
Abstract
In this chapter we discuss higher-order methods for optimization problems in machine learning applications. We also present underlying theoretical background as well as detailed experimental results for each of these higher order methods and also provide their in-depth comparison with respect to competing methods in the context of real-world datasets. We show that higher-order methods, contrary to popular understanding, can achieve significantly superior results compared to state-of-the-art competing methods in shorter wall-clock times yielding orders of magnitude of relative speedup for typical real-world datasets.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sub-sampled Newton-type methods
- ADMM based optimization framework
- Natural gradient
- Kronecker Factored Approximated Curvature (KFAC)
- Trust-region framework
Increasing processing power of the bare metal hardware has motivated significant interest in the development of optimization techniques for machine learning problems on massively large datasets. Applications such as autonomous vehicles, artificial intelligence (Google’s GO system [38], IBM’s Deep Blue [34], IBM’s Project Debator [35]), image classification, and cybersecurity have been enabled by developments in optimization techniques and their parallel implementations. Many current applications mentioned above are modeled as either convex or non-convex optimization problems. These problems have rich theoretical foundations, as well as algorithmic (both serial and parallel) contributions. In this chapter, we focus on finite-sum minimization problems in the context of convex and non-convex formulations. We discuss state-of-the-art optimization methods for these problems under real-world assumptions on parallel platforms. We also highlight the need for hardware accelerators, such as GPUs, in significantly accelerating solutions.
1 Introduction and Motivation
Finite-sum optimization problems can be written in the form:

Here, each f i(x) is a smooth convex function, representing a loss (or misfit) corresponding to the ith observation (or measurement). These problems are well studied in the machine learning community [7, 27, 66]. In such applications, F in Eq. (1) corresponds to the empirical risk [65], and the goal of solving Eq. (1) is to obtain a solution with small generalization error, i.e., high predictive accuracy on “unseen” data. We consider Eq. (1) at scale, where the values of n and d are large. In such settings, the mere computation of the first- and second-order statistics (gradient and the Hessian, respectively) of F increases linearly in n. In large-scale settings, operations involving these statistics constitute the main computational bottleneck. In such cases, randomized sub-sampling has been shown to be highly successful in reducing computational and memory costs of the state-of-the-art optimizers to be effectively independent of n.
The most commonly used optimization technique in machine learning is gradient descent and its stochastic version, stochastic gradient descent (SGD). Gradient descent is a simple iterative procedure that takes steps in the direction of the negative gradient of the function, evaluated at the current point, using a step-size that is chosen to satisfy appropriate descent conditions. The stochastic variant of gradient descent estimates the gradient using mini-batches, as opposed to the entire training dataset. Algorithms such as gradient descent, that solely rely on gradient information, are often referred to as first-order methods. In typical problem settings, gradient descent does not offer good convergence results owing to a number of limitations: (1) approximating proper learning rate (a.k.a. step-size), (2) same learning rate schedule is applied to all components of the parameters, when in most cases some components of parameters change frequently compared to other components that change slowly, and (3) minimizing highly non-convex error functions associated with deep learning problems, like neural networks, are known to be dominated by saddle points surrounded by high error plateaus, which make it very hard to escape from these regions for methods like SGD [22]. To address these challenges, several first-order alternatives have been proposed in recent literature such as SGD with Momentum (henceforth referred to as Momentum) [60], Adam [42], Adagrad [24], Adadelta [81], and RMSProp [32, 68]. However the hyper-parameter space for these methods becomes large and the methods become difficult to tune.
Compared with first-order alternatives, second-order methods use additional curvature information in the form of the Hessian matrix. As a result of incorporating such information, in addition to faster convergence rates, second-order methods offer a variety of, rather more subtle, benefits. For example, unlike first-order methods, Newton-type methods have been shown to be highly resilient to increasing problem ill-conditioning [63, 64, 72]. Furthermore, second-order methods typically require fewer parameters (e.g., inexactness tolerance for the subproblem solver and line-search parameters) and are less sensitive to their specific settings [4, 71]. By using curvature information at each iteration, these methods scale the gradient so that it is a more suitable direction to follow. Consequently, they typically require much fewer iterations, as compared to first-order counterparts.
A key challenge in optimization for machine learning problems is the large, often, distributed nature of the training dataset. It may be infeasible to collect the entire training set at a single node and process it serially because of resource constraints (the training set may be too large for a single mode), privacy (data may be constrained to specific locations), or the need for reducing optimization time. In each of these cases, there is a need for optimization methods that are suitably adapted to the parallel and distributed computing environments.
Distributed optimization solvers adopt one of the two strategies: (1) executing each operation in conventional solvers (e.g., SGD or (quasi) Newton) in a distributed environment, e.g., [15, 18, 20, 23, 29, 40, 61, 69, 83]; or (2) executing an ensemble of local optimization procedures that operate on their own data, with a coordinating procedure that harmonizes the models over iterations, e.g., [74, 75]. The trade-offs between these two methods are relatively well understood in the context of existing solvers—namely that the communication overhead of methods in the first class is higher, whereas the convergence rate of the second class of methods is compromised. For this reason, methods in the first class are generally preferred in tightly coupled data-center type environments, whereas methods in the latter class are preferred for wide area deployments.
A method that occupies the middle ground between first- and second-order methods relies on the natural gradient [36, 37, 76], proposed by Shun-chin Amari. This work posits that in fitting probabilistic models, the underlying parametric distributions can be thought of as belonging to a manifold, whose geometry is governed by the Fisher information matrix. Under this hypothesis, scaling the gradient using the Fisher information matrix can result in more effective directions for navigating the manifold of the parametric probability densities. However, in high-dimensional settings, using the exact Fisher matrix can be intractable. To remedy this, Martens et al. [30, 52] proposed a method, called Kronecker Factored Approximated Curvature (KFAC), to approximate the Fisher information matrix and its inverse-vector product, and applied it to applications in neural networks and reinforcement learning. It was shown that KFAC can significantly outperform many of the first-order alternatives.
Deep learning models such as convolution neural networks, residual neural networks, and LSTM [33] have millions of parameters for state-of-the-art network architectures and training such networks is a time-consuming proposition particularly when massively large datasets are used for training. Higher-order solvers that use higher-order statistics of the underlying networks are often prohibitively expensive at scale. In this context more effective solvers which would yield better, if not similar, results in same number of epochs, as well as speedup in processing the mini-batches are critical to the performance of the optimizer. Higher-order solvers like Newton-type methods and KFAC methods have been shown to achieve significantly better results compared to first-order solvers for convex and non-convex optimization problems. GP-GPUs provide powerful platforms for realizing these results in practice. With thousands of compute cores, associated high performance memory architecture, single-instruction-multiple-thread (SIMT), and programming semantics, GPUs are capable of handling large compute intensive tasks with significant performance gains over traditional CPU cores. In fact without hardware accelerators, like GPUs, training state-of-the-art deep learning networks is often not possible in practice.
The rest of this chapter is organized as follows: Section 2 provides an overview of the existing methods for convex and non-convex problems in machine learning. Section 4 provides a discussion of higher-order methods for convex optimization; Sect. 6 extends these results to distributed settings, dealing with massively large datasets. Finally, in Sect. 5 we provide an in-depth analysis of a hybrid method, which uses Fisher information matrix of the objective function, in the context of deep convolution neural networks. These developments have motivated the development of distributed optimizers for non-convex applications, which involve deep networks with millions of model parameters and trained on massively large datasets.
