
Abstract
We propose the novel implementation of a depth variable importance score in a classification tree method designed for the precision medicine setting. The goal is to identify clinically meaningful subgroups to better inform personalized treatment decisions. In the proposed Depth Importance in Precision Medicine (DIPM) method, a random forest of trees is first constructed at each node. Then, a depth variable importance score is used to select the best split variable. This score makes use of the observation that more important variables tend to be selected closer to root nodes of trees. In particular, we aim to outperform an existing method designed for the analysis of high-dimensional data with continuous outcome variables. The existing method uses an importance score based on weighted misclassification of out-of-bag samples upon permutation. Overall, our method is favorable because of its comparable and sometimes superior performance, simpler importance score, and broader pool of candidate splits. We use simulations to demonstrate the accuracy of our method and apply the method to a clinical dataset.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Improving the field of medicine using personalized health data has become a primary focus for researchers. Instead of the traditional focus on average responses to interventions, precision medicine recognizes the heterogeneity that exists between individuals and aims to find the optimal treatment for each person [7, 13]. With the increasing number of large datasets available for analysis, identifying which features are important is a challenge. Ultimately, the development of more sophisticated methodology to match the development of these kinds of data is important to help improve the health outcomes and quality of life experienced by each individual patient.
The classification tree is an attractive method for precision medicine due to its flexibility and relatively simple structure. Multiple candidate features may be considered simultaneously, and the final result is an easily interpretable tree. In general, a classification tree is a method that divides the overall sample into smaller and smaller subgroups using optimized subdivisions of the data. The subdivisions, or splits, are based on a predetermined list of candidate split variables. Traditionally, classification trees are used to identify homogenous subgroups of the sample and classify each subject’s membership in a predetermined list of categories. In the context of precision medicine, the method is modified to identify subgroups of patients that perform especially well or especially poorly in a treatment group and determine which treatment is best for each subject.
Currently, there are multiple existing tree-based methods designed for the precision medicine setting. Existing methods include: an extension of the RECursive Partition and Amalgamation (RECPAM) algorithm [12], model-based partitioning (MOB) [14, 19], interaction trees (IT) [15,16,17], the simultaneous threshold interaction modelling algorithm (STIMA) [4], virtual twins (VT) [6], subgroup identification based on differential effect search (SIDES) [9], an extension to SIDES known as SIDEScreen [8], qualitative interaction trees (QUINT) [5], generalized, unbiased, interaction detection and estimation (GUIDE) trees [10, 11], a relative-effectiveness based method [18, 20], and an importance index based method [22].
Although multiple methods already exist, the type of outcome as well as other features of the data determine which subset of methods the user may choose from. For instance, the method developed by Zhang et al. [20] only applies to clinical data with a binary outcome and two treatment groups. Meanwhile, IT, QUINT, STIMA, and the method developed by Zhu et al. [22] apply to data with a continuous outcome. In addition, RECPAM, IT, MOB, SIDES, GUIDE, and the method developed by Zhu et al. [22] have been extended to analyze survival data with right-censored survival times. To date, only IT and GUIDE have an extension for data with longitudinal outcomes. Furthermore, a problem across methods is weakened performance as the number of candidate covariates increases. As noted in Tsai et al. [18], having more candidate covariates decreases the “signal-to-noise ratio” which can lower the chance of finding the most important variables. These concerns are especially problematic given the increased availability of higher dimensional data.
One method of particular interest is the weighted classification tree developed by Zhu et al. [22]. This method aims to achieve better performance in cases of high dimensionality and is designed for data with a continuous outcome variable and two treatment groups. A variable importance score based on weighted misclassification is used to find the best split variable at each node. However, as no method uniformly outperforms all other methods in this setting, there are several drawbacks. In particular, we find that the weighted method’s variable importance score misses important signals in the presence of correlations between variables and that the method is unnecessarily complex overall. Instead, we propose the usage of the depth variable importance score developed by Chen et al. [3], and Zhang and Singer [21]. Adapting this measure for usage within a tree and within the precision medicine framework is novel. Here, we make the case that the proposed Depth Importance in Precision Medicine (DIPM) method is favorable to the aforementioned method because of the proposed method’s comparable and sometimes superior performance, simpler importance score, and broader pool of candidate splits. Developing an importance score that is intuitive and convenient to compute that yields comparable or even better results will set the stage for outright superior performance with more complex data scenarios to be demonstrated in future work. The overall goal is to identify variables that are important in the context of precision medicine. Note that the proposed method is an exploratory method as opposed to a confirmatory model. Thus, here we focus on introducing our new importance score and demonstrate its advantage by using datasets with continuous outcome variables for the easy comparison with an existing method.
