
Abstract
Secondary acute myeloid leukemia (s-AML) is recognized as AML arising from prior hematological disease, usually myelodysplastic syndrome, myeloproliferative neoplasm, or aplastic anemia or has myelodysplasia related changes or develops in context of prior cytotoxic or radiation therapy. Understanding the biology of s-AML is of utmost importance as this portends a high-risk disease including a potential for lower responses to chemotherapy. Some therapeutic developments in recent times have been CPX-351 as well as venetoclax-containing combinations. Of particular ongoing and forthcoming interest is the role of targeted therapy itself or in combination with other agents. Lastly, the role of allogeneic stem cell transplantation remains crucial although debated, and with improvement in treatment strategies provides opportunities to use the two in combination for best results.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction: What Is Secondary AML?
Acute myeloid leukemia (AML) is the most common type of acute leukemia, the incidence of which increases with advancing age. The etiology remains elusive for the most part, but development following a prior cytotoxic agent or as a consequence of an antecedent myeloid disorder has been widely recognized. In the 1997 World Health Organization (WHO) classification of neoplastic disease of the hematopoietic and lymphoid tissues, AML with multilineage dysplasia, defined as dysplastic changes in two or more cell lines, and therapy-related AML were recognized as distinct entities due to morphological differences from de novo AML, characteristic cytogenetic abnormalities, and worse prognosis [1].
Secondary acute myeloid leukemia (s-AML) in the current era informally refers to the AML that evolves from an antecedent hematological disease (AML-AHD), usually a myeloid malignancy such as myelodysplastic syndrome (MDS), myeloproliferative neoplasms (MPN), or aplastic anemia; has myelodysplasia-related changes (AML-MRC); or develops after exposure to a cytotoxic chemotherapy or radiation treatment for a prior neoplasm (t-AML) (Table 3.1). Per the 2016 WHO classification, AML with MRC is defined as AML meeting at least one of the following criteria: (a) presence of 50% of more dysplastic cells in at least two cell lines and in the absence of favorable-risk mutations of NPM1 or biallelic CEBPA or del(9q) [2]; (b) an antecedent hematologic disorder (MDS or MDS/MPN); or (c) presence of an MDS-related cytogenetic abnormality.
t-AML has been associated with prior chemotherapy, typically alkylating agents and DNA topoisomerase II inhibitors, as well as prior radiation therapy. Alkylating agents such as melphalan, cyclophosphamide, and nitrogen mustard may lead to dysplastic changes similar to MDS, bi- or tri-lineage cytopenias, abnormalities of chromosomes 5 and 7 or both, and complex karyotype, typically with a latency of over 5 years from exposure [3,4,5,6]. These karyotypes have been reported to comprise 76% of all abnormal karyotype in one series of patients with t-AML or therapy-related MDS [7]. t-AML associated with topoisomerase II inhibitors, for example, doxorubicin, etoposide, and mitoxantrone, is characterized by shorter interval (around 2–3 years) between exposure and diagnosis, absence of preceding MDS, and a genetic abnormality involving translocation of MLL gene on chromosome 11, band q23, and RUNX1/AML1 gene on chromosome 21, band q22 [7,8,9,10]. t-AML has also been described following the use of other chemotherapy agents such as azathioprine, 5-flourouracil, methotrexate, 6-mercaptopurine, and fludarabine [7, 11, 12]).
2 Why Does It Matter?
Two large population studies reported that s-AML constituted approximately one in every four cases of AML diagnosed between 1997–2006 and 2000–2013 in these respective studies [13, 14]). More specifically, AML-AHD has been reported in approximately 16–19% and t-AML in approximately 7% of all AML diagnoses in several studies [13,14,15,16]. This number may reasonably be expected to rise due to improved survival following chemotherapy for prior malignancies.