2 Related Research
SGD [6] is the most commonly used first-order method in machine learning, owing to its simplicity and inexpensive per-iteration cost. Iterations in SGD require computation of the gradient on a mini-batch scaled by a predetermined learning schedule and possibly Nesterov-accelerated momentum [55]. It has been argued that high-dimensional non-convex functions such as those arising in deep learning are riddled with undesirable saddle points [2, 21, 22, 39]. For instance, convolutional neural networks, CNNs, display structural symmetry in their parameter space, which leads to an abundance of saddle points [3, 30, 51]. First-order methods, such as SGD, are known to “zig-zag” in high curvature areas and “stagnate” in low curvature regions [3, 22]. In these regions step-size (or learning rate) plays a critical role. Perturbed gradient based methods [28, 39, 45], where random noise is injected in the gradient computation have been proposed and shown to converge to second-order stationary points. However, their computational cost is often worse compared to second-order methods.
One of the primary reasons for the susceptibility of first-order methods to getting trapped in saddle points or nearly flat regions is their reliance on gradient information. Indeed, navigating around saddle points and plateau-like regions can become a challenge for these methods because the gradient is close to zero in most directions [22]. To this end, a number of alternate methods have been proposed in recent times, which using history of gradients aim to approximate curvature information, and hence maintaining the simplicity of SGD. Such methods include Adam [42] and Adagrad [24]. However, such approximations of the Hessian do not always scale the gradient according to the entire curvature information. Hence, these methods suffer from similar deficiencies near saddle points and flat regions. More effective variants of these curvature approximations are those in quasi-Newton methods such as SR1 [58], DFP [58], and BFGS [48, 58], which use rank-1 and rank-2 updates to iteratively approximate the Hessian. Aided by line-search methods, typically satisfying Strong-Wolfe [58] conditions, these methods yield good results compared to first-order methods for convex problems [43]. However, these methods remain as topics of active investigation in the non-convex regime.
Newton-type optimizers have been developed as alternatives to first-order methods. These optimizers can effectively navigate the steep and flat regions of the optimization landscape. By incorporating curvature information in the form of the Hessian matrix, e.g., negative curvature directions, these methods can escape saddle points [2, 21, 70, 73, 77, 79, 80]. Nocedal and Wright [58] propose the use of absolute Hessian matrix, H, to update parameters. Dauphin et al. [21] propose a saddle-free Newton method that optimizes first-order Taylor series approximation of the objective function in a trust-region framework constrained by the distance between successive updates measured by the curvature, |H| of the objective function. In order to make this computationally tractable, the Lanczos-method is used to compute the eigenvectors corresponding to few highest eigenvalues as an approximation to H. Negative curvature descent methods, where the eigenvector corresponding to the least eigenvalue is used to traverse past the parameter manifold around saddle points, have been proposed by Yaodong Yu et al. [80]. Negative curvature can be embedded in gradient descent based methods, which upon encountering saddle points injects random perturbations in the gradient to navigate past the saddle points (perturbed gradient). Neon [2, 73] and Flash [79] methods also use negative curvature direction in a novel form in stochastic methods to navigate past the saddle points. However, such methods need to compute the least eigen-pair for each iteration, which is computationally expensive. To avoid explicitly forming the Hessian matrices, Hessian-free methods [14, 50, 54, 82] have been proposed, which only require Hessian-vector products. Arguably, a highly effective among these methods is the trust-region based method that comes with attractive theoretical guarantees and is relatively easy to implement [16, 70, 71, 77].
Several distributed optimization solvers have been developed recently [15, 18, 20, 23, 29, 40, 61, 69, 83]. Among these, [15, 23, 29, 40] are classified as first-order methods. Although they incur low computational costs, they have higher communication costs due to a large number of messages exchanged per mini-batch and high total iteration counts. Second-order variants [18, 20, 61, 69, 83] are designed to improve convergence rate, as well as to reduce communication costs (because of more accurate descent direction leading to fewer epochs to reach convergence). DANE [20] and the accelerated scheme AIDE [61] use SVRG [41] as the subproblem solver to approximate the Newton direction. These methods are often sensitive to the fine-tuning of SVRG. DiSCO [83] uses distributed preconditioned conjugate gradient (PCG) to approximate the Newton direction. The number of communications across nodes per PCG call is proportional to the number of PCG iterations. In contrast to DiSCO, GIANT [69] executes CG at each node and approximates the Newton direction by averaging the solution from each CG call. Empirical results have shown that GIANT outperforms DANE, AIDE, and DiSCO. The solver of Dunner et al. [25] is shown to outperform GIANT; however, it is restricted to sparse datasets. More recently, DINGO [18] has been developed, which unlike GIANT can be applied to a class of non-convex functions, namely invex [19], which includes convexity as a special sub-class. However, in the absence of invexity, the method can converge to undesirable stationary points.
A popular choice in distributed settings is ADMM [9], which combines dual ascent method and the method of multipliers. ADMM only requires one round of communication per iteration. However, ADMM’s performance is affected by the selection of the penalty parameter [74, 75], as well as the choice of local subproblem solvers.
Lying on the spectrum between first- and second-order methods is Amari’s natural gradient method [36, 37]. This method provided a new direction in the context of high-dimensional optimization of probabilistic models. In this work, Amari showed that natural gradient descent yields Fisher efficient estimate of the parameters; he subsequently applied the method to multi-layer perceptrons for solving blind source detection problems. However, computing Fisher matrix and its inverse in high-dimensional settings is computationally expensive both in terms of memory and computational resources. RMSProp [32, 68] methods use a diagonal approximation of Fisher matrix of the objective function to compute the descent direction. These methods incur little overhead with regard to diagonal approximation but nevertheless fail to make progress relative to SGD in some cases. Martens et al. [30, 51, 52] proposed the KFAC method, which approximates the natural gradient using Kronecker products of smaller matrices formed during back-propagation. KFAC method and its distributed counterpart [3] have been shown to outperform well-tuned SGD in many applications.
For non-convex optimization, we discuss an optimizer that couples the advantages of trust-region and KFAC methods and propose a stochastic optimization framework involving trust region objective computed on a mini-batch, constrained to directions that are aligned with those obtained from KFAC. Major computational tasks in updating the parameters in our method are Hessian-vector products involving the solution of the trust region subproblem, as well as finding the KFAC direction. Our Hessian-vector products can be computed at a similar cost as that of gradient computation using back-propagation. Furthermore, the Fisher matrix approximation and its inverse are only needed once every few mini-batches, thus reducing average iteration cost significantly. Invariance to re-parameterization, as well as immunity to large batch sizes, makes this method a suitable alternative to first-order methods for practitioners.
3 Notation and Assumptions
In the rest of this chapter, vectors are denoted by bold lowercase letters, e.g., v, and matrices or random variables are denoted by bold uppercase letters, e.g., V. For a vector v and a matrix V, \( \left \| \mathbf {v} \right \| \) and \( \left \| \mathbf {V} \right \| \) denote the vector ℓ 2 norm and matrix spectral norm, respectively, while \( \left \| \mathbf {V} \right \|{ }_{F} \) is the matrix Frobenius norm. ∇f(x) and ∇2f(x) are the gradient and the Hessian of f evaluated at x, respectively, and \( \mathbb {I} \) denotes the identity matrix. For two symmetric matrices A and B, A ≽B indicates that A −B is symmetric positive semi-definite. The superscript, e.g., x (k), denotes iteration counter and ln(x) is the natural logarithm of x. \( \mathcal {S} \) denotes a collection of indices from {1, 2, ⋯ , n}, with potentially repeated items and its cardinality is denoted by \( | \mathcal {S} | \).