The remainder of this paper is structured as follows. First, details of the proposed DIPM method and the weighted classification tree method are provided. Then, simulation scenarios assessing and comparing the methods are presented. Next, results of an application to a real-world dataset are described. Lastly, the discussion section includes closing remarks and directions for future work.
2 Methods
2.1 Overview
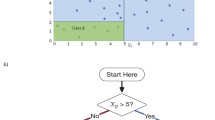
We begin with a brief overview of our method. The proposed DIPM method is designed for the analysis of clinical datasets with a continuous outcome variable Y and two treatment assignments A and B. Without loss of generality, higher values of Y denote better health outcomes. Candidate split variables may be binary, ordinal, or nominal. All of the learning data are said to be in the first or root node, and nodes may be split into two child nodes. Borrowing the terminology used in Zhu et al. [22], at each node in the tree, a random forest of “embedded” trees is grown to determine the best variable to split the node. Once the best variable is identified, the best split of the best variable is the split that maximizes the difference in response rates between treatments A and B. Note that “the best variable” is “best” in a narrow sense as defined below. In addition, a flowchart outlining the general steps of our method’s algorithm is provided in Fig. 16.1.

Overview of DIPM method classification tree algorithm. A flowchart outlining the general steps of the proposed method’s algorithm is depicted in the figure above
2.2 Depth Variable Importance Score
The depth variable importance score is used to find the best split variable at a node. In general, the score incorporates two pieces of information: the depth of a node within a tree and the magnitude of the relevant effect. Depth information is used because more important variables tend to be selected closer to the root node. Meanwhile, the strength of a split is also taken into account. This second component of the variable importance score is a statistic. The statistic that is used depends on the context of the analysis at hand.
Recall that at each node in the overall classification tree, a random forest is constructed to find the best split variable at the node. Once the forest is fit, for each tree T in this forest, the following sum is calculated for each covariate j:
\(T_j\) is the set of nodes in tree T split by variable j. L(t) is the depth of node t. The root node has depth 1, the left and right child nodes of the root node have depth 2, etc. \(G_t\) captures the magnitude of the effect of splitting node t. Since the outcome is continuous, \(G_t\) is set equal to the \(t^2\) statistic from testing the significance of \(\beta _3\) in the model:
This model is fit using the pertinent within-node data. The test statistic t is squared because the magnitude of the interaction is of interest, while there is no preference in the effect’s direction. Note that this \(t^2\) statistic is identical to the statistic used at each node split in the interaction tree method [16].
Next, a “G replacement” feature is implemented that potentially alters the variable importance scores score(T, j). For each tree T in the forest, the G at each split is replaced with the highest G value of any of its descendant nodes if this maximum exceeds the value at the current split. This replacement step is performed because a variable that yields a split with a large effect of interest further down in the tree is certainly important even if its importance is not captured right away. By “looking ahead” at the G values of future splits, a variable’s importance is reinforced.
Lastly, the final variable importance scores are averaged across all M trees in the forest f:
The best split variable is the variable with the largest value of score(f, j).
2.3 Split Criteria
To identify the best split at a node t, the squared difference in response rates between treatments A and B at node t is first assessed:
Then, among the list of candidate splits, only splits with child nodes with at least nmin subjects are considered. Of the splits with a sufficient number of subjects, the best split maximizes the weighted sum of the squared difference in response rates of the child nodes:
Node t is split only when the best split yields two child nodes with a greater difference in treatment response rates than at the current node:
Splitting stops when there are not enough subjects in any candidate node splits or when no remaining \(DIFF(t_L, t_R)\) values exceed DIFF(t).