More importantly, numerous studies over the years have shown inferior survival in patients with s-AML compared to de novo AML, which makes it imperative to recognize this as a prognostic factor, and indeed an unmet need for treatment options [13,14,15,16]. Response to standard chemotherapy in patients with s-AML has also traditionally been reported to be lower than response rates seen with de novo AML. This is likely a result of a higher incidence of high-risk genetic and molecular features, genetic alterations, or selection of chemotherapy-resistant clones in cases of prior treatment of the AHD. The high-risk cytogenetic and mutational profile in s-AML, such as mutations in tumor protein (TP53) and complex karyotype, may render leukemia cells less responsive to chemotherapy and a poorer overall survival [17]. Mechanism of resistance and resulting lower response to chemotherapy in s-AML have been attributed to an over-expression of multidrug resistance gene 1 (MDR1) which results in increase in efflux pumps such as p-glycoprotein leading to a decreased intracellular concentrations and overall exposure to anthracycline chemotherapy, hence rendering the resistance to drugs [18, 19]. Other plausible mechanisms of resistance include expression of proteins conferring multidrug resistance such as multidrug resistance-associated protein 1 (MRP1) and lung resistance protein (LRP) [20, 21]. Of these, MDR1 expression and the resultant drug efflux has been reported to increase with increasing age from 17% in age <35 years while being 39% in age >50 years, which at least partly contributes to decreased responses to chemotherapy in s-AML which is commonly seen in older patients [19]. Additionally, patients with s-AML are older and may have been treated previously with cytotoxic agents, both of which can possibly limit the ability to utilize high dose or cytotoxic chemotherapy to treat s-AML [16, 22].
To summarize, understanding the biology as well as management strategies for s-AML is important in treatment planning and risk-stratification as the incidence of this diagnosis is anticipated to rise, response as well as survival in these patients is inferior compared to de novo AML, and treatment options can be limited by age or other patient factors.
3 Biology and Genomics of Secondary AML: Are They Different and How?
Clonal hematopoiesis plays an important role in the development of s-AML from both an AHD and in t-AML [23, 24]. It is now being realized that clonal hematopoiesis exists early, likely even prior to exposure to cytotoxic therapy in t-AML. Subsequent exposure to chemotherapy results in selection of a drug resistant preleukemic clone. Mutant TP53 clones have been found years prior to chemotherapy exposure or diagnosis of t-AML [25, 26]. Recent studies have also shown that both hematopoietic and stromal compartments of the bone marrow are involved in the malignant transformation of hematopoiesis and that mesenchymal stem cells undergo remodeling upon exposure to MDS cells [27].
3.1 AML-AHD from Preceding MDS
Transformation of MDS to AML (AML-MDS) is mediated by clonal evolution or increase in the number of mutations as well as expansion of existing mutant clones. A variety of cytogenetic abnormalities and mutations in genes affecting splicing machinery and chromatin modifiers have been reported more commonly in AML-MDS than in de novo AML, including SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, and STAG2 [28]. Paired sampling of MDS and AML-MDS bone marrow samples suggested that at least one new driver mutation occurs in most (59% in the study) patients in the process of transformation, except in patients with TP53 mutation [28]. Most of these mutations were in genes encoding myeloid transcription factors (RUNX1, CEBPA, GATA2) and signal transduction proteins (FLT3 or RAS). Another study comparing samples of patients with s-AML versus high-risk MDS showed enrichment of NRAS, FLT3, WT1, NPM1, IDH1/2, PTPN11 genes in s-AML [29]. Whole exome sequencing studies have demonstrated that mutations in signaling pathways (such as NRAS and PTPN11) occur or expand significantly during transformation of disease [30]. This was, however, not observed for mutations associated with DNA methylation or splicing machinery, which were noted to expand at the initial stages of MDS but not at the time of progression to s-AML. Another study using a comprehensive transcriptome sequencing in CD34+ bone marrow cells identified two subgroups of MDS by gene expression profiling: one with increased expression of genes in the erythroid/megakaryocytic lineage and the second with upregulation of genes related to immature progenitor cells. The later demonstrated upregulation of various signaling pathways and downregulation of pathways related to metabolism and DNA repair and was exclusively associated with leukemic transformation and shorter survival [31].
