other clinical variables, so that there are more dimensions through which to differentiate the samples. Future directions will be to evaluate the confidence metric and search for other mathematical formulae to instill a single value upon samples as a means of determining how well that sample lends itself towards classification. We would also like to test our methodology on other independent datasets as well as incorporating other clinical information. Still, we have found results that display the utility of integrating clinical covariates with microarray information as well as the strength of using case-based reasoning for cancer classification.
References
Anaissi, A.: Case-base retrieval of childhood leukaemia patients using gene expression data, January 2013
Anaissi, A., Goyal, M., Catchpoole, D.R., Braytee, A., Kennedy, P.J.: Case-based retrieval framework for gene expression data. Cancer Inform. 14, 21–31 (2015). https://doi.org/10.4137/CIN.S22371
Ayyad, S.M., Saleh, A.I., Labib, L.M.: Gene expression cancer classification using modified k-nearest neighbors technique. Biosystems 176, 41–51 (2019). https://doi.org/10.1016/j.biosystems.2018.12.009
Bell, J.T., et al.: Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 8(4) (2012). https://doi.org/10.1371/journal.pgen.1002629
Boriah, S., Chandola, V., Kumar, V.: Similarity measures for categorical data: A comparative evaluation. In: Proceedings of the 2008 SIAM International Conference on Data Mining (2008). https://doi.org/10.1137/1.9781611972788.22
Colaprico, A., et al.: Tcgabiolinks: An R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. (2015). https://doi.org/10.1093/nar/gkv1507
Flanagan, J.M., et al.: Platinum-based chemotherapy induces methylation changes in blood dna associated with overall survival in patients with ovarian cancer. Clin. Cancer Res. 23(9), 2213–2222 (2016). https://doi.org/10.1158/1078-0432.ccr-16-1754
Hall, M., Frank, E., Holmes, G., Pfahringer, B., Reutemann, P., Witten, I.H.: The WEKA data mining software: An update. SIGKDD Explor. 11(1), 10–18 (2009)
Horvath, S., et al.: Aging effects on DNA methylation modules in human brain and blood tissue. Gen. Biol. 13(10) (2012). https://doi.org/10.1186/gb-2012-13-10-r97
Lamy, J.B., Sekar, B., Guezennec, G., Bouaud, J., Séroussi, B.: Explainable artificial intelligence for breast cancer: A visual case-based reasoning approach. Artif. Intell. Med. 94, 42–53 (2019). https://doi.org/10.1016/j.artmed.2019.01.001
Li, S., Harner, E.J., Adjeroh, D.A.: Random KNN feature selection - A fast and stable alternative to random forests. BMC Bioinforma. 12(1), 450 (2011). https://doi.org/10.1186/1471-2105-12-450, http://www.biomedcentral.com/1471-2105/12/450
Song, M.A., et al.: Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics 10(12), 1177–1187 (2015). https://doi.org/10.1080/15592294.2015.1121362
van Vliet, M.H., Horlings, H.M., van de Vijver, M.J., Reinders, M.J., Wessels, L.F.: Integration of clinical and gene expression data has a synergetic effect on predicting breast cancer outcome. PLoS ONE 7(7) (2012). https://doi.org/10.1371/journal.pone.0040358
Yang, G.S., et al.: Differential DNA methylation following chemotherapy for breast cancer is associated with lack of memory improvement at one year. Epigenetics, 1–12 (2019). https://doi.org/10.1080/15592294.2019.1699695
Yao, B., Li, S.: ANMM4CBR: A case-based reasoning method for gene expression data classification. Algorithm. Mol. Biol. 5(1), 1–11 (2010). https://doi.org/10.1186/1748-7188-5-14
Zhu, B., et al.: Integrating clinical and multiple omics data for prognostic assessment across human cancers. Sci. Rep. 7(1), 1–13 (2017). https://doi.org/10.1038/s41598-017-17031-8
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this paper

Cite this paper
Bartlett, C.L., Liu, G., Bichindaritz, I. (2020). Classifying Breast Cancer Tissue Through DNA Methylation and Clinical Covariate Based Retrieval. In: Watson, I., Weber, R. (eds) Case-Based Reasoning Research and Development. ICCBR 2020. Lecture Notes in Computer Science(), vol 12311. Springer, Cham. https://doi.org/10.1007/978-3-030-58342-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-58342-2_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-58341-5
Online ISBN: 978-3-030-58342-2
eBook Packages: Computer ScienceComputer Science (R0)