This split criterion was first proposed by Zhang for data with binary outcomes [18, 20]. Since the proposed method uses continuous outcomes, the mean of Y is used in place of \(Pr(Y=1)\).
2.4 Random Forest
A random forest is grown at each node in the overall tree and then used to select the best split variable. Once this variable is identified, all possible splits of the variable are considered, and the best split is found using the criteria described in Sect. 16.2.3.
The forest is constructed as follows. The forest contains a total of M embedded trees, and the recommended value of M is 1000. Each embedded tree is grown using a bootstrap sample. The bootstrap sample contains the same number of subjects as the original sample size at the current node. Then, at each node in the embedded trees, either: (1) all possible splits of all variables are considered, or (2) all possible splits of a certain number, mtry, of randomly selected variables are considered. The best split is again found using the criteria described in Sect. 16.2.3.
A recommended value of mtry for a dataset with p variables is \(floor(\sqrt{p})\). This value is the default value of mtry used in the randomForest R package implementing Breiman’s random forest method for classification. The aim is to use a value that balances the strength of each tree by being large enough while minimizing the correlation between trees by being small enough [2].
Also, note that the minimum number of subjects in nodes of the overall classification tree does not have to equal the minimum number of subjects in nodes of the embedded trees. Put another way, nmin is the minimum node size of the overall tree, while nmin2 is the minimum node size of trees in the random forest. nmin and nmin2 do not have to be equivalent.
2.5 Best Predicted Treatment Class
The best predicted treatment class of a node is the treatment group that performs best based on the subjects within the given node. In the proposed method, the means of the response values Y are compared by treatment group. Recall that higher values of Y denote greater benefit for patients. Therefore, if \(\bar{Y}_A > \bar{Y}_B\) within a node, then treatment A is the best predicted treatment at that node. If \(\bar{Y}_B > \bar{Y}_A\), then treatment B is the best predicted treatment. If \(\bar{Y}_A = \bar{Y}_B\), then neither treatment is best.
2.6 Splits by Variable Type
The list of possible splits for a candidate split variable depends on the variable’s type. For a binary variable, the variable has only two possible values: 0 and 1. Therefore, there is only one possible split: the left child node subsets the data with subjects whose values equal 0, and the right child node contains the rest of the subjects whose values equal 1.
For an ordinal variable, each unique value is a candidate split point. For each candidate split point s, the left child node considers the subjects with values less than or equal to s, and the right child node contains the rest of the subjects with values greater than s. Note that considering the largest unique value is redundant, since every subject takes values less than or equal to the maximum, and no one has values exceeding the maximum.
For a nominal variable, all combinations of all possible subsets of the categories are considered as candidate splits. For example, consider a nominal variable with three categories A, B, and C. One possible split is that the left child node subsets the data with subjects in category A, and the right child node contains the remaining subjects in categories B and C. The two other possible splits are A, B with complement C, and A, C with complement B. In general, for a nominal variable with k categories, the total number of possible splits is \(2^{k-1} - 1\) [21].
2.7 Comparison Method
The weighted classification tree method by Zhu et al. [22] also uses a forest of M embedded trees to find the best split variable at each node. Again, once the best split variable is found, the best split of all possible splits of that variable is used. However, the forest used in the weighted classification tree method is a forest of extremely randomized trees. These trees select one random split for each variable. The best of these splits is used to split a node, and the split criteria is a weighted Gini impurity score. In addition, each tree uses bootstrap samples that consist of randomly sampling 80\(\%\) of the node data without replacement.
Before the overall classification tree is constructed, mean estimates for each subject are predicted using a random forest of regression trees. These estimates are used to: (1) construct subject specific weights to be used when calculating the variable importance scores, and (2) perform “treatment flipping”. One way to construct the weights is to take the absolute value of the difference between outcome variable Y and the estimated mean for each subject. Next, if a subject’s Y value is smaller than the subject’s estimated mean, then that subject is placed in the other treatment group. In other words, the treatment is “flipped”. Note that treatment flipping does not affect the best predicted treatment at any terminal node of the tree and is done to solve the problem of greater bias for splits near the boundary of a variable.