Identification of the above-mentioned mutations in patients with MDS may herald emerging AML subclones and potentially identify patients at risk for transformation to s-AML.
3.2 AML-AHD from MPN
MPN are another set of myeloid disorders that can potentially transform into AML (AML-MPN) with a rather dismal prognosis [32]. In a single-center study of 91 patients, a clonal abnormality was identified in 91% patients including complex karyotype (54%), core binding factor (CBF) gene mutations (3%), and chromosome 5 or 7 abnormalities (32%) [32]. The three known driver mutations in myelofibrosis, JAK2V617F, CALR or MPL, also impact time to leukemia transformation. Mutations in CALR were associated with lesser risk of transformation when compared with “triple-negative” (absence of all three mutations) and JAK2V617F, but with no difference compared to mutations in MPL [33]. Subsequently, genomic profiling of samples from patients with AML-MPN has delineated differences from genetics patterns of de novo AML. Recurrent point mutations in TET2, ASXL1, SRSF2, TP53, and IDH1/2 have been reported to be more common in AML-MPN than in de novo AML [34,35,36,37]. Single-nucleotide polymorphism (SNP) array analysis of 88 chronic phase MPN and 71 MPN-blast phase samples showed three-times higher genomic changes in the latter [38]). Aberrations in chromosomes 3p (FOXP1), 4q (TET2), 7p (IKZF1), 7q (CUX1), 8q (MYC), 12p (ETV6), 17p (TP53), 21q (RUNX1) were seen more commonly in AML-MPN samples (MPN-blast phase) than in chronic phase [38, 39]. Genomic profiling of AML-MPN samples demonstrated a higher frequency of somatic TP53 mutations accompanying JAK2V617F mutations, in contrast to chronic phase MPN samples where TP53 mutations were less frequent [40]. TP53 loss in combination with JAK2V617F mutation led to expansion of blasts, in both the bone marrow and the peripheral blood, and fully penetrant AML in murine models, thus biologically validating these genomic observations.
Collectively, these data demonstrate that recurrent mutations in epigenetic regulators, transcription factors, spliceosome complex members as well as TP53 have been associated with the leukemia transformation of MPNs. This mutational spectrum appears to differ from that observed in de novo AML, thus potentially explaining biological differences and clinical behavior of AML-MPN.
3.3 t-AML
t-AML has a high representation of high-risk or unfavorable cytogenetic abnormalities and somatic mutations. In a series of 306 patients with t-MDS/t-AML from a single center, over 70% patients had del(5q), del(7q), or monosomy 7 [7]. Of these, chromosome 7 abnormalities [del(7q) and monosomy 7] occur in almost half of these patients and is associated with mutations that activate the RAS pathway [41, 42]. TP53 mutations occur in up to 37% of patients with t-AML and can be associated with del(5q) as well as complex karyotype [28, 43]. Around one-third patients with t-AML can harbor mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 [28]. Additionally, association of MLL rearrangements and exposure to topoisomerase II has been described above.
Overall, the genetic composition of t-AML is notable for a higher frequency of unfavorable mutations that are associated with poor response to therapy and inferior outcomes.
4 Management of s-AML: How to Think of Treatment Options?
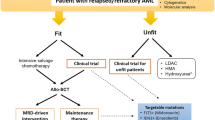
The overall prognosis of patients with s-AML remains poor whether they arise from an antecedent MDS, MPN, or t-AML; and treatment options have not changed significantly in over four decades. A proposed treatment schema with contemporary options is depicted in Fig. 3.1.

Treatment schema for secondary AML
4.1 Induction and Chemotherapy: Past, Present, and Future
Anthracycline-based therapy, in combination with cytarabine, in the historic “7+3” regimen has remained the traditional therapy for s-AML, although response rates have been lower than that with de novo AML [13, 14, 44]. While both AML-AHD and t-AML show inferior outcomes compared to de novo AML, s-AML arising from AHD other than MDS was associated with an even lower survival in patients over 60 years of age with adverse karyotype [13].