We assume that each f i is twice-differentiable, smooth, and convex, i.e., for some 0 < K i < ∞ and \( \forall \mathbf {x} \in \mathbb {R}^{ p } \)
We also assume that F is smooth and strongly convex, i.e., 0 < γ ≥ K < ∞ and \( \forall \mathbf {x}\in \mathbb {R}^{ p } \)
Note that assumption (2b) implies uniqueness of the minimizer, x ∗, which is assumed to be attained. The quantity
is known as the condition number of the problem.
For an integer 1 ≤ q ≤ n, let \( \mathcal {Q} \) be the set of indices corresponding to q largest K i’s and define the “sub-sampling” condition number as
where
It is easy to see that for any two integers q and r such that 1 ≤ q ≤ r ≤ n, we have κ ≤ κ r ≤ κ q. Finally, define
4 Convex Optimization Problems
The standard deterministic or full gradient method, which dates back to Cauchy [13], for minimizing (1) uses iterates of the form:
Here, α k is the step-size at iteration k. However, when n ≫ 1, the full gradient method can be inefficient because its iteration cost scales linearly in n. In addition, when p ≫ 1 or when individual functions f i are complicated (e.g., evaluating each f i may require the solution of a partial differential equation), the mere evaluation of the gradient can be computationally prohibitive. Consequently, a stochastic variant of full gradient descent, stochastic gradient descent (SGD) was developed [5, 6, 8, 17, 46, 62]. In such methods a subset \( \mathcal {S} \subset \{1, 2, \cdots , n \} \) is chosen at random and the update is obtained by
When \( |\mathcal {S}| \ll n \) (e.g., \( |\mathcal {S}| = 1 \) for simple SGD), the iteration cost of stochastic gradient methods is independent of n and can be much cheaper than the full gradient methods, making them suitable for modern problems with large n. This class of methods is referred to as first-order methods, since only the gradient information is used at each iteration. By incorporating curvature information (e.g., Hessian) as a form of scaling the gradient, i.e.,
we can significantly improve convergence rate. This class of methods, which take curvature information into account, are known as second-order methods. Compared to first-order methods, they enjoy superior convergence rate in theory, as well as in real application scenarios. This is because of implicit local scaling of components at a given x, which is determined by the local curvature of F. This local curvature determines the condition number of a F at x. Consequently, second-order methods can rescale the gradient direction so that it is a better direction to traverse. Second-order methods have long been used in many machine learning applications [6, 11, 12, 47, 50, 78].
The canonical example of second-order methods, Newton’s method [10, 55, 57], uses a step-size of one and scales the gradient by the inverse of the Hessian, i.e.,
It is well known that for a smooth and strongly convex function F, the Newton direction is always a descent direction and with a suitable step-size, α
k, global convergence is guaranteed. In addition, for cases when F is not strongly convex, Levenberg–Marquardt type regularization [44, 49] of the Hessian can be used to obtain a globally convergent algorithm. Newton’s method exhibits scale invariance, i.e., for some new parameterization \( \tilde { \mathbf {x} } = \mathbf {A} \mathbf {x} \) for invertible matrix A, optimal search direction in the new coordinate system is \( \tilde { \mathbf {p} } = \mathbf {A } \mathbf {p} \), where p is the original optimal search direction. In contrast the search direction produced by gradient descent methods behaves in an opposite fashion  . This scale invariance property is important for effectively optimizing poorly scaled parameters; see [50] for an intuitive explanation of this phenomenon. However, when n, p ≫ 1, the per-iteration cost of this algorithm is significantly higher than that of first-order methods.
. This scale invariance property is important for effectively optimizing poorly scaled parameters; see [50] for an intuitive explanation of this phenomenon. However, when n, p ≫ 1, the per-iteration cost of this algorithm is significantly higher than that of first-order methods.
We now discuss a sub-sampling based method that approximates the gradient and Hessian of the objective function and present analyses of bounds on sample sizes. We then present results for an accelerated sub-sampled Newton’s method over a range of real-world datasets and show that such methods can be highly competitive for machine learning applications.
For the optimization problem Eq. (1), in each iteration, consider selecting two sample sets of indices from {1, 2, …, n}, uniformly at random with or without replacement. Let \(\mathcal {S}_{ \mathbf {g} }\) and \(\mathcal {S}_{\mathbf {H}}\) denote the sample collections, and define g and H as


to be the sub-sampled gradient and Hessian, respectively.
Lemma 1 (Uniform Hessian Sub-sampling)
Given any 0 < 𝜖 H < 1, 0 < δ < 1, \( \mathbf {x} \in \mathbb {R}^{p} \), and assumption (2a) holds, if
then for H(x) defined in (6b), we have
where γ and κ 1 are defined in (2b) and (4), respectively.
Using random matrix concentration inequalities, Roosta et al., [63, 64] derive lower bounds on the sample sizes for gradient and Hessian computation to probabilistically guarantee their utility in sub-sampled Newton-type methods. Depending on κ 1, the sample size \( | \mathcal {S}_{\mathbf {H}} | \) can be smaller than n. In addition, we can always guarantee that the sub-sampled Hessian is uniformly positive definite and, consequently, the direction given by it, indeed, yields a direction of descent. Note that the sample size \( | \mathcal {S}_{\mathbf {H}} | \) here grows only linearly in κ 1 compared to quadratically as in [63, 64].
Lemma 2 (Uniform Gradient Sub-Sampling)
For a given \( \mathbf {x} \in \mathbb {R}^{p} \) , let:
For any 0 < 𝜖 g < 1 and 0 < δ < 1, if
then for g(x) defined in (6a), we have
Lemma 2 assumes that sampling preserves as much first-order information from the full gradient as possible. Note that in each iteration, G(x) is required to guarantee the theoretical bounds on the gradient sample size, \( | \mathcal {S}_{ \mathbf {g} } | \). Fortunately this can be estimated for most of the well-known objective functions [63].
With the bounds in Lemmas 1 and 2 on the size of the samples, \( |\mathcal {S}_{ \mathbf {g} }| \) and \( |\mathcal {S}_{\mathbf {H}}| \), one can, with high probability, ensure that g and H are “suitable” approximations to the full gradient and Hessian, in an algorithmic sense [63, 64]. For each iterate x (k), using the corresponding sub-sampled approximations of the full gradient, g(x (k)), and the full Hessian, H(x (k)), we consider inexact Newton-type iterations of the form
where p k is a search direction satisfying
for some inexactness tolerance 0 < θ < 1 and α k is the largest α ≤ 1 such that:
for some β ∈ (0, 1).