Once a forest f containing M trees is fit at a node, a weighted variable importance score is calculated for each variable j to find the best split variable. This importance score uses the out-of-bag (OOB) samples at a node to calculate the weighted ratio of misclassified treatments when values are randomly permuted to the amount of misclassification when values are left the same:
\(\widehat{f}_{m}\) denotes the predicted best treatment classes of the mth tree in the forest. \(L_{m,o}\) is the OOB data for the mth tree. For the OOB samples, \(w_i\) is the ith subject’s weight, \(A_i\) is the ith subject’s treatment assignment after treatment flipping, \(\mathbf{x} _i^{(-j)}\) is the ith subject’s vector of data without variable j, \(x_i^{(j)}\) is the ith subject’s value of variable j, and \(\tilde{x}_i^{(j)}\) is an independent, randomly permuted copy of variable j. I is the indicator function. The best split variable is the variable with the largest value of \({score}^*_{cla}(f,j)\).
2.8 Implementation
The proposed method is implemented using an R program. The R code calls a C program to generate the final classification tree. The C backend is used to take advantage of C’s higher computational speed in comparison to R. Meanwhile, the weighted classification method developed by Zhu et al. [22] is implemented using their RLT package on CRAN. All computations for the simulation studies and data analysis are implemented in R.
3 Simulation Studies
3.1 Methods
In addition to the weighted classification tree and proposed DIPM methods, two other methods are compared in our simulation studies. These additional methods do not use a random forest at each node. Instead, the additional methods are tree methods that consider all possible splits of all candidate variables at each node. One of these methods uses the weighted Gini impurity score to compare all splits, while the other uses the “DIFF” score described in Sect. 16.2.3. These methods act as controls to further study the effect of using a broader pool of candidate splits.
3.2 Scenarios
The following scenarios assess the proposed DIPM method and compare it to the weighted classification tree method. The overall strategy is to design scenarios with known, underlying signals and then measure how often each method accurately detects these signals. This strategy allows us to compare the variable importance scores of the weighted and proposed methods. Recall that the DIPM method is an exploratory method as opposed to a confirmatory model, and the primary goal is to identify important variables in the context of precision medicine. Therefore, measuring correct variable selection alone is sufficient.
In particular, the simulations are designed to assess how each method performs with increasing amounts of correlation. Altogether, we expect each method to perform worse with greater amounts of correlation, while we are interested in assessing how each method performs in comparison with the others. Note that in all simulations, treatment assignments are randomly generated from \(\{A, B\}\) with equal probability. \(I_A\) and \(I_B\) denote the indicators for assignments to treatments A and B respectively. Furthermore, the error term \(\varepsilon \) in each scenario is normally distributed, i.e., \(\varepsilon \sim N(0,1)\).
Scenarios 1 through 4 assess method performance as the magnitude of correlation between so-called Z variables and truly important variables increases. In scenarios 1 through 4, there are 250 X variables in the data that are all ordinal. In addition to the X variables, 50 Z variables are part of the data. Each Z is highly correlated with truly important variables as specified for each scenario below. The formulas used to calculate the correlated Z variables include a random term \(\varepsilon _i\) that is normally distributed, i.e., \(\varepsilon _i \sim N(0,sd=\sigma )\). When generating the Z variables, decreasing values of \(\sigma \) are used. As \(\sigma \) decreases, the correlation between the Z variables and the important variables increases. For each value of \(\sigma \), 1000 simulations are run for sample sizes of 250 subjects. Overall, we expect method performance to decrease as \(\sigma \) decreases, i.e., as the correlation level between each Z variable and a truly important variable increases. When the correlation level is greater, the probability that each method erroneously selects a correlated Z variable instead of a truly important variable is greater as well.
Scenario 1: The first scenario consists of an underlying linear model containing the treatment and one important continuous variable. The formula for the outcome variable Y is:
The 250 X variables in the data are all independent and normally distributed, i.e., N(0, 1). The 50 Z variables in the data are each highly correlated with variable \(X_1\) and calculated as follows: \(Z_i=0.8X_1 + 0.1X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\).