CPX-351 is a novel agent, recently approved by Food and Drug Administration (FDA) and European Medicine Agency (EMA), for treatment of newly diagnosed t-AML or AML-MRC. It is the dual-drug liposomal encapsulation of cytarabine and daunorubicin at a fixed 5:1 molar ratio at which maximal synergistic activity had been demonstrated in preclinical models [45,46,47]. The encapsulated liposomal formulation improves pharmacokinetic characteristics and allows for intracellular delivery which is enhanced in leukemia cells compared to normal cells. The particular benefit of CPX-351 in patients with s-AML was seen in a randomized phase 2 study conducted in patients with newly diagnosed AML of ages 60–75 years comparing CPX-351 with the standard 7+3 regimen [48]. While a statistical significant advantage in overall survival or event-free survival was not seen for the overall study population, a planned subgroup analysis in high-risk patients including those with s-AML showed a significantly superior overall survival in this subgroup (HR 0.46, p = 0.01). These findings were confirmed in a phase 3 trial of patients with ages 60–75 years with newly diagnosed t-AML or AML-MRC. The study showed significantly superior overall survival compared to 7+3 induction (9.56 vs 5.95 months, HR 0.69, p = 0.003) along with higher remission rates (47.7% vs 33.3%, p = 0.016) and improved event-free survival (2.53 vs 1.31 months, HR 0.74, p = 0.021) [49]. An exploratory landmark analysis in patients who underwent allogeneic stem cell transplantation (HCT) after induction was notable for a significantly favorable overall survival with CPX-351 (median not reached vs 10.25 months, HR 0.46, p = 0.009). Although this analysis was influenced by confounding factors such as that the decision to undergo HCT was not randomized, these results do establish CPX-351 as a suitable treatment option as a bridge to HCT.
Azanucleosides are pyrimidine analogs which at lower doses have established themselves as potent inhibitors of DNA methylation and are commonly referred to as hypomethylating agents. Due to the noted significance of DNA methylation in transformation of MDS to AML, hypomethylating agents have an established role in treatment of patients with s-AML especially those who are not candidates for high dose chemotherapy [50, 51]. The two commonly used hypomethylating agents are 5-azacytadine and 5-aza-2′-deoxycytidine (or decitabine). Azacitidine was FDA approved for use in MDS based on data from the AZA-001 trial which included patients with up to 30% blasts, which would now be classified as AML for >20% blasts, and showed improved survival compared to best supportive care [52]. Subsequently, a phase 3 study (AZA-AML-001) was conducted in patients with AML, including AML-AHD and AML-MRC, who were ≥65 years old and continued to show improved overall survival with azacitidine versus best supportive therapy (median, 8.9 vs 4.9 months, HR 0.74) [53]. Similarly, decitabine was compared to conventional therapy in older patients with AML, resulting improved response rate (17.8% vs 7.8%, OR 2.5, p = 0.001) although no statistical improvement in survival was reported in the primary analysis (7.7 vs 5 months, p = 0.108) until an unplanned analysis a year later (HR 0.82, p = 0.037) [54]. Both azacitidine and decitabine have been reported to have responses in AML-MPN although most reports are relatively small series. Decitabine showed responses in 6/21 (29%) patients with AML-MPN, including one partial response, with the response lasting a median of 7 months [55]. Azacitidine therapy resulted in an overall response in 10 (38%) with complete response in 2 (8%) of the 26 patients with AML-MPN, with median duration of response of 9 months [56]. Ruxolitinib, a selective JAK1 and JAK2 inhibitor, as a single agent also has moderate activity with responses seen in 3/18 (17%) patients with AML-MPN, of which 2 were complete responses [57]. Given the individual activity with these agents and the synergistic activity demonstrated in murine models of JAK2V617-driven AML, a phase 1 trial was conducted using a combination of decitabine and ruxolitinib in patients with accelerated phase (10–19% blasts in peripheral blood or bone marrow) and blast phase (>20% blasts in peripheral blood and bone marrow) [58]. No dose-limiting toxicity was observed, and overall responses were seen in 5/13 (39%) patients with AML-MPN.