The requirement in Eq. (9c) is often referred to as Armijo-type line-search [58], and (9b) is the θ-relative error approximation condition of the exact solution to the linear system:
which is similar to that arising in classical Newton’s method. Note that in (strictly) convex settings, where the sub-sampled Hessian matrix is symmetric positive definite (SPD), conjugate gradient (CG) with early stopping can be used to obtain an approximate solution to Eq. (10) satisfying Eq. (9b). It has also been shown [63, 64] that to inherit the convergence properties of the rather expensive algorithm that employs the exact solution to Eq. (10), the inexactness tolerance, θ, in Eq. (9b) can be chosen in the order of the inverse of the square root of the problem condition number. As a result, even for ill-conditioned problems, only a relatively moderate tolerance for CG ensures that we maintain convergence properties of the exact update (see also examples in Sect. 4.1). Putting all of these together, we obtain Algorithm 1, which under specific assumptions has been shown [63, 64] to be globally linearly convergentFootnote 1 with problem-independent local convergence rate.Footnote 2
Algorithm 1: Sub-Sampled Newton Method

Theorem 1 (Global Convergence of Algorithm: 1: Inexact Update)
Let Assumptions 2 hold. Also let 0 < θ < 1 be given. For any \( \mathbf {x} ^{ k } \in \mathbb {R}^{p} \), using Algorithm 1 with \( \epsilon _{\mathbf {H}} < \frac {1}{2} \), the “inexact” update direction 6b, and
we have the following with probability 1 − δ:
-
1.
if “STOP,” then
$$\displaystyle \begin{aligned} \left \| \nabla F( \mathbf{x} ^{(k)} ) \right \| < ( 1 + \sigma ) \epsilon_{ \mathbf{g} }, \end{aligned} $$(11) -
2.
otherwise, global convergence results for Hessian sample size hold where
-
(a)
if
$$\displaystyle \begin{aligned} \theta \le \sqrt{\frac{(1 - \epsilon_{ \mathbf{H} })}{4 \tilde{ \kappa }}}, \end{aligned} $$(12)then \( \rho = 4 \alpha _{ k } \beta / 9 \tilde { \kappa } \),
-
(b)
otherwise \( \rho = 4 \alpha _{ k } \beta ( 1 - \theta )( 1 - \epsilon _{\mathbf {H}}) / 9 \tilde { \kappa }^{ 2 } \), with \( \tilde { \kappa } \) defined as in (6). Moreover, for both cases, the step-size is at least
$$\displaystyle \begin{aligned} \alpha_{ k } \ge \frac{( 1 - \theta )( 1 - \beta )( 1 - \epsilon_{\mathbf{H}} )}{ \kappa }, \end{aligned} $$(13)where κ is defined as in (3).
-
(a)
Theorem 1 says that, in order to guarantee a faster convergence rate, the linear system needs to be solved to a “high-enough” accuracy, which is in the order of \( \mathcal {O}(\sqrt {1 / \tilde { \kappa }}) \).
4.1 Experimental Results
We compare our methods to state-of-the-art methods—SGD with momentum (henceforth referred to as Momentum) [67], Adagrad [24], Adadelta [81], Adam [42], and RMSProp [68] as implemented in TensorFlow [1].
Table 1 presents the datasets used, along with the Lipschitz continuity constant of ∇F(x), denoted by L. Recall that, an over-estimate of the condition number of the problem, as defined in [63], can be obtained by (L + λ)∕λ. As it is often done in practice, we first normalize the datasets such that each column of the data matrix \( \mathbf {A} \in \mathbb {R}^{n \times p}\) has Euclidean norm one. This helps with the conditioning of the problem. The resulting dataset is, then, split into training and testing sets, as shown in Table 1.
We present results for two implementations of second-order methods: (a) FullNewton, the classical Newton-CG algorithm [58], which uses the exact gradient and Hessian, and (b) SubsampledNewton-20 \(, |\mathcal {S}_{ \mathbf {g} }| = 0.2 n \), and SubsampledNewton-100 \(, |\mathcal {S}_{ \mathbf {g} }| = n \), are compared against first-order methods using batch sizes 128 and 20%, respectively. These methods use \( |\mathcal {S}_{\mathbf {H}}| = 0.05 n \). CG-tolerance is set to 10−4. Maximum number of CG iterations is 10 for all datasets except Drive Diagnostics and Gisette, for which it is 1000. λ is set to 10−3 and we perform 100 iterations (epochs) for each dataset.
Tables 2 and 3 present the performance results of the proposed Newton-type methods in comparison with first-order methods for batch sizes 128 and 20%, respectively. In each of these tables we show the plots for cumulative time vs. test accuracy in column 1 and cumulative time vs. objective function (training) in column 2. Please note that x-axis in all the plots is in “log-scale.”
4.1.1 Drive Diagnostics Dataset
Row 2 of Tables 2 and 3 shows the results for the Drive Diagnostics dataset for batch sizes 128 and 20% (of the dataset), respectively. We notice that all Newton-type methods achieve lower objective function in the initial few iterations compared to first-order counterparts. When the batch size is larger, we notice that first-order methods take longer to achieve the same objective function value compared to smaller batch sized counterparts. Note that smaller gradient sample size yields similar results (objective function value and generalization error) throughout the simulations.
4.1.2 MNIST and CIFAR-10 Datasets
Rows 3 and 4 in Tables 2 and 3 present plots for MNIST and CIFAR-10 datasets, respectively. Regardless of the batch size, Newton-type methods clearly outperform first-order methods for these two datasets. When larger batch size is used for first-order methods, we notice that these methods take more epochs compared to their smaller batch sized counterparts in reaching same objective function value and generalization error. This behavior is more prominent in CIFAR-10 dataset, which represents a relatively ill-conditioned problem. As a result, in terms of lowering the objective function on CIFAR-10, first-order methods are negatively impacted by problem ill-conditioning, whereas all Newton-type methods show excellent robustness. (Note that, for CIFAR-10, our proposed methods are ≈1000 × faster than first-order alternatives irrespective of the mini-batch size.)
4.1.3 Newsgroups20 Dataset
Plots in row 5 of Tables 2 and 3 represent Newsgroups20 dataset, which is a sparse dataset, and the Hessian is ≈1e6 × 1e6. We clearly notice SubsampledNewton-100 yields superior training accuracy compared to all methods (column 1). However, SubsampledNewton-20 takes more epochs to achieve the same objective function value as its full gradient counterpart, as seen in column 4. This can be attributed to a smaller gradient sample size and the sparse nature of this dataset.
4.2 Sensitivity to Hyper-Parameter Tuning
A major consideration for first-order methods is that of fine-tuning of various underlying hyper-parameters, most notably, the step-size [4, 71]. Indeed, the success of most such methods is strongly determined by many trial-and-error steps to find proper parameter settings. In contrast, second-order optimization methods involve much less parameter tuning and are less sensitive to specific choices of their hyper-parameters [4, 71].
To further highlight these issues, we demonstrate the sensitivity of several first-order methods with respect to their learning rate. Figure 1 shows the results of multiple runs of SGD with Momentum, Adagrad, RMSProp and Adam on Newsgroups20 dataset with several choices of step-size. Each method is run 13 times using step-sizes in the range 10−6∕L to 106∕L, in increments of 10, where L is the Lipschitz constant; see Table 1. It is clear that small step-sizes can result in stagnation, whereas large step-sizes can cause the method to diverge. Only if the step-size is within a particular and often narrow range, which greatly varies across various methods, does one see reasonable performance.