Scenario 2: The second scenario consists of an underlying model with an exponential term containing the treatment and two important continuous variables. The formula for the outcome variable Y is:
The 250 X variables in the data are all independent and normally distributed, i.e., N(0, 1). The first 25 Z variables are highly correlated with variable \(X_2\) and calculated as follows: \(Z_i=0.1\,X_1 + 0.8\,X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\). The last 25 Z variables are highly correlated with \(X_{10}\) and calculated as follows: \(Z_i=0.1X_1 + 0.8X_{10} + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\).
Scenario 3: The third scenario consists of an underlying tree model containing the treatment and two important binary variables. The formula for the outcome variable Y is:
The first 230 X variables in the data are from the Discrete Uniform distribution, i.e., Discrete Uniform[0, 2]. These variables are meant to simulate SNP data that have possible values of 0, 1, or 2. The next 10 X variables are Poisson distributed with mean 1, i.e., Poisson(1). The final 10 X variables are Poisson distributed with mean 2, i.e., Poisson(2). The Poisson distributed variables are meant to simulate ordinal count data that could be collected in a clinical trial. In addition, the first 25 Z variables are highly correlated with variable \(X_2\) and calculated as follows: \(Z_i=X_2+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\). The last 25 Z variables are highly correlated with \(X_{10}\) and calculated as follows: \(Z_i=X_{10}+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\). All 50 Z variables are rounded to the nearest integer. To continue simulating SNP data, values less than 0 are set to 0, and values exceeding 2 are set to 2.
Scenario 4: The fourth scenario consists of an underlying tree model containing the treatment and three important binary variables. The formula for the outcome variable Y is:
The first 230 X variables in the data are from the Discrete Uniform distribution, i.e., Discrete Uniform[0, 2]. These variables are meant to simulate SNP data that have possible values of 0, 1, or 2. The next 10 X variables are Poisson distributed with mean 1, i.e., Poisson(1). The final 10 X variables are Poisson distributed with mean 2, i.e., Poisson(2). The Poisson distributed variables are meant to simulate ordinal count data that could be collected in a clinical trial. In addition, the 50 Z variables in the data are each highly correlated with variable \(X_1\) and calculated as follows: \(Z_i=X_1+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=\sigma )\). All 50 Z variables are rounded to the nearest integer. To continue simulating SNP data, values less than 0 are set to 0, and values exceeding 2 are set to 2.
Scenarios 5 through 8 assess method performance as the number of variables correlated with truly important variables increases. In scenarios 5 through 8, there are 100 X variables in the data that are all ordinal and independent and normally distributed, i.e., N(0, 1). In addition to the X variables, a varying number of Z variables are part of the data. Each Z is highly correlated with truly important variables and calculated as specified for each scenario below. For each varying number of Z variables, 1000 simulations are run for sample sizes of 250 subjects. Overall, we expect method performance to decrease as the number of Z variables in the data increases. As the number of Z variables increases, the chance of selecting a correlated Z variable instead of a truly important variable also increases.
Scenario 5: The fifth scenario consists of an underlying linear model containing the treatment and one important continuous variable. The formula for the outcome variable Y is:
Each Z variable in the data is highly correlated with \(X_1\) and calculated as follows: \(Z_i=0.8X_1 + 0.1X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=0.5)\).
Scenario 6: The sixth scenario consists of an underlying model with an exponential term containing the treatment and two important continuous variables. The formula for the outcome variable Y is:
Each Z variable in the data is highly correlated with \(X_2\) and calculated as follows: \(Z_i=0.1X_1 + 0.8X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=0.5)\).
Scenario 7: The seventh scenario consists of an underlying tree model containing the treatment and two important binary variables. The formula for the outcome variable Y is:
Each Z variable in the data is highly correlated with \(X_2\) and calculated as follows: \(Z_i=0.1X_1 + 0.8X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=0.5)\).
