
Abstract
Adenosine, deriving from ATP released by dying cancer cells and then degradated in the tumor environment by CD39/CD73 enzyme axis, is linked to the generation of an immunosuppressed niche favoring the onset of neoplasia. Signals delivered by extracellular adenosine are detected and transduced by G-protein-coupled cell surface receptors, classified into four subtypes: A1, A2A, A2B, and A3. A critical role of this nucleoside is emerging in the modulation of several immune and nonimmune cells defining the tumor microenvironment, providing novel insights about the development of novel therapeutic strategies aimed at undermining the immune-privileged sites where cancer cells grow and proliferate.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Adenosine
- Adenosine receptors
- Adenosine deaminase
- Nucleoside transporters
- CD73
- CD39
- Immune cells
- Myeloid cells
- Lymphocytes
- Angiogenesis
- Neoplasia
- Tumor microenvironment
- Metastasis
- Apoptosis
- Cell proliferation
9.1 Introduction
Until a few years ago, the term cancer defined an abnormal growth of cells which tend to proliferate in an uncontrolled way and, in some cases, to metastasize [1]. It has been for long conceived that hallmarks of cancer were intrinsic genetic features eliciting tumor development, proliferation, and progression and that targeting such cell pathways could be sufficient to reach a therapeutic cancer control [1].
This simplistic view of the neoplastic disease has been radically changed since the introduction of the concept of tumor microenvironment (TME) [2]. Basically, the neoplastic microenvironment is a dynamic niche consisting of multiple cell types, including several immune cell populations (i.e., macrophages, neutrophils, mast cells, myeloid-derived suppressor cells, dendritic cells, NK cells, T and B lymphocytes), cytokines, blood vessels, cancer cells, and their surrounding stroma (which consists of fibroblasts, endothelial cells, pericytes, and mesenchymal cells) all collaborating to shape tumor progression [2]. The TME is an immunosuppressed milieu, characterized by the presence of mediators able to defuse the immune surveillance, impairing T-cell infiltration and favoring the accrual and activation of regulatory cells and thus contributing to cancer dissemination [3].
At the end of the 1990s, Blay et al. provided the first evidence about a marked increase of adenosine levels, a metabolite resulting from the degradation of ATP, in the cancer microenvironment [4, 5]. This sparked the interest of the scientific community about the role of this nucleoside in tumor onset and progression. Indeed, the concentrations of adenosine, physiologically present at low levels in the interstitial fluids of unstressed tissues, increase rapidly in response to pathophysiological conditions, such as hypoxia, ischemia, inflammation, or trauma, in order to generate a variety of cellular responses aimed at restoring tissue homeostasis [6, 7]. The extracellular adenosine accumulation following an acute injury exerts protective effects, shielding cells and tissues from an excessive inflammatory response and immune-mediated damage and promoting the healing processes [6, 8,9,10,11]. However, the persistence of increased adenosine concentrations beyond the acute injury phase can become detrimental to tissues paving the way to the generation of an immunosuppressed niche, an ideal condition for the onset and development of neoplasia [12]. Interestingly, further investigations allowed to observe that elevated adenosine concentrations within neoplastic environment exerted an immunosuppressant activity and counteracted the effector T-cell functions, mainly via the engagement of the adenosine A2A receptors [13, 14].
It needs to be emphasized that the release of adenosine is actively increased as a result of specific genetic alterations that occur during tumor progression [15]. Several tumors are characterized by an altered purine metabolism, which facilitates the production of adenosine or reduces its degradation resulting in a “protective adenosine halo,” spurring the cancer development and progression [15]. In this regard, it has observed that the purine metabolism undergoes a marked reorganization within the cancer niche, with a marked increase in the expression and activity of the main adenosine-generating enzyme ecto-5′-nucleotidase, also designated CD73 [16]. Several types of neoplasia display a high expression of CD73, which quickly dephosphorylates the AMP to adenosine, leading to the generation of an immunosuppressive and pro-angiogenic environment, which facilitates the development of neoplasia [17]. Following these studies, the CD73-adenosine axis has emerged as one of the most promising pathways in immuno-oncology. Indeed, several preclinical studies highlighted the value of CD73 as a potential therapeutic target for cancer management [17]. The pharmacological blockade of CD73, via monoclonal antibodies (mAbs) or small molecules, was effective in counteracting cancer cell growth and proliferation and angiogenesis and in curbing neoplastic cell spreading [18,19,20,21]. In parallel, several authors reported a significant downregulation of the ecto-adenosine deaminase and its cofactor CD26 (also known as dipeptidyl peptidase 4) and/or nucleoside transporters in several tumor cells, which determines the accumulation of adenosine within the intratumoral milieu [15, 22,23,24].
9.2 Adenosine Signaling: Receptors, Enzymes, and Transporters
Adenosine signaling strictly depends on the levels of extracellular concentrations reached in the receptor biophase [25, 26]. The extracellular levels of this nucleoside are regulated by a complex ecto-enzyme machinery and uptake system, aimed at shaping the purinergic signaling based on the variations of tissue health condition [16, 25, 27].
Physiologically, the main source of adenosine is the S-adenosylhomocysteine [28]. Following the synthesis, adenosine is released in the extracellular space via nucleoside transporters [28]. Based on their molecular and functional characteristics, these transporters are classified into: (1) equilibrative nucleoside transporters (ENTs), which carry nucleosides across cell membranes in either directions, based on their concentration gradient. They include four subtypes, named as ENT1, ENT2, ENT3, and ENT4 [7, 29]; (2) concentrative nucleoside transporters (CNTs), comprising CNT1, CNT2, and CNT3, which allow the intracellular influx of nucleosides against their concentration gradient, exploiting the sodium ion gradient across cellular membranes as source of energy [7].
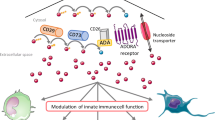
Under adverse conditions, comprising inflammation, hypoxia, ischemia, trauma, or neoplastic microenvironment, the extracellular levels of adenosine increase significantly, reaching micromolar range [30, 31]. In these contexts, adenosine accumulation stems from increased extracellular dephosphorylation of ATP, mediated in a sequential manner by ecto-nucleoside triphosphate diphosphohydrolase-1 (also named CD39) and by ecto-5′-nucleotidase (CD73) (Fig. 9.1) [9]. A number of studies identified CD73 as a critical checkpoint in regulating the duration and the magnitude of the “adenosine halo” surrounding immune cells [32]. Of note, adenosine can be generated through an alternative catabolic pathway, which is initiated by the nicotinamide adenine dinucleotide (NAD+) glycohydrolases/CD38 enzyme axis that converts extracellular NAD+ into adenosine diphosphate ribose (ADPR) [33]. ADPR is then metabolized by CD203a into AMP and then converted by CD73 into adenosine [33].

The purinergic pathway. Once released into the extracellular environment, ATP exerts its effects via P2 receptor engagement (P2X and P2Y). ATP is catabolized via a set of nucleotidases [ecto-ATPase, ecto-apyrase (CD39), and ecto-5’nucleotidase (CD73)], leading to the sequential dephosphorylation of ATP to ADP and AMP and subsequent production of the active metabolite adenosine, which stimulates P1 (A1, A2A, A2B and A3) receptors. The CD38-CD203a enzyme axis on the cell surface, operating independently or in synergy with the conventional CD39/CD73 pathway, also contributes to the generation of adenosine. Several cell types express nucleoside transporters (NT) and adenosine deaminase, which mediate the uptake or deamination of extracellular adenosine, respectively, thus actively participating in the termination of adenosine signaling. ADP adenosine diphosphate, ADPR ADP-ribose, AMP adenosine monophosphate, ATP adenosine triphosphate, NAD+ nicotinamide adenine dinucleotide
As described under physiological conditions, adenosine massively released under pathological conditions can be re-up taken into the cells through the nucleoside transporter or converted into inosine via adenosine deaminase both inside and outside the cell [1, 34, 35] and then into the stable end product uric acid via xanthine oxidase activity [36].
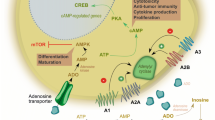
Most of the biological actions of adenosine are mediated by G-protein-coupled cell surface receptors, classified into A1, A2A, A2B, and A3 [37]. A1 and A3 receptors are coupled with pertussis toxin-sensitive G proteins of the Gi, Gq. and Go family [17]. Of note, the engagement of A1 and A3 receptors can also elicit the intracellular Ca2+ mobilization [17]. The signaling mechanism of A2A and A2B receptors relies on the activation of adenylyl cyclase via Gs or Golf. Moreover, A2B receptors can also stimulate phospholipase C via Gq [17].
In addition, all adenosine receptors couple to mitogen-activated protein kinase (MAPK) pathways, including extracellular signal-regulated kinase 1 (ERK1), ERK2, and p38 MAPK [38]. Adenosine also shows receptor-independent effects, since it can cross the cell membrane, acting on less well-defined intracellular mechanisms, comprising the S-adenosylhomocysteine hydrolase systems [39], AMP-activated protein kinase (AMPK) [40], and adenosine kinase [41].
9.3 The Adenosine System and Immune Cell Interactions in Neoplastic Microenvironment
9.3.1 Endothelial Cells
Endothelial cells constitute the inner layer, commonly termed as endothelium, of blood and lymphatic vessels. These cells play a crucial role in the control of hemostasis, blood fluidity, platelet aggregation, and vascular tone, as well as lymphocytes trafficking and vascular permeability [42].
Into the TME, many soluble factors, including vascular endothelial growth factors (VEGFs), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGFs), and chemokines derived from cancer cells or inflammatory cells, stimulate the proliferation and migration of endothelial cells, which form new tumor blood vessels surrounded by perivascular stromal cells, also known as pericytes [43]. Aberrant tumor vascularization promotes tumor progression and metastasis because tumor endothelial cells (TECs) overexpress matrix metalloproteinases (MMPs) and secrete cytokines and growth factors that facilitate tumor cells migration, enhancing their invasive behavior [44].
Adenosine has emerged as modulator factor of endothelium functions, acting on endothelial cells through several mechanisms. The stimulation of adenosine receptors on endothelial cells modulates positively the expression of many proteins, such as CD73 [45], VEGF [46,47,48], IL-8 [48], and basic fibroblast growth factor [48, 49]while inhibits the production of the antiangiogenic factor thrombospondin-1 [50, 51]. Furthermore, adenosine causes the downregulation of adhesion molecules, such as vascular cell adhesion molecule (VCAM)-1, intercellular adhesion molecule (ICAM)-1, and E-selectin on endothelial cells, impairing leukocyte extravasation [52,53,54,55].
Tumor endothelial cells express both CD39 and CD73, and several studies have demonstrated the critical role of these ecto-nucleotidases in promoting tumor angiogenesis. At this regard, it has been observed that deficiency of CD39 or pharmacological inhibition of CD39 activity reduced significantly tumor growth and metastasis, and these effects are associated with reduced tumor angiogenesis [56,57,58]. The expression of CD73 on endothelial cells promotes both growth and migration of endothelial cells and can also stimulate the formation of tube-like structures [59]. Indeed, in mice lacking CD73 tumor size and angiogenesis were reduced when compared to wild-type animals, and CD73+/+ pulmonary microvascular endothelial cells (PMECs) showed an increased capability to form capillary-like structures than CD73−/− PMECs, especially when cultured in cancer-conditioned medium. Interestingly, it emerged that this effect was related both to enzymatic and nonenzymatic activity of CD73 [59]. Later Allard and collaborators clearly demonstrated the role of CD73 and the effects of anti-CD73 therapy on angiogenesis in a mouse model of breast cancer [18]. In particular, they showed that CD73 on cancer cells induces the release of VEGF, via adenosine release, whereas the expression of CD73 on endothelial cells reduced the tube formation and the migration of endothelial cells independently on its enzymatic activity [18]. Therefore, the inhibition of enzymatic and nonenzymatic effects of CD73 reduces the levels of VEGF and angiogenesis in the TME in mice [18]. Another study by Wang et al. showed that CD73 on tumor endothelial cells can also inhibit the adhesion and transmigration of T cells into tumor lesions, through the reduction of ICAM-1 expression [60].