The more recent success story in the treatment paradigm of AML is venetoclax, an orally administered B-cell leukemia/lymphoma-2 (bcl-2) inhibitor, that has been studied in various combinations with hypomethylating agents as well as low-dose cytarabine in older patients with AML who are not candidates for intensive therapy. Approximately one quarter to half of the patients in these studies have s-AML and have shown responses ranging from 8% to 35% in these patients when utilized as upfront therapy [59,60,61]. It should be noted though that in these studies with venetoclax/hypomethylating agent combinations, no patients with prior hypomethylating agent exposure were included (Table 3.2).
4.2 Transplantation or No Transplantation? The Perpetual Enigma
Prospective or randomized studies to compare outcomes with versus without HCT do not exist and are unlikely to be conducted. Studies to date show variable outcomes regarding this, which is likely the result of patient selection in these single institution or retrospective studies, but do favor improved outcomes in patients who undergo HCT after achieving a complete response [62,63,64,65,66,67]. While HCT remains the only potential curative option, s-AML has been identified as an independent risk factor for poorer outcomes after HCT [68]. Hence, careful consideration to the various clinical and genetic factors that impact HCT outcomes is warranted to identify who would benefit most from an HCT. Data from the Center for International Bone Marrow Transplant Research (CIBMTR) using HCT data between 1990 and 2004 for t-AML or t-MDS and from European Society for Blood and Bone Marrow Transplantation (EBMT) of HCTs performed between 2000 and 2016 for s-AML have demonstrated inferior survival with advancing age at HCT, poor risk cytogenetics, active disease at HCT, and alternative donors (other than HLA-identical sibling or partially or well-matched unrelated donor) [69, 70]. Among genetic markers, a study from Japan Marrow Donor Program, that included 24% patients with s-AML showed that patients with mutations in NRAS (HR 1.64, p = 0.0075), TP53 (HR 1.49, p = 0.0096), CBL (HR 1.55, p = 0.024) as well as complex karyotype (HR 1.45, p = 0.046) had particularly inferior outcomes post-HCT [68].
Emergence of therapeutic options prior to HCT increases the optimism for ability to achieve deeper remissions and possibly improving the outcomes from HCT. The possibility of incorporation of the novel therapies as maintenance strategies after HCT and availability of these drugs for treatment of relapse post-HCT further pave the way to utilize the graft-versus-leukemia in combination with the targeted agents.
5 Future Directions
Conventional therapeutic options have demonstrated limited activity in s-AML. Identification of novel recurrent somatic mutations in MDS, MPN, as well as post-MPN AML have helped to improve our understanding of disease biology, as well as to elucidate novel therapeutic possibilities. An example is the presence of IDH1/2 mutations that is seen in over 20% patients with AML-MPN (versus around 4% in chronic phase MPN) [71, 72]. Ivosidenib and enasidenib have shown promising activity in relapsed/ refractory AML with IDH1 and IDH2 mutations, respectively [73, 74]. Whether these agents can be used in combination with other active agents in patients with s-AML as front-line therapy is being actively studied with encouraging early results. As mentioned above, mutations in TP53 are also enriched in patients with progression to AML following MDS or MPN, which presents a particularly challenging problem. The role of investigational drugs such as APR-246, with the potential ability to restore the function of point mutant TP53 in tumor cells, is currently being explored in TP53 mutated myeloid malignancies in combination with azacitidine (NCT 03072043). Additionally, novel venetoclax-based or CPX-351-based combination studies with targeted therapies are in progress. Collectively, these recent biological and clinical insights have the potential to alter the historically poor clinical course of patients with s-AML, and hopefully yield better options and outcomes.
Abbreviations
- AHD:
-
Antecedent hematological disorder
- AML:
-
Acute myeloid leukemia
- CBF:
-
Core binding factor
- CIBMTR:
-
Center for International Bone Marrow Transplant Research
- EBMT:
-
European Society for Blood and Bone Marrow Transplantation
- HCT:
-
Hematopoietic cell transplantation
- JAK:
-
Janus kinase
- MDS:
-
Myelodysplastic syndrome
- MPN:
-
Myeloproliferative neoplasm
- SNP:
-
Single-nucleotide polymorphism
- TP:
-
Tumor protein
- WHO:
-
World Health Organization
References
Harris NL, Jaffe ES, Diebold J, et al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835–49.
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Friedberg JW, Neuberg D, Stone RM, et al. Outcome in patients with myelodysplastic syndrome after autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Oncol. 1999;17:3128–35.
Koontz MZ, Horning SJ, Balise R, et al. Risk of therapy-related secondary leukemia in Hodgkin lymphoma: the Stanford University experience over three generations of clinical trials. J Clin Oncol. 2013;31:592–8.
Le Beau MM, Albain KS, Larson RA, et al. Clinical and cytogenetic correlations in 63 patients with therapy-related myelodysplastic syndromes and acute nonlymphocytic leukemia: further evidence for characteristic abnormalities of chromosomes no. 5 and 7. J Clin Oncol. 1986;4:325–45.
Travis LB, Holowaty EJ, Bergfeldt K, et al. Risk of leukemia after platinum-based chemotherapy for ovarian cancer. N Engl J Med. 1999;340:351–7.
Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102:43–52.
Pedersen-Bjergaard J, Philip P. Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA-topoisomerase II. Blood. 1991;78:1147–8.
Rowley JD, Olney HJ. International workshop on the relationship of prior therapy to balanced chromosome aberrations in therapy-related myelodysplastic syndromes and acute leukemia: overview report. Genes Chromosomes Cancer. 2002;33:331–45.
Takeyama K, Seto M, Uike N, et al. Therapy-related leukemia and myelodysplastic syndrome: a large-scale Japanese study of clinical and cytogenetic features as well as prognostic factors. Int J Hematol. 2000;71:144–52.
Ertz-Archambault N, Kosiorek H, Taylor GE, et al. Association of therapy for autoimmune disease with myelodysplastic syndromes and acute myeloid leukemia. JAMA Oncol. 2017;3:936–43.
Offman J, Opelz G, Doehler B, et al. Defective DNA mismatch repair in acute myeloid leukemia/myelodysplastic syndrome after organ transplantation. Blood. 2004;104:822–8.
Granfeldt Ostgard LS, Medeiros BC, Sengelov H, et al. Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33:3641–9.
Hulegardh E, Nilsson C, Lazarevic V, et al. Characterization and prognostic features of secondary acute myeloid leukemia in a population-based setting: a report from the Swedish Acute Leukemia Registry. Am J Hematol. 2015;90:208–14.
Boddu P, Kantarjian HM, Garcia-Manero G, et al. Treated secondary acute myeloid leukemia: a distinct high-risk subset of AML with adverse prognosis. Blood Adv. 2017a;1:1312–23.
Kayser S, Dohner K, Krauter J, et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117:2137–45.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19:1405–13.
Leith CP, Kopecky KJ, Godwin J, et al. Acute myeloid leukemia in the elderly: assessment of multidrug resistance (MDR1) and cytogenetics distinguishes biologic subgroups with remarkably distinct responses to standard chemotherapy. A Southwest Oncology Group study. Blood. 1997;89:3323–9.
Leith CP, Kopecky KJ, Chen IM, et al. Frequency and clinical significance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia: a Southwest Oncology Group Study. Blood. 1999;94:1086–99.
Cole SP, Bhardwaj G, Gerlach JH, et al. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–4.
Scheffer GL, Wijngaard PL, Flens MJ, et al. The drug resistance-related protein LRP is the human major vault protein. Nat Med. 1995;1:578–82.
Head DR. Revised classification of acute myeloid leukemia. Leukemia. 1996;10:1826–31.
Gillis NK, Ball M, Zhang Q, et al. Clonal haemopoiesis and therapy-related myeloid malignancies in elderly patients: a proof-of-concept, case-control study. Lancet Oncol. 2017;18:112–21.
Takahashi K, Wang F, Kantarjian H, et al. Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid neoplasms: a case-control study. Lancet Oncol. 2017;18:100–11.
Schulz E, Kashofer K, Heitzer E, et al. Preexisting TP53 mutation in therapy-related acute myeloid leukemia. Ann Hematol. 2015;94:527–9.
Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–5.
Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14:824–37.
Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–76.
Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49:204–12.
Kim T, Tyndel MS, Kim HJ, et al. The clonal origins of leukemic progression of myelodysplasia. Leukemia. 2017;31:1928–35.
Shiozawa Y, Malcovati L, Galli A, et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood. 2017;130:2642–53.
Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105:973–7.
Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124:2507–13; quiz 615
Abdel-Wahab O, Manshouri T, Patel J, et al. Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res. 2010;70:447–52.
Green A, Beer P. Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med. 2010;362:369–70.
Harutyunyan A, Klampfl T, Cazzola M, Kralovics R. p53 lesions in leukemic transformation. N Engl J Med. 2011;364:488–90.
Zhang SJ, Rampal R, Manshouri T, et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood. 2012;119:4480–5.
Thoennissen NH, Krug UO, Lee DH, et al. Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms. Blood. 2010;115:2882–90.
Klampfl T, Harutyunyan A, Berg T, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood. 2011;118:167–76.
Rampal R, Ahn J, Abdel-Wahab O, et al. Genomic and functional analysis of leukemic transformation of myeloproliferative neoplasms. Proc Natl Acad Sci U S A. 2014;111:E5401–10.
McNerney ME, Brown CD, Peterson AL, et al. The spectrum of somatic mutations in high-risk acute myeloid leukaemia with −7/del(7q). Br J Haematol. 2014;166:550–6.
Side LE, Curtiss NP, Teel K, et al. RAS, FLT3, and TP53 mutations in therapy-related myeloid malignancies with abnormalities of chromosomes 5 and 7. Genes Chromosomes Cancer. 2004;39:217–23.
Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45.
Schoch C, Kern W, Schnittger S, Hiddemann W, Haferlach T. Karyotype is an independent prognostic parameter in therapy-related acute myeloid leukemia (t-AML): an analysis of 93 patients with t-AML in comparison to 1091 patients with de novo AML. Leukemia. 2004;18:120–5.
Lim WS, Tardi PG, Dos Santos N, et al. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine:daunorubicin formulation, in bone marrow xenografts. Leuk Res. 2010;34:1214–23.
Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5:1854–63.
Tardi P, Johnstone S, Harasym N, et al. In vivo maintenance of synergistic cytarabine:daunorubicin ratios greatly enhances therapeutic efficacy. Leuk Res. 2009;33:129–39.
Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123:3239–46.
Lancet JE, Uy GL, Cortes JE, et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36:2684–92.
Jiang Y, Dunbar A, Gondek LP, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113:1315–25.
Ruter B, Wijermans PW, Lubbert M. DNA methylation as a therapeutic target in hematologic disorders: recent results in older patients with myelodysplasia and acute myeloid leukemia. Int J Hematol. 2004;80:128–35.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32.
Seymour JF, Dohner H, Butrym A, et al. Azacitidine improves clinical outcomes in older patients with acute myeloid leukaemia with myelodysplasia-related changes compared with conventional care regimens. BMC Cancer. 2017;17:852.
Kantarjian HM, Thomas XG, Dmoszynska A, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol. 2012;30:2670–7.
Badar T, Kantarjian HM, Ravandi F, et al. Therapeutic benefit of decitabine, a hypomethylating agent, in patients with high-risk primary myelofibrosis and myeloproliferative neoplasm in accelerated or blastic/acute myeloid leukemia phase. Leuk Res. 2015;39:950–6.
Thepot S, Itzykson R, Seegers V, et al. Treatment of progression of Philadelphia-negative myeloproliferative neoplasms to myelodysplastic syndrome or acute myeloid leukemia by azacitidine: a report on 54 cases on the behalf of the Groupe Francophone des Myelodysplasies (GFM). Blood. 2010;116:3735–42.
Eghtedar A, Verstovsek S, Estrov Z, et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood. 2012;119:4614–8.
Rampal RK, Mascarenhas JO, Kosiorek HE, et al. Safety and efficacy of combined ruxolitinib and decitabine in accelerated and blast-phase myeloproliferative neoplasms. Blood Adv. 2018;2:3572–80.
DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17.
Maiti A, DiNardo CD, Cortes JE, et al. Interim analysis of phase II study of venetoclax with 10-day decitabine (DEC10-VEN) in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2018;132:286.
Wei AH, Strickland SA Jr, Hou JZ, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37:1277–84.
Boddu PC, Kantarjian HM, Ravandi F, et al. Characteristics and outcomes of older patients with secondary acute myeloid leukemia according to treatment approach. Cancer. 2017b;123:3050–60.
Cherington C, Slack JL, Leis J, et al. Allogeneic stem cell transplantation for myeloproliferative neoplasm in blast phase. Leuk Res. 2012;36:1147–51.
Kennedy JA, Atenafu EG, Messner HA, et al. Treatment outcomes following leukemic transformation in Philadelphia-negative myeloproliferative neoplasms. Blood. 2013;121:2725–33.
Oosterveld M, Muus P, Suciu S, et al. Chemotherapy only compared to chemotherapy followed by transplantation in high risk myelodysplastic syndrome and secondary acute myeloid leukemia; two parallel studies adjusted for various prognostic factors. Leukemia. 2002;16:1615–21.
Oosterveld M, Suciu S, Verhoef G, et al. The presence of an HLA-identical sibling donor has no impact on outcome of patients with high-risk MDS or secondary AML (sAML) treated with intensive chemotherapy followed by transplantation: results of a prospective study of the EORTC, EBMT, SAKK and GIMEMA Leukemia Groups (EORTC study 06921). Leukemia. 2003;17:859–68.
Shin SH, Yahng SA, Yoon JH, et al. Survival benefits with transplantation in secondary AML evolving from myelodysplastic syndrome with hypomethylating treatment failure. Bone Marrow Transplant. 2013;48:678–83.
Yoshizato T, Nannya Y, Atsuta Y, et al. Genetic abnormalities in myelodysplasia and secondary acute myeloid leukemia: impact on outcome of stem cell transplantation. Blood. 2017;129:2347–58.
Litzow MR, Tarima S, Perez WS, et al. Allogeneic transplantation for therapy-related myelodysplastic syndrome and acute myeloid leukemia. Blood. 2010;115:1850–7.
Sengsayadeth S, Labopin M, Boumendil A, et al. Transplant outcomes for secondary acute myeloid leukemia: acute leukemia working party of the European Society for blood and bone marrow transplantation study. Biol Blood Marrow Transplant. 2018;24:1406–14.
Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A. IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia. 2010;24:1146–51.
Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24:1302–9.
DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98.
Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jain, T., Rampal, R.K. (2021). Insights into the Pathobiology of Secondary AML. In: Faderl, S.H., Kantarjian, H.M., Estey, E. (eds) Acute Leukemias. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-53633-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-53633-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-53632-9
Online ISBN: 978-3-030-53633-6
eBook Packages: MedicineMedicine (R0)