Scenario 8: The final scenario consists of an underlying tree model containing the treatment and three important binary variables. The formula for the outcome variable Y is:
Each Z variable in the data is highly correlated with \(X_1\) and calculated as follows: \(Z_i=0.8X_1 + 0.1X_2 + 0.1X_3+\varepsilon _i\), where \(\varepsilon _i \sim N(0,sd=0.5)\).
3.3 Results
All simulation results are presented in Table 16.1. As expected, across all of the simulation scenarios, as the amount of correlation between the Z variables and truly important variables increases, method performance decreases. Method performance is assessed by measuring each method’s ability to select the correct relevant variables at early splits. In general, the forest based methods tend to outperform the control methods in scenarios with non-tree models, i.e., scenarios 1 and 5 which contain a linear term and scenarios 2 and 6 which contain an exponential term. Meanwhile, the control methods tend to outperform the forest based methods in scenarios with underlying tree models, i.e., scenarios 3, 4, 7, and 8.
When comparing the DIPM method that selects mtry variables at each node in embedded trees with the weighted classification tree method, the weighted method slightly outperforms the DIPM method in scenarios 1, 5, and 7. However, in scenarios 2, 4, 6, and 8, the DIPM method outperforms the weighted method. Finally, in scenario 3, the weighted method outperforms the DIPM method until \(\sigma =0.3\). Based on these simulation scenarios, the DIPM method demonstrates comparable and sometimes superior performance in comparison to the more complex weighted method. Although the DIPM method does not consistently outperform the weighted method, recall that our goal is to demonstrate how our intuitive and easy-to-compute importance score can still yield generally comparable performance to the weighted method. These initial developments will then set the stage for consistently better performance in data of greater complexity in future work.

Results of CCLE data application from Zhu et al. [22]. Boxplots comparing the two treatments are in each node, and paired t-test p-values are beneath terminal nodes. For the first split using tissue, S1 is the set of categories: autonomic ganglia, large intestine, pancreas, skin, biliary tract, oesophagus, stomach, thyroid, and urinary tract

Results of CCLE data application using the DIPM method. Boxplots comparing the two treatments are in each node, and paired t-test p-values are beneath terminal nodes. For the first split using tissue, S2 is the set of categories: autonomic ganglia, large intestine, pancreas, and skin
4 Analysis of CCLE Data
The DIPM method is applied to a real-world dataset. The data used are a product of the Cancer Cell Line Encyclopedia (CCLE) project by the Broad Institute and the Novartis Institutes for Biomedical Research [1]. The data consist of genetic information and pharmacologic outcomes for more than 1,100 human cancer cell lines. The data are publicly available online (https://portals.broadinstitute.org/ccle/) and are also used by Zhu et al. in their paper [22].
Drug activity measures of multiple drugs are recorded for each cell line. Following the analysis by Zhu et al., two drugs, RAF265 and PD-0325901, are selected for the present analysis. Although Zhu et al. pre-screen the gene expressions and use only the top 500 genes, we use all available gene expressions. For the two selected drugs, there are 447 cell lines, 18,988 gene expressions, and 3 clinical variables available for analysis. The clinical variables are gender, tissue type, and histology. Since the outcome variable is measured for each cell line for each of the two treatments, the final dataset contains 894 observations and 18,991 candidate split variables. All in all, the application of the proposed method to these data produce useful insights. We can use the DIPM method to search for genetic and/or clinical subgroups with varying drug activity levels across the two selected drugs. Moreover, the application presents us with the opportunity to apply the proposed method to a dataset with a large number of candidate split variables.
The constructed tree for the DIPM method is compared to the final tree presented by Zhu et al. [22]. The two trees are depicted in Figs. 16.2 and 16.3. Since their final tree has a maximum depth of 4, we also present the results with a maximum depth of 4. Terminal node pairs with different optimal treatments are pruned. This simple pruning strategy removes redundant splits and is proposed in Tsai et al. [18]. Furthermore, paired t-test p-values comparing the mean drug activity levels of each treatment are reported beneath the terminal nodes of the two trees. This is done to help quantify how different the two drugs are with respect to drug activity levels within each subgroup. Note that the paired t-test is used since the outcome variable is recorded for both drugs for each cell line.
Both methods identify tissue type as the best split variable at the root node. Though the first split variable is the same, the split values are slightly different. The weighted method places tissue categories autonomic ganglia, large intestine, pancreas, skin, biliary tract, oesophagus, stomach, thyroid, and urinary tract in the child node that identifies PD-0325901 as the optimal treatment. Meanwhile, the DIPM method places only autonomic ganglia, large intestine, pancreas, and skin in the child node that identifies PD-0325901 as the optimal treatment. Despite these differences, ultimately, the t-test p-values comparing the two treatments in these nodes are both approximately equal, i.e., p-value \(=\) 2.2e-16.
Meanwhile, the subsequent splits of the DIPM method tree differ from those in Zhu et al.’s final tree. The other splits in Zhu et al.’s final tree use the PLA2G4A and COL5A2 genes. By contrast, the other splits in the proposed method’s tree use KCNK2, DCLK2, PARP14, and OXTR. Although neither method clearly outperforms the other in this data application, overall, these results point to the robustness of the effect of tissue type as a potentially useful subgroup indicator. The identified gene expression variables by both methods are also potentially useful subgroup indicators that would have to be examined further for true biological relevance.
5 Discussion
In this article, we present the novel DIPM method. The DIPM method is an exploratory method designed to search through existing clinical data for variables that are important in the context of precision medicine. We demonstrate how the proposed method performs well and, in particular, how it compares to the weighted classification tree developed by Zhu et al. [22]. In our simulations, the depth variable importance score demonstrates comparable and sometimes better performance than the variable importance score of the weighted method. The DIPM method achieves this level of performance as a simpler method overall. The DIPM method has no subject specific weights, has no treatment flipping, and considers all possible splits instead of one random split per variable at the nodes of embedded trees. Searching through all splits strengthens the proposed method and better ensures that signals are not missed by sheer chance as in the weighted method. Furthermore, calculating the depth variable importance score is simpler than randomly permuting each variable and counting the misclassifications of out-of-bag samples at each node. In short, the proposed method is less complicated and easier to understand.
Although the presently proposed DIPM method is restricted to the analysis of datasets with continuous outcome variables, the flexibility of the depth variable importance score makes the method readily extendable to other outcome variable types. One useful extension of the DIPM method will be the application to censored survival outcomes. To achieve this application, we will redefine the split criteria and the G statistic in the depth variable importance score accordingly. Note that Zhu et al. have already extended their weighted classification tree method to the analysis of right-censored survival data. It would be interesting to discover whether our markedly simpler method can in fact outperform the weighted method for data with survival endpoints. Also, it would be useful to extend the DIPM method to data with longitudinal outcomes. As mentioned in the introduction, to date, only IT and GUIDE have an extension for data with longitudinal outcomes. It would be interesting to create and assess the performance of the DIPM method when adapted to longitudinal data as well.
A topic of interest for future consideration is covariate selection bias. When searching for the best split at a node, covariates with a greater number of possible splits tend to be selected more often than covariates with fewer splits. The concern is that this phenomenon occurs even when the covariate is not relevant. In this research setting, only Loh et al. have directly addressed this bias by developing a two-step approach [10, 11]. Though we are aware of this bias, we do not directly address covariate selection bias with the proposed method. We aim to continue to consider this issue while developing tree-based methodology moving forward.
References
Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A.A., et al.: The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012)
Breiman, L.: Random forests. Mach. Learn. 45, 5–32 (2001)
Chen, X., Liu, C.T., Zhang, M., Zhang, H.: A forest-based approach to identifying gene and gene-gene interactions. Proc. Natl. Acad. Sci. U.S.A. 104, 19199–19203 (2007)
Dusseldorp, E., Conversano, C., Van Os, B.J.: Combining an additive and tree-based regression model simultaneously: STIMA. J. Comput. Graph. Stat. 19, 514–530 (2010)
Dusseldorp, E., Van Mechelen, I.: Qualitative interaction trees: a tool to identify qualitative treatment-subgroup interactions. Stat. Med. 33, 219–237 (2014)
Foster, J.C., Taylor, J.M.G., Ruberg, S.J.: Subgroup identification from randomized clinical trial data. Stat. Med. 30, 2867–2880 (2011)
Hamburg, M.A., Collins, F.S.: The path to personalized medicine. N. Engl. J. Med. 363, 301–304 (2010)
Lipkovich, I., Dmitrienko, A.: Strategies for identifying predictive biomarkers and subgroups with enhanced treatment effect in clinical trials using sides. J. Biopharm. Stat. 24, 130–153 (2014)
Lipkovich, I., Dmitrienko, A., Denne, J., Enas, G.: Subgroup identification based on differential effect search-a recursive partitioning method for establishing response to treatment in patient subpopulations. Stat. Med. 30, 2601–2621 (2011)
Loh, W.Y., Fu, H., Man, M., Champion, V., Yu, M.: Identification of subgroups with differential treatment effects for longitudinal and multiresponse variables. Stat. Med. 35, 4837–4855 (2016)
Loh, W.Y., He, X., Man, M.: A regression tree approach to identifying subgroups with differential treatment effects. Stat. Med. 34, 1818–1833 (2015)
Negassa, A., Ciampi, A., Abrahamowicz, M., Shapiro, S., Boivin, J.F.: Tree-structured subgroup analysis for censored survival data: validation of computationally inexpensive model selection criteria. Stat. Comput. 15, 231–239 (2005)
Ruberg, S.J., Chen, L., Wang, Y.: The mean does not mean as much anymore: finding sub-groups for tailored therapeutics. Clin. Trials 7, 574–583 (2010)
Seibold, H., Zeileis, A., Hothorn, T.: Model-based recursive partitioning for subgroup analyses. Int. J. Biostat. 12, 45–63 (2016)
Su, X., Meneses, K., McNees, P., Johnson, W.O.: Interaction trees: Exploring the differential effects of an intervention programme for breast cancer survivors. J. R. Stat. Soc. (Appl. Stat.) 60, 457–474 (2011)
Su, X., Tsai, C.L., Wang, H., Nickerson, D.M., Li, B.: Subgroup analysis via recursive partitioning. J. Mach. Learn. Res. 10, 141–158 (2009)
Su, X., Zhou, T., Yan, X., Fan, J., Yang, S.: Interaction trees with censored survival data. Int. J. Biostat. 4, 1–26 (2008)
Tsai, W.M., Zhang, H., Buta, E., O’Malley, S., Gueorguieva, R.: A modified classification tree method for personalized medicine decisions. Stat. Interface 9, 239–253 (2016)
Zeileis, A., Hothorn, T., Hornik, K.: Model-based recursive partitioning. J. Comput. Graph. Stat. 17, 492–514 (2008)
Zhang, H., Legro, R.S., Zhang, J., Zhang, L., Chen, X., et al.: Decision trees for identifying predictors of treatment effectiveness in clinical trials and its application to ovulation in a study of women with polycystic ovary syndrome. Hum. Reprod. 25, 2612–2621 (2010)
Zhang, H., Singer, B.: Recursive Partitioning and Applications. Springer, New York (2010)
Zhu, R., Zhao, Y.Q., Chen, G., Ma, S., Zhao, H.: Greedy outcome weighted tree learning of optimal personalized treatment rules. Biometrics 73, 391–400 (2017)
Acknowledgements
This work was supported in part by NIH Grants T32MH14235, R01 MH116527, and NSF grant DMS1722544. We thank an anonymous referee for their invaluable comments. The Cancer Cell Line Encyclopedia (CCLE) data used in this article are obtained from the CCLE of the Broad Institute. Their database is available publicly online, and they did not participate in the analysis of the data or the writing of this report.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Chen, V., Zhang, H. (2020). Depth Importance in Precision Medicine (DIPM): A Tree and Forest Based Method. In: Fan, J., Pan, J. (eds) Contemporary Experimental Design, Multivariate Analysis and Data Mining. Springer, Cham. https://doi.org/10.1007/978-3-030-46161-4_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-46161-4_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-46160-7
Online ISBN: 978-3-030-46161-4
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)