The adenosine-induced effects on tumor angiogenesis are mainly dependent on A2A and A2B receptors expressed on endothelial cells, cancer cells, and immune cells [61,62,63]. Indeed, mice lacking A2B receptors [62] or treated with a selective A2B receptors antagonist [63, 64] displayed a reduced release of VEGF in the tumor lesions as well as an inhibition of tumor growth. Furthermore, Sitkovsky’s group demonstrated that the inhibition of hypoxia in the TME, by using supplemental oxygen therapy, can induce tumor regression in mice [65]. Tumor levels of VEGF and vascularization were inhibited as consequence of reduced intratumoral hypoxia and hypoxia inducible factor (HIF)- 1α. In other words, oxygenation is able to downregulate the expression of adenosine-generating enzymes CD39 and CD73 and thus the accumulation of extracellular adenosine in the TME, which are driven by HIF-1α [65]. Altogether these studies indicate a clear role of adenosine in promoting vascularization and release of angiogenic factors such as VEGF into the TME.
9.3.2 Pericytes
Pericytes are an heterogeneous cell population distinct in different subtypes located within the basement membrane of capillaries [66]. Each subtype, besides to exert common functions, can play different roles within the microcirculation [67]. In particular, the common functions of pericytes include vessel formation and stabilization as well as the regulation of blood flow, via relaxation/contraction mechanisms [68,69,70]. Pioneering studies reported that such cells display macrophage-like characteristics, suggesting a possible participation of these cells also in immune responses [71,72,73]. Increasing evidences described the pericytes in the TME as important modifiers of disease progression, contributing directly or indirectly to tumor angiogenesis, growth, and metastasis, despite, the mechanisms still remain to be clarified.
During the angiogenic process, the endothelial cells release several signals able to induce pericyte recruitment in order to provide physical and chemical support for new blood vessels. Of note, in TME, the vascular architecture does not accomplish complete maturation, resulting in several structural and functional abnormalities [74,75,76,77]. Indeed, it has been reported that an excessive pericyte coverage along tumor microvessels improves the vasculature stability and perfusion, paving the way to the tumor growth. By contrast, low pericyte coverage compromises vessel structure integrity, which becomes leaky, facilitating tumor cell invasion/extravasion [78,79,80,81]. In this regard, several authors demonstrated that the blockade of pericyte recruitment, via PDGF𝛽 pathway inhibition, induced an epithelial cell loss with subsequent regression of tumor vessels [82, 83]. Interestingly, a recent study, performed by Birbrair and colleagues, allowed to demonstrate that only type 2 pericytes, a subtype located within both large blood vessels and small capillaries, participate in new blood vessel formation during tumor angiogenesis, thus representing an intriguing target for antiangiogenic cancer therapy [84].
At present, the evidence about the role of adenosine in the modulation of pericyte functions are scanty. In this regard, it has been reported that adenosine is able to promote pericyte relaxation, triggering an increment of capillary diameter [85]. Indeed, adenosine is able to activate ATP-sensitive potassium (KATP) currents in pericytes through the activation of both A1 and A2A adenosine receptors [86]. By contrast, extracellular ATP triggers an increase in intracellular calcium levels, through the activation of P2X7 receptors, thus determining the pericyte contraction [87, 88].
The pericytes are endowed with CD39 and CD73, which undergo to a rapid increase in their expression and activity under hypoxic conditions, thus leading to a reduction of extracellular concentration of ATP and to a marked presence of adenosine levels [89]. The high extracellular adenosine concentrations activate A2A adenosine receptors on pericytes, determining an increase in KATP channel activity and promoting the dilatation of microvessels with a marked influx of nutrient and oxygen delivery [86]. In parallel, other authors reported a critical role of CD39 in regulating pericyte activity in new vessel formation [56]. The CD39-null mice displayed a reduced angiogenesis characterized by a poor recruitment of surrounding pericytes, thus supporting the evidence about an involvement of this enzyme in the process of neovascularization [56]. A recent study performed by Zhu et al. focused their attention on the role played by the adenosine deaminase protein cat eye syndrome critical region protein 1 (CECR1) in tumor angiogenesis [90]. Such protein, highly expressed in the macrophage lineage, beyond to regulate the differentiation of monocytes into macrophages and to stimulate T helper and macrophage proliferation, is involved in tumor cell proliferation and migration [91]. In this context, Zhu and colleagues reported that CECR1, produced by tumor-associated macrophage, promotes pericyte recruitment and migration, thus supporting the tumor angiogenesis [90].
9.3.3 Myeloid Cells (Monocytes/Macrophages, DCs, and MDSCs)
Mononuclear phagocytes family comprises cells originated in the bone marrow from myeloid precursor cells, which firstly differentiate in blood monocytes. These latter circulate in bloodstream and turn into macrophages or dendritic cells (DCs) when they enter tissue. Mononuclear phagocytes play a critical role during inflammation, wound healing, and immune response by producing several molecules, including cytokines, prostanoids, angiogenic factors, and antimicrobial peptides.
Macrophages can infiltrate the TME, where they exhibit an anti-inflammatory phenotype (M2-like) and in this context are referred to as tumor-associated macrophages (TAMs) [92]. Macrophage infiltration into the tumor stroma is driven by chemokines (CCL2, CCL5, CCL5, and CX3CL1), VEGF, and macrophage colony-stimulating factor (M-CSF) [93, 94], and accumulation of these cells in the TME can strongly favor tumor progression. Several studies demonstrated that TAMs potently suppress the antitumor immune response, stimulate angiogenesis, and promote metastasis dissemination, through a facilitation of the extracellular matrix (ECM) remodeling [95,96,97,98,99].
Adenosine has an important role in regulating the differentiation and maturation of mononuclear cells, as well as their recruitment into TME [100]. Interestingly, adenosine promotes M2 differentiation of macrophages, through activation of A2B receptors and A2A receptors [9, 100, 101]. In particular, the engagement of A2A receptors reduces the production of interleukin (IL)-12, tumor necrosis factor (TNF) [102], nitric oxide [103], and superoxide [104], while it increases the production of VEGF [105].
In support of a role exerted by adenosine in controlling macrophage functions into the tumor, Yegutkin and collaborators demonstrated that CD73-deficient mice showed a reduced tumor infiltration of M2-like macrophages [106]. Later, an elegant work by Cekic and collaborators, by using myeloid-specific A2A receptors deficient mice, clearly reported the role of A2A receptors in myeloid cells in tumor-bearing mice. Indeed, this study showed that signaling of A2A receptor in myeloid cells favors the polarization to M2-like macrophages that in turn suppress CD8+T cells and NK cell-mediated antitumor response [107]. In human ovarian ascites, Montalban Del Barrio et al observed that TAMs highly express both CD39 and CD73, and adenosine generated by ovarian cancer cells promotes the differentiation of M2 macrophages, able in turn to produce adenosine that impaired T-cell responses [108].
A large number of evidence indicate that adenosine signaling can also markedly affect the features of dendritic cells (DCs), which are specialized antigen-presenting cells (APCs) fundamental in orchestrating adaptive immune responses. Under physiological conditions, mature DCs expressing high levels of major histocompatibility complex (MHC) class II and co-stimulatory molecules, such as CD80/86, modulate the activation and the differentiation of T cells into Th1-like cells [109]. Conversely, in pathological conditions, including cancer, the maturation and differentiation of DCs can be altered becoming tolerogenic and thus able to induce T-cell anergy, T- cell death, or regulatory T-cell (Treg) expansion [110].
Within TME, adenosine can critically impair the maturation of DCs favoring the differentiation of a tolerogenic DC subset that produce transforming growth factor (TGF)-β, arginase 2, VEGF, IL-8, IL-10, and IL-6, while the secretion of IL-12, TNF, and chemokines was downregulated [111,112,113,114,115,116]. A study from Novitskiy et al. demonstrated that in the absence of adenosine deaminase, the adenosine levels into microenvironments were increased, resulting in an enhanced tolerogenic function of DCs [113]. These results were confirmed by Naval-Macabuhay et al. [117]. In this study, the authors observed that the addition of adenosine deaminase to DC:T cell cultures reduced the adenosine levels and promoted the priming of effector T cells, while the induction of Tregs was suppressed [117].
Adenosine affects the maturation of DCs through the activation of A2B receptors [114, 118]. In tumor-bearing hosts, the blockade of A2B receptors inhibited the tumor growth by promoting DC activation and consequent T-cell activation [119]. When maturing as tolerogenic, DCs lose their ability to induce T-cell proliferation thus contributing to tumor growth, as observed in several cancer models [119, 120].
Interestingly, in a colon cancer mouse model, the inhibition of CD39 enhances the recruitment of DCs into the tumor lesion together with T cells, restoring the efficacy of chemotherapeutic agents against autophagy-deficient cancer cells [121]. These effects depend on increased concentrations of extracellular ATP as consequence of CD39 inhibition that otherwise would be degraded to adenosine. In support, other studies have demonstrated that inhibition of adenosine generation from ATP increases infiltration of CD103+DCs and enhanced CD8+T cell/Treg ratio, improving radiation therapy-induced tumor control [122, 123].
In tumor-bearing hosts, immature myeloid cells (IMCs) can generate an immature subset of myeloid cells with regulatory functions called myeloid-derived suppressor cells (MDSCs) [124,125,126,127]. MDSCs can differentiate into TAMs and DCs, or they cannot differentiate at all [128, 129]. This cell population, together with TAMs and tolerogenic DCs, contribute to strongly induce immunosuppression and angiogenesis in the TME.
MDSCs limit antitumor immune response through multiple mechanisms, including arginase 1 (Arg 1), nitric oxide synthase 2 (NOS2), NADPH oxidase 2 (NOX2), indoleamine 2,3-dioxygenase, suppressive cytokines, and pro-angiogenic factors. Moreover, MDSCs that express high levels of CD39 and CD73 produce adenosine that contributes to MDSC-mediated immunosuppression in the TME [120]. Adenosine can stimulate MDSCs expansion by binding the A2B receptors, expressed by myeloid precursors [130]. In this study, Ryzhov and collaborators observed that tumor infiltration of MDSCs that express high levels of CD73 is reduced in mice deficient of A2B receptor [130]. Accordingly, in a mouse melanoma model, the blockade of A2B receptors with a selective antagonist leads to a significant decrease of the number of tumor-infiltrating MDSCs [63, 131, 132]. These studies clearly indicate a role for A2B receptor in promoting expansion and accumulation of MDSCs in the tumor lesions, where these cells contribute to promote tumor progression by suppressing T- cell-mediated response and favoring tumor angiogenesis [34, 63, 120, 131, 132]. TGF-β signaling could have a critical role in promoting the recruitment of CD39 and CD73 expressing MDSCs in the tumor lesions [120]. In line to this, Li et al. demonstrated that tumor-derived TGF-β activates HIF-1α that stimulates the expression of CD39 and CD73 on MDSCs isolated from tumor tissues and peripheral blood of patients with non-small cell lung cancer [133]. In the same study, it has been observed that the accumulation of CD39+CD73+MDSCs is closely correlated with tumor progression in cancer patients and with their response to chemotherapy [133]. An additional study from Li et al. reported that metformin can reduce the expression and the activity of CD39 and CD73 in MDSCs of diabetic patients with ovarian cancer, through activation of the protein kinase α (AMPKα) which suppresses HIF-1α [134]. These patients treated with metformin show lower circulating CD39+CD73+ MDSCs and longer overall survival than patients not receiving metformin [134]. Elevated levels of CD39/CD73-expressing MDSCs have also been reported in colorectal cancer patients [135]. To note, other studies have also demonstrated that CD73-derived adenosine can promote resistance to targeted therapy including anti ErbB2 mAb in cancer patients [136]. At this regard, it has been shown that inhibition of CD73 can potentiate the therapeutic efficacy of an anti-ErbB2 mAb in a mouse model of breast cancer, by decreasing the number of tumor-infiltrating MDSCs, and this effect is associated with an increased number of CD8+T cells [136].
9.3.4 Lymphocytes
Lymphocytes are white blood cells originated from the bone marrow-derived progenitors and include three main families: T cells, B cells, and natural killer (NK) cells, all involved in immune response against pathogens and tumor cells.
Cancer patients with high frequency of infiltrating lymphocytes into the TME generally show enhanced survival rates [137]. Analysis of several solid tumors has pointed out that two different phenotypes exist: T-cell infiltrated tumors and non-T-cell infiltrated tumors [138]. In the first case, chemokines promote the influx of CD8+ effector T cells [138], but these get functionally inhibited by many inhibitory stimuli, including regulatory cells and/or inhibitory mediators, such as IDO, anti-inflammatory cytokines, and adenosine. In the second case, a lack of chemokines leads to reduced infiltration of T cells [138]. Interestingly, it has been observed that the type, density, and location of immune cells within the tumor lesions could predict survival of patients affected by colorectal cancer [139]. Of note, it has been observed that an elevated frequency of Tregs in TME can weaken an efficient immune response against cancer [138, 140].
Adenosine plays a critical role in suppressing lymphocytes activity within TME. The enzymes involved in the adenosine production, CD39 and CD73, as well as the adenosine receptors are widely expressed on lymphocyte populations [141,142,143,144]. Most of the adenosine-induced suppressive effects on T cells are dependent on the predominantly expressed A2A receptors. The stimulation of A2A receptors on lymphocytes negatively regulates the production of cytokines and proliferation. Specifically, A2A receptor activation on cytotoxic CD8+T cells results in reduced secretion of IL-2, TNF-α, and interferon (IFN)-γ [145]; likewise its stimulation on NK cells inhibits the production of IFN-γ [146] and TNF-α [147, 148]. On T cells, A2A receptor stimulation also reduces the expression of CD25 and CD40 ligand (CD40L) while increases the expression of programmed cell death protein (PD)-1 and cytotoxic T-lymphocyte antigen 4 (CTLA)-4 [149]. Additionally, this receptor is involved in promoting peripheral tolerance by inducing T-cell anergy [143]. A2A receptor has been observed to be involved in driving naive CD4+ T cell differentiation toward regulatory CD4+FOXP3+ T cells, which also express CD73 and CD39 [150]. Furthermore, Tregs cultured with A2A receptor agonists showed a stronger immunosuppressive activity [150]. All these suppressive effects on T cells are dependent on A2A receptor-induced accumulation of intracellular cAMP, which causes the activation of the protein kinase A (PKA) and the exchange protein directly activated by cAMP (EPAC) [151]. PKA phosphorylates the transcription factors cAMP response element-binding protein (CREB) and the nuclear factor of activated T cells (NF-AT) [152,153,154]. Additionally, EPAC exerts its action by sequestering Raf-1, causing thus the inhibition of T- cell receptor (TCR)-induced MEK-ERK activation [155, 156]. Furthermore, activation of PKA in T cells inhibits the TCR/CD28 transduction signaling during activation by reducing the phosphorylation of ZAP-70 [157]. This effect strongly impairs the production of IL-2, proliferation, and release of effector cytokines such as IFN-γ and granzyme B [157, 158].
The first evidence for a role of A2A receptors in controlling antitumor T-cell responses was provided by Ohta and collaborators in 2006 [61] . In this work, the authors demonstrated that mice lacking A2A receptor rejected immunogenic tumors in a CD8+T-cell-dependent manner. Accordingly, blockade of this receptor with a selective antagonist delayed tumor growth [61]. Later many studies have been focused on the therapeutic potential of targeting A2A receptors with selective antagonists in many types of tumors in mice. Selective blockade of this receptor increases the tumor infiltration of CD8+T cells, NK cells, and the tumor levels of cytokines while reducing the expression of T- cell co-inhibitory receptors (reviewed in [159]). Interestingly, other studies have also demonstrated that pharmacological inhibition of A2A receptor can enhance the efficacy of immunotherapeutic agents, including inhibitors of immune checkpoint PD-1 [160, 161], PD-L1 [162], and CTLA-4 [162, 163], by improving T-cell-mediated antitumor response. Because blockade of A2A receptors decreases the expression of multiple checkpoint pathways, including PD-1 and LAG-3, on both CD8+ effector T cells (Teff) and Tregs in the TME, A2A receptor antagonists synergistically enhance also the antitumor immune response of adoptively transferred T cells [164,165,166].
While the effects of A2A stimulation have been clearly investigated, the role of A2B receptor on lymphocytes is still not completely understood. Antagonists of A2B receptors, as well as A2A, reduced the expression of FOXP3 in Tregs and the production of IL-10 [167]. In addition, A2B antagonists have showed inhibitory effects on Tregs differentiation [167]. In some mouse tumor models, antagonism of A2B receptors is able to inhibit tumor growth by enhancing the number of tumor-infiltrating CD8+ T cells [63, 119, 131], most likely as consequence of reduced expansion/recruitment of MDSCs.
As well as adenosine receptors, also CD73 plays an important role in controlling functions and behavior of T cells. It was observed that this nucleotidase is increased in anergic T cells and impairs the responsivity of these cells in vitro [168]. Interestingly, tumor CD73 has been shown to impair T-cell activity, while its inhibition improves the therapeutic antitumor effects of transferred tumor-specific T cells [169]. Based on the crucial role of CD73 in producing adenosine in the TME, targeting CD73 may inhibit tumor growth and improve antitumor CD8+T-cell response [20, 170, 171], especially in combination with chemotherapeutic agents [172], radiotherapy [122, 123], and inhibitors of immune checkpoints [18, 163]. A recent paper by Young and collaborators (2016) showed that the simultaneous inhibition of CD73 and A2A receptor in a mouse metastasis models decreased the metastatic burden enhancing the number and efficacy of infiltrating effector T cells [173]. These data demonstrated definitively that these two targets in the adenosine signaling pathway are nonredundant, but it is possible to reach an optimal therapeutic response by inhibiting both the A2A receptor-mediated adenosine effects and enzymatic and nonenzymatic activity of CD73 [173].
Underlying the crucial tumor-promoting role of CD39+ Treg cells, it has been observed that adoptive transfer of Tregs from CD39-deficient mice into immunodeficient mice enhanced NK cell functions, conversely of what happened in wild-type animals [57]. In line with these findings, Stagg and co-workers demonstrated that also CD73 expression on Tregs was critical for Treg-mediated tumor growth [170]. Specifically, Treg depletion in wild-type mice abrogated significantly tumor growth, while Treg depletion in CD73-deficient mice showed no effect [170].
Very recently, a critical role of B cells in the adenosine-mediated pro-tumor effects also emerged [174]. In particular, this study demonstrated that B cells are able to produce CD19+ extracellular vesicles (EVs), which express both CD73 and CD39 and can hydrolyze ATP into adenosine [174]. Treatment with these EVs can reduce the chemotherapy efficacy in tumor-bearing mice, while pretreatment of EVs with a CD39 inhibitor, a CD73 inhibitor, or an anti-CD73 mAb prevented this effect [174]. Furthermore, Zhang et al. extended their investigation in gastric and colon cancer patients, whose progression-free survival resulted to be negatively correlated with EV levels in serum [174]. These interesting results give evidence of the contribution of B cells in immunosuppression and will be certainly the starting point for further studies in the characterization of adenosine-producing EVs.
9.4 Adenosine System and Neoplastic Cells
9.4.1 Apoptosis
Cell apoptosis is defined as a death program pivotally involved in the maintenance of tissue homeostasis [175]. Two major triggers can activate this process: the extrinsic pathway, which is mediated by extracellular signals, and the intrinsic pathway, promoted by the recruitment of signals of intracellular origin [176, 177]. Of note, cancer cells can be able to evade programmed cell death, representing another key mechanism, along with deranged proliferation, promoting their uncontrolled growth [175]. Malignant cells can become resistant to apoptosis through several mechanisms, including disruption of proapoptotic/antiapoptotic protein balance, reduced caspase activity, and impaired signaling of receptors involved in death program [178]. In this regard, the adenosine system is one of the pathways involved in the modulation of such mechanisms.
Adenosine can promote the apoptotic process in cancer cells through the modulation of extrinsic signaling pathways [33, 179,180,181,182,183]. In particular, this nucleoside displayed to increase the expression of TNF receptor 1 (TNFR1) and the TNF-related apoptosis-inducing ligand receptor-2 (TRAILR2, also known as DR5 and TNFRSF10B) in the human hepatoma cell line HepG2. Moreover, adenosine was also able to modulate the expression of downstream intracellular signaling proteins, including TNFR associated with a death domain (TRADD), receptor-interacting protein kinase 1 (RIPK1), and Fas-associated death domain (FADD). Globally, these events trigger the activation of caspase-8 and caspase-9 followed by the activation of the effector caspase-3 [33].
The majority of the extrinsic proapoptotic actions of adenosine are likely to be mediated by A3 receptors [33, 179,180,181,182,183]. In in vitro experiments, A3 receptor activation upregulated the extrinsic pathway by enhancing the expression of CD95 (Fas) [181], as well as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in leukemia cells [182]. In addition, such receptors increased the expression of CD95, caspase-8, and caspase-3 in a human hepatoma cell line [183].
In a thyroid cancer cell line, the activation of A3 receptors elicited an increase in apoptosis through the activation of the proapoptotic cytokine TRAIL via stimulation of NF-κB and by potentiating the TRAILR2 expression [34]. Likewise, in thyroid cancer cells, A3 receptor stimulation was associated with a reduced expression of the antiapoptotic proteins BCL-2 and cellular FLICE protein (FLIP), as well as phosphorylated AKT [34].
The activation of A2A receptors resulted in the activation of intrinsic apoptotic process in human Caco-2 cancer cell line as well as in HepG2 cells [32, 184], through the reduction of mitochondrial membrane potential and the increase in cytochrome c efflux from mitochondria into cytosol. This effect results in the activation of caspase-9 and caspase-3 [32, 184]. These receptors can also reduce the expression of the antiapoptotic protein BCL-XL and upregulate the proapoptotic factor BID in hepatocellular carcinoma cells, thereby disrupting mitochondrial membrane potential [32].
A2B receptors also elicited the proapoptotic effects in osteosarcoma cells through the intrinsic pathway, by downregulating BCL-2 and BCL-XL [28]. Interestingly, the study also showed that cisplatin incubation induced an increment of extracellular adenosine levels and p53-dependent expression of A2B receptors, thus suggesting the occurrence of a p53-induced priming mechanism for cisplatin that uses A2B receptor signaling as an additional death-inducing pathway [28].
A3 receptors also mediate the activation of the intrinsic apoptotic pathway in cancer. Indeed, the stimulation of such receptors in human lung cancer cells elicited the upregulation of the proapoptotic gene NOXA through the activation of p53 [185]. Furthermore, A3 receptor stimulation induced the loss of mitochondrial membrane integrity and caspase-3 activation, along with a decrease in the expression level of BCL-2 and an increase in BAX expression [30, 186]. Several lines of evidence suggest that adenosine can also induce the apoptotic process independently to adenosine receptor activation. In particular, it has been observed that apoptosis of astrocytoma cells can be activated by the internalization of adenosine into cancer cells, followed by its phosphorylation to AMP via adenosine kinase [31, 35, 187,188,189]. The subsequent raise in intracellular AMP activates the AMPK complex, resulting in apoptosis mediated by caspase-9 and caspase-3 [189]. However, the underlying mechanisms by which AMPK activation leads to caspase-9 activation remain unclear. The adenosine-AMP-AMPK pathway is also able to induce apoptosis in a human hepatocarcinoma cell line by the dissipation of mitochondrial membrane potential occurring through BCL-XL phosphorylation and then its inactivation. This was followed by mitochondrial damage and release of several mitochondrial proteins, such as SMAC (also known as DIABLO) into the cytosol, eventually leading to the activation of caspase-3 [188]. In the same cancer cell line, the adenosine-mediated increment of AMP concentration triggered apoptosis by reducing the expression of FLIP, a specific inhibitor of caspase-8 [31]. In another study, the increase of intracellular AMP levels elicited an enhancement of p53 transcription, which in turn activated caspase-independent apoptosis in pleural mesothelioma cells [35].
The apoptotic process can also occur in a programmed fashion but independently from caspase activation [190]. Indeed, the apoptotic-inducing factor (AIF), which is physiologically located into the mitochondrial intermembrane space, can be released in the cytosol in response to certain mitochondrial damaging insults. Then, the AIF, translocating into the nucleus and binds to nuclear DNA, induced a caspase-independent chromatin condensation [191]. In this context, the A3 receptor activation in a human bladder cell line elicited caspase-independent apoptosis through increased nuclear accumulation of AIF [192]. Likewise, adenosine was found to induce the translocation into the nucleus of mitochondrion-associated inducer of death (AMID) [36, 193, 194], a homologous of AIF molecule [195]. This event was followed by the occurrence of caspase-independent apoptosis in several types of human cancer cell lines [36, 193, 194, 196], which was, in some cases, dependent from the recruitment of A3 receptors [36, 193, 194].
Overall, it is likely that adenosine can induce cancer cell apoptosis mostly by the activation of A3 receptors. Moreover, A2A and A2B receptors, as well as intracellular targeting of adenosine are also involved in promoting the apoptotic process.
9.4.2 Cell Proliferation
Cancer cells are characterized by an altered cell cycle, caused by genetic and epigenetic deregulation of genes, which, in turn, can lead to the progression of quiescent cells into S phase [197,198,199]. In this context, adenosine pathway plays dual and opposite actions on cell growth, both promoting and counteracting tumor cell proliferation. Such opposite effects were ascribed to the engagement of different adenosine receptors in particular tumors [200]. For instance, A1 adenosine receptor subtype has been found to regulate cell growth in breast cancer. Indeed, mRNA and protein expression of this adenosine receptor subtype significantly increased in various breast cancer cell lines and in primary breast tumor sections, and its activation promoted cancer cell proliferation through the downregulation of p27, a cyclin-dependent kinase (CDK) inhibitor, and the upregulation of CDK4 [201]. In addition, it has been reported that breast cancer cell proliferation induced by estradiol could involve A1 adenosine receptor activation, since estradiol has been found to induce transcriptional upregulation of A1 receptor [202]. In support of this view, both gene deletion and pharmacological blockade of A1 receptors decreased MCF-7 breast cancer cell proliferation through the inhibition of estrogen receptor α (ERα)-dependent transcriptional activity [202]. These findings suggest that estradiol, ERα, and the A1 adenosine receptor create a loop involved in the modulation of breast cancer cell proliferation [202].
Besides A1 adenosine receptors, A2A receptors expressed in MCF-7 cells have been found to promote ERα transcription and, in turn, stimulate cancer cell proliferation [203], thus suggesting that A2A receptor subtype also regulates cell proliferation through the activation of estrogen signaling.
Of interest, an involvement of A2B receptors in tumor cell proliferation has been also described. In particular, Ma et al. (2010) observed an increased expression of A2B receptors in neoplastic colorectal tissues as well as in several human colorectal adenocarcinoma-derived cell lines [204]. The authors also demonstrated that the pharmacological blockade of A2B receptors with the selective antagonist MRS1754 counteracted the neoplastic growth in colonic carcinoma cells, thus indicating that A2B receptors have cancer-promoting properties [204]. Wei et al. (2013) showed that A2B receptor activation increased cell proliferation in both androgen-dependent and independent-prostate cancer cell lines [205]. However, the mechanisms underlying these effects need to be clarified.
Increasing evidence also suggest that adenosine contributes to tumor cell proliferation through A3 receptors [206,207,208,209,210,211,212,213,214,215]. In particular, it has been observed that the A3 receptor activation inhibited the lymphoma cell proliferation, through the blockade of telomerase activity as well as cell cycle in G0/G1 phase [211]. In support of this view, the stimulation of A3 receptors elicited the arrest of G0/G1 cell cycle in prostate cancer cell lines, through the downregulation of CDK4 and cyclin D1 and upregulation of p53 [186]. In addition, A3 receptor stimulation counteracted cell proliferation in human melanoma cells by inhibiting G1 phase of cell cycle [207]. Moreover, the proliferation of melanoma and colonic cancer cell lines was found to be inhibited by A3 receptor activation [208, 210, 216]. The mechanisms underlying this antiproliferative effect were ascribed to the ability of A3 receptor of downregulating the expression of protein kinase A catalytic subunit (PKAc), which, in turn, decreased the expression of protein kinase B (PKB). Such an effect was associated with a decrease in the phosphorylation of glycogen synthase kinase (GSK)-3β while increasing β- catenin phosphorylation and ubiquitination, with consequent inhibition of MYC and cyclin D1 transcription [208, 210, 216].
Of note, treatment with selective A3 receptor agonists inhibited cell proliferation in breast cancer cell lines, through the downregulation of ERBB2 expression and reduction of the activity of its downstream effector, ERK, thus corroborating the antiproliferative effect of A3 receptor subtype [217, 218]. Likewise, A3 receptor activation inhibited the proliferation in human papillary carcinoma cells via the reduction of ERK phosphorylation and the inhibition of the G1 phase of cell cycle [219].
Overall, these findings suggest that adenosine pathways through the modulation of its receptors play a dual role in cancer cell proliferation. In particular, A1, A2A, and A2B receptors have proliferative effects, while A3 receptors decrease cancer cell proliferation.
9.4.3 Metastatic Process
Metastasis is a cancer expansion process where tumor cells can spread throughout the body, increase, and flourish in other tissues [220]. 90% of all cancer-related death is associated with the development of tumor metastases [221]. In this context, increasing evidence highlight a pivotal involvement of adenosine pathway in the dissemination of several cancer cells [56, 57, 222, 223]. In a previous paper, Jackson et al. (2007) showed that CD39 knockout animals, inoculated intravascularly with melanoma cells, displayed fewer and smaller pulmonary metastases than wild-type mice [56]. In addition, Sun et al. (2010) observed that the expression of CD39 in Tregs inhibited NK cell activity and promoted hepatic metastatic tumor growth in mice, thus suggesting the involvement of CD39 enzymatic activity in metastatic processes [57].
Of interest, besides the relevance of CD39 in metastasis, a pivotal involvement of CD73 has been also described [224]. Indeed, several studies have shown a correlation between the expression and activity of this enzyme in cancer cells and their ability to spread, invade, and adhere to extracellular matrix (ECM) in different tissues [106, 170, 225,226,227]. Zhi et al. (2007) observed that silencing CD73 with small interfering RNA prevented human breast cancer cell adhesion to ECM and inhibited their migration [228]. In addition, the forced increase in CD73 expression enhanced invasiveness and adhesiveness to the ECM in breast cancer cell lines. Such an increase was counteracted by pharmacological blockade of CD73 activity and reproduced upon incubation of exogenous adenosine [225, 226]. These findings indicate a role of CD73-derived adenosine in the metastatic potential of cancer cells [225].
Of note, the employment of CD73 knockout mice allowed to better characterize the role of this enzyme in metastatic processes. Indeed, the CD73 deletion in mice protected against the development of melanoma metastases as compared with wild-type mice [106, 170]. The mechanisms underlying the pro-metastatic effect of CD73 were ascribed to its ability of supporting transendothelial migration of cancer cells [170]. However, the role of extracellular adenosine in mediating the effect of CD73 in in vivo models needs further investigations.
A pioneering study by Stagg et al. (2010) reported that adenosine increased tumor cell migration in vitro and metastasis in vivo, through the activation of A2B receptors, recently regarded as a target of the metastasis-inducing transcription factor FOS-related antigen-1 (FRA-1) in breast cancer cells [20, 229]. Indeed, gene deletion as well as the pharmacological blockade of A2B receptors inhibited the survival and metastatic potential of breast cancer cells in vitro [229]. In addition, treatment with theophylline and a chemotherapeutic agent suppressed breast cancer colonization in mouse lungs via the blockade of A2B receptors [229], thus confirming the role of adenosine A2B receptors in the promotion of metastasis. In support of this view, Ntantie et al. (2013) showed that the activation of A2B receptors elicited an increase in cell scattering through a reduction of cell-cell adhesion and impairments of adherens cell junctions via the suppression of the small GTPase RAP1 prenylation [230].
Besides A2B receptors, an involvement in cancer metastasis for A2A receptor subtype has been identified [231, 232]. Ohta et al. (2006) reported that adoptively transferred CD8+ T cells in mice treated with A2A receptor antagonist decreased metastasis in a melanoma model [61]. Shi et al. (2019) observed that an increased expression of A2A receptors in gastric cancer tissues correlated positively with disease stage and promoted metastatic processes [233]. In particular, adenosine through A2A receptors promoted gastric cancer cell migration and invasion via activation of PI3K-AKT-mTOR signaling [233].
In addition, in a pioneering study, Young et al. (2016) observed that the combination of A2A receptor antagonist and antibody anti-CD73 counteracted tumor development and metastasis, thus suggesting a role of adenosine via A2A receptor in the promotion of metastasis, and, that the concomitant inhibition of CD73 and A2A could represent an innovative pharmacological strategy in cancer therapy [173].
Of interest, over the last years, a crucial role in metastatic processes has been ascribed to exosomes, vesicles with a diameter of 30–100 nm secreted by neoplastic cells. They promote the survival and outgrowth of disseminated tumor cells through the formation of pre-metastatic niches [234]. One of the mechanisms underlying the metastatic effects of the cancer exosomes was ascribed to their ability of increasing the adenosine levels within the tumor microenvironment, through the CD39 and CD73 enzymes. These results indicate that exosome participate to cancer immune evasion via the CD39/CD73/adenosine signaling [235]
However, the cellular and molecular mechanisms underlying the role of the adenosine system in regulating metastasis remain to be clarified. For instance, the involvement of adenosine in regulating neoplastic cell motility cycle as well as epithelial-to-mesenchymal transition needs to be investigated. In addition, data on the role of adenosine on lysis oxidases and proteins involved in promoting metastasis are still lacking.