Sensitivity of various first-order methods with respect to the choice of the step-size, i.e., learning rate. It is clear that too small a step-size can lead to slow convergence, while larger step-sizes cause the method to diverge. The range of step-sizes for which some of these methods perform reasonably can be very narrow. This is in contrast with Newton-type, which comes with a priori “natural” step-size, i.e., α = 1, and only occasionally requires the line-search to intervene. (a) SGD with Momentum. (b) Adagrad. (c) RMSProp. (d) Adam. (e) Step-sizes
Remark 1
For some first-order methods, e.g., momentum based, line-search type techniques simply cannot be used. For others, the starting step-size for line-search is, almost always, a priori unknown. This is in sharp contrast with randomized Newton-type methods considered here, which come with a priori “natural” step-size, i.e., α = 1, and furthermore, only occasionally require the line-search to intervene; see [63, 64] for theoretical guarantees in this regard.
5 Non-convex Optimization
With the goal of avoiding being trapped at saddle points, many first-order alternatives such as Adam and Adagrad, and quasi-Newton methods that use low-rank updates, approximate underlying curvature of the objective function. These methods either require a large number of iterations for convergence (first-order alternatives) or are unstable in practice. Natural gradient based methods were proposed in the early 1960s and have been shown to yield efficient parameter estimates for non-convex applications, but were computationally expensive because of high-dimensionality of deep learning problems. Recently, Martens et al. [30, 50,51,52] proposed approximation methods to efficiently estimate Fisher matrix (and associated natural gradient direction) and proved that natural gradient based learning methods can yield superior results for non-convex applications. In this section, we describe a stochastic trust-region based method and validate it using real-world datasets for learning convolution neural networks (CNNs).
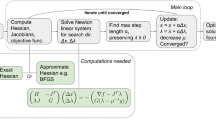
We describe a technique called Fisher Informed Trust-REgion (FITRE ) method, which is inspired by Martens and Grosse [52], Xu et al. [70], and Yao et al. [77] and is formalized in Algorithm 2. At the heart of FITRE lies the stochastic trust-region method using the local quadratic approximation:
We adopt the approach of [77] and use a stochastic estimate of the gradient g t and Hessian H t. [70, 71]. The step-length which is governed by the trust-region radius Δt is automatically adjusted based on the quality of the quadratic approximation and the amount of descent in the objective function. In practice, (14) is approximated by restricting the problem to lower dimensional spaces, e.g., Cauchy condition, which amounts to searching in a one-dimensional space spanned by the gradient. Here, we do the same, however by restricting the subproblem to the space spanned by the direction derived from the Kronecker factorization of the Fisher matrix, or its combination with the gradient.
Our choice is motivated by the following observation: when the objective function involves probabilistic models, as is the case in many deep learning applications, natural gradient direction amounts to the steepest descent direction among all possible directions inside a ball measured by KL-divergence between the underlying parametric probability densities. On the contrary, the (standard) gradient represents the direction of steepest descent among all directions constrained in a ball measured by the Euclidean metric [30], which is less informative than the former, though much easier to compute. To alleviate the computational burden of working with the Fisher information matrix and its inverse, Kronecker-product based approximations [51, 52] have shown success in simultaneously preserving desirable properties of the exact Fisher matrix such as invariance to re-parameterization and resilience to large batch sizes. Indeed, many empirical studies have confirmed that the natural gradient provides an effective descent direction for optimization of neural networks [30, 50,51,52].
5.1 Natural Gradient Computation
We present an overview of the approximations involved in estimating the natural gradient direction. We refer readers to [30, 52] for a detailed discussion on estimation of Fisher information matrix and approximations used in deriving the natural gradient direction.
We define
where \( \mathcal {D} { \boldsymbol \theta } \) is the gradient of the loss function, which is computed using the conventional back-propagation algorithm. Since the network defines a conditional distribution p(y|x, θ), its associated Fisher information matrix is given by

Natural gradient is defined as F −1(θ)∇h(θ). It defines the direction in parameter space that gives the largest change in the objective function per unit change in the model, as measured by the KL-divergence, which is measured between the model output distribution and the true label distribution. In the context of this discussion, for simplicity, we drop the dependence of F and h on θ.
5.2 Natural Gradient Using Kronecker Factored Approximate Curvature Matrix:
We define:

where Ψ l−1 and Γ l denote the second moment matrices of the activation and pre-activation derivatives, respectively.
To invert F̆, we use the fact that: (1) we can invert a block-diagonal matrix by inverting each of the blocks and (2) the Kronecker product satisfies the identity \( \left ( \mathbf {A} \otimes \mathbf {B} \right )^{-1} = {\mathbf {A}}^{-1} \otimes {\mathbf {B}}^{-1} \):
The approximate natural gradient F̆−1∇h can be computed as follows:
A common multiple of the identity matrix is added to F for two reasons: First, as a regularization parameter, which corresponds to a penalty of  . This translates to F + λI to approximate the curvature of the regularized objective function. The second reason is to use it as a damping parameter to account for multiple approximations used to derive F̆, which corresponds to adding γI to the approximate curvature matrix. Therefore, we aim to compute: \( \left [ \breve { \mathbf {F} } + \left ( \lambda + \gamma \right ) \mathbf {I} \right ]^{-1} \nabla h \).
. This translates to F + λI to approximate the curvature of the regularized objective function. The second reason is to use it as a damping parameter to account for multiple approximations used to derive F̆, which corresponds to adding γI to the approximate curvature matrix. Therefore, we aim to compute: \( \left [ \breve { \mathbf {F} } + \left ( \lambda + \gamma \right ) \mathbf {I} \right ]^{-1} \nabla h \).
Since adding the term (λ + γ)I breaks the Kronecker factorization structure, an approximated version is used for computational purposes, which is as follows:
for some π ℓ.
Algorithm 2: FITRE

Algorithm 2 describes a realization of our proposed method in trust-region settings. First, the natural gradient direction, p t is computed and used in determining the step-size using the quadratic approximation of the objective function at p t, whose closed-form solution is \( ( \Delta / \left \Vert \mathbf {H} _{t} \mathbf {p} _{t} + \mathbf {g} _{t} \right \Vert ) \left ( \mathbf {H} _{t} \mathbf {p} _{t} + \mathbf {g} _{t} \right ) \) (note that gradient, g t, can also be used to estimate the step-size and may yield a better descent direction in some cases). Once the step-size, η is determined, ρ t is computed over the same mini-batch to determine the trust-region radius as well as the iterate update. These steps are repeated until desired generalization is achieved. Note that we can compare the efficiency of natural gradient direction with that of the standard gradient and use the appropriate one at each iteration, this is referred to as “KFAC + gradient” in this algorithm.
5.3 Updating KFAC Block Matrices
Block matrices, Ψ l and Γ l, are typically updated using a momentum term to capture the variance in input samples across successive mini-batches. If sample points across the dataset are well correlated, with little variance among the sample points, the inverse block matrices, \( \boldsymbol {\Psi }^{ -1 }_{l} \) and \( \boldsymbol {\Gamma }^{ -1 }_{l} \), need not be updated for every mini-batch. “KFAC Update Frequency,” the frequency with which the inverse block matrices are updated, is typically decided based on the size of the input dataset as well as the correlatio n among the sample points. For boot strapping the optimizer, we either use a larger sample of the dataset, like 5 × the mini-batch size, or use the very first mini-batch itself for computing the block inverses.
5.4 Experimental Results
Tables 4 and 5 present a comparison of our solver, FITRE with other state-of-the-art methods on the ImageNet dataset using VGG11 convolutional neural networks (CNNs) and Tables 6 and 7 show the results for VGG16 CNN. In these tables, we show the generalization errors plotted against wall-clock time and against number of epochs in Columns 3 and 4, respectively, and negative log-likelihood (NLL) using softmax cross-entropy loss function against wall-clock time and against number of epochs in Columns 1 and 2, respectively. KFAC update frequency is set to 5 (mini-batches) for the first row and for the second row, it is set to 25. Plots in Tables 4 and 6 use default initialization, as defined in pyTorch, which is a uniform distribution. Corresponding results using Kaiming initialization [31] (this initialization is based on random Gaussian distribution) are shown in Tables 5 and 7.
The following conclusions can be made from the plots for VGG11 (as shown in Tables 4 and 5) and VGG16 (as shown in Tables 6 and 7). (1) FITRE minimizes the likelihood function to a significantly smaller value compared to well-tuned SGD, and at any given wall-clock instant (FITRE yields better NLL value compared to SGD), (2) Kaiming initialization yields superior generalization errors compared to default initialization of the CNNs, (3) contrary to expectations KFAC update frequency of 25 yields better generalization errors relative to more frequent updates, (4) with increasing network complexity, VGG16 compared to VGG11, FITRE yields significantly better generalization errors compared to SGD, showcasing its superior scaling characteristics compared to SGD, and (5) default initialization is relatively immune to ℓ 2 regularization compared to Kaiming initialization.
For VGG16 network with Kaiming initialization and KFAC update frequency of 25 we observe that to attain 50% test accuracy FITRE (with 1e-6 regularization) takes ≈6500 s compared to ≈20,500 s for SGD (for all regularizations used); a speedup of 3.2 over SGD. Furthermore, when regularization is set to 1e−4 FITRE achieves 53.5% test accuracy, whereas SGD fails to obtain similar accuracy. Similar arguments can be made for the VGG11 network as well. This shows that even though FITRE is computationally more expensive on a per-iteration basis, it yields significantly better results in shorter time compared to SGD. This can be attributed to better descent direction (SGD’s gradient vs. FITRE ’s natural gradient) and an adaptive second-order approximated learning rate computation within the trust-region framework used by the FITRE .
Contrary to expectation, we notice that for VGG11 CNN and default initialization, FITRE’s execution of 50 epochs takes less time compared to SGD for KFAC update frequency 25. FITRE makes two passes over the network (one forward and backward pass for gradient computation and another pass for Hessian-vector product computation used to compute the learning rate in the trust-region framework). One would expect that SGD is at least twice as fast as FITRE on the wall-clock time (on a per-iteration basis). We note that SGD’s pyTorch implementation uses auto-differentiation to compute the gradient of the given network, whereas our implementation of the FITRE is R-operator based (as proposed by Perlmutter et al. [59]). We note that GPU memory management in pyTorch is not efficient [26, 43]. pyTorch allocates and frees memory very often and tends to persist very little information on the device. Even though FITRE makes two passes over the network and computes inverses of smaller matrices at each layer of the network (for computing the inverse of the KFAC block matrices) our implementation persists relevant information on the GPU memory. Coupled with our efficient implementation of the R-operator based Hessian-vector product, we can significantly reduce the computation cost associated with each mini-batch. In addition, our proposed method is a true stochastic online method in which there is no dependence on any part of the dataset other than the current mini-batch during its entire execution, compared to state-of-the-art existing second-order methods [53, 56].
We also note that default initialization is immune to regularization for both networks (VGG11 and VGG16) and for both methods (FITRE and SGD). These two methods show negligible changes in NLL function values (as well as generalization errors) while the FITRE yields superior results compared to SGD for significant part of the execution. At the end of the execution, SGD tends to achieve similar generalization errors compared to FITRE but on minimizing the NLL function FITRE always achieves superior results. However, when using Kaiming initialization, based on random Gaussian distribution, for both the networks, we notice that regularization helps in achieving superior generalization errors for FITRE (with VGG11 network, KFAC update frequency set to 25 and regularization of 1e−6) compared to SGD. But in all cases, FITRE yields superior results when the underlying model does not use any regularization. Compared to FITRE , SGD is relatively invariant to Kaiming initialization as well, as shown in plots in columns 1 and 2 of Tables 5 and 7. Notice that there is very little change in objective function value throughout the simulations.
KFAC update frequency is a hyper-parameter used to control the frequency with which the block matrix inverses are computed at each layer of the network. These block inverses are used to compute the natural gradient direction eventually for each mini-batch. Since these blocks approximate the Fisher matrix of the loss function, they are updated once every few mini-batches. Martens et al. [30, 52] argue that more frequent updates of these block inverses make them too rigid and may lead to overfitting. Using larger values for this update frequency has the effect of a regularizer on the underlying model and helps in avoiding overfitting. As an added advantage, this dependence of the FITRE reduces its computation cost (note also that the computation of block inverses can be delegated to slave processing units, if available, further reducing the computation cost thereby decreasing the time for processing each mini-batch). This is also one of the reasons why our proposed method scales well with increasing network complexity. We note that for VGG16 (with Kaiming initialization), a larger and more complex network compared to VGG11, FITRE yields superior generalization errors as well as minimizing objective function compared to SGD.
6 Distributed Higher-Order Methods
Typically, machine learning problems are associated with massively large datasets during the training phase for learning model parameters. In such scenarios, one must resort to distributed/parallel methods for training models, due to resource constraints on individual nodes. Even in stochastic settings, where only a small part of the dataset is processed at any point in time, time spent in training is a critical parameter contributing to the use of distributed procedures in deep learning. Furthermore, it may be infeasible to accumulate the dataset at a single physical location either due to privacy or resource constraints of the underlying application or system. In such applications communication-efficient optimizers can significantly reduce the training time while optimally using the compute resources. The need for such methods is even more pressing when nodes in distributed systems are connected through high latency networks.
Several distributed optimization techniques have been developed in the recent past to address these concerns. Distributed methods, which are direct adaptations of existing first-order or quasi-Newton methods (i.e., those that parallelize kernel operations such as matrix–vector and dot products), suffer from high communication overhead because of the exchange of model parameters at least once in each iteration among the compute nodes, in addition to inherent communication overhead of the optimizer itself. Ensemble methods in which local optimization procedures compute local solutions (using only locally available data) and a coordinating consensus procedure, which harmonizes local solutions to form a global solution, are more efficient in high latency environments.
In this section, we discuss a communication-efficient method, called Newton-ADMM, based on Alternating Direction Methods of Multipliers (ADMM) framework coupled with sub-sampled Newton-type methods for local optimization as discussed in Sect. 4, along with results using real-world datasets in the context of convex optimization problems of the form (1).
Let \( \mathcal {N} \) denote the number of nodes (compute elements) in the distributed environment. Assume that the input dataset \( \mathcal {D} \) is split among the \( \mathcal {N} \) nodes as \( \mathcal {D} = \mathcal {D}_{1} \cup \mathcal {D}_{2} \ldots \cup \mathcal {D}_{\mathcal {N}} \). Using this notation, (1) can be written as:
where z represents a global variable enforcing consensus among x i’s at all the nodes. In other words, the constraint enforces a consensus among the nodes so that all the local variables, x i, agree with global variable z. This formulation (20) is often referred to as a global consensus problem. ADMM is based on an augmented Lagrangian framework; it solves the global consensus problem by alternating iterations on primal/dual variables. In doing so, it inherits the benefits of decomposability of dual ascent and the superior convergence properties of the method of multipliers. For a detailed discussion on ADMM method, we refer the readers to [9].
ADMM methods introduce a penalty parameter ρ, which is the weight on the measure of disagreement between x i’s and global consensus variable, z. The most common adaptive penalty parameter selection is Residual Balancing [9], which tries to balance the dual norm and residual norm of ADMM. Recent empirical results using Spectral Penalty Selection (SPS) [75], which is based on the estimation of the local curvature of subproblem at each node, yield significant improvement in the efficiency of ADMM. Using the SPS strategy for penalty parameter selection, ADMM iterates can be written as follows:
With ℓ 2 −regularization, i.e., g(x) = λ∥x∥2∕2, (21b) has a closed-form solution given by
where λ is the regularization parameter.
Algorithm 3: ADMM method (outer solver)

Algorithm 3 presents a distributed optimization method incorporating the above formulation of ADMM. Steps 1 and 2 initialize the multipliers, y, and consensus vectors, z, to zeros. In each iteration, Single Node Newton method, Algorithm 1, is executed with local x i, y i, and global z vectors. Upon termination of Algorithm 1 at all nodes, resulting local Newton directions, \( \mathbf {x} _{i}^{k}\), are gathered at the master node, which generates the next iterates for vectors y and z using spectral step-sizes described in [75]. These steps are repeated until convergence.
Remark 2
Note that in each ADMM iteration only one round of communication is required (a “gather” and a “scatter” operation), which can be executed in \(\mathcal {O}(\log (\mathcal {N}))\) time. Further, the application of the GPU-accelerated inexact Newton-CG Algorithm 1 at each node significantly speeds up the local computation per epoch. The combined effect of these algorithmic choices contributes to the high overall efficiency of the proposed Newton-ADMM Algorithm 3 when applied to large datasets.
6.1 ADMM Residuals and Stopping Criteria:
The consensus problem (20) can be solved by iterating ADMM subproblems (21a), (21c), and (21b). To monitor the convergence of ADMM, we can check the norm of primal and dual residuals, r k and d k, which are defined as follows:
where \(\forall i \in \{1,2,\dots ,\mathcal {N}\}\),
As k →∞, z k →z ∗ and \(\forall i, \mathbf {x} _{i}^{k}\rightarrow \mathbf {z} ^*\). Therefore, the norm of primal and dual residuals, ||r k|| and ||d k||, converges to zero. In practice, we do not need the solution to high precision, thus ADMM can be terminated as \(|| \mathbf {r} _{i}^k || \leq \epsilon ^{pri}\) and \(|| \mathbf {d} _{i}^k || \leq \epsilon ^{dual}\). Here, 𝜖 pri and 𝜖 dual can be chosen as:
The choice of absolute tolerance 𝜖 abs depends on the chosen problem and the choice of relative tolerance 𝜖 rel for the stopping criteria is, in practice, set to 10−3 or 10−4.
6.2 Experimental Results
In this section, we evaluate the performance of Newton-ADMM as compared with several state-of-the-art alternatives. In these experiments, pyTorch is used as the software platform and nodes are equipped with NVIDIA P100 GPU accelerators. Table 8 describes the datasets that are used for validation purposes.
6.3 Comparison with Distributed First-Order Methods
While the per-iteration cost of first-order methods (synchronous SGD) is relatively low, they require larger number of iterations, increasing associated communication overhead, and CPU–GPU transactions (because of resource constraints on GPUs, data must be swapped back to the CPU for temporarily releasing global memory on the GPUs, partly because of the pyTorch’s execution model). In this experiment, we demonstrate that these drawbacks of first-order methods are significant, in the context of MNIST, CIFAR-10, HIGGS, and E18 datasets using four workers for Newton-ADMM and synchronous SGD, both with the GPUs enabled and GPUs disabled. The results are shown in Table 9. We note that GPU-accelerated Newton-ADMM method with minimal communication overhead yields significantly better results—over an order of magnitude faster in most cases, when compared to synchronous SGD.
We present the ratio of CPU time to GPU time for Newton-ADMM and SGD in Table 10. We observe that for both Newton-ADMM and SGD, the CPU–GPU time ratio is proportional to the dimension of datasets. For example, on the dataset with the lowest dimension (HIGGS), the CPU–GPU time ratio is the least for both Newton-ADMM and SGD, whereas on the dataset with the highest dimension (E18), the CPU–GPU time ratios are the highest for both Newton-ADMM and SGD. In all cases, the use of GPUs results in highest speedup for Newton-ADMM. The gain in GPU utilization is compromised by large number of CPU–GPU memory transfers for SGD. As a result, SGD shows meaningful GPU acceleration only for the E18 dataset.
Second, we observe that Newton-ADMM has much lower communication cost, compared to SGD. This can be observed from Table 9. In all cases, SGD takes longer than Newton-ADMM with GPUs enabled. This is mainly because SGD requires a large number of gradient communications across nodes. As a result, we observe that Newton-ADMM is 4.9x, 6.3x, 22.6x, and 17.8x times faster than SGD on MNIST, CIFAR-10, HIGGS, and E18 datasets, respectively.
Finally, we observe that Newton-ADMM has superior convergence properties compared to SGD. This is demonstrated in Table 9 for the HIGGS dataset. We observe that Newton-ADMM converges to low objective function values in just a few iterations. On the other hand, the objective function value, even at 100-th epoch for SGD, is still higher than Newton-ADMM.
6.4 Comparison with Distributed Second-Order Methods
We compare Newton-ADMM against DANE [20], AIDE [61], and GIANT [69], which have been shown in recent results to perform well. In each iteration, DANE [20] requires an exact solution of its corresponding subproblem at each node. This constraint is relaxed in an inexact version of DANE, called InexactDANE [61], which uses SVRG [41] to approximately solve the subproblems. Another version of DANE, called Accelerated Inexact DanE (AIDE), uses techniques for accelerating convergence while still using InexactDANE to solve individual subproblems [61]. However, using SVRG to solve subproblems is computationally inefficient due to its double loop formulation, with the outer loop requiring full gradient recalculation and several stochastic gradient calculations in inner loop. Figure 2 shows the comparison between these methods on the MNIST dataset with λ = 10−5. Although InexactDANE and AIDE start at lower objective function values, the average epoch time compared to Newton-ADMM and GIANT is orders of magnitude higher (order of 1000x). For instance, to reach an objective function value less than 0.25 on the MNIST dataset, Newton-ADMM takes only 2.4 s, whereas InexactDANE consumes an hour and a half.

Training objective function and test accuracy comparison over time for Newton-ADMM, GIANT, InexactDANE, and AIDE on MNIST dataset with λ = 10−5. We run both Newton-ADMM and GIANT for 100 epochs. Since the computation times per epoch for InexactDANE and AIDE are high, we only run 10 epochs for these methods. (a) Time vs. Test Accuracy. (b) Time vs. Objective Function (training)
7 Concluding Remarks
Optimization techniques for training machine learning models are of significant current interest. Common machine learning models lead to convex or non-convex optimization problems defined on very large datasets. This necessitates the development of efficient (in terms of optimization time), effective (in terms of generalization error), and parallelizable methods.
Most current machine learning applications rely on stochastic gradient descent to solve the underlying optimization problems. In this chapter, we have discussed the use of higher-order methods that rely on curvature, in addition to gradient information for computing the descent direction. We rely on two important concepts: the use of sampling in dealing with the dense Hessian matrix and the use of natural gradient in non-convex optimization. We show that second-order methods are fast (in terms of iteration counts), can be made efficient (in terms of per-iteration computation cost), result in solutions that are generalizable (as determined by test accuracy), are robust to problem ill-conditioning, and do not require extensive hyper-parameter tuning. Finally, we show how these methods can be parallelized using ADMM and formulated to efficiently use GPUs to deliver accurate and scalable solvers.
Notes
- 1.
It converges linearly to the optimum, starting from any initial guess x (0).
- 2.
If the iterates are close enough to the optimum, it converges with a constant linear rate independent of the problem-related quantities.
References
M. ABADI, P. BARHAM, J. CHEN, Z. CHEN, A. DAVIS, J. DEAN, M. DEVIN, S. GHEMAWAT, G. IRVING, M. ISARD, ET AL., TensorFlow: A system for large-scale machine learning., in OSDI, vol. 16, 2016, pp. 265–283.
Z. ALLEN-ZHU AND Y. LI, Neon2: Finding local minima via first-order oracles, in Advances in Neural Information Processing Systems, 2018, pp. 3720–3730.
J. BA, R. GROSSE, AND J. MARTENS, Distributed second-order optimization using Kronecker-factored approximations, ICLR, (2017).
A. S. BERAHAS, R. BOLLAPRAGADA, AND J. NOCEDAL, An Investigation of Newton-Sketch and Subsampled Newton Methods, arXiv preprint arXiv:1705.06211, (2017).
D. P. BERTSEKAS AND J. N. TSITSIKLIS, Neuro-dynamic Programming, Athena Scientific, 1996.
L. BOTTOU, Large-scale machine learning with stochastic gradient descent, in Proceedings of COMPSTAT’2010, Springer, 2010, pp. 177–186.
L. BOTTOU, F. E. CURTIS, AND J. NOCEDAL, Optimization methods for large-scale machine learning, arXiv preprint arXiv:1606.04838, (2016).
L. BOTTOU AND Y. LECUN, Large scale online learning, Advances in neural information processing systems, 16 (2004), p. 217.
S. BOYD, N. PARIKH, E. CHU, B. PELEATO, J. ECKSTEIN, ET AL., Distributed optimization and statistical learning via the alternating direction method of multipliers, Foundations and Trends® in Machine learning, 3 (2011), pp. 1–122.
S. BOYD AND L. VANDENBERGHE, Convex optimization, Cambridge university press, 2004.
R. H. BYRD, G. M. CHIN, W. NEVEITT, AND J. NOCEDAL, On the use of stochastic Hessian information in optimization methods for machine learning, SIAM Journal on Optimization, 21 (2011), pp. 977–995.
R. H. BYRD, G. M. CHIN, J. NOCEDAL, AND Y. WU, Sample size selection in optimization methods for machine learning, Mathematical programming, 134 (2012), pp. 127–155.
A. CAUCHY, Méthode générale pour la résolution des systemes d’équations simultanées, Comp. Rend. Sci. Paris, 25 (1847), pp. 536–538.
O. CHAPELLE AND D. ERHAN, Improved preconditioner for Hessian free optimization, in In NIPS Workshop on Deep Learning and Unsupervised Feature Learning, 2011.
J. CHEN, X. PAN, R. MONGA, S. BENGIO, AND R. JOZEFOWICZ, Revisiting distributed synchronous SGD, arXiv preprint arXiv:1604.00981, (2016).
A. R. CONN, N. I. GOULD, AND P. L. TOINT, Trust region methods, vol. 1, SIAM, 2000.
A. COTTER, O. SHAMIR, N. SREBRO, AND K. SRIDHARAN, Better mini-batch algorithms via accelerated gradient methods, in Advances in neural information processing systems, 2011, pp. 1647–1655.
R. CRANE AND F. ROOSTA, DINGO: Distributed Newton-Type Method for Gradient-Norm Optimization, in Proceedings of the Advances in Neural Information Processing Systems, 2019. Accepted.
B. CRAVEN, Invex functions and constrained local minima, Bulletin of the Australian Mathematical society, 24 (1981), pp. 357–366.
H. DANESHMAND, A. LUCCHI, AND T. HOFMANN, DynaNewton-Accelerating Newton’s Method for Machine Learning, arXiv preprint arXiv:1605.06561, (2016).
Y. DAUPHIN, H. DE VRIES, AND Y. BENGIO, Equilibrated adaptive learning rates for non-convex optimization, in Advances in Neural Information Processing Systems, 2015, pp. 1504–1512.
Y. N. DAUPHIN, R. PASCANU, C. GULCEHRE, K. CHO, S. GANGULI, AND Y. BENGIO, Identifying and attacking the saddle point problem in high-dimensional non-convex optimization, arXiv:1406.2572v1, (2014).
J. DEAN, G. CORRADO, R. MONGA, K. CHEN, M. DEVIN, M. MAO, A. SENIOR, P. TUCKER, K. YANG, Q. V. LE, ET AL., Large scale distributed deep networks, in Advances in neural information processing systems, 2012, pp. 1223–1231.
J. DUCHI, E. HAZAN, AND Y. SINGER, Adaptive subgradient methods for online learning and stochastic optimization, The Journal of Machine Learning Research, 12 (2011), pp. 2121–2159.
C. DüNNER, A. LUCCHI, M. GARGIANI, A. BIAN, T. HOFMANN, AND M. JAGGI, A distributed second-order algorithm you can trust, arXiv preprint arXiv:1806.07569, (2018).
C.-H. FANG, S. B. KYLASA, F. ROOSTA-KHORASANI, M. W. MAHONEY, AND A. GRAMA, Distributed second-order convex optimization, arXiv preprint arXiv:1807.07132, (2018).
J. FRIEDMAN, T. HASTIE, AND R. TIBSHIRANI, The elements of statistical learning, vol. 1, Springer series in statistics Springer, Berlin, 2001.
R. GE, F. HUANG, C. JIN, AND Y. YUAN, Escaping from saddle points – online stochastic gradient for tensor decomposition, arXiv preprint: arXiv:1503.02101v1, (2015).
P. GOYAL, P. DOLLáR, R. GIRSHICK, P. NOORDHUIS, L. WESOLOWSKI, A. KYROLA, A. TULLOCH, Y. JIA, AND K. HE, Accurate, large minibatch SGD: training ImageNet in 1 hour, arXiv preprint arXiv:1706.02677, (2017).
R. GROSSE AND J. MARTENS, A Kronecker-factored approximate fisher matrix for convolution layers, arXiv:1602.01407v2, (2016).
K. HE, X. ZHANG, S. REN, AND J. SUN, Delving deep into rectifiers: Surpassing human-level performance on ImageNet classification, arXiv preprint arXiv:1502.01852, (2015).
G. HINTON, Neural networks for machine learning, Coursera, video lectures. 307, (2012).
S. HOCHREITER AND J. SCHMIDHUBