
Abstract
Tissue-resident memory T (TRM) cells are strategically positioned within the epithelial layers of many tissues to provide enduring site-specific immunological memory. This unique T-cell lineage is endowed with the capacity to rapidly respond to tissue perturbations and has a well-documented role in eradicating pathogens upon reexposure. Emerging evidence has highlighted a key role for TRM cells in cancer immunity. Single-cell approaches have identified TRM cells among other CD8+ tumor-infiltrating lymphocyte (TIL) subsets, and their presence is a positive indicator of clinical outcome in cancer patients. Furthermore, recent preclinical studies have elegantly demonstrated that TRM cells are a critical component of the antitumor immune response. Given their unique functional abilities, TRM cells have emerged as a potential immunotherapeutic target. Here, we discuss TRM cells in the framework of the cancer-immunity cycle and in the context of the T cell- and non-T cell-inflamed tumor microenvironments (TME). We highlight how their core features make TRM cells uniquely suited to function within the metabolically demanding TME. Finally, we consider potential therapeutic avenues that target TRM cells to augment the antitumor immune response.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Tissue-resident memory T cells
- Cancer
- Immunity
- Microenvironment
- Immune exclusion
- T cell dysfunction
- Antigen-presenting cells
- Immunotherapy
3.1 Introduction
While the immune response to cancer is incompletely understood, cytotoxic CD8+ T lymphocytes are thought to be the fundamental antitumor effector cells. The presence of a CD8+T-cell infiltrate is a positive prognostic marker in most types of solid cancer [1,2,3] and can predict clinical response to immune checkpoint blockade therapy [4, 5]. Most of these therapies aim to increase the frequency and function of tumor-infiltrating T lymphocytes (TILs) by inhibiting the negative regulatory pathways present in the tumor microenvironment (TME). Despite recent advances in immunotherapy to harness the power of these cells, cures remain rare, and only a subset of patients exhibit durable responses. At present, a fundamental objective in the field of immunotherapy is to understand the biological mechanisms behind the lack of clinical response and to develop new therapeutic approaches to overcome these obstacles.
Research over the past 20 years has revealed a network of checkpoints impeding effective T cell-mediated tumor destruction [6]. This checkpoint network can be framed into two generalized phases: T cell homing to and entry into the tumor site and overcoming the immunosuppressive TME. Once within a tumor tissue, a T cells’ ability to function requires adaptation to a challenging environment. Furthermore, different cancer tissues exhibit distinct local microenvironments. Even different metastases within the same patient may create distinct milieus capable of influencing T-cell function [7]. As such, T cells must be adaptable to survive and operate in diverse and difficult environments. The availability of nutrients and oxygen, local cytokine and chemokine concentrations, extracellular matrix components, adhesion molecules, and commensal microbials are among many other physiological variables. Within this framework, a recently discovered T-cell subset, called tissue-resident memory T (TRM) cells, has emerged as a critical T-cell lineage in the fight against cancer, in part because its most distinctive feature is its ability to persist and function within demanding tissue microenvironments.
Memory T cells enter tissues in response to external stimuli (for example, infection or inflammation) and a subset remain as long-lived permanent residents in that tissue. In this process, after pathogen encounter, naïve T cells are primed in the draining lymph node by dendritic cells carrying antigen from the site of infection. Naïve T cells differentiate into effector T cells, which then migrate to the infected tissues to clear the pathogen. After clearance of the infection, the majority of effector T cells die or leave the tissue, but some differentiate into TRM cells that impart long-term localized T-cell immunity with the capacity to rapidly respond to subsequent infection. TRM cells reside in most major organs and are abundant in epithelial barrier tissues such as the skin, lung, kidneys, and gastrointestinal and reproductive tracts [8,9,10]. Commitment to tissue of residence distinguishes TRM from effector T (TEFF) cells, effector memory T (TEM) cells, and central memory T (TCM) cell subsets. It also differentiates TRM cells functionally; they can persist in tissues for years [11, 12], display heightened response kinetics [13, 14], exhibit a unique metabolism [15], and are transcriptionally distinct compared to other T-cell memory subsets [10, 16]. These characteristics are a reflection of an adaptation to survive and function in the local environment and highlight TRM cells as a potentially targetable population for the development of novel immunotherapies in the fight against cancer. In this chapter, we will discuss the fundamental properties of TRM cells in the context of cancer and approaches to target TRM cells as a cancer therapy.
3.2 Core Features of TRM Cells
Bunkered in barrier tissue at the interface between the host and environment, TRM cells are strategically positioned to react quickly to tissue perturbations, such as infection, injury, or cancer. TRM cells are not immobile within tissues: instead, they migrate locally along components of the extracellular matrix and blood capillary to patrol resident tissue for signs of barrier disruption, such as cellular distress signals produced in response to pathogen insult or from cellular death or injury. TRM cells display a wide range of migratory behavior, which is likely dictated by the local anatomical properties. For instance, in the dense regions of the epidermis, TRM cells take on a dendritic morphology and patrol at a slower rate (0.5–1.5 μm min−1) [17,18,19] compared to TRM cells in the myometrium of the female reproductive tract (10 μm min−1) [20] or liver sinusoids [21]. This localized surveillance by TRM cells in tissues after pathogen encounter is a critical feature of immune memory and anamnestic protection against repeat pathogen exposure.
The commitment to remain in tissue after pathogen clearance is a defining feature of TRM cells and has been elegantly demonstrated in transplantation and parabiosis experiments. In one set of foundational studies, tissues from virally infected mice containing TRM cells were transplanted into naïve congenic recipients, where they were restrained within the graft [13, 22, 23] and exhibited protective functions upon reinfection [13, 23]. In another set of experiments, by fusing the circulatory system of a virus-immune mouse with a virus-naïve parabiont, TRM cells were found in the tissues of the immune parabiont but not the naïve parabiont, whereas T memory cells equilibrated between parabionts in secondary lymphoid organs (SLO, e.g., spleen and lymph nodes) [13, 22, 24, 25].
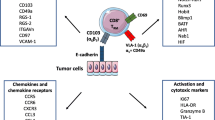
Tissue retention is maintained by two main mechanisms: expression of adhesion molecules, and a lack of responsiveness to cytokines and chemokines that direct cells back into circulation. Important for the capacity to remain in tissues are cell surface receptors that also function as useful markers to define and identify TRM cells. In this role are an array of adhesion molecules, some upregulated rapidly after T-cell activation (e.g., CD44) that are important for tissue entry, while others are tissue-specific and are only expressed once T cells gain access to the tissue. While the exact mechanistic roles adhesion molecules play are still being investigated, especially in the case of cancer, evidence indicates that they are critical for TRM formation but are not always required for maintenance [26]. Two well-studied adhesion molecules, the αE integrin CD103, which pairs with β7, and the α1 integrin CD49a, which pairs with β1 and together are called VLA-1, are found at variable frequencies on epithelium-localized TRM cells across many tissues [19, 26,27,28,29,30]. CD8+ and CD4+ TRM cells can be found in the dermal and basement membrane regions but are more likely to be CD103 negative [19, 30]. While CD103 is critical for TRM cell formation in epithelial tissues, it was found to be dispensable for TRM maintenance in models of intestinal infection [31, 32]. Intriguingly, CD103 expression was found to be enriched on CD8+ TILs in epithelial-derived tumors [33,34,35] and is important for antitumor activities of CD8+ TILs [33, 36, 37]. Studies inhibiting VLA-1 or CD103 by using blocking antibodies or gene-deficient T cells highlighted the importance of these integrins for generating TRM cells against tumors [38] and led to a loss of tumor control [39].
Aiding in tissue retention is the refractory nature of TRM cells to mediators that signal T cell egress from tissues into the afferent lymphatics. Egress is mainly dictated by sphingosine-1-phosphate (S1P) gradients, which are established by vascular endothelial cells. Accordingly, TRM cells display minimal levels of the S1P receptor (S1P1, SIPR1) [40]. Continued unresponsiveness to S1P is promoted by CD69, a C-type lectin that interferes with the function of S1P1, and is expressed on many TRM cells, especially those in the skin [28]. Moreover, TRM cells lack surface expression CCR7, a lymphoid tissue homing chemokine receptor, and CD62L (L-selectin), responsible for tethering to high endothelial venules for entry into lymph nodes (LN). Caution should be exercised for solely using cell surface markers, the most common being CD103 and CD69, for the identification of TRM cells. CD103 is expressed on only a subset of TRM cells and is more enriched in epithelial sites compared to other organs in the body [28]. While CD69 seems to be expressed on the majority of epidermal-localized TRM cells, this does not hold true for other sites such as the kidney and liver [28]. Furthermore, CD69+ T cells were found to recirculate in the LNs of mice [41]. Therefore, in addition to phenotypic characterization, residency should be tested functionally by complementary methods including parabiosis, transplantation, in vivo intravascular antibody staining, and in situ labeling of cells [42].
An important feature of TRM cells is their ability to provide accelerated protection against repeatedly encountered pathogens. This function relies on their strategic location in barrier tissues, where pathogens are most likely to be encountered, and where epithelial cancers originate. When a disruption in homeostasis is recognized by TRM cells, they rapidly produce effector molecules that not only impede the spread of the pathogen but also alarm and mobilize the surrounding tissue into an anti-pathogen state. In response to local secretion of alarmins such as ATP and IL-33, TRM cells are reactivated and induce a strong effector cytokine program [40, 43]. Reactivated TRM cells can upregulate perforin and granzyme B and efficiently kill target cells [8, 30], secrete IFN-γ, TNF-α, and IL-2, and activate dendritic cells (DCs), NK cells, and other resident CD4+ and CD8+ T cells [44]. The role of IFN-γ is particularly important for protection. In response to IFN-γ, nearby cells upregulate interferon-stimulated genes, which are critical for resisting pathogen spread and also activate the endothelium to express adhesion molecules that support recruitment of immune cells. Furthermore, it was found that TNF-α plus IFN-γ do the latter synergistically, which induced the recruitment of TCM and TEM cells from peripheral blood [45]. Reactivated TRM cells also contribute directly to the recruitment of other immune cells by producing chemokines [14, 30]. These mechanisms by which TRM perform their protective functions against pathogens naturally engender the question of whether these same functions are also used against neoplasms. This question remains largely uninvestigated, although recent reports in addition to circumstantial evidence point to an important role for TRM cells in antitumor immunity. In the following sections, we will first focus on the similarities and differences between the TRM lineage and CD8+ TILs. In particular, we review how TRM cells may be generated against tumor antigens, the phenotypic features of the TRM cells compared to TILs, and the transcriptional and metabolic profiles of T cells within tumors. We explore how TRM cells fit within the framework of the T cell- and non-T cell-inflamed TME and finally, we end with current immunotherapeutic strategies under investigation to target or augment the antitumor TRM cell response.
3.3 Immunologic Memory to Tumors
The immune system plays a critical role in protecting the host from cancer [46]. The innate sensing of tumor cell-derived factors can lead to an adaptive T-cell response through the presentation of tumor-associated antigens generated from genetic mutations and epigenetic changes that occur during carcinogenesis [47]. Spontaneously primed CD8+ T cells can home to tumor sites and accumulate there, even if tumors are not completely eliminated [48, 49]. In fact, an extensive body of work over the past 20 years suggests that T cells frequently prune neoplastic cells from healthy tissue throughout life [46, 50, 51]. Under this premise, it follows that tumor antigen-specific TRM cells likely form in response to tumors.
Early evidence of immunologic memory against cancer was uncovered in mice that received autologous tumor transplantation. After resection of 3-methylcholanthrene (MCA)-induced tumors, mice were protected against a subsequent challenge with the same tumor cells but not those derived from a different MCA-induced tumor [52, 53]. These studies were followed by a conceptual proposal by Burnet and Thomas, who independently posited that an evolutionary-driven feature of the immune system is to detect and eliminate incipient tumors that arise in tissues susceptible to genetic mutations, such as the highly proliferative epithelium [54, 55]. This theory evolved into what is known today as “cancer immunoediting” [56]. However, it is still unclear as to the relative involvement of different effector and memory T-cell subsets in the antitumor immune response.
Circumstantial evidence from human tumor biopsies pointed toward TRM cells as a major T-cell population found within tumors. CD8+ TILs isolated from biopsies of human solid tumors often express markers characteristic of residency, including CD103 [33, 34, 57], VLA-1 [39], and CD69 [33, 57]. Although phenotypic similarity alone is not adequate to identify TRM cells, transcriptional profiling of CD8+ TILs from human tumors has revealed a TRM cell gene signature, in at least a subpopulation of CD8+ TILs [33, 34, 58]. The detailed study of the TRM cell response to cancer has been hampered by a lack of robust mouse models. Mice are kept in abnormally hygienic Specific Pathogen Free (SPF) barrier facilities, the most common mouse husbandry practice, and hence lack key characteristics of the human immune systems due to a lack of exposure to common pathogens. Perhaps most importantly, SPF laboratory mice contain fewer TRM cells and have a naïve immune phenotype more similar to the immune profile of human neonates rather than adults [59, 60]. SPF facilities also prevent exposure to environmental carcinogens, which may be important for the natural pruning of neoplastic cells and the formation of TRM cells against future cancers. Several studies have circumvented this limitation by populating the skin with TRM cells before tumor challenge by vaccination [61, 62], resection of a growing tumor [38], or epicutaneous engraftment of tumor cells [63]. These studies revealed that TRM cells can indeed form against tumors and if the tumor is successfully eradicated, the resulting TRM cells display antitumor properties that are distinct from those of circulating memory T cells.
It is important to keep in mind that the successful initiation of an immune response against an established tumor will inevitably lead to a heterogenous CD8+ TIL population, which includes TRM and circulating effector and memory cells. This heterogeneity makes understanding the biology of a given CD8+ TIL subset very difficult, as there is no current way to identify TRM cells versus newly recruited T cells expressing TRM-like phenotypic markers. In addition, it remains unknown whether bona-fide TRM cells can form within an established tumor, as structural and molecular features within tumors differ from normal tissues. Nevertheless, it is clear that many tumors are populated by CD8+ TILs that display a tissue-resident phenotype, and recent studies in mice clearly demonstrate a role for TRM cells in antitumor immunity.
3.4 Tumor-Antigen Specificity
The three tenets of cancer immunoediting describe different phases of the antitumor immune response: elimination, equilibrium, and escape [64]. During the elimination phase, nascent transformed cells are effectively pruned by the immune system from otherwise healthy tissue. This presents a dichotomy regarding the formation of TRM cells against neoplastic tissue: those that successfully eliminate nascent transformed cells thus preventing tumor development, and those that form after a tumor is established, during the equilibrium or escape phases. In the latter situation, the cues within the TME influencing TRM cell formation are likely to be different from those experienced in normal tissue. The immunological mechanisms pertinent to TRM cell formation in these two scenarios remain to be elucidated.
Certain steps and features in the cancer-immune cycle are required regardless of the environmental cues influencing TRM differentiation, and most important of these is tumor antigen reactivity.
At the core of the endogenous antitumor immune response are T cells that have the ability to recognize tumor-specific antigens. Tumor antigens can fall under three main classifications: tumor-specific, tumor-associated, and cancer-testis antigens [65]. Tumor-specific antigens, also referred to as neoantigens, are absent from normal cells and recognized as foreign by the host immune system. They are derived from non-synonymous driver or passenger mutations and viral genes [66]. Since tumors are derived from normal self-tissues, how neoantigens arise and how the innate immune system initiates an effector rather than a tolerogenic adaptive immune response had been unclear. Recent data have indicated that many tumor-specific antigens are derived from mutational processes that also drive oncogenesis. Defects in DNA repair machinery, exposure to mutagens (e.g., UV light and tobacco smoking), and abnormalities in enzymes that modify DNA can lead to somatic mutations, genome translocations, and alterations in gene expression as part of the process of carcinogenesis [47]. These processes lead to diverse mutational landscapes, with some commonly mutated oncogenes or tumor-suppressor genes that are characteristics of certain cancer types but also defined by a spectrum of unique mutations specific to an individual tumor [67].
It is clear that multiple cancer types can be infiltrated by CD8+ T cells, and in many cases, a proportion of these T cells are tumor-antigen specific. However, recent evidence questions this central dogma by suggesting that tumor residency does not always translate to tumor-specificity. Profiling T-cell reactivity among CD8+ TILs in human tumors with MHCI tetramers or cloning TCRα/β pairs from intratumoral T cells revealed that nearly all tumors analyzed were infiltrated by both virus-specific and tumor-specific CD8+ T cells [68,69,70,71]. To distinguish bystander from tumor-reactive T cells, expression of the ATP catabolizing ectonucleoside, CD39, was found to be a promising marker for tumor reactivity. Expression of CD39 correlated with TRM genetic signatures and with co-expression of other resident markers, namely CD69 and CD103 [34, 69, 72]. Deciphering whether a TIL expressing TRM phenotypic markers is a recent immigrant or resident cell remains a challenging problem in the field, especially with human tumor samples. Nevertheless, it is clear that many tumors are populated with tumor-reactive CD8+ TILs that display a tissue-resident phenotype.
3.5 Innate Immune Factors Regulating Antitumor T-Cell Responses
Local activation of APCs is a required initiating step for a productive adaptive T-cell response against tumor antigens. The innate signaling pathways involved in this activation step were first hinted in transcriptome profiling of human tumors, where a type I IFN gene signature was found to correlate with a T-cell infiltrate [48, 73]. Mice deficient in genes involved in IFN signaling, IFNAR, and STAT1, could not control immunogenic tumors [49, 74]. Ultimately, the required APC cell population receiving the type I IFN signals were mapped to a rare population of CD8α positive classical dendritic cell (cDC1). cDC1s are known for their ability to cross-present antigens and are developmentally dependent on the transcription factors Batf3 and IRF8 [75, 76]. cDC1s are also important for the generation of TRM cells in the skin and lung. Batf3-deficient mice exhibit blunted TRM development in the skin after intradermal vaccinia virus (VACV) immunization [77]. Given the essential role cDC1s play in priming CD8+ T cells against tumor antigens, it is likely that they are also critical for TRM formation against tumors; however, detailed studies addressing this question remain to be performed.
The functional role for type I IFNs prompted the next level question regarding the nature of the damage-associated molecular pattern (DAMP) that could induce type I IFN production in a sterile tumor without pathogen exposure. Early studies identified several DAMPs that could be released by stressed or dying tumor cells that subsequently could lead to productive T-cell priming. For example, high-mobility group protein B1 (HMGB1) binding to TLR4 and extracellular ATP binding to the P2X7 purinergic receptor triggering activation of the NLRP3 inflammasome were both reported to induce DC maturation and subsequent activation of anti-tumor T cells [78, 79]. In apparent contrast to inducing DC maturation, extracellular ATP can also impact TRM cell maintenance. TRM cells can express P2RX7, which induces cell death when triggered [80]. However, attrition of TRM cells can be fine-tuned by their ability to regulate local ATP concentrations through the action of the ectoenzyme CD39. Beyond HMGB1 and ATP, tumor-derived DNA was found to be a potent initiator of the endogenous antitumor immune response [81, 82]. DCs recruited to the TME were found to take up tumor-derived DNA leading to stimulator of interferon genes (STING)-dependent production of type I IFNs [83]. Besides DCs, endothelial cells of the tumor vasculature were also reported to produce type I IFNs in response to STING activation [84]. STING signaling not only activates DCs but also induces the upregulation of adhesion molecules on endothelial cells of the tumor-associated vasculature, a critical step for T-cell extravasation into the tumor [85]. STING is an endoplasmic reticulum adaptor that is activated by cyclic dinucleotides generated by cGAMP synthase [86]. The mechanism by which tumor-derived DNA can gain access to the cytosol to activate the STING pathway has yet to be elucidated. But consistent with this mechanism, immunogenic tumors fail to be rejected and grow progressively in mice lacking STING and spontaneous priming of CD8+ T cells against tumor antigens is nearly ablated [81, 82, 87]. The cellular cues alerting the immune system of a nascent tumor are just starting to be uncovered. Mouse models highlight an important role for sensing tumor-derived DNA. Whether this occurs in the absence of spontaneous tumor cell death remains to be determined.
3.6 Antigen-Presenting Cells: The Gatekeepers of the Antitumor T-Cell Response
The process by which DCs are initially recruited to the tumor site is not fully understood and likely depends on the chemokine repertoire produced by tumor cells or the surrounding tissue. Alternatively, a subset of DCs exists at steady-state in barrier tissues, such as the CD103+ variety found in the skin [88], and therefore local activation may not require DC recruitment from the periphery. In support of this notion, it was found that CD103+ DCs were uniquely capable in the uptake of tumor antigens and trafficking to the lymph node to prime CD8+ T cells (Fig. 3.1) [89]. Regardless of the mode of DC recruitment, tumor cells can acquire the ability to produce chemokines that can contribute to their own growth, survival, and metastasis [90]. Genomic aberrations such as oncogenic pathways can impact the array of chemokines expressed. In a melanoma model, B-Raf pathway activation led to the production of the chemokine CCL4, which contributed to the recruitment of cDC1s in a CCR5-dependent manner [87]. Taken together, it is likely that both tissue-localized and circulatory DCs can effectively prime CD8+ T cells, indicating another layer of control for effective T-cell responses. Nevertheless, delineating the anatomical logistics of DC activation will be important for determining which DC populations can be effectively targeted as a cancer therapy.

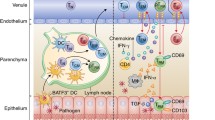
TRM cells in the context of T cell-inflamed, T cell-excluded, and non-T cell-inflamed tumors. DCs acquire tumor antigens and traffic to the LN where they prime T cells. Studies in mice indicate that the Batf3-lineage DC is particularly critical in this process. TGF-β signaling in the LN can precondition T cells to become TRM cells upon entering their target tissue prior to tumor development. This suggests the possibility that preexisting TRM cells are present in many tumors, despite the failure at a later time to recruit new T cells. TRM cells may be an actionable target to disrupt the non-T cell-inflamed phenotype. T cell infiltration into tumors is regulated by many factors. A lack of T cell priming can occur when DCs are not recruited to the TME, for instance in the case of tumor-intrinsic β-catenin activation (non-T cell-inflamed). The tumor vasculature can also diminish T cell influx when adhesion molecules are not upregulated or induce T cell apoptosis via Fas-FasL interaction (both T cell-excluded and non-T cell-inflamed). TGF-β in the TME can act on cancer-associated fibroblasts to exclude T cells to the marginal area (T cell-excluded). In response to proper homing signals, such as CXCL9 and CXCL10 and adhesion molecule expression on vascular endothelium, T cells migrate into the tumor leading to the T cell-inflamed phenotype. This phenotype exhibits an IFN-γ-gene signature, which correlates with responsiveness to checkpoint blockade therapy
After DC activation by type I IFN at the site of tumor formation, DCs traffic to the draining lymph node to prime tumor-antigen-specific naïve T cells. T-cell priming encompasses a complex series of spatial, biochemical, transcriptional, proliferative, and differentiation events that engender clonal populations of activated T cells with effector and tissue-homing programs [91]. The tissue-homing program is characterized by expression of specific chemokine receptors responsible for correctly trafficking T cells to the site of an infection or tumor and aid in entry into the tissue. The CXC-chemokine receptor 3 (CXCR3) has an intriguing role, as it was shown to be important for T-cell entry into many inflamed peripheral sites [92]. Adoptively transferred antigen-specific CXCR3-deficient CD8+ T cells failed to traffic to infected skin [26], vaginal epithelium [93, 94], and melanoma [95]. Antibody-mediated blockade of CXCR3 prevented T-cell infiltration in a model of pancreatic ductal carcinoma [96]. It must be noted that CXCR3 was not required for tissue infiltration in all experimental systems. For instance, CXCR3-deficient mice displayed similar numbers of skin-infiltrating T cells after cutaneous VACV infection and B16F10 [97] tumor engraftment [98], suggesting the existence of compensatory mechanisms. Beyond its role in aiding T-cell entry into tissues and tumors, CXCR3 was also shown to play an active role in TRM generation within tissues [26], possibly through influencing intra-tissue migration [99] and cell–cell interactions [100, 101]. Consistent with this latter role, CD8+ TIL interactions with cDC1s were mediated by CXCR3, which was critical for the effectiveness of anti-PD-1 blockade in mice [98].
A strong correlation between the presence of CD8+ T cells and expression of the CXCR3 ligands, CXCL9 and CXCL10, has been observed across a range of tumor types [48, 102, 103]. The source of these chemokines in early recruitment of CD8+ T cells was initially attributed to tumor cells [48, 104] or activated keratinocytes [26]; however, in a murine model of oncogene-induced melanoma, cDC1s were identified to be the major source of CXCL9 and CXCL10 within the TME and those DCs were required to recruit activated CD8+ T cells to the tumor site in a CXCR3-dependent manner [87]. Similar results were found in an engraftable tumor model using a dual reporter for CXCL9 and CXCL10, where CD11b+ DCs were also producing these chemokines [98]. In summary, DCs play a critical role in regulating the antitumor immune response, acting as sentries for detecting the initial cellular cues provided by nascent tumors. The cDC1 subset is critical for priming and recruiting CD8+ T cells to the tumor site, and for providing a stimulus to T cells within the tumor.
3.7 TRM Cell Commitment, Maintenance, and Function
T-cell memory encompasses not just the antigen for which a TCR is specific, but also the anatomic site of T-cell activation. The initial site of T-cell priming imprints chemokine receptors and adhesion molecules that biases migration to a specific tissue or organ where the pathogen is first encountered [105]. Upregulation of tissue-specific chemokine and adhesion molecules occurs after T-cell stimulation in coordination with molecular cues encountered in secondary lymphoid organs (SLOs) [106]. For example, in conjunction with TGF-β and retinoic acid, DCs that emigrate from intestinal tissue induce expression of α4β7 and CCR9 on T cells within the mesenteric lymph nodes, guiding T cells to inflamed sites within the intestine [107].
Migratory potential seems to be lost over time as TEFF cells isolated from the spleen 7 days after LCMV infection failed to generate TRM cells [22]. This raises the question of whether TRM cells are restricted to only populate sites of infection. In this scenario, the TRM-arm of immunological memory would be compromised if the pathogen is re-encountered elsewhere in the body. Addressing this question, it was found that TRM cells populated distant non-infected skin sites in response to skin-localized vaccinia virus infection [24]. In addition, overlapping TCR repertoires were found between TCM and TRM cell populations after skin immunization, which points to a common naive T-cell precursor and suggests that TCM cells can serve as a reservoir for the formation of TRM cells upon re-challenge [108]. In support of this notion, TCM cells possess stem-like properties [109], a feature shared with TRM cells, and after transfer into a naïve host, TCM cells can differentiate into TRM cells upon re-challenge [61]. These findings revealed additional pathways for the formation of TRM cells, providing protection at sites secondary to the initial pathogen encounter. It also suggested that at least part of the TRM genetic program is initiated in SLOs, while the final commitment steps occur in tissue. This is supported by a recent study proposing a mechanism where naïve T cells are preconditions to become TRM cells after interacting with cognate antigen, cDCs, and TGF-β in the LN [110]. The full extent of the genetic determinants permissive for TRM development initiated in the LN remains to be elucidated.
In the early phases after T-cell priming and entry into target tissue, T cells are endowed with the capacity to patrol and eliminate infected or cancerous cells. Signals within the peripheral tissue environment aid in this transformation by heightening cytotoxic capacity and cellular motility. One example is TGF-β, which has a well-documented role in the induction of the TRM phenotype. In response to TGF-β, T cells entering epithelial tissue upregulate CD103 and downregulate the transcription factor KLF2, a promoter of S1PR1, which together enforce tissue residence [40]. CD103 is not only important for retaining T cells within tissues, it contributes to T-cell movement toward tumor regions [36], enhances cytotoxic functions against tumor cells [111], and can convey survival signals [26, 29]. These functional roles may help explain why the intratumoral expression of CD8 and CD103 is a more robust prognostic indicator of overall survival and predictor of response to anti-PD-1 therapy than CD8 alone [33, 34, 112]. However, an alternative possibility for this observation is that CD103 may identify the critical cDC1 population, which has been linked to the efficacy of anti-PD-1 therapy [98, 113].
After successful elimination of infected cells, T cells undergo a contraction phase where they can die by apoptosis, enter the circulation to become TCM or TEM cells, or remain in the tissue to become TRM cells. The selection of cells entering the TRM-lineage may not entirely be stochastic, but likely depends on the differentiation state, expression of pro-survival cytokine receptors and adhesion molecules, and transcriptional regulation. For example, homeostatic cytokines, namely IL-7 and IL-15, are important for TRM formation. However, their requirements are heterogenous among different tissues. IL-15 and IL-7 signaling is critical for TRM formation in the skin, kidney, and liver, but not for the female reproductive tract, pancreas, or small intestine [114,115,116,117].
During the T-cell response to a pathogen, it remains unclear when commitment to the TRM lineage occurs. Highly differentiated cells that are marked by high KLRG-1 and low IL-7R expression fail to differentiate into TRM cells [26, 32, 117]. However, using a KLRG-1 lineage tracing mouse model, it was found that T cells which downregulated KLRG-1 but retained IL-7R expression during the contraction phase were able to differentiate into TRM cells [117]. Thus, expression of KLRG-1 does not exclude cells from entering the TRM cell fate and its downregulation before TRM-formation may indicate escape from a terminally differentiated state. Consistent with this notion, TRM cells do not express KLRG-1 at steady state and local antigen persistence is not required for TRM maintenance in some tissues [29, 118]. On the other hand, KLRG-1 is upregulated after antigen stimulation [119]; therefore, it is intriguing that CD8+ TILs generally do not express KLRG-1 despite the presence of local cognate antigens [120, 121]. However, in response to checkpoint blockade therapy or agonistic antibodies against co-stimulatory receptors, KLRG-1 is upregulated, which correlates with greater antitumor activity and indicates a transition into a more effector-like state [122, 123]. In summary, TRM differentiation may be initiated in the LN and finalized in the tissue. The steps toward TRM commitment involve local cytokine signals, which can vary among different tissues. The exact mechanism driving differentiation and commitment, as well as markers identifying TRM-precursors, remains to be elucidated.
3.8 TRM Cells and Tumor Immune Exclusion
Despite the ability of the immune system to recognize cancer cells, not all patients respond to checkpoint blockade therapy. Anti-PD-1/PD-L1 therapy exhibits an almost bimodal response. Some patients experience complete eradication of tumors, while a majority derive little or no clinical benefit. To explain this dichotomy, gene expression profiling across all cancer types has revealed that tumors can be classified into three major subsets, the T cell-inflamed, T cell-excluded, and non-T cell-inflamed based on the relative abundance of T cell-related transcripts (e.g., CD8A, GZMA, PRF1, and IFNG) and location of T cells relative to the tumor core (Fig. 3.1) [4, 124,125,126,127]. By segregating tumors this way, it was found that the majority of patients responding to checkpoint blockade therapy contained a T cell-inflamed tumor phenotype [4], suggesting that in these patients, the immune system has been restrained while remaining primed for reinvigoration. However, some patients within this subset fail to respond to checkpoint blockade therapy, indicating that additional resistance mechanisms must be overcome to achieve effective clinical responses [128, 129].
The non-T cell-inflamed subtype of tumor is remarkably devoid of immune cell signatures, including the negative regulatory pathways normally seen in the T cell-inflamed tumor. Exclusion of T cells from the tumor may result from the breakdown of key events required for successful T-cell recruitment. These include innate immune activation, chemotaxis, and extravasation into the tumor or surrounding tissue, and penetration from peritumoral space into the tumor bed. The mechanisms behind the evolution of non-T cell-inflamed tumors are currently under intense investigation and recent observations have helped to understand this phenotype. Genetic events with oncogenic potential include those that increase immune evasion. For example, activation of the Wnt/β-catenin pathway led to a loss of chemokines critical for the recruitment of the Batf3-DC lineage to the tumor site, and thus a failure to activate the innate immune system [127, 130]. Inactivating mutations or deletions of PTEN led to activation of the PI3K-AKT pathway and subsequent decrease in tumor cell autophagy, thereby diminishing innate immune activation and T-cell priming [131]. The non-T cell-inflamed TME appears to be independent of nonsynonymous mutation load, indicating that a lack of immunogenic T-cell antigens does not drive exclusion [132]. However, tumor evolution in response to immune pressure can lead to tumor cells with defects in antigen presentation machinery. Many of these cases were documented from patients that developed acquired resistance to immunotherapies through loss of function mutations in B2M and HLA genes [133,134,135,136,137]. Neoantigen loss also contributes to immune resistance through selective killing of tumor subclones or gene silencing via epigenetic processes and chromosomal deletions can lead to an overall decrease in tumor immunogenicity [138, 139].
The tumor-associated vasculature is also a critical barrier regulating T-cell infiltration [140]. A network of arterioles, capillaries, and postcapillary venules provide avenues for T cells to enter the tumor peritumorally though the tumor stroma or intratumorally through the tumor parenchyma. Unlike peritumoral blood vessels, which can be derived from exiting normal endothelium, intratumoral vessels are often found to be immature [141]. This immaturity is driven by rapid angiogenesis when the metabolic demands of the tumor surpass the supply of the local vasculature. In response, tumors produce angiogenic factors, including vascular endothelial growth factor (VEGF), angiopoietins, and thrombospondins to induce the formation of new blood vessels. These angiogenic factors contribute to leaky and chaotically organized vessels, which often express low levels of adhesion molecules (e.g., E/P-selectin, ICAM-1/2, VCAM-1, and VAP-1) [141,142,143,144,145,146], fail to respond to inflammatory stimuli [147, 148], and can express FasL to directly kill antitumor T cells [149]. Additional players in tumor angiogenesis are pericytes, which surround blood vessels and contribute to new blood vessel formation and immune cell trafficking. Pericyte phenotype and coverage along the tumor vasculature are often found to be abnormal when compared to normal adjacent vasculature [150]. Interestingly, immune cells can interact with pericytes in a positive feedback loop resulting in normalization of blood vessels and immune-favorable changes in the TME such as hypoxia mitigation [151]. Similarly, genetic deletion of the G-protein signaling component Rsg5 induced pericyte-mediated vasculature normalization and increased T-cell recruitment [152]. These studies indicated that pericytes associated with the tumor vasculature have abnormal activity and distribution. Normalization of pericyte function to promote T-cell infiltration into the TME may be a potential therapeutic approach. Entry into the TME via the peritumoral route also contains hurdles for T cells. Stroma surrounding the tumor often contains immune-suppressive cell populations such as cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells, and tumor-associated macrophages. CAFs sterically inhibit T-cell ingress into the tumor through the synthesis of a dense extracellular matrix [153, 154]. Overall, endothelial cell immaturity, anergy, and pericyte abnormality diminish T-cell infiltration directly into the tumor parenchyma, diverting T cells to enter via perivascular routes, where the dense ECM of the tumor stroma can border T cells.
When considering immune exclusion in the context of TRM cells, there is the possibility that TRM cell localization at tumor sites precedes tumor formation. Given the patrolling nature of TRM cells and their relative abundance within tissues (a recent study found around 500 TRM cells per mm3 in healthy human skin [19]), the question arises as to how some tumors can apparently develop without T cells present. One possibility lies in how T-cell infiltration is calculated. While methods may differ, quantification of T-cell infiltrations from RNASeq data is generally scored based on relative expression of a T cell-related gene signature. Therefore, non-T cell-inflamed samples may contain RNA transcripts below a defined threshold, but this does not translate to a complete lack of T cell-related transcripts. Thus, T cells may be present at low frequency in non-T cell-inflamed tumors, which may represent a preexisting TRM cell population. This is consistent with recent findings that in some T cell-containing tumors the majority of T cells are specific for commonly encountered viruses (e.g., EBV, CMV) and not tumor antigens [69, 70]. However, studies quantifying T-cell infiltration by histology clearly identify tumors that are devoid of T cells, termed “immune-desert”, or T cells that are retained to the peritumoral area, termed “immune-excluded” [6]. These phenotypes suggest that tumors may actively exclude T cells.
Active exclusion of T cells may involve sequestering T cells to peritumoral regions via coinhibitory receptor interactions such as PD-1:PD-L1. Biopsies taken before and after anti-PD-1/PD-L1 therapy show an increase in tumor penetration after treatment [155]. Another possibility is adhesion molecule-mediated retention of T cells in the stroma. Recent studies provide evidence for a role of TGF-β in driving peritumoral T-cell retention. While TGF-β exerts positive immune effects on TRM cell differentiation and function, the abundance of TGFB transcripts in the tumor also correlates with poor prognosis in multiple cancer types [156,157,158]. Indeed, the role of TGF-β in cancer immunity is complex and contextual, exhibiting pleotropic effects on cancer, stromal, and immune cells within the tumor [159]. TGF-β can be co-opted by cancers to promote their progression by evading the growth-inhibitory effects through inactivating mutations in the TGF-β signaling pathway and maintaining the immune suppressive effects on surrounding stroma and immune cells. In particular, TGF-β signaling in CAFs was associated with poor prognosis in colorectal cancer [160, 161]. Furthermore, transcriptome analysis of tumors from patients with metastatic urothelial cancer refractory to the PD-L1 antagonist atezolizumab had an enrichment for genes involved in the TGF-β signaling pathway. This enrichment correlated an immune-excluded phenotype [162]. Similar evidence was found in a genetically engineered mouse model of colorectal cancer. In this model, combinatorial oncogenic mutations led to metastatic tumors with an immune excluded phenotype and TGF-β transcriptional signature. Interfering with TGF-β via blocking antibodies or a small molecule inhibitor for TGFBR1 redistributed T cells into the intratumoral zone and sensitized mice to PD-L1 blockade therapy [163]. Taken together, an interesting relationship emerges between the TRM-promoting and the immune-excluding effects of TGF-β, where the sum of the effects results in retainment of immune cells to peritumoral regions.
3.9 T-Cell Dysfunction
The defining features of TRM cells: tissue residency, tissue patrol, and rapid response to stimulus, have mainly been described under conditions of tissue homeostasis after pathogen clearance. In the context of chronic infection or persistent antigen exposure, less is known about TRM differentiation or how the TRM genetic program is influenced. However, it is well known that T cells isolated from tumors or from secondary lymphoid organs during chronic viral infections are dysfunctional or exhausted. Much of the knowledge surrounding T-cell dysfunction is derived from in vivo models of chronic viral infections, in particular clone 13 LCMV. In this model, antigen is continuously present, which drives the breakdown of immunological memory formation and pushes responding T cells into a state termed exhaustion, which is characterized by a gradual and sequential loss of effector functions [164,165,166]. Furthermore, continuous TCR signaling induces an NFAT-driven transcriptional program, promoting the expression of inhibitory receptors, including PD-1 [167, 168], which in turn blunts CD28 co-stimulatory receptor signaling in a SHP2-dependent manner [169]. Other in vivo studies found that SHP2 was dispensable for promoting exhaustion; thus, similar phosphatase-recruiting inhibitory receptors may compensate [170]. In fact, both tumor-antigen specific TILs and exhausted virus-specific CD8+ T cells upregulate and maintain expression of an array of co-inhibitory receptors, including CTLA-4, TIM-3, TIGIT, and LAG-3, in addition to PD-1 [120, 121, 171,172,173]. Engagement of these receptors has been shown to blunt proliferation and cytokine production by T cells, and blocking interactions between these receptors and their corresponding ligands can restore T-cell function [120, 174, 175]. Due to these key features that parallel chronic infection and cancer, persistence of antigen and expression of inhibitory receptors on T cells, it has long been proposed that dysfunctional CD8+ TILs resemble virally exhausted CD8+ T cells. Some studies have found similarities between these two cellular states [121, 176]. Other studies have found significant differences. For example, despite expression of inhibitory receptors, CD8+ TILs were found to not be functionally inert and retained the capacity to proliferate, produce cytokines, and lyse target cells [120, 174, 177]. Under the latter premise, it is unclear why a tumor is not controlled by the immune system despite a tumor-reactive T-cell infiltrate with tumoricidal properties. This question and the discrepancy surrounding T-cell functionality between chronic viruses and cancer have been looming in the background for many years [178] and not until recently, with the technological advances of single-cell genomic analyses, has a more encompassing picture emerged.
Investigations into CD8+ TIL biology using single-cell RNA sequencing (scRNA Seq) technology have revealed previously unappreciated transcriptional heterogeneity. Clustering of single cells based on the expression of core genetic signatures suggests a developmental continuum, at least for some T-cell subsets. This type of analysis has revealed similar CD8+ TIL subsets across many human cancers including human lung cancer [179,180,181], breast cancer [58, 182], liver cancer [183], colorectal cancer [184], and human and mouse melanoma [176, 185, 186]. In addition , the immune cell infiltrate appears to differ significantly depending on the tumor tissue type, the individual patient, and even among different metastasis sites within the same host [7]. Host genetics and environmental influences such as the composition of microbiota can have a profound impact on the transcriptional landscape of tumor-infiltrating immune cell populations [187, 188]. Despite much observed heterogeneity, these studies revealed several key CD8+ TIL populations that are commonly found across many tumor types. One of the most abundant T-cell types are dysfunctional CD8+ TILs, characterized by expression of inhibitory (e.g., Pdcd1, Havrc2), co-stimulatory (e.g., Tnfrsf9), effector cytokines (e.g., Ifng, Gzmb), and cell cycle genes. This population usually contains many expanded clones, suggesting tumor specificity, and is often actively proliferating. Generally, within this population is a subset with a transcriptional signature similar to TRM cells that has been described in mice and humans [58, 179, 180, 186]. However, the precise relationship between TRM cells and infiltrating T cells is not well characterized. Memory T-cell populations, including TCM and TEM were identified as having lower expression of inhibitory receptors while retaining expression of effector molecules (e.g., Gzmk and Prf1) and in the case of TCM, expressing genes important for circulating among secondary lymphoid organs (e.g., Sell and Ccr7). Finally, a new stem-like T-cell population was found, which was characterized by the expression of genes that promote self-renewal properties (e.g., Tcf1) [189].
A primary goal of checkpoint blockade is to reinvigorate T cells into a state of potent effector function. It follows that determining the T-cell populations responding to checkpoint blockade can have important clinical implications for selecting patients who are more likely to respond. An analysis of immune cell infiltrates responding to anti-PD-1 therapy revealed that the presence of the CD8+ TCF7+ stem-like TIL population indicated a greater probability of a clinical response [185]. Furthermore, studies have suggested an interplay between the stem-like and dysfunctional CD8+ TIL populations, where the stem-like cells act as long-lived progenitors for the dysfunctional population [176, 190]. Since the stem-like population expresses intermediate levels of PD-1, it is thought that they are antigen experienced. Furthermore, PD-1 was found to promote survival of this population by preventing overstimulation [191]. This population expresses SLAM6 and CXCR5 and has antigen-independent self-renewal properties [176, 192]. In response to anti-PD-1 therapy, T cells from the stem-like population differentiate and expand into effector cells with a dysfunctional phenotype [176, 185, 192]. Integration of these datasets indicates that transitional response to anti-PD-1 therapy first gives rise to potent cytotoxic T cells, which overtime enter into a dysfunctional state [192,193,194]. In line with this evidence, TCR sequence analysis revealed an overlap between the dysfunctional and stem-like T-cell populations, suggesting a clonal relationship. In addition, adoptive transfer of the stem-like TIL population into tumor-bearing mice further demonstrated their transition into the dysfunctional phenotype [176, 185]. Importantly, dysfunctional CD8+ TILs can also respond to checkpoint blockade. In one proposed mechanism, it was found that CD8+ TILs undergo a futile cycle of proliferation and apoptosis at steady state, which was reversed by agonistic anti-4-1BB plus anti-PD-L1 antibody treatment or by inhibiting Fas–FasL interactions [174, 175]. Similarly, targeting co-stimulatory receptors such as 4-1BB, GITR, and OX40, or other co-inhibitory receptors like LAG-3, both of which are absent or lowly expressed on stem-like TILs, can restore CD8+ TIL function and induce tumor control [195]. Taken together, blocking PD-1 may stimulate PD-1+ TCF7+ stem-like CD8+ TILs, giving rise to potent effector cells. Although these cells may eventually enter into a dysfunctional state, dysfunctional cells can be reinvigorated in response to the same anti-PD-1/PD-L1 therapy. Developing a more comprehensive understanding of the clonal and functional relationships between the stem-like population, TRM cells, and dysfunctional CD8+ TILs is an investigational priority with considerable therapeutic implications.
In response to cognate antigen recognition, T cells undergo a metabolic switch from oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) to aerobic glycolysis and glutaminolysis. While energetically less efficient, this allows for the rapid production of biosynthetic molecules such as nucleotides, amino acids, and lipids that are required for clonal expansion and the acquisition of effector functions [196]. Similarly, cancer cells use more aerobic glycolysis and glutaminolysis compared to normal cells to support their rapid growth. This, in combination with poor angiogenesis, nearly depletes exogenous glucose and fills the tumor with hypoxic regions. In addition, uncontrolled cell growth and necrosis can lead to a buildup of byproducts such as lactate and extracellular potassium that can interfere with T-cell function [197, 198]. Accordingly, after entering the TME, T cells undergo profound metabolic changes in response to competition with tumor cells for nutrients. For instance, in melanoma and renal cell carcinoma, CD8+ TILs exhibited severely diminished glycolysis and mitochondrial function [199, 200]. In a preclinical model, CD8+ TILs in tumors exhibiting low glycolytic activity maintained tumoricidal activity, while those in high glycolytic tumors did not. These data highlight a local competition for glucose that can impede the antitumor functions of T cells and impair immunotherapy [201, 202]. Due to these metabolic constraints, CD8+ TILs developed the altered metabolism necessary to support survival and function. For example, in response to hypoxia and hypoglycemia, CD8+ TILs upregulate PPAR-α signaling and increase FAO of exogenous lipids and decrease glycolysis. When treated with a PPAR-α agonist, CD8+ T cells displayed enhanced cytolytic function after adoptive transfer into a tumor-bearing host [203]. A shift to FAO after entering the TME is also partially due to PD-1 signaling, which can inhibit glycolysis and promote FAO [204]. In the face of a metabolically hostile TME, TRM cells may therefore be ideally suited to function within a tumor. As TRM cells differentiate, they adapt to the metabolic constraints and available energy resources in their residing tissue. For example, TRM cells that reside in the skin epidermis, which is avascular and relies on diffusion for nutrients [205, 206], have altered their metabolism to function with less oxygen and glucose. This is accomplished through mitochondrial beta oxidation using exogenous FAs scavenged from the surrounding environment [15, 207]. Reliance on FAs could benefit TRM cells inside solid tumors as the lipid content is generally higher compared to normal tissue [208]. In summary, TIL function is impeded by the immunosuppressive and metabolically challenging TME, which can push TILs into a state of dysfunction. Checkpoint blockade therapy can reinvigorate TILs and understanding which TIL populations respond is critical for designing new therapies. Evidence suggests that both the stem-like and dysfunctional TIL populations contain the capacity to respond. TRM cells may be a component of the responding populations; however, further investigation is needed to characterize the nature of TRM cell responses. Nonetheless, TRM cells are prime targets for checkpoint blockade therapy due to their ability to function under the metabolic constraints within their tissue of residence.
3.10 Targeting TRM Cells in Cancer Immunotherapy
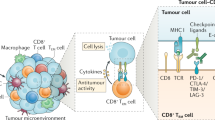
The magnitude of the T-cell infiltrate in tumors is a major determinant of effective immunotherapy, including checkpoint blockade. Patients with low or no T-cell infiltrate are generally less likely to respond. Designing new therapeutic interventions to augment the chances of a response is a principal goal for researchers and clinicians. Targeting TRM cells may provide new therapeutic avenues by either directly augmenting TRM cell function or inducing the recruitment of peripheral T cells (Fig. 3.2). In fact, TRM cells may be a component of the T-cell pool that is reinvigorated in response to checkpoint blockade. In their core genetic signature, TRM cells can express a range of co-inhibitory receptors, such as PD-1, TIM-3, and LAG-3, as well as costimulatory receptors, such as 4-1BB and ICOS [10, 15, 26, 209]. Since TRM cells are poised to rapidly respond, expression of these co-inhibitory receptors on TRM cells is thought to limit unwarranted activation. In a mouse model of contact hypersensitivity, antibody blockade of PD-1 and TIM-3 exacerbated TRM-driven skin inflammation in response to allergen rechallenge. Further, in response to viral challenge, TRM cells were found to proliferate in situ generating a secondary pool of TRM cells [17, 210]. These observations suggest that TRM functions can be augmented in response to checkpoint blockade.

Immunotherapeutic strategies that target TRM cells to induce tumor control. TRM cells may be one of the responding intratumoral T cell populations to checkpoint blockade, such as anti-PD-1, which augments their tumoricidal functions. Interactions between T cells and cDCs via CXCR3 are important for anti-PD-1 efficacy. In situ tumor vaccination with tumor antigens may activate preexisting TRM cells and infiltrating TEFF cells. In situ vaccination could also be used to activate anti-viral TRM cells to induce positive immune changes within the TME. Such changes may include production of chemokines to recruit TEFF cells or cytokines that support TEFF cell functions. The “prime and pull” strategy, which has been successfully used to recruit T cells into tissues, could be adapted to recruit T cells to the tumor. In this strategy, the frequency of circulating tumor antigen-specific TEFF cells is increased by immunization. Chemokines or inflammatory mediators are then injected into the tumor tissue to recruit these TEFF cells. Finally, T cells extracted from the patient could be modified to express TRM genes, for example, by promoting Runx3 activity, which may improve T cell infiltration and function after adoptive transfer back into the patient
TRM cells can also be targeted by vaccination. Cancer vaccine therapies can provoke two different TRM responses by (i) priming new T-cell infiltration into the tumor and (ii) activating TRM cells already present in the tumor at the time of vaccination. The recruitment of new effector T cells was the goal of many cancer vaccine trials, which have only shown limited efficacy [211]. One possible explanation may involve the route of administration. It is well documented that intramuscular vaccination induces the formation of circulating memory T cells, but only weakly induces TRM cells in tissues [212, 213]. It is now evident that manipulation of the target tissue is needed to induce the proper homing and inflammatory signaling required for TRM formation. Vaccine administration to mucosal sites, for instance, through intranasal, cervicovaginal, or skin scarification routes, more robustly generates TRM cells at the site of vaccination [214,215,216]. In a preclinical model of orthotopic head and neck or lung cancer, intranasal, but not intramuscular, vaccination protected nearly all mice from tumor growth when given prophylactically, and inhibited tumor growth in the therapeutic setting [216, 217]. Similarly, vaccination by skin scarification was sufficient to slow tumor growth and synergized with circulating memory T cells [61]. Site-specific vaccination may also provide a means to disperse T cells to other unmanipulated sites, such as in the case of skin scarification [24]. This phenomenon may provide a means to promote T-cell infiltration when in situ vaccination is not possible and may help explain cases of abscopal tumor regression [218].
Other approaches combine systemic immunization with tissue-specific stimulation. In these “prime and pull” strategies, the TRM precursor frequency is increased either by adoptive transfer of activated antigen-specific T cells or by subcutaneous vaccination, which is followed by antigen-independent stimulation of the target tissue to recruit and promote TRM cell formation. For example, after subcutaneous vaccination to induce a circulating memory T-cell response, intravaginal injection of CXCL9 and CXCL10 resulted in the recruitment and formation of TRM cells in the vaginal tissue [94]. Likewise, adoptively transferred activated T cells were effectively recruited to the skin by topical application of the contact sensitizer dinitrofluorobenzene (DNFB) [17].
While cancer vaccination strategies show great promise, they are only possible when tumor antigens are known. Identifying neo-antigens and formulating personalized cancer vaccines remain logistically challenging and expensive. An alternative approach is to target virus-specific T cells within the tumor to incite favorable changes in other host cells found within the TME. This approach may be more feasible because for many common pathogens, immunogenic peptides are known and tumors often contain virus-specific T cells. Indeed, a recent study found that T-cell immunity against commensal papillomavirus was critical for controlling development of skin tumors in response to chemically- or UV-induced carcinogenesis [219]. Further, activating pre-existing antiviral immunity can enhance antitumor immunity. Rosato et al. [71] showed that reactivation of VSV-specific CD8+ T cells by intratumoral peptide injections could delay tumor growth and synergized with anti-PD-L1 antibody therapy.
One key to therapeutic strategies designed to enhance T-cell infiltration into tumors may lie in understanding and promoting the specific DC populations and attendant T-cell transcriptional programs that imprint TRM precursor behavior. Recent data suggest that T cells in secondary lymphoid organs can be conditioned by migratory DCs to become TRM cells in a TGF-β-dependent manner [110]. Promoting TRM precursor characteristics can also be accomplished by manipulating transcriptional activity directly in T cells. Runx3 was found to program CD8+ T cells for tissue residency and adoptive transfer of T cells overexpressing Runx3 augmented T-cell accumulation in the tumor while enhancing their anti-tumor activity [220]. These studies suggest that peripheral T cells can be programed to become TRM cells by promoting genes and cellular pathways that regulate TRM development. This may be an attractive approach to potentiate adoptive cell transfer therapies.
3.11 Concluding Remarks
TRM cells are a unique lineage of T cells with specialized functions endowing them with the capacity to adapt and survive in their tissue of residence. Their high abundance in most peripheral tissues and ability to rapidly respond to stimuli make them prime targets for cancer immunotherapies. Studies in mice have clearly demonstrated a role for both peripheral T cells and TRM cells in antitumor immunity. However, a lack of cellular markers defining TRM cells from other infiltrating effector T-cell subsets has hindered determining their composition within human tumors. Recent single-cell transcriptome analyses have revealed that most immune-infiltrated tumors contain T cells with a TRM-like genetic profile. Further, the abundance of T cells with TRM cell characteristics often correlates with a favorable outcome and several lines of evidence suggest that TRM cells may be an important population activated by anti-PD-1 therapy.
Cancer vaccination is one type of therapy that can activate intratumoral TRM cells. However, the route of administration is a critical component influencing the effectiveness of this approach. While in situ vaccination has shown great potential, the immune-suppressive TME can diminish its effect. Furthermore, it remains to be determined how the TME affects TRM differentiation and whether newly infiltrated T cells can become bona fide TRM cells with their specific functional qualities. Understanding these influences, including which immune-inhibitory pathways are active in the TME, will be important for deciding which therapy will best synergize with in situ vaccination. Vaccinating against common viral antigens can activate preexisting TRM cells to induce positive immune changes in the TME and sensitize the tumor to checkpoint blockade therapy. On balance, TRM cells possess the desired functional characteristics that can be harnessed to eliminate tumors, and the study of TRM cell biology in the context of cancer is nascent and a worthy endeavor.
References
Galon J, Pagès F, Marincola FM, Angell HK, Thurin M, Lugli A, Zlobec I, Berger A, Bifulco C, Botti G, Tatangelo F, Britten CM, Kreiter S, Chouchane L, Delrio P, Arndt H, Asslaber M, Maio M, Masucci GV, Mihm M, Vidal-Vanaclocha F, Allison JP, Gnjatic S, Hakansson L, Huber C, Singh-Jasuja H, Ottensmeier C, Zwierzina H, Laghi L, Grizzi F, Ohashi PS, Shaw PA, Clarke BA, Wouters BG, Kawakami Y, Hazama S, Okuno K, Wang E, O’Donnell-Tormey J, Lagorce C, Pawelec G, Nishimura MI, Hawkins R, Lapointe R, Lundqvist A, Khleif SN, Ogino S, Gibbs P, Waring P, Sato N, Torigoe T, Itoh K, Patel PS, Shukla SN, Palmqvist R, Nagtegaal ID, Wang Y, D’Arrigo C, Kopetz S, Sinicrope FA, Trinchieri G, Gajewski TF, Ascierto PA, Fox BA (2012) Cancer classification using the Immunoscore: a worldwide task force. J Transl Med 10:205. https://doi.org/10.1186/1479-5876-10-205
Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman WH, Galon J (2005) Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353:2654–2666. https://doi.org/10.1056/NEJMoa051424
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman WH, Pagès F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964. https://doi.org/10.1126/science.1129139
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, Piha-Paul SA, Yearley J, Seiwert TY, Ribas A, McClanahan TK (2017) IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127:2930–2940. https://doi.org/10.1172/JCI91190
Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, Zhang C, Lunceford JK, Joe A, Cheng J, Webber AL, Ibrahim N, Plimack ER, Ott PA, Seiwert TY, Ribas A, McClanahan TK, Tomassini JE, Loboda A, Kaufman D (2018) Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362:eaar3593. https://doi.org/10.1126/science.aar3593
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer–immune set point. Nature 541:321–330. https://doi.org/10.1038/nature21349
Horton BL, Fessenden TB, Spranger S (2019) Tissue site and the cancer immunity cycle. Trends Cancer 5:593–603. https://doi.org/10.1016/j.trecan.2019.07.006
Masopust D, Vezys V, Marzo AL, Lefrançois L (2001) Preferential localization of effector memory cells in nonlymphoid tissue. Science 291:2413–2417. https://doi.org/10.1126/science.1058867
Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K-I, Dowgiert RK, Kupper TS (2006) The vast majority of CLA+ T cells are resident in normal skin. J Immunol 176:4431–4439. https://doi.org/10.4049/jimmunol.176.7.4431
Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho S-H, Lerner H, Friedman AL, Shen Y, Farber DL (2017) Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep 20:2921–2934. https://doi.org/10.1016/j.celrep.2017.08.078
Clark RA (2015) Resident memory T cells in human health and disease. Sci Transl Med 7:269rv1–269rv1. https://doi.org/10.1126/scitranslmed.3010641
Lian CG, Bueno EM, Granter SR, Laga AC, Saavedra AP, Lin WM, Susa JS, Zhan Q, Chandraker AK, Tullius SG, Pomahac B, Murphy GF (2014) Biomarker evaluation of face transplant rejection: association of donor T cells with target cell injury. Mod Pathol 27:788–799. https://doi.org/10.1038/modpathol.2013.249
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR (2009) Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10:524–530. https://doi.org/10.1038/ni.1718
Schenkel JM, Fraser KA, Vezys V, Masopust D (2013) Sensing and alarm function of resident memory CD8+ T cells. Nat Immunol 14:509–513. https://doi.org/10.1038/ni.2568
Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, Divito SJ, Fuhlbrigge R, Puigserver P, Krueger JG, Hotamisligil GS, Clark RA, Kupper TS (2017) Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543:252–256. https://doi.org/10.1038/nature21379
Mackay LK, Kallies A (2017) Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol 38:94–103. https://doi.org/10.1016/j.it.2016.11.004
Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, Mackay LK (2018) Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol 19:183–191. https://doi.org/10.1038/s41590-017-0027-5
Ariotti S, Beltman JB, Chodaczek G, Hoekstra ME, van Beek AE, Gomez-Eerland R, Ritsma L, van Rheenen J, Marée AFM, Zal T, de Boer RJ, Haanen JBAG, Schumacher TN (2012) Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci U S A 109:19739–19744. https://doi.org/10.1073/pnas.1208927109
Dijkgraaf FE, Matos TR, Hoogenboezem M, Toebes M, Vredevoogd DW, Mertz M, van den Broek B, Song J-Y, Teunissen MBM, Luiten RM, Beltman JB, Schumacher TN (2019) Tissue patrol by resident memory CD8 + T cells in human skin. Nat Immunol 20:756–764. https://doi.org/10.1038/s41590-019-0404-3
Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, Fonseca R, Burbach BJ, Hickman HD, Vezys V, Fife BT, Masopust D (2018) Intravital mucosal imaging of CD8 + resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol 19:173–182. https://doi.org/10.1038/s41590-017-0029-3
McNamara HA, Cai Y, Wagle MV, Sontani Y, Roots CM, Miosge LA, O’Connor JH, Sutton HJ, Ganusov VV, Heath WR, Bertolino P, Goodnow CG, Parish IA, Enders A, Cockburn IA (2017) Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol 2:eaaj1996. https://doi.org/10.1126/sciimmunol.aaj1996
Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R (2010) Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207:553–564. https://doi.org/10.1084/jem.20090858
Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR (2008) Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319:198–202. https://doi.org/10.1126/science.1151869
Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS (2012) Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483:227–231. https://doi.org/10.1038/nature10851
Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrançois L (2004) Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity 20:551–562. https://doi.org/10.1016/s1074-7613(04)00103-7
Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon M-L, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M, Carbone FR, Gebhardt T (2013) The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14:1294–1301. https://doi.org/10.1038/ni.2744
Gebhardt T, Mackay LK (2012) Local immunity by tissue-resident CD8+ memory T cells. Front Immunol. https://doi.org/10.3389/fimmu.2012.00340
Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, Masopust D (2015) Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161:737–749. https://doi.org/10.1016/j.cell.2015.03.031
Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, Vezys V, Masopust D (2012) Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188:4866–4875. https://doi.org/10.4049/jimmunol.1200402
Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, Höög C, Tjernlund A, Michaëlsson J, Folkersen L, Mjösberg J, Blomqvist L, Ehrström M, Ståhle M, Bryceson YT, Eidsmo L (2017) CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 46:287–300. https://doi.org/10.1016/j.immuni.2017.01.009
Bergsbaken T, Bevan MJ (2015) Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8+ T cells responding to infection. Nat Immunol 16:406–414. https://doi.org/10.1038/ni.3108
Sheridan BS, Pham Q-M, Lee Y-T, Cauley LS, Puddington L, Lefrançois L (2014) Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40:747–757. https://doi.org/10.1016/j.immuni.2014.03.007
Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, Woo E, Friedmann PS, King EV, Thomas GJ, Sanchez-Elsner T, Vijayanand P, Ottensmeier CH (2017) Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Publ Group 122:3491. https://doi.org/10.1038/ni.3775
Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, Chang S-C, Grunkemeier G, Leidner R, Bell RB, Weinberg AD (2018) Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9:2724. https://doi.org/10.1038/s41467-018-05072-0
Webb JR, Milne K, Nelson BH (2015) PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res 3:926–935. https://doi.org/10.1158/2326-6066.CIR-14-0239
Boutet M, Gauthier L, Leclerc M, Gros G, de Montpreville V, Théret N, Donnadieu E, Mami-Chouaib F (2016) TGFβ signaling intersects with CD103 integrin signaling to promote T-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res 76:1757–1769. https://doi.org/10.1158/0008-5472.CAN-15-1545
Komdeur FL, Prins TM, van de Wall S, Plat A, Wisman GBA, Hollema H, Daemen T, Church DN, de Bruyn M, Nijman HW (2017) CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Onco Targets Ther 6:e1338230. https://doi.org/10.1080/2162402X.2017.1338230
Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, Molodtsov AK, Bowers JS, Angeles CV, Paulos CM, Huang YH, Turk MJ (2017) Resident memory T cells in skin mediate durable immunity to melanoma. Sci Immunol 2:eaam6346. https://doi.org/10.1126/sciimmunol.aam6346
Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, Donda A, Romero P, Verdeil G, Speiser DE (2016) Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol 7:573. https://doi.org/10.3389/fimmu.2016.00573
Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC (2013) Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14:1285–1293. https://doi.org/10.1038/ni.2745
Park SL, Gebhardt T, Mackay LK (2019) Tissue-resident memory T cells in cancer immunosurveillance. Trends Immunol 40:735–747. https://doi.org/10.1016/j.it.2019.06.002
Masopust D, Soerens AG (2019) Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol 37:521–546. https://doi.org/10.1146/annurev-immunol-042617-053214
da Silva HB, Beura LK, Wang H, Hanse EA, Gore R, Scott MC, Walsh DA, Block KE, Fonseca R, Yan Y, Hippen KL, Blazar BR, Masopust D, Kelekar A, Vulchanova L, Hogquist KA, Jameson SC (2018) The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature 559:1–21. https://doi.org/10.1038/s41586-018-0282-0
Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song J-Y, Jacobs H, Haanen JB, Schumacher TN (2014) T cell memory. Skin-resident memory CD8+ T cells trigger a state of tissue-wide pathogen alert. Science 346:101–105. https://doi.org/10.1126/science.1254803
Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D (2014) Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346:1254536–1254101. https://doi.org/10.1126/science.1254536
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29:235–271. https://doi.org/10.1146/annurev-immunol-031210-101324
Gajewski TF, Schreiber H, Fu Y-X (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14:1014–1022. https://doi.org/10.1038/ni.2703
Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF (2009) Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69:3077–3085. https://doi.org/10.1158/0008-5472.CAN-08-2281
Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med 208:2005–2016. https://doi.org/10.1084/jem.20101159
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–907. https://doi.org/10.1038/nature06309
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen Y-S, Shea LK, Hundal J, Wendl MC, Demeter R, Wylie T, Allison JP, Smyth MJ, Old LJ, Mardis ER, Schreiber RD (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482:400–404. https://doi.org/10.1038/nature10755
Klein G, Sjögren HO, Klein E, HELLSTROM KE (1960) Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res 20:1561–1572
Prehn RT, MAIN JM (1957) Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst 18:769–778. https://doi.org/10.1093/jnci/18.6.769
Burnet FM (1967) Immunological aspects of malignant disease. Lancet 1:1171–1174. https://doi.org/10.1016/s0140-6736(67)92837-1
Thomas L (1982) On immunosurveillance in human cancer. Yale J Biol Med 55:329–333
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998. https://doi.org/10.1038/ni1102-991
Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, Ariyan S, Narayan D, Kluger H, Deng Y, Verma R, Das R, Bacchiocchi A, Halaban R, Sznol M, Dhodapkar MV, Dhodapkar KM (2016) Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight 1:e88955. https://doi.org/10.1172/jci.insight.88955
Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, Salgado R, Byrne DJ, Teo ZL, Dushyanthen S, Byrne A, Wein L, Luen SJ, Poliness C, Nightingale SS, Skandarajah AS, Gyorki DE, Thornton CM, Beavis PA, Fox SB, Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Darcy PK, Speed TP, Mackay LK, Neeson PJ, Loi S (2018) Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med 24:986–993. https://doi.org/10.1038/s41591-018-0078-7
Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, Thompson EA, Fraser KA, Rosato PC, Filali-Mouhim A, Sekaly RP, Jenkins MK, Vezys V, Haining WN, Jameson SC, Masopust D (2016) Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532:512–516. https://doi.org/10.1038/nature17655
Reese TA, Bi K, Kambal A, Filali-Mouhim A, Beura LK, Bürger MC, Pulendran B, Sekaly R-P, Jameson SC, Masopust D, Haining WN, Virgin HW (2016) Sequential infection with common pathogens promotes human-like immune gene expression and altered vaccine response. Cell Host Microbe 19:713–719. https://doi.org/10.1016/j.chom.2016.04.003
Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, Mejías-Pérez E, Esteban M, Melero I, Hidalgo A, Sancho D (2017) Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun 8:16073. https://doi.org/10.1038/ncomms16073
Menares E, Gálvez-Cancino F, Cáceres-Morgado P, Ghorani E, López E, Díaz X, Saavedra-Almarza J, Figueroa DA, Roa E, Quezada SA, Lladser A (2019) Tissue-resident memory CD8+ T cells amplify anti-tumor immunity by triggering antigen spreading through dendritic cells. Nat Commun 10:4401. https://doi.org/10.1038/s41467-019-12319-x
Park SL, Buzzai A, Rautela J, Hor JL, Hochheiser K, Effern M, McBain N, Wagner T, Edwards J, McConville R, Wilmott JS, Scolyer RA, Tüting TTX, Palendira U, Gyorki D, Mueller SN, Huntington ND, Bedoui S, Hölzel MHX, Mackay LK, Waithman J, Gebhardt T (2019) Tissue-resident memory CD8+ T cells promote melanoma–immune equilibrium in skin. Nature 565:1–23. https://doi.org/10.1038/s41586-018-0812-9
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360. https://doi.org/10.1146/annurev.immunol.22.012703.104803
Zamora AE, Crawford JC, Thomas PG (2018) Hitting the target: how T cells detect and eliminate tumors. J Immunol 200:392–399. https://doi.org/10.4049/jimmunol.1701413
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 14:135–146. https://doi.org/10.1038/nrc3670
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Imielinsk M, Jäger N, Jones DTW, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt ANJ, Valdés-Mas R, van Buuren MM, van’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR (2013) Signatures of mutational processes in human cancer. Nature 500:415–421. https://doi.org/10.1038/nature12477
Erkes DA, Smith CJ, Wilski NA, Caldeira-Dantas S, Mohgbeli T, Snyder CM (2017) Virus-specific CD8+ T cells infiltrate melanoma lesions and retain function independently of PD-1 expression. J Immunol 198:2979–2988. https://doi.org/10.4049/jimmunol.1601064
Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, Duan K, Ang N, Poidinger M, Lee YY, Larbi A, Khng AJ, Tan E, Fu C, Mathew R, Teo M, Lim WT, Toh CK, Ong B-H, Koh T, Hillmer AM, Takano A, Lim TKH, Tan E-H, Zhai W, Tan DSW, Tan IB, Newell EW (2018) Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557:575–579. https://doi.org/10.1038/s41586-018-0130-2
Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, Kluin RJC, Snaebjornsson P, Milne K, Nelson BH, Zijlmans H, Kenter G, Voest EE, Haanen JBAG, Schumacher TN (2019) Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med 25:89–94. https://doi.org/10.1038/s41591-018-0266-5
Rosato PC, Wijeyesinghe S, Stolley JM, Nelson CE, Davis RL, Manlove LS, Pennell CA, Blazar BR, Chen CC, Geller MA, Vezys V, Masopust D (2019) Virus-specific memory T cells populate tumors and can be repurposed for tumor immunotherapy. Nat Commun 10:567. https://doi.org/10.1038/s41467-019-08534-1
Li B, Liu L, Zhang J, Chen J, Ye J, Filatenkov A, Shukla S, Qiao J, Zhan X, Wu C, Fu Y-X (2018) The landscape of antigen-specific T cells in human cancers. bioRxiv:1–42. https://doi.org/10.1101/459842
Wolf DM, Lenburg ME, Yau C, Boudreau A, van’t Veer LJ (2014) Gene co-expression modules as clinically relevant hallmarks of breast cancer diversity. PLoS One 9(2):e88309
Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208:1989–2003. https://doi.org/10.1084/jem.20101158
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100. https://doi.org/10.1126/science.1164206
Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC, Belardelli F, Gabriele L (2002) ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 196:1415–1425. https://doi.org/10.1084/jem.20021263
Iborra S, Martínez-López M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, Del Fresno C, Sancho D (2016) Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity 45:847–860. https://doi.org/10.1016/j.immuni.2016.08.019
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira J-P, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L (2007) Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 13:1050–1059. https://doi.org/10.1038/nm1622
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini J-L, Schlemmer F, Tasdemir E, Uhl M, Génin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, André F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178. https://doi.org/10.1038/nm.2028
Stark R, Wesselink TH, Behr FM, Kragten NAM, Arens R, Koch-Nolte F, van Gisbergen KPJM, van Lier RAW (2018) TRM maintenance is regulated by tissue damage via P2RX7. Sci Immunol 3:eaau1022. https://doi.org/10.1126/sciimmunol.aau1022
Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre M-L, Gajewski TF (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41:830–842. https://doi.org/10.1016/j.immuni.2014.10.017
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, Huang X, Gajewski TF, Chen ZJ, Fu Y-X, Weichselbaum RR (2014) STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41:843–852. https://doi.org/10.1016/j.immuni.2014.10.019
Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, Fan Y, Yang Y, Wang J, Glisson BS, Wistuba II, Sage J, Heymach JV, Gibbons DL, Byers LA (2019) Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov 9:646–661. https://doi.org/10.1158/2159-8290.CD-18-1020
Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, Martinon F, Modlin RL, Speiser DE, Gilliet M (2015) STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc Natl Acad Sci U S A 112:15408–15413. https://doi.org/10.1073/pnas.1512832112
Yang H, Lee WS, Kong SJ, Kim CG, Kim JH, Chang SK, Kim S, Kim G, Chon HJ, Kim C (2019) STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest 129:4350–4364. https://doi.org/10.1172/JCI125413
Ng KW, Marshall EA, Bell JC, Lam WL (2018) cGAS–STING and cancer: dichotomous roles in tumor immunity and development. Trends Immunol 39:44–54. https://doi.org/10.1016/j.it.2017.07.013
Spranger S, Dai D, Horton B, Gajewski TF (2017) Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 31:711–723.e4. https://doi.org/10.1016/j.ccell.2017.04.003
Kashem SW, Haniffa M, Kaplan DH (2017) Antigen-presenting cells in the skin. Annu Rev Immunol 35:469–499. https://doi.org/10.1146/annurev-immunol-051116-052215
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, Ginhoux F, Merad M (2016) Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44:924–938. https://doi.org/10.1016/j.immuni.2016.03.012
Chow MT, Luster AD (2014) Chemokines in cancer. Cancer Immunol Res 2:1125–1131. https://doi.org/10.1158/2326-6066.CIR-14-0160
Qi H, Kastenmüller W, Germain RN (2014) Spatiotemporal basis of innate and adaptive immunity in secondary lymphoid tissue. Annu Rev Cell Dev Biol 30:141–167. https://doi.org/10.1146/annurev-cellbio-100913-013254
Groom JR, Luster AD (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89:207–215. https://doi.org/10.1038/icb.2010.158
Nakanishi Y, Lu B, Gerard C, Iwasaki A (2009) CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 462:510–513. https://doi.org/10.1038/nature08511
Shin H, Iwasaki A (2012) A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491:463–467. https://doi.org/10.1038/nature11522
Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, Luster AD, Evans SS (2015) Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6:298. https://doi.org/10.1038/ncomms8458
Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJ, Balli D, Hay CA, Sela Y, Merrell AJ, Liudahl SM, Gordon N, Norgard RJ, Yuan S, Yu S, Chao T, Ye S, Eisinger-Mathason TSK, Faryabi RB, Tobias JW, Lowe SW, Coussens LM, Wherry EJ, Vonderheide RH, Stanger BZ (2018) Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49:178–193.e7. https://doi.org/10.1016/j.immuni.2018.06.006
Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, Yewdell JW (2015) CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity 42:524–537. https://doi.org/10.1016/j.immuni.2015.02.009
Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, Luster AD (2019) Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity. https://doi.org/10.1016/j.immuni.2019.04.010
Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, Liu AJ, Hunter CA (2012) Generalized Lévy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486:545–548. https://doi.org/10.1038/nature11098
Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, Kaech SM (2014) CD4+ T cell help guides formation of CD103+ lung-resident memory CD8+ T cells during influenza viral infection. Immunity 41:633–645. https://doi.org/10.1016/j.immuni.2014.09.007
Slütter B, Pewe LL, Kaech SM, Harty JT (2013) Lung airway-surveilling CXCR3(hi) memory CD8(+) T cells are critical for protection against influenza A virus. Immunity 39:939–948. https://doi.org/10.1016/j.immuni.2013.09.013
Musha H, Ohtani H, Mizoi T, Kinouchi M, Nakayama T, Shiiba K, Miyagawa K, Nagura H, Yoshie O, Sasaki I (2005) Selective infiltration of CCR5+CXCR3+ T lymphocytes in human colorectal carcinoma. Int J Cancer 116:949–956. https://doi.org/10.1002/ijc.21135
Mulligan AM, Raitman I, Feeley L, Pinnaduwage D, Nguyen LT, O’Malley FP, Ohashi PS, Andrulis IL (2013) Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the Ontario familial breast cancer registry. Clin Cancer Res 19:336–346. https://doi.org/10.1158/1078-0432.CCR-11-3314
Hong M, Puaux A-L, Huang C, Loumagne L, Tow C, Mackay C, Kato M, Prévost-Blondel A, Avril M-F, Nardin A, Abastado J-P (2011) Chemotherapy induces intratumoral expression of chemokines in cutaneous melanoma, favoring T-cell infiltration and tumor control. Cancer Res 71:6997–7009. https://doi.org/10.1158/0008-5472.CAN-11-1466
Olson MR, McDermott DS, Varga SM (2012) The initial draining lymph node primes the bulk of the CD8 T cell response and influences memory T cell trafficking after a systemic viral infection. PLoS Pathog 8:e1003054. https://doi.org/10.1371/journal.ppat.1003054
Liu L, Fuhlbrigge RC, Karibian K, Tian T, Kupper TS (2006) Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity 25:511–520. https://doi.org/10.1016/j.immuni.2006.06.019
Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, von Andrian UH (2003) Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424:88–93. https://doi.org/10.1038/nature01726
Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, Robins H, Krueger JG, Clark RA, Kupper TS (2015) Common clonal origin of central and resident memory T cells following skin immunization. Nat Med 21:647–653. https://doi.org/10.1038/nm.3860
Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, Drexler I, Höfer T, Riddell SR, Busch DH (2014) Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity 41:116–126. https://doi.org/10.1016/j.immuni.2014.05.018
Mani V, Bromley SK, Äijö T, Mora-Buch R, Carrizosa E, Warner RD, Hamze M, Sen DR, Chasse AY, Lorant A, Griffith JW, Rahimi RA, McEntee CP, Jeffrey KL, Marangoni F, Travis MA, Lacy-Hulbert A, Luster AD, Mempel TR (2019) Migratory DCs activate TGF-β to precondition naïve CD8+ T cells for tissue-resident memory fate. Science 366:eaav5728. https://doi.org/10.1126/science.aav5728
Le Floc’h A, Jalil A, Vergnon I, Le Maux CB, Lazar V, Bismuth G, Chouaib S, Mami-Chouaib F (2007) Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med 204:559–570. https://doi.org/10.1084/jem.20061524
Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, Ferguson A, Chen J, Hewavisenti R, Hersey P, Gebhardt T, Weninger W, Britton WJ, Saw RPM, Thompson JF, Menzies AM, Long GV, Scolyer RA, Palendira U (2018) CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti–PD-1 treatment. Clin Cancer Res 24:3036–3045. https://doi.org/10.1158/1078-0432.CCR-17-2257
Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, Freeman GJ, Warren SE, Ong S, Browning E, Twitty CG, Pierce RH, Le MH, Algazi AP, Daud AI, Pai SI, Zippelius A, Weissleder R, Pittet MJ (2018) Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-γ and IL-12. Immunity 49:1148–1161.e7. https://doi.org/10.1016/j.immuni.2018.09.024
Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, Saya H, Amagai M, Nagao K (2015) Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med 21:1272–1279. https://doi.org/10.1038/nm.3962
Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR (2015) T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43:1101–1111. https://doi.org/10.1016/j.immuni.2015.11.008
Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, Masopust D (2016) IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J Immunol 196:3920–3926. https://doi.org/10.4049/jimmunol.1502337
Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, Lietzenmayer M, Kroehling L, Takumi A, Kometani K, Inoue T, Kluger Y, Kaech SM, Kurosaki T, Okada T, Flavell RA (2018) KLRG1+ effector CD8+ T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity 48:716–729.e8. https://doi.org/10.1016/j.immuni.2018.03.015
Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T (2012) Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109:7037–7042. https://doi.org/10.1073/pnas.1202288109
Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ (2012) Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol 86:8161–8170. https://doi.org/10.1128/JVI.00889-12
Williams JB, Horton BL, Zheng Y, Duan Y, Powell JD, Gajewski TF (2017) The EGR2 targets LAG-3 and 4-1BB describe and regulate dysfunctional antigen-specific CD8+ T cells in the tumor microenvironment. J Exp Med 214:381–400. https://doi.org/10.1084/jem.20160485
Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, Rufer N, Speiser DE (2011) Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest 121:2350–2360. https://doi.org/10.1172/JCI46102
Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP (2011) Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One 6:e19499. https://doi.org/10.1371/journal.pone.0019499
Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD, Newell EW (2017) Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat Commun 8:562. https://doi.org/10.1038/s41467-017-00627-z
Trujillo JA, Sweis RF, Bao R, Luke JJ (2018) T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res 6:990–1000. https://doi.org/10.1158/2326-6066.CIR-18-0277
Spranger S, Gajewski TF (2018) Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 18:139–147. https://doi.org/10.1038/nrc.2017.117
Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160:48–61. https://doi.org/10.1016/j.cell.2014.12.033
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 523:231–235. https://doi.org/10.1038/nature14404
Larkin J, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373:1270–1271. https://doi.org/10.1056/NEJMc1509660
Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V (2011) An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 61:1019–1031. https://doi.org/10.1007/s00262-011-1172-6
Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF (2019) WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 25:3074–3083. https://doi.org/10.1158/1078-0432.CCR-18-1942
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres Cabala CA, Cooper ZA, Chen P-L, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget M-A, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL, Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P (2016) Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6:202–216. https://doi.org/10.1158/2159-8290.CD-15-0283
Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, Gajewski AP, Andrade J, Gajewski TF (2016) Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. PNAS 113:E7759–E7768. https://doi.org/10.1073/pnas.1609376113
Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA (1996) Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst 88:100–108. https://doi.org/10.1093/jnci/88.2.100
Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, Du VY, Schlessinger J, Goldberg SB, Chiang A, Sanmamed MF, Melero I, Agorreta J, Montuenga LM, Lifton R, Ferrone S, Kavathas P, Rimm DL, Kaech SM, Schalper K, Herbst RS, Politi K (2017) Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov 7:1420–1435. https://doi.org/10.1158/2159-8290.CD-17-0593
Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, Frederick DT, Hazar-Rethinam M, Nadres BA, Seventer EE, Shukla SA, Yizhak K, Ray JP, Rosebrock D, Livitz D, Adalsteinsson V, Getz G, Duncan LM, Li B, Corcoran RB, Lawrence DP, Stemmer-Rachamimov A, Boland GM, Landau DA, Flaherty KT, Sullivan RJ, Hacohen N (2017) Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8:1–10. https://doi.org/10.1038/s41467-017-01062-w
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TNM, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829. https://doi.org/10.1056/NEJMoa1604958
Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, Nowak JA, Nishihara R, Qian ZR, Inamura K, Morikawa T, Nosho K, Abril-Rodriguez G, Connolly C, Escuin-Ordinas H, Geybels MS, Grady WM, Hsu L, Hu-Lieskovan S, Huyghe JR, Kim YJ, Krystofinski P, Leiserson MDM, Montoya DJ, Nadel BB, Pellegrini M, Pritchard CC, Puig-Saus C, Quist EH, Raphael BJ, Salipante SJ, Shin DS, Shinbrot E, Shirts B, Shukla S, Stanford JL, Sun W, Tsoi J, Upfill-Brown A, Wheeler DA, Wu CJ, Yu M, Zaidi SH, Zaretsky JM, Gabriel SB, Lander ES, Garraway LA, Hudson TJ, Fuchs CS, Ribas A, Ogino S, Peters U (2018) Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8:730–749. https://doi.org/10.1158/2159-8290.CD-17-1327
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman W, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf RB, Brahmer JR, Karchin R, Pardoll DM, Velculescu VE (2017) Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 7:264–276. https://doi.org/10.1158/2159-8290.CD-16-0828
Williams JB, Li S, Higgs EF, Cabanov A, Wang X, Huang H, Gajewski TF (2020) Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat Commun 11:602. https://doi.org/10.1038/s41467-020-14290-4
Sackstein R, Schatton T, Barthel SR (2017) T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab Investig 97:669–697. https://doi.org/10.1038/labinvest.2017.25
Clark RA, Huang SJ, Murphy GF, Mollet IG, Hijnen D, Muthukuru M, Schanbacher CF, Edwards V, Miller DM, Kim JE, Lambert J, Kupper TS (2008) Human squamous cell carcinomas evade the immune response by down-regulation of vascular E-selectin and recruitment of regulatory T cells. J Exp Med 205:2221–2234. https://doi.org/10.1084/jem.20071190
Weishaupt C, Munoz KN, Buzney E, Kupper TS, Fuhlbrigge RC (2007) T-cell distribution and adhesion receptor expression in metastatic melanoma. Clin Cancer Res 13:2549–2556. https://doi.org/10.1158/1078-0432.CCR-06-2450
Enarsson K, Johnsson E, Lindholm C, Lundgren A, Pan-Hammarström Q, Strömberg E, Bergin P, Baunge E-L, Svennerholm A-M, Quiding-Järbrink M (2006) Differential mechanisms for T lymphocyte recruitment in normal and neoplastic human gastric mucosa. Clin Immunol 118:24–34. https://doi.org/10.1016/j.clim.2005.08.001
Forster-Horváth C, Döme B, Paku S, Ladányi A, Somlai B, Jalkanen S, Tímár J (2004) Loss of vascular adhesion protein-1 expression in intratumoral microvessels of human skin melanoma. Melanoma Res 14:135–140. https://doi.org/10.1097/00008390-200404000-00010
Yoong KF, McNab G, Hübscher SG, Adams DH (1998) Vascular adhesion protein-1 and ICAM-1 support the adhesion of tumor-infiltrating lymphocytes to tumor endothelium in human hepatocellular carcinoma. J Immunol 160:3978–3988
Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH (1995) Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med 181:811–816. https://doi.org/10.1084/jem.181.2.811
Dirkx AEM, oude Egbrink MGA, Castermans K, van der Schaft DWJ, Thijssen VLJL, Dings RPM, Kwee L, Mayo KH, Wagstaff J, Bouma-ter Steege JCA, Griffioen AW (2006) Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J 20:621–630. https://doi.org/10.1096/fj.05-4493com
Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang W-C, Gollnick SO, Dewhirst MW, Rose-John S, Repasky EA, Baumann H, Evans SS (2011) IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest 121:3846–3859. https://doi.org/10.1172/JCI44952
Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G (2014) Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 20:607–615. https://doi.org/10.1038/nm.3541
Birbrair A, Zhang T, Wang Z-M, Messi ML, Olson JD, Mintz A, Delbono O (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307:C25–C38. https://doi.org/10.1152/ajpcell.00084.2014
Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim I, Welte T, Sheng K, Dobrolecki LE, Zhang X, Putluri N, Phung TL, Mani SA, Stossi F, Sreekumar A, Mancini MA, Decker WK, Zong C, Lewis MT, Zhang XH-F (2017) Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 544:250–254. https://doi.org/10.1038/nature21724
Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Gröne H-J, Hämmerling GJ, Arnold B, Ganss R (2008) Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature 453:410–414. https://doi.org/10.1038/nature06868
Turley SJ, Cremasco V, Astarita JL (2015) Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol 15:669–682. https://doi.org/10.1038/nri3902
Kaur A, Ecker BL, Douglass SM, Kugel CH III, Webster MR, Almeida FV, Somasundaram R, Hayden J, Ban E, Ahmadzadeh H, Franco-Barraza J, Shah N, Mellis IA, Keeney F, Kossenkov A, Tang H-Y, Yin X, Liu Q, Xu X, Fane M, Brafford P, Herlyn M, Speicher DW, Wargo JA, Tetzlaff MT, Haydu LE, Raj A, Shenoy V, Cukierman E, Weeraratna AT (2019) Remodeling of the collagen matrix in aging skin promotes melanoma metastasis and affects immune cell motility. Cancer Discov 9:64–81. https://doi.org/10.1158/2159-8290.CD-18-0193
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571. https://doi.org/10.1038/nature13954
Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E, Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S (2015) The consensus molecular subtypes of colorectal cancer. Nat Med 21:1350–1356. https://doi.org/10.1038/nm.3967
Lin T-H, Shao Y-Y, Chan S-Y, Huang C-Y, Hsu C-H, Cheng A-L (2015) High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res 21:3678–3684. https://doi.org/10.1158/1078-0432.CCR-14-1954
Massagué J (2008) TGFbeta in cancer. Cell 134:215–230. https://doi.org/10.1016/j.cell.2008.07.001
Batlle E, Massagué J (2019) Transforming growth factor-β signaling in immunity and cancer. Immunity 50:924–940. https://doi.org/10.1016/j.immuni.2019.03.024
Isella C, Brundu F, Bellomo SE, Galimi F, Zanella E, Porporato R, Petti C, Fiori A, Orzan F, Senetta R, Boccaccio C, Ficarra E, Marchionni L, Trusolino L, Medico E, Bertotti A (2017) Selective analysis of cancer-cell intrinsic transcriptional traits defines novel clinically relevant subtypes of colorectal cancer. Nat Commun 8:15107. https://doi.org/10.1038/ncomms15107
Calon A, Lonardo E, Berenguer-Llergo A, Espinet E, Hernando-Momblona X, Iglesias M, Sevillano M, Palomo-Ponce S, Tauriello DVF, Byrom D, Cortina C, Morral C, Barceló C, Tosi S, Riera A, Attolini CS-O, Rossell D, Sancho E, Batlle E (2015) Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 47:320–329. https://doi.org/10.1038/ng.3225
Ganesh K, Massagué J (2018) TGF-beta inhibition and immunotherapy: checkmate. Immunity 48:626–628. https://doi.org/10.1016/j.immuni.2018.03.037
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, Byrom D, Matarin JA, Calon A, Rivas EI, Nebreda AR, Riera A, Attolini CS-O, Batlle E (2018) TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554:538–543. https://doi.org/10.1038/nature25492
Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R (2003) Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol 77:4911–4927
Wherry EJ, Ha S-J, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27:670–684. https://doi.org/10.1016/j.immuni.2007.09.006
Wherry EJ (2011) T cell exhaustion. Nat Publ Group 131:492–499. https://doi.org/10.1038/ni.2035
Martinez GJ, Pereira RM, Äijö T, Kim EY, Marangoni F, Pipkin ME, Togher S, Heissmeyer V, Zhang YC, Crotty S, Lamperti ED, Ansel KM, Mempel TR, Lähdesmäki H, Hogan PG, Rao A (2015) The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42:265–278. https://doi.org/10.1016/j.immuni.2015.01.006
Oestreich KJ, Yoon H, Ahmed R, Boss JM (2008) NFATc1 regulates PD-1 expression upon T cell activation. J Immunol 181:4832–4839. https://doi.org/10.4049/jimmunol.181.7.4832
Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD (2017) T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 355:1428–1433. https://doi.org/10.1126/science.aaf1292
Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, Morgado L, Zehn D, Birchmeier W, Vivier E, Guarda G (2018) Shp-2 is dispensable for establishing T cell exhaustion and for PD-1 signaling in vivo. Cell Rep 23:39–49. https://doi.org/10.1016/j.celrep.2018.03.026
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DAA, Wherry EJ (2009) Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Publ Group 10:29–37. https://doi.org/10.1038/ni.1679
Anderson AC, Ha S-J, Araki K, Freeman GJ, Kuchroo VK, Ahmed R (2010) Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A 107:14733–14738. https://doi.org/10.1073/pnas.1009731107
Chen L, Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–242. https://doi.org/10.1038/nri3405
Horton BL, Williams JB, Cabanov A, Spranger S, Gajewski TF (2018) Intratumoral CD8+ T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol Res 6:14–24. https://doi.org/10.1158/2326-6066.CIR-17-0249
Zhu J, Powis de Tenbossche CG, Cané S, Colau D, van Baren N, Lurquin C, Schmitt-Verhulst A-M, Liljeström P, Uyttenhove C, Van den Eynde BJ (2017) Resistance to cancer immunotherapy mediated by apoptosis of tumor-infiltrating lymphocytes. Nat Commun 8:1404. https://doi.org/10.1038/s41467-017-00784-1
Miller BC, Sen DR, Abosy Al R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, Haining WN (2019) Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20:326–336. https://doi.org/10.1038/s41590-019-0312-6
Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, Tsai K, Nosrati A, Nardo L, Alvarado MD, Algazi AP, Pampaloni MH, Lobach IV, Hwang J, Pierce RH, Gratz IK, Krummel MF, Rosenblum MD (2016) Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest 126:3447–3452. https://doi.org/10.1172/JCI87324
Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, Schietinger A, Schumacher TN, Schwartzberg PL, Sharpe AH, Speiser DE, Wherry EJ, Youngblood BA, Zehn D (2019) Defining “T cell exhaustion”. Nat Rev Immunol 14:1–10. https://doi.org/10.1038/s41577-019-0221-9
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, Kang B, Liu Z, Jin L, Xing R, Gao R, Zhang L, Dong M, Hu X, Ren X, Kirchhoff D, Roider HG, Yan T, Zhang Z (2018) Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24:978–985. https://doi.org/10.1038/s41591-018-0045-3
Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, Chee SJ, Eschweiler S, King EV, Awad AS, Hanley CJ, McCann KJ, Bhattacharyya S, Woo E, Alzetani A, Seumois G, Thomas GJ, Ganesan A-P, Friedmann PS, Sanchez-Elsner T, Ay F, Ottensmeier CH, Vijayanand P (2019) Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med 216:2128–2149. https://doi.org/10.1084/jem.20190249
Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, Novellis P, Veronesi G, Lugli E (2018) High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J Exp Med 215:2520–2535. https://doi.org/10.1084/jem.20180684
Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, Choi K, Fromme RM, Dao P, McKenney PT, Wasti RC, Kadaveru K, Mazutis L, Rudensky AY, Pe’er D (2018) Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174:1293–1308.e36. https://doi.org/10.1016/j.cell.2018.05.060
Zheng C, Zheng L, Yoo J-K, Guo H, Zhang Y, Guo X, Kang B, Hu R, Huang JY, Zhang Q, Liu Z, Dong M, Hu X, Ouyang W, Peng J, Zhang Z (2017) Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169:1342–1356.e16. https://doi.org/10.1016/j.cell.2017.05.035
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, Gao R, Kang B, Zhang Q, Huang JY, Konno H, Guo X, Ye Y, Gao S, Wang S, Hu X, Ren X, Shen Z, Ouyang W, Zhang Z (2018) Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564:268–272. https://doi.org/10.1038/s41586-018-0694-x
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani A-C, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane J-P, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, Hacohen N (2018) Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175:998–1013.e20. https://doi.org/10.1016/j.cell.2018.10.038
Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen JBAG, Blank CU, Horlings HM, David E, Baran Y, Bercovich A, Lifshitz A, Schumacher TN, Tanay A, Amit I (2019) Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176:775–789.e18. https://doi.org/10.1016/j.cell.2018.11.043
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, Chang EB, Gajewski TF (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350:1084–1089. https://doi.org/10.1126/science.aac4255
Houlahan KE, Shiah Y-J, Gusev A, Yuan J, Ahmed M, Shetty A, Ramanand SG, Yao CQ, Bell C, O’Connor E, Huang V, Fraser M, Heisler LE, Livingstone J, Yamaguchi TN, Rouette A, Foucal A, Espiritu SMG, Sinha A, Sam M, Timms L, Johns J, Wong A, Murison A, Orain M, Picard V, Hovington H, Bergeron A, Lacombe L, Lupien M, Fradet Y, Têtu B, McPherson JD, Pasaniuc B, Kislinger T, Chua MLK, Pomerantz MM, van der Kwast T, Freedman ML, Mani RS, He HH, Bristow RG, Boutros PC (2019) Genome-wide germline correlates of the epigenetic landscape of prostate cancer. Nat Med 25:1615–1626. https://doi.org/10.1038/s41591-019-0579-z
Held W, Siddiqui I, Schaeuble K, Speiser DE (2019) Intratumoral CD8+ T cells with stem cell-like properties: implications for cancer immunotherapy. Sci Transl Med 11:eaay6863. https://doi.org/10.1126/scitranslmed.aay6863
Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, Hu J, Hocker JD, Hawk NV, Kapoor V, Telford WG, Gurusamy D, Yu Z, Bhandoola A, Xue H-H, Roychoudhuri R, Higgs BW, Restifo NP, Bender TP, Ji Y, Gattinoni L (2019) The transcription factor c-Myb regulates CD8+ T cell stemness and antitumor immunity. Nat Immunol 20:337–349. https://doi.org/10.1038/s41590-018-0311-z
Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ (2015) Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med 439:1125–1137. https://doi.org/10.1084/jem.20142237
Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, Luther SA, Speiser DE, Held W (2019) Intratumoral Tcf1+PD-1+CD8+ T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50:195–211.e10. https://doi.org/10.1016/j.immuni.2018.12.021
Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, Prince SS, Wiese M, Lardinois D, Ho P-C, Klein C, Karanikas V, Mertz KD, Schumacher TN, Zippelius A (2018) A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 24:1–17. https://doi.org/10.1038/s41591-018-0057-z
Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Bürger MC, Shan Q, Hale JS, Lee J, Nasti TH, Sharpe AH, Freeman GJ, Germain RN, Nakaya HI, Xue H-H, Ahmed R (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537:417–421. https://doi.org/10.1038/nature19330
Kean LS, Turka LA, Blazar BR (2017) Advances in targeting co-inhibitory and co-stimulatory pathways in transplantation settings: the Yin to the Yang of cancer immunotherapy. Immunol Rev 276:192–212. https://doi.org/10.1111/imr.12523
MacIver NJ, Michalek RD, Rathmell JC (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31:259–283. https://doi.org/10.1146/annurev-immunol-032712-095956
Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, Guittard GC, Yu Z, Carbonaro V, Okkenhaug K, Schrump DS, Linehan WM, Roychoudhuri R, Restifo NP (2016) Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537:539–543. https://doi.org/10.1038/nature19364
Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha N-H, Lee P-H, Shin M, Patel SJ, Yu Z, Palmer DC, Kruhlak MJ, Liu X, Locasale JW, Huang J, Roychoudhuri R, Finkel T, Klebanoff CA, Restifo NP (2019) T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363:eaau0135–eaau0114. https://doi.org/10.1126/science.aau0135
Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM (2016) The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45:374–388. https://doi.org/10.1016/j.immuni.2016.07.009
Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang Y-CJ, Corona AL, Gemta LF, Vincent BG, Wang RC, Kim B, Hong J, Chen C-L, Bullock TN, Irish JM, Rathmell WK, Rathmell JC (2017) Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2:95. https://doi.org/10.1172/jci.insight.93411
Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162:1217–1228. https://doi.org/10.1016/j.cell.2015.08.012
Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJW, Tonc E, Schreiber RD, Pearce EJ, Pearce EL (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162:1229–1241. https://doi.org/10.1016/j.cell.2015.08.016
Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ (2017) Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 32:377–391.e9. https://doi.org/10.1016/j.ccell.2017.08.004
Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, Li L, Boussiotis VA (2015) PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun 6:6692. https://doi.org/10.1038/ncomms7692
Carreau A, Hafny-Rahbi El B, Matejuk A, Grillon C, Kieda C (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med 15:1239–1253. https://doi.org/10.1111/j.1582-4934.2011.01258.x
Rao G, Guy RH, Glikfeld P, LaCourse WR, Leung L, Tamada J, Potts RO, Azimi N (1995) Reverse iontophoresis: noninvasive glucose monitoring in vivo in humans. Pharm Res 12:1869–1873. https://doi.org/10.1023/a:1016271301814
Pan Y, Kupper TS (2018) Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol 9:1347. https://doi.org/10.3389/fimmu.2018.01347
Munir R, Lisec J, Swinnen JV, Zaidi N (2019) Lipid metabolism in cancer cells under metabolic stress. Br J Cancer 120:1–9. https://doi.org/10.1038/s41416-019-0451-4
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, Validire P, Besse B, Mami-Chouaib F (2015) CD8+CD103+ tumor–infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194:3475–3486. https://doi.org/10.4049/jimmunol.1402711
Mlecnik B, Bindea G, Angell HK, Sasso MS, Obenauf AC, Fredriksen T, Lafontaine L, Bilocq AM, Kirilovsky A, Tosolini M, Waldner M, Berger A, Fridman WH, Rafii A, Valge-Archer V, Pagès F, Speicher MR, Galon J (2014) Functional network pipeline reveals genetic determinants associated with in situ lymphocyte proliferation and survival of cancer patients. Sci Transl Med 6:228ra37–228ra37. https://doi.org/10.1126/scitranslmed.3007240
Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, Gilboa E, Hammond SA, Hershberg R, Korman AJ, Kvistborg P, Melief C, Mellman I, Palucka AK, Redchenko I, Robins H, Sallusto F, Schenkelberg T, Schoenberger S, Sosman J, Türeci Ö, Van den Eynde B, Koff W, Coukos G (2016) The human vaccines project: a roadmap for cancer vaccine development. Sci Transl Med 8:334ps9. https://doi.org/10.1126/scitranslmed.aaf0685
Nizard M, Roussel H, Tartour E (2016) Resident memory T cells as surrogate markers of the efficacy of cancer vaccines. Clin Cancer Res 22:530–532. https://doi.org/10.1158/1078-0432.CCR-15-2364
Park CO, Kupper TS (2015) The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med 21:688–697. https://doi.org/10.1038/nm.3883
Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS (2010) Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med 16:224–227. https://doi.org/10.1038/nm.2078
Sun Y-Y, Peng S, Han L, Qiu J, Song L, Tsai Y, Yang B, Roden RBS, Trimble CL, Hung C-F, Wu T-C (2016) Local HPV recombinant vaccinia boost following priming with an HPV DNA vaccine enhances local HPV-specific CD8+ T-cell-mediated tumor control in the genital tract. Clin Cancer Res 22:657–669. https://doi.org/10.1158/1078-0432.CCR-15-0234
Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, Marcheteau E, Fabre E, Mandavit M, Terme M, Blanc C, Escudie J-B, Gibault L, Barthes FLP, Granier C, Ferreira LCS, Badoual C, Johannes L, Tartour E (2017) Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun 8:15221. https://doi.org/10.1038/ncomms15221
Sandoval F, Terme M, Nizard M, Badoual C, Bureau M-F, Freyburger L, Clement O, Marcheteau E, Gey A, Fraisse G, Bouguin C, Merillon N, Dransart E, Tran T, Quintin-Colonna F, Autret G, Thiebaud M, Suleman M, Riffault S, Wu T-C, Launay O, Danel C, Taieb J, Richardson J, Zitvogel L, Fridman WH, Johannes L, Tartour E (2013) Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med 5:172ra20. https://doi.org/10.1126/scitranslmed.3004888
Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 6:226ra32. https://doi.org/10.1126/scitranslmed.3008095
Strickley JD, Messerschmidt JL, Awad ME, Li T, Hasegawa T, Ha DT, Nabeta HW, Bevins PA, Ngo KH, Asgari MM, Nazarian RM, Neel VA, Jenson AB, Joh J, Demehri S (2019) Immunity to commensal papillomaviruses protects against skin cancer. Nature 575:519–522. https://doi.org/10.1038/s41586-019-1719-9
Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, Wang D, Getzler AJ, Nguyen T, Crotty S, Wang W, Pipkin ME, Goldrath AW (2017) Runx3 programs CD8+ T cell residency in non-lymphoid tissues and tumours. Nature 552:253–257. https://doi.org/10.1038/nature24993
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Williams, J.B., Kupper, T.S. (2020). Resident Memory T Cells in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1273. Springer, Cham. https://doi.org/10.1007/978-3-030-49270-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-49270-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-49269-4
Online ISBN: 978-3-030-49270-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)