References
Pitot HC (1993) The molecular biology of carcinogenesis. Cancer 72(3 Suppl):962–970
Witz IP, Levy-Nissenbaum O (2006) The tumor microenvironment in the post-paget era. Cancer Lett 242(1):1–10
Tuccitto A, Shahaj E, Vergani E, Ferro S, Huber V, Rodolfo M, Castelli C, Rivoltini L, Vallacchi V (2019) Immunosuppressive circuits in tumor microenvironment and their influence on cancer treatment efficacy. Virchows Arch Int J Pathol 474(4):407–420
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57(13):2602–2605
Hoskin DW, Butler JJ, Drapeau D, Haeryfar SM, Blay J (2002) Adenosine acts through an a3 receptor to prevent the induction of murine anti-cd3-activated killer t cells. Int J Cancer Journal international du cancer 99(3):386–395
Hasko G, Linden J, Cronstein B, Pacher P (2008) Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 7(9):759–770
Antonioli L, Fornai M, Colucci R, Ghisu N, Tuccori M, Del Tacca M, Blandizzi C (2008) Regulation of enteric functions by adenosine: pathophysiological and pharmacological implications. Pharmacol Ther 120(3):233–253
Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S (2013) Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med 91(2):165–172
Hasko G, Cronstein BN (2004) Adenosine: an endogenous regulator of innate immunity. Trends Immunol 25(1):33–39
Csoka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, Ledent C, Deitch EA, Spolarics Z, Nemeth ZH, Hasko G (2008) Adenosine a2a receptor activation inhibits t helper 1 and t helper 2 cell development and effector function. FASEB J 22(10):3491–3499
Himer L, Csoka B, Selmeczy Z, Koscso B, Pocza T, Pacher P, Nemeth ZH, Deitch EA, Vizi ES, Cronstein BN, Hasko G (2010) Adenosine a2a receptor activation protects cd4+ t lymphocytes against activation-induced cell death. FASEB J 24(8):2631–2640
Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A (2008) Hypoxia-adenosinergic immunosuppression: tumor protection by t regulatory cells and cancerous tissue hypoxia. Clin Cancer Res 14(19):5947–5952
Ohta A (2016) A metabolic immune checkpoint: Adenosine in tumor microenvironment. Front Immunol 7:109
Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A (2014) Hostile, hypoxia-a2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res 2(7):598–605
Linden J (2006) Adenosine metabolism and cancer. Focus on "adenosine downregulates dppiv on ht-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing erk1/2 activity via a novel pathway". Am J Physiol Cell Physiol 291(3):C405–C406
Antonioli L, Novitskiy SV, Sachsenmeier KF, Fornai M, Blandizzi C, Hasko G (2017) Switching off cd73: a way to boost the activity of conventional and targeted antineoplastic therapies. Drug Discov Today 22:1686
Antonioli L, Blandizzi C, Pacher P, Hasko G (2013) Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer 13(12):842–857
Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J (2014) Anti-cd73 therapy impairs tumor angiogenesis. Int J Cancer 134(6):1466–1473
Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, Hammond SA, Rothstein R, Rios-Doria J, Poon E, Holoweckyj N et al (2016) Targeting cd73 in the tumor microenvironment with medi9447. Onco Targets Ther 5(8):e1208875
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, Dwyer KM, Smyth MJ (2010) Anti-cd73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 107(4):1547–1552
Bavaresco L, Bernardi A, Braganhol E, Cappellari AR, Rockenbach L, Farias PF, Wink MR, Delgado-Canedo A, Battastini AM (2008) The role of ecto-5′-nucleotidase/cd73 in glioma cell line proliferation. Mol Cell Biochem 319(1–2):61–68
Tan EY, Richard CL, Zhang H, Hoskin DW, Blay J (2006) Adenosine downregulates dppiv on ht-29 colon cancer cells by stimulating protein tyrosine phosphatase(s) and reducing erk1/2 activity via a novel pathway. Am J Physiol Cell Physiol 291(3):C433–C444
Pennycooke M, Chaudary N, Shuralyova I, Zhang Y, Coe IR (2001) Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem Biophys Res Commun 280(3):951–959
Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE (2003) Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 22(47):7524–7536
Antonioli L, Colucci R, La Motta C, Tuccori M, Awwad O, Da Settimo F, Blandizzi C, Fornai M (2012) Adenosine deaminase in the modulation of immune system and its potential as a novel target for treatment of inflammatory disorders. Curr Drug Targets 13(6):842–862
Antonioli L, Fornai M, Colucci R, Ghisu N, Blandizzi C, Del Tacca M (2006) A2a receptors mediate inhibitory effects of adenosine on colonic motility in the presence of experimental colitis. Inflamm Bowel Dis 12(2):117–122
Antonioli L, Fornai M, Colucci R, Awwad O, Ghisu N, Tuccori M, Del Tacca M, Blandizzi C (2011) Differential recruitment of high affinity a1 and a2a adenosine receptors in the control of colonic neuromuscular function in experimental colitis. Eur J Pharmacol 650(2–3):639–649
Long JS, Crighton D, O’Prey J, Mackay G, Zheng L, Palmer TM, Gottlieb E, Ryan KM (2013) Extracellular adenosine sensing-a metabolic cell death priming mechanism downstream of p53. Mol Cell 50(3):394–406
Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD (2004) The equilibrative nucleoside transporter family, slc29. Pflugers Arch Eur J Physiol 447(5):735–743
Aghaei M, Karami-Tehrani F, Panjehpour M, Salami S, Fallahian F (2012) Adenosine induces cell-cycle arrest and apoptosis in androgen-dependent and -independent prostate cancer cell lines, lncap-fgc-10, du-145, and pc3. Prostate 72(4):361–375
Yang D, Yaguchi T, Yamamoto H, Nishizaki T (2007) Intracellularly transported adenosine induces apoptosis in huh-7 human hepatoma cells by downregulating c-flip expression causing caspase-3/−8 activation. Biochem Pharmacol 73(10):1665–1675
Tamura K, Kanno T, Fujita Y, Gotoh A, Nakano T, Nishizaki T (2012) A(2a) adenosine receptor mediates hepg2 cell apoptosis by downregulating bcl-x(l) expression and upregulating bid expression. J Cell Biochem 113(5):1766–1775
Yang D, Yaguchi T, Lim CR, Ishizawa Y, Nakano T, Nishizaki T (2010) Tuning of apoptosis-mediator gene transcription in hepg2 human hepatoma cells through an adenosine signal. Cancer Lett 291(2):225–229
Morello S, Sorrentino R, Porta A, Forte G, Popolo A, Petrella A, Pinto A (2009) Cl-ib-meca enhances trail-induced apoptosis via the modulation of nf-kappab signalling pathway in thyroid cancer cells. J Cell Physiol 221(2):378–386
Nogi Y, Kanno T, Nakano T, Fujita Y, Tabata C, Fukuoka K, Gotoh A, Nishizaki T (2012) Amp converted from intracellularly transported adenosine upregulates p53 expression to induce malignant pleural mesothelioma cell apoptosis. Cell Physiol Biochem 30(1):61–74
Yang D, Yaguchi T, Nagata T, Gotoh A, Dovat S, Song C, Nishizaki T (2011) Amid mediates adenosine-induced caspase-independent huh-7 cell apoptosis. Cell Physiol Biochem 27(1):37–44
Antonioli L, Colucci R, Pellegrini C, Giustarini G, Tuccori M, Blandizzi C, Fornai M (2013) The role of purinergic pathways in the pathophysiology of gut diseases: pharmacological modulation and potential therapeutic applications. Pharmacol Ther 139(2):157–188
Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J (2008) Inhibition of t cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (review). Int J Oncol 32(3):527–535
Mato JM, Martinez-Chantar ML, Lu SC (2008) Methionine metabolism and liver disease. Annu Rev Nutr 28:273–293
da Silva CG, Jarzyna R, Specht A, Kaczmarek E (2006) Extracellular nucleotides and adenosine independently activate amp-activated protein kinase in endothelial cells: involvement of p2 receptors and adenosine transporters. Circ Res 98(5):e39–e47
Boison D (2013) Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 65(3):906–943
Félétou M (2011) The endothelium: part 1: multiple functions of the endothelial cells—focus on endothelium-derived vasoactive mediators. Morgan & Claypool Life Sciences, San Rafael
Hida K, Maishi N, Annan DA, Hida Y (2018) Contribution of tumor endothelial cells in cancer progression. Int J Mol Sci 19(5):1272
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Narravula S, Lennon PF, Mueller BU, Colgan SP (2000) Regulation of endothelial cd73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J Immunol 165(9):5262–5268
Acurio J, Herlitz K, Troncoso F, Aguayo C, Bertoglia P, Escudero CJPS (2017) Adenosine a2a receptor regulates expression of vascular endothelial growth factor in feto-placental endothelium from normal and late-onset pre-eclamptic pregnancies. Purinergic Signal 13(1):51–60
Khoa ND, Montesinos MC, Williams AJ, Kelly M, Cronstein BN (2003) Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol 171(8):3991–3998
Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T, Biaggioni I (2002) Differential expression of adenosine receptors in human endothelial cells. Circ Res 90(5):531–538
Grant MB, Tarnuzzer RW, Caballero S, Ozeck MJ, Davis MI, Spoerri PE, Feoktistov I, Biaggioni I, Shryock JC, Belardinelli L (1999) Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ Res 85(8):699–706
Canale FP, Ramello MC, Núñez N, Furlan CLA, Bossio SN, Serrán MG, Boari JT, del Castillo A, Ledesma M, Sedlik C, Piaggio E et al (2018) Cd39 expression defines cell exhaustion in tumor-infiltrating cd8<sup>+</sup> t cells. Cancer Res 78(1):115–128
Katamura K, Shintaku N, Yamauchi Y, Fukui T, Ohshima Y, Mayumi M, Furusho K (1995) Prostaglandin e2 at priming of naive cd4+ t cells inhibits acquisition of ability to produce ifn-gamma and il-2, but not il-4 and il-5. J Immunol 155(10):4604–4612
Henttinen T, Jalkanen S, Yegutkin GG (2003) Adherent leukocytes prevent adenosine formation and impair endothelial barrier function by ecto-5′-nucleotidase/cd73-dependent mechanism. J Biol Chem 278(27):24888–24895
Bouma MG, Wildenberg FAvd, Buurman WA (1996) Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physio 270(2):C522–C529
Grünewald JKG, Ridley AJ (2010) Cd73 represses pro-inflammatory responses in human endothelial cells. J Inflamm 7(1):10
Walker G, Langheinrich AC, Dennhauser E, Bohle RM, Dreyer T, Kreuzer J, Tillmanns H, Braun-Dullaeus RC, Haberbosch W (1999) 3-deazaadenosine prevents adhesion molecule expression and atherosclerotic lesion formation in the aortas of c57bl/6j mice. Arterioscler Thromb Vasc Biol 19(11):2673–2679
Jackson SW, Hoshi T, Wu Y, Sun X, Enjyoji K, Cszimadia E, Sundberg C, Robson SC (2007) Disordered purinergic signaling inhibits pathological angiogenesis in cd39/entpd1-null mice. Am J Pathol 171(4):1395–1404
Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, Murakami T, Robson SC (2010) Cd39/entpd1 expression by cd4+foxp3+ regulatory t cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 139(3):1030–1040
Feng L, Sun X, Csizmadia E, Han L, Bian S, Murakami T, Wang X, Robson SC, Wu Y (2011) Vascular cd39/entpd1 directly promotes tumor cell growth by scavenging extracellular adenosine triphosphate. Neoplasia (New York, NY) 13(3):206–216
Wang L, Tang S, Wang Y, Xu S, Yu J, Zhi X, Ou Z, Yang J, Zhou P, Shao Z (2013) Ecto-5′-nucleotidase (cd73) promotes tumor angiogenesis. Clin Exp Metastasis 30(5):671–680
Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, Zhang B (2011) Cd73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest 121(6):2371–2382
Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF et al (2006) A2a adenosine receptor protects tumors from antitumor t cells. Proc Natl Acad Sci U S A 103(35):13132–13137
Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, Dikov MM, Feoktistov I (2008) Host a(2b) adenosine receptors promote carcinoma growth. Neoplasia (New York, NY) 10(9):987–995
Sorrentino C, Miele L, Porta A, Pinto A, Morello S (2015) Myeloid-derived suppressor cells contribute to a2b adenosine receptor-induced vegf production and angiogenesis in a mouse melanoma model. Oncotarget 6(29):27478–27489
Wilkat M, Bast H, Drees R, Dünser J, Mahr A, Azoitei N, Marienfeld R, Frank F, Brhel M, Ushmorov A, Greve J et al Adenosine receptor 2b activity promotes autonomous growth, migration as well as vascularization of head and neck squamous cell carcinoma cells. n/a(n/a)
Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, Abbott R, Philbrook P, Thayer M, Shujia D, Rodig S et al (2014) Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med 92(12):1283–1292
Yamazaki T, Mukouyama YS (2018) Tissue specific origin, development, and pathological perspectives of pericytes. Front Cardiovasc Med 5:78
Dias Moura Prazeres PH, Sena IFG, Borges IDT, de Azevedo PO, Andreotti JP, de Paiva AE, de Almeida VM, de Paula Guerra DA, Pinheiro Dos Santos GS, Mintz A, Delbono O et al (2017) Pericytes are heterogeneous in their origin within the same tissue. Dev Biol 427(1):6–11
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 7(4):452–464
Ribatti D, Nico B, Crivellato E (2011) The role of pericytes in angiogenesis. Int J Dev Biol 55(3):261–268
Gerhardt H, Betsholtz C (2003) Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res 314(1):15–23
Balabanov R, Washington R, Wagnerova J, Dore-Duffy P (1996) Cns microvascular pericytes express macrophage-like function, cell surface integrin alpha m, and macrophage marker ed-2. Microvasc Res 52(2):127–142
Pieper C, Marek JJ, Unterberg M, Schwerdtle T, Galla HJ (2014) Brain capillary pericytes contribute to the immune defense in response to cytokines or lps in vitro. Brain Res 1550:1–8
Pieper C, Pieloch P, Galla HJ (2013) Pericytes support neutrophil transmigration via interleukin-8 across a porcine co-culture model of the blood-brain barrier. Brain Res 1524:1–11
Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G et al (2003) Endothelial pdgf-b retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 17(15):1835–1840
Huang FJ, You WK, Bonaldo P, Seyfried TN, Pasquale EB, Stallcup WB (2010) Pericyte deficiencies lead to aberrant tumor vascularizaton in the brain of the ng2 null mouse. Dev Biol 344(2):1035–1046
Raza A, Franklin MJ, Dudek AZ (2010) Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol 85(8):593–598
Sun H, Guo D, Su Y, Yu D, Wang Q, Wang T, Zhou Q, Ran X, Zou Z (2014) Hyperplasia of pericytes is one of the main characteristics of microvascular architecture in malignant glioma. PLoS One 9(12):e114246
Ribeiro AL, Okamoto OK (2015) Combined effects of pericytes in the tumor microenvironment. Stem Cells Int 2015:868475
Cao Y, Zhang ZL, Zhou M, Elson P, Rini B, Aydin H, Feenstra K, Tan MH, Berghuis B, Tabbey R, Resau JH et al (2013) Pericyte coverage of differentiated vessels inside tumor vasculature is an independent unfavorable prognostic factor for patients with clear cell renal cell carcinoma. Cancer 119(2):313–324
Zhang L, Nishihara H, Kano MR (2012) Pericyte-coverage of human tumor vasculature and nanoparticle permeability. Biol Pharm Bull 35(5):761–766
Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, Duncan MB, Xie L, Maeda G, Vong S, Sugimoto H et al (2012) Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 21(1):66–81
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153(3):543–553
Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in pdgf-b-deficient mice. Science 277(5323):242–245
Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O (2014) Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol 307(1):C25–C38
Sweeney MD, Ayyadurai S, Zlokovic BV (2016) Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 19(6):771–783
Li Q, Puro DG (2001) Adenosine activates atp-sensitive k(+) currents in pericytes of rat retinal microvessels: role of a1 and a2a receptors. Brain Res 907(1–2):93–99
Sugiyama T, Kawamura H, Yamanishi S, Kobayashi M, Katsumura K, Puro DG (2005) Regulation of p2x7-induced pore formation and cell death in pericyte-containing retinal microvessels. Am J Physiol Cell Physiol 288(3):C568–C576
Kawamura H, Sugiyama T, Wu DM, Kobayashi M, Yamanishi S, Katsumura K, Puro DG (2003) Atp: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J Physiol 551(Pt 3):787–799
Ceruti S, Colombo L, Magni G, Vigano F, Boccazzi M, Deli MA, Sperlagh B, Abbracchio MP, Kittel A (2011) Oxygen-glucose deprivation increases the enzymatic activity and the microvesicle-mediated release of ectonucleotidases in the cells composing the blood-brain barrier. Neurochem Int 59(2):259–271
Zhu C, Chrifi I, Mustafa D, van der Weiden M, Leenen PJM, Duncker DJ, Kros JM, Cheng C (2017) Cecr1-mediated cross talk between macrophages and vascular mural cells promotes neovascularization in malignant glioma. Oncogene 36(38):5356–5368
Zhu C, Mustafa DAM, Krebber MM, Chrifi I, Leenen PJM, Duncker DJ, Dekker L, Luider TM, Kros JM, Cheng C (2018) Comparative proteomic analysis of cat eye syndrome critical region protein 1- function in tumor-associated macrophages and immune response regulation of glial tumors. Oncotarget 9(71):33500–33514
Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, Ouyang GF, Okada M, Balazs M, Adany R, Shibata T et al (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear m1- and m2-type characteristics. J Leukoc Biol 83(5):1136–1144
Xuan W, Qu Q, Zheng B, Xiong S, Fan GH (2015) The chemotaxis of m1 and m2 macrophages is regulated by different chemokines. J Leukoc Biol 97(1):61–69
Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM (2012) Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol 227(1):17–28
Sidibe A, Ropraz P, Jemelin S, Emre Y, Poittevin M, Pocard M, Bradfield PF, Imhof BA (2018) Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat Commun 9(1):355
Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, Tazzyman S, Danson S, Addison C, Clemons M, Gonzalez-Angulo AM et al (2015) Perivascular m2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res 75(17):3479–3491
Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6(3):1670–1690
Ojalvo LS, Whittaker CA, Condeelis JS, Pollard JW (2010) Gene expression analysis of macrophages that facilitate tumor invasion supports a role for wnt-signaling in mediating their activity in primary mammary tumors. J Immunol 184(2):702–712
Sica A, Schioppa T, Mantovani A, Allavena P (2006) Tumour-associated macrophages are a distinct m2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur J Cancer (Oxford, England : 1990) 42(6):717–727
Haskó G, Pacher P (2012) Regulation of macrophage function by adenosine. Arterioscler Thromb Vasc Biol 32(4):865–869
Csoka B, Selmeczy Z, Koscso B, Nemeth ZH, Pacher P, Murray PJ, Kepka-Lenhart D, Morris SM Jr, Gause WC, Leibovich SJ, Hasko G (2012) Adenosine promotes alternative macrophage activation via a2a and a2b receptors. FASEB J 26(1):376–386
Haskó G, Kuhel DG, Chen J-F, Schwarzschild MA, Deitch EA, Mabley JG, Marton A, Szabó C (2000) Adenosine inhibits il-12 and tnf-α production via adenosine a2a receptor-dependent and independent mechanisms. FASEB J 14(13):2065–2074
Costales MG, Alam MS, Cavanaugh C, Williams KM (2018) Extracellular adenosine produced by ecto-5′-nucleotidase (cd73) regulates macrophage pro-inflammatory responses, nitric oxide production, and favors salmonella persistence. Nitric Oxide 72:7–15
Si QS, Nakamura Y, Kataoka K (1997) Adenosine inhibits superoxide production in rat peritoneal macrophages via elevation of camp level. Immunopharmacology 36(1):1–7
Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ (2007) Synergistic up-regulation of vascular endothelial growth factor (vegf) expression in macrophages by adenosine a2a receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the vegf promoter. Mol Biol Cell 18(1):14–23
Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemela J, Laurila JP, Elima K, Jalkanen S, Salmi M (2011) Altered purinergic signaling in cd73-deficient mice inhibits tumor progression. Eur J Immunol 41(5):1231–1241
Cekic C, Day Y-J, Sag D, Linden J (2014) Myeloid expression of adenosine a<sub>2a</sub> receptor suppresses t and nk cell responses in the solid tumor microenvironment. Cancer Res 74(24):7250–7259
Montalban Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wockel A, Dietl J, Lutz MB, Mittelbronn M, Wischhusen J, Hausler SFM (2016) Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, cd39- and cd73-dependent mechanism for tumor immune escape. J Immunother Cancer 4:49
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392(6673):245–252
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL (2015) Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol 194(7):2985–2991
Dickenson JM, Reeder S, Rees B, Alexander S, Kendall D (2003) Functional expression of adenosine a2a and a3 receptors in the mouse dendritic cell line xs-106. Eur J Pharmacol 474(1):43–51
Challier J, Bruniquel D, Sewell AK, Laugel B (2013) Adenosine and camp signalling skew human dendritic cell differentiation towards a tolerogenic phenotype with defective cd8(+) t-cell priming capacity. Immunology 138(4):402–410
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, Dikov MM (2008) Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112(5):1822–1831
Addi AB, Lefort A, Hua X, Libert F, Communi D, Ledent C, Macours P, Tilley SL, Boeynaems J-M, Robaye B (2008) Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the a2b receptor. Eur J Immunol 38(6):1610–1620
Panther E, Idzko M, Herouy Y, Rheinen H, Gebicke-Haerter PJ, Mrowietz U, Dichmann S, Norgauer J (2001) Expression and function of adenosine receptors in human dendritic cells. FASEB J 15(11):1963–1970
Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, Girolomoni G, Norgauer J (2003) Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the t-cell stimulatory capacity of human dendritic cells. Blood 101(10):3985–3990
Naval-Macabuhay I, Casanova V, Navarro G, Garcia F, Leon A, Miralles L, Rovira C, Martinez-Navio JM, Gallart T, Mallol J, Gatell JM et al (2016) Adenosine deaminase regulates treg expression in autologous t cell-dendritic cell cocultures from patients infected with hiv-1. J Leukoc Biol 99(2):349–359
Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, Linden J, Ernst PB (2009) The a2b adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol 182(8):4616–4623
Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J (2012) Adenosine a2b receptor blockade slows growth of bladder and breast tumors. J Immunol 188(1):198–205
Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, Feoktistov I, Moses HL, Novitskiy SV (2014) Role of tgf-beta signaling in generation of cd39+cd73+ myeloid cells in tumors. J Immunol 193(6):3155–3164
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S et al (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334(6062):1573–1577
Wennerberg E, Kawashima N, Demaria SJJfIoC (2015) Adenosine regulates radiation therapy-induced anti-tumor immunity. J Immunother Cancer 3(2):P378
Wennerberg E, Cronstein B, Formenti SC, Demaria S (2017) Adenosine generation limits radiation-induced tumor immunogenicity by abrogating recruitment and activation of cd103<sup>+</sup> dcs. J Immunol 198(1 Supplement):154.156–154.156
Jordan KR, Kapoor P, Spongberg E, Tobin RP, Gao D, Borges VF, McCarter MD (2017) Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients. Cancer Immunol Immunother CII 66(4):503–513
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12(4):253–268
Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, Restifo NP (1999) Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits cd8+ t cell responses by dysregulating antigen-presenting cell maturation. J Immunol 162(10):5728–5737
Morales JK, Kmieciak M, Knutson KL, Bear HD, Manjili MH (2010) Gm-csf is one of the main breast tumor-derived soluble factors involved in the differentiation of cd11b-gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res Treat 123(1):39–49
Kusmartsev S, Gabrilovich DI (2003) Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol 74(2):186–196
Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, Cho HI, Celis E, Quiceno DG, Padhya T, McCaffrey TV et al (2010) Hif-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207(11):2439–2453
Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, Dikov MM, Feoktistov I (2011) Adenosinergic regulation of the expansion and immunosuppressive activity of cd11b+gr1+ cells. J Immunol 187(11):6120–6129
Iannone R, Miele L, Maiolino P, Pinto A, Morello S (2013) Blockade of a2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia 15(12):1400–1409
Morello S, Miele L (2014) Targeting the adenosine a2b receptor in the tumor microenvironment overcomes local immunosuppression by myeloid-derived suppressor cells. Oncoimmunology. 2014 Feb 14;3:e27989. https://doi.org/10.4161/onci.27989. eCollection 2014.
Li J, Wang L, Chen X, Li L, Li Y, Ping Y, Huang L, Yue D, Zhang Z, Wang F, Li F et al (2017) Cd39/cd73 upregulation on myeloid-derived suppressor cells via tgf-beta-mtor-hif-1 signaling in patients with non-small cell lung cancer. Onco Targets Ther 6(6):e1320011
Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, Zhang C, Yue D, Qin G, Zhang T, Li F et al (2018) Metformin-induced reduction of cd39 and cd73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res 78(7):1779–1791
Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, Boidot R, Végran F, Bonnefoy N, Vincent J, Bengrine-Lefevre L et al (2016) Accumulation of mdsc and th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a folfox–bevacizumab drug treatment regimen. Cancer Res 76(18):5241–5252
Turcotte M, Allard D, Mittal D, Bareche Y, Buisseret L, Jose V, Pommey S, Delisle V, Loi S, Joensuu H, Kellokumpu-Lehtinen PL et al (2017) Cd73 promotes resistance to her2/erbb2 antibody therapy. Cancer Res 77(20):5652–5663
Oelkrug C, Ramage JM (2014) Enhancement of t cell recruitment and infiltration into tumours. Clin Exp Immunol 178(1):1–8
Gajewski TF, Schreiber H, Fu Y-X (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14(10):1014–1022
Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman WH, Pages F, Galon J (2011) Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol 29(6):610–618
Crimeen-Irwin B, Scalzo K, Gloster S, Mottram PL, Plebanski M (2005) Failure of immune homeostasis -- the consequences of under and over reactivity. Curr Drug Targets Immune Endocr Metabol Disord 5(4):413–422
Allard B, Longhi MS, Robson SC, Stagg J (2017) The ectonucleotidases cd39 and cd73: novel checkpoint inhibitor targets. Immunol Rev 276(1):121–144
Antonioli L, Pacher P, Vizi ES, Hasko G (2013) Cd39 and cd73 in immunity and inflammation. Trends Mol Med 19(6):355–367
Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, Drake CG, Powell JD (2008) A2a receptor signaling promotes peripheral tolerance by inducing t-cell anergy and the generation of adaptive regulatory t cells. Blood 111(1):251–259
Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, Franco R (1999) Expression of a2b adenosine receptors in human lymphocytes: their role in t cell activation. J Cell Sci 112(Pt 4):491–502
Erdmann AA, Gao ZG, Jung U, Foley J, Borenstein T, Jacobson KA, Fowler DH (2005) Activation of th1 and tc1 cell adenosine a2a receptors directly inhibits il-2 secretion in vitro and il-2-driven expansion in vivo. Blood 105(12):4707–4714
Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J (2006) Adenosine a2a receptor activation reduces hepatic ischemia reperfusion injury by inhibiting cd1d-dependent nkt cell activation. J Exp Med 203(12):2639–2648
Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J (1999) Purine metabolites suppress proliferation of human nk cells through a lineage-specific purine receptor. J Immunol 162(12):7376–7382
Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E (2006) Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res 66(15):7758–7765
Sevigny CP, Li L, Awad AS, Huang L, McDuffie M, Linden J, Lobo PI, Okusa MD (2007) Activation of adenosine 2a receptors attenuates allograft rejection and alloantigen recognition. J Immunol 178(7):4240–4249
Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M (2012) The development and immunosuppressive functions of cd4(+) cd25(+) foxp3(+) regulatory t cells are under influence of the adenosine-a2a adenosine receptor pathway. Front Immunol 3:190
Gerlo S, Verdood P, Hooghe-Peters EL, Kooijman RJC, Sciences ML (2005) Multiple camp-induced signaling cascades regulate prolactin expression in t cells. Cell Mol Life Sci 63(1):92
Jimenez JL, Punzon C, Navarro J, Munoz-Fernandez MA, Fresno M (2001) Phosphodiesterase 4 inhibitors prevent cytokine secretion by t lymphocytes by inhibiting nuclear factor-kappab and nuclear factor of activated t cells activation. J Pharmacol Exp Ther 299(2):753–759
Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, Gorelik EJCI (2008) Immunotherapy: cooperation of adenosine and prostaglandin e2 (pge2) in amplification of camp–pka signaling and immunosuppression. Cancer Immunol Immunother 57(11):1611–1623
Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, Jackson EK, Gorelik E (2007) Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific cd8+ and cd4+ t lymphocytes by adenosine-protein kinase a type i signaling. Cancer Res 67(12):5949–5956
Carey KD, Dillon TJ, Schmitt JM, Baird AM, Holdorf AD, Straus DB, Shaw AS, Stork PJS (2000) Cd28 and the tyrosine kinase lck stimulate mitogen-activated protein kinase activity in t cells via inhibition of the small g protein rap1. Mol Cell Biol 20(22):8409–8419
Bivona TG, Wiener HH, Ahearn IM, Silletti J, Chiu VK, Philips MR (2004) Rap1 up-regulation and activation on plasma membrane regulates t cell adhesion. J Cell Biol 164(3):461–470
Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, Lacher S, Müller CE, Frey J, Simeoni L, Schraven B et al (2009) Adenosine regulates cd8 t-cell priming by inhibition of membrane-proximal t-cell receptor signalling. Immunology 128(1 Suppl):e728–e737
Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Osborne BA, Minter LM, Miele L, Morello S (2019) Adenosine a2a receptor stimulation inhibits tcr-induced notch1 activation in cd8+t-cells. Front Immunol 10:162–162
Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P, Coukos G (2019) Targeting adenosine in cancer immunotherapy to enhance t-cell function. Front Immunol 10:925
Mittal D, Young A, Stannard K, Yong M, Teng MWL, Allard B, Stagg J, Smyth MJ (2014) Antimetastatic effects of blocking pd-1 and the adenosine a2a receptor. Cancer Res 74(14):3652–3658
Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, Kershaw MH, Stagg J, Darcy PK (2015) Adenosine receptor 2a blockade increases the efficacy of anti-pd-1 through enhanced antitumor t-cell responses. Cancer Immunol Res 3(5):506–517
Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, Liu L, Buggy JJ, McCaffery I, Miller RA (2018) A2ar antagonism with cpi-444 induces antitumor responses and augments efficacy to anti-pd-(l)1 and anti-ctla-4 in preclinical models. Cancer Immunol Res 6(10):1136–1149
Iannone R, Miele L, Maiolino P, Pinto A, Morello S (2014) Adenosine limits the therapeutic effectiveness of anti-ctla4 mab in a mouse melanoma model. Am J Cancer Res 4(2):172–181
Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, Powell JD (2018) Inhibition of the adenosine a2a receptor modulates expression of t cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and act in murine cancer models. Cancer Immunology Immunother CII 67(8):1271–1284
Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M (2018) A(2a) adenosine receptor gene deletion or synthetic a(2a) antagonist liberate tumor-reactive cd8(+) t cells from tumor-induced immunosuppression. J Immunol 201(2):782–791
Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, Davenport AJ, John LB, Mardiana S, Slaney CY, Johnstone RW et al (2017) Targeting the adenosine 2a receptor enhances chimeric antigen receptor t cell efficacy. J Clin Invest 127(3):929–941
Nakatsukasa H, Tsukimoto M, Harada H, Kojima S (2011) Adenosine a2b receptor antagonist suppresses differentiation to regulatory t cells without suppressing activation of t cells. Biochem Biophys Res Commun 409(1):114–119
Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA, Mueller DL (2012) Arthritogenic self-reactive cd4+ t cells acquire an fr4hicd73hi anergic state in the presence of foxp3+ regulatory t cells. J Immunol 188(1):170–181
Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, Shin T, Curiel TJ, Zhang B (2010) Cd73 on tumor cells impairs antitumor t-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res 70(6):2245–2255
Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MWL, Darcy PK, Smyth MJ (2011) Cd73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71(8):2892–2900
Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, Smyth MJ (2012) Cd73-deficient mice are resistant to carcinogenesis. Cancer Res 72(9):2190–2196
Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, Stagg J (2013) Cd73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci U S A 110(27):11091–11096
Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, Huang Q, Liu J, Takeda K, Teng MWL, Sachsenmeier K et al (2016) Co-inhibition of cd73 and a2ar adenosine signaling improves anti-tumor immune responses. Cancer Cell 30(3):391–403
Zhang F, Li R, Yang Y, Shi C, Shen Y, Lu C, Chen Y, Zhou W, Lin A, Yu L, Zhang W et al (2019) Specific decrease in b-cell-derived extracellular vesicles enhances post-chemotherapeutic cd8(+) t cell responses. Immunity 50(3):738–750.e737
Melet A, Song K, Bucur O, Jagani Z, Grassian AR, Khosravi-Far R (2008) Apoptotic pathways in tumor progression and therapy. Adv Exp Med Biol 615:47–79
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Tait SW, Green DR (2010) Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11(9):621–632
Wong RS (2011) Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res 30:87
Saito M, Yaguchi T, Yasuda Y, Nakano T, Nishizaki T (2010) Adenosine suppresses cw2 human colonic cancer growth by inducing apoptosis via a(1) adenosine receptors. Cancer Lett 290(2):211–215
Wen LT, Knowles AF (2003) Extracellular atp and adenosine induce cell apoptosis of human hepatoma li-7a cells via the a3 adenosine receptor. Br J Pharmacol 140(6):1009–1018
Kim SG, Ravi G, Hoffmann C, Jung YJ, Kim M, Chen A, Jacobson KA (2002) P53-independent induction of fas and apoptosis in leukemic cells by an adenosine derivative, cl-ib-meca. Biochem Pharmacol 63(5):871–880
Mlejnek P, Dolezel P, Kosztyu P (2012) P-glycoprotein mediates resistance to a3 adenosine receptor agonist 2-chloro-n6-(3-iodobenzyl)-adenosine-5′-n-methyluronamide in human leukemia cells. J Cell Physiol 227(2):676–685
Cohen S, Stemmer SM, Zozulya G, Ochaion A, Patoka R, Barer F, Bar-Yehuda S, Rath-Wolfson L, Jacobson KA, Fishman P (2011) Cf102 an a3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J Cell Physiol 226(9):2438–2447
Yasuda Y, Saito M, Yamamura T, Yaguchi T, Nishizaki T (2009) Extracellular adenosine induces apoptosis in caco-2 human colonic cancer cells by activating caspase-9/−3 via a(2a) adenosine receptors. J Gastroenterol 44(1):56–65
Otsuki T, Kanno T, Fujita Y, Tabata C, Fukuoka K, Nakano T, Gotoh A, Nishizaki T (2012) A3 adenosine receptor-mediated p53-dependent apoptosis in lu-65 human lung cancer cells. Cell Physiol Biochem 30(1):210–220
Aghaei M, Panjehpour M, Karami-Tehrani F, Salami S (2011) Molecular mechanisms of a3 adenosine receptor-induced g1 cell cycle arrest and apoptosis in androgen-dependent and independent prostate cancer cell lines: involvement of intrinsic pathway. J Cancer Res Clin Oncol 137(10):1511–1523
Yang D, Yaguchi T, Nakano T, Nishizaki T (2010) Adenosine-induced caspase-3 activation by tuning bcl-xl/diablo/iap expression in huh-7 human hepatoma cells. Cell Biol Toxicol 26(4):319–330
Yang D, Yaguchi T, Nakano T, Nishizaki T (2011) Adenosine activates ampk to phosphorylate bcl-xl responsible for mitochondrial damage and diablo release in huh-7 cells. Cell Physiol Biochem 27(1):71–78
Sai K, Yang D, Yamamoto H, Fujikawa H, Yamamoto S, Nagata T, Saito M, Yamamura T, Nishizaki T (2006) A(1) adenosine receptor signal and ampk involving caspase-9/−3 activation are responsible for adenosine-induced rcr-1 astrocytoma cell death. Neurotoxicology 27(4):458–467
Broker LE, Kruyt FA, Giaccone G (2005) Cell death independent of caspases: a review. Clin Cancer Res 11(9):3155–3162
Cande C, Cecconi F, Dessen P, Kroemer G (2002) Apoptosis-inducing factor (aif): key to the conserved caspase-independent pathways of cell death? J Cell Sci 115(Pt 24):4727–4734
Kanno T, Gotoh A, Fujita Y, Nakano T, Nishizaki T (2012) A(3) adenosine receptor mediates apoptosis in 5637 human bladder cancer cells by g(q) protein/pkc-dependent aif upregulation. Cell Physiol Biochem 30(5):1159–1168
Nagaya H, Gotoh A, Kanno T, Nishizaki T (2013) A3 adenosine receptor mediates apoptosis in in vitro rcc4-vhl human renal cancer cells by up-regulating amid expression. J Urol 189(1):321–328
Kanno T, Nakano T, Fujita Y, Gotoh A, Nishizaki T (2012) Adenosine induces apoptosis in sbc-3 human lung cancer cells through a(3) adenosine receptor-dependent amid upregulation. Cell Physiol Biochem 30(3):666–677
Wu M, Xu LG, Li X, Zhai Z, Shu HB (2002) Amid, an apoptosis-inducing factor-homologous mitochondrion-associated protein, induces caspase-independent apoptosis. J Biol Chem 277(28):25617–25623
Tsuchiya A, Kanno T, Saito M, Miyoshi Y, Gotoh A, Nakano T, Nishizaki T (2012) Intracellularly transported adenosine induces apoptosis in [corrected] mcf-7 human breast cancer cells by accumulating amid in the nucleus. Cancer Lett 321(1):65–72
Ciardiello F, Tortora G (2008) Egfr antagonists in cancer treatment. N Engl J Med 358(11):1160–1174
Sadikovic B, Al-Romaih K, Squire JA, Zielenska M (2008) Cause and consequences of genetic and epigenetic alterations in human cancer. Curr Genomics 9(6):394–408
Johnson DG, Walker CL (1999) Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol 39:295–312
Ohana G, Bar-Yehuda S, Barer F, Fishman P (2001) Differential effect of adenosine on tumor and normal cell growth: focus on the a3 adenosine receptor. J Cell Physiol 186(1):19–23
Mirza A, Basso A, Black S, Malkowski M, Kwee L, Pachter JA, Lachowicz JE, Wang Y, Liu S (2005) Rna interference targeting of a1 receptor-overexpressing breast carcinoma cells leads to diminished rates of cell proliferation and induction of apoptosis. Cancer Biol Ther 4(12):1355–1360
Lin Z, Yin P, Reierstad S, O’Halloran M, Coon VJ, Pearson EK, Mutlu GM, Bulun SE (2010) Adenosine a1 receptor, a target and regulator of estrogen receptoralpha action, mediates the proliferative effects of estradiol in breast cancer. Oncogene 29(8):1114–1122
Etique N, Grillier-Vuissoz I, Lecomte J, Flament S (2009) Crosstalk between adenosine receptor (a2a isoform) and eralpha mediates ethanol action in mcf-7 breast cancer cells. Oncol Rep 21(4):977–981
Ma DF, Kondo T, Nakazawa T, Niu DF, Mochizuki K, Kawasaki T, Yamane T, Katoh R (2010) Hypoxia-inducible adenosine a2b receptor modulates proliferation of colon carcinoma cells. Hum Pathol 41(11):1550–1557
Wei Q, Costanzi S, Balasubramanian R, Gao ZG, Jacobson KA (2013) A(2b) adenosine receptor blockade inhibits growth of prostate cancer cells. Purinergic Signal 9:271
Panjehpour M, Karami-Tehrani F (2004) An adenosine analog (ib-meca) inhibits anchorage-dependent cell growth of various human breast cancer cell lines. Int J Biochem Cell Biol 36(8):1502–1509
Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, Leung E, Baraldi PG, Borea PA (2002) Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol 119(4):923–933
Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P (2003) A3 adenosine receptor activation in melanoma cells: association between receptor fate and tumor growth inhibition. J Biol Chem 278(43):42121–42130
Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA (2005) A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in a375 human melanoma cells. J Biol Chem 280(20):19516–19526
Fishman P, Bar-Yehuda S, Ohana G, Barer F, Ochaion A, Erlanger A, Madi L (2004) An agonist to the a3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of gsk-3 beta and nf-kappa b. Oncogene 23(14):2465–2471
Fishman P, Bar-Yehuda S, Ohana G, Pathak S, Wasserman L, Barer F, Multani AS (2000) Adenosine acts as an inhibitor of lymphoma cell growth: a major role for the a3 adenosine receptor. Eur J Cancer (Oxford, England : 1990) 36(11):1452–1458
Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, Silberman D, Slosman G, Fishman P (2003) Inhibition of primary colon carcinoma growth and liver metastasis by the a3 adenosine receptor agonist cf101. Br J Cancer 89(8):1552–1558
Panjehpour M, Karami-Tehrani F (2007) Adenosine modulates cell growth in the human breast cancer cells via adenosine receptors. Oncol Res 16(12):575–585
Bar-Yehuda S, Barer F, Volfsson L, Fishman P (2001) Resistance of muscle to tumor metastases: a role for a3 adenosine receptor agonists. Neoplasia (New York, NY) 3(2):125–131
Fishman P, Bar-Yehuda S, Barer F, Madi L, Multani AS, Pathak S (2001) The a3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp Cell Res 269(2):230–236
Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K (2002) Evidence for involvement of wnt signaling pathway in ib-meca mediated suppression of melanoma cells. Oncogene 21(25):4060–4064
Lu J, Pierron A, Ravid K (2003) An adenosine analogue, ib-meca, down-regulates estrogen receptor alpha and suppresses human breast cancer cell proliferation. Cancer Res 63(19):6413–6423
Chung H, Jung JY, Cho SD, Hong KA, Kim HJ, Shin DH, Kim H, Kim HO, Shin DH, Lee HW, Jeong LS et al (2006) The antitumor effect of lj-529, a novel agonist to a3 adenosine receptor, in both estrogen receptor-positive and estrogen receptor-negative human breast cancers. Mol Cancer Ther 5(3):685–692
Morello S, Petrella A, Festa M, Popolo A, Monaco M, Vuttariello E, Chiappetta G, Parente L, Pinto A (2008) Cl-ib-meca inhibits human thyroid cancer cell proliferation independently of a3 adenosine receptor activation. Cancer Biol Ther 7(2):278–284
Chiang AC, Massague J (2008) Molecular basis of metastasis. N Engl J Med 359(26):2814–2823
Spano D, Heck C, De Antonellis P, Christofori G, Zollo M (2012) Molecular networks that regulate cancer metastasis. Semin Cancer Biol 22(3):234–249
Kunzli BM, Bernlochner MI, Rath S, Kaser S, Csizmadia E, Enjyoji K, Cowan P, d’Apice A, Dwyer K, Rosenberg R, Perren A et al (2011) Impact of cd39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal 7(2):231–241
Arab S, Hadjati J (2019) Adenosine blockage in tumor microenvironment and improvement of cancer immunotherapy. Immune Netw 19(4):e23
Leone RD, Emens LA (2018) Targeting adenosine for cancer immunotherapy. J Immunother Cancer 6(1):57
Zhou P, Zhi X, Zhou T, Chen S, Li X, Wang L, Yin L, Shao Z, Ou Z (2007) Overexpression of ecto-5′-nucleotidase (cd73) promotes t-47d human breast cancer cells invasion and adhesion to extracellular matrix. Cancer Biol Ther 6(3):426–431
Wang L, Zhou X, Zhou T, Ma D, Chen S, Zhi X, Yin L, Shao Z, Ou Z, Zhou P (2008) Ecto-5′-nucleotidase promotes invasion, migration and adhesion of human breast cancer cells. J Cancer Res Clin Oncol 134(3):365–372
Cappellari AR, Rockenbach L, Dietrich F, Clarimundo V, Glaser T, Braganhol E, Abujamra AL, Roesler R, Ulrich H, Battastini AM (2012) Characterization of ectonucleotidases in human medulloblastoma cell lines: Ecto-5’nt/cd73 in metastasis as potential prognostic factor. PLoS One 7(10):e47468
Zhi X, Chen S, Zhou P, Shao Z, Wang L, Ou Z, Yin L (2007) Rna interference of ecto-5′-nucleotidase (cd73) inhibits human breast cancer cell growth and invasion. Clin Exp Metastasis 24(6):439–448
Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, Smit MA, Geiger TR, Laoukili J, Iskit S, Rodenko B et al (2013) Identification of a pharmacologically tractable fra-1/adora2b axis promoting breast cancer metastasis. Proc Natl Acad Sci U S A 110(13):5139–5144
Ntantie E, Gonyo P, Lorimer EL, Hauser AD, Schuld N, McAllister D, Kalyanaraman B, Dwinell MB, Auchampach JA, Williams CL (2013) An adenosine-mediated signaling pathway suppresses prenylation of the gtpase rap1b and promotes cell scattering. Sci Signal 6(277):ra39
Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK (2013) Blockade of a2a receptors potently suppresses the metastasis of cd73+ tumors. Proc Natl Acad Sci U S A 110(36):14711–14716
Mao L, Fan TF, Wu L, Yu GT, Deng WW, Chen L, Bu LL, Ma SR, Liu B, Bian Y, Kulkarni AB et al (2017) Selective blockade of b7-h3 enhances antitumour immune activity by reducing immature myeloid cells in head and neck squamous cell carcinoma. J Cell Mol Med 21(9):2199–2210
Shi L, Wu Z, Miao J, Du S, Ai S, Xu E, Feng M, Song J, Guan W (2019) Adenosine interaction with adenosine receptor a2a promotes gastric cancer metastasis by enhancing pi3k-akt-mtor signaling. Mol Biol Cell 30(19):2527–2534
Alderton GK (2012) Metastasis. Exosomes drive premetastatic niche formation. Nat Rev Cancer 12(7):447
Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z (2011) Cancer exosomes express cd39 and cd73, which suppress t cells through adenosine production. J Immunol 187(2):676–683
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Antonioli, L. et al. (2021). Adenosine Signaling in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1270. Springer, Cham. https://doi.org/10.1007/978-3-030-47189-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-030-47189-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-47188-0
Online ISBN: 978-3-030-47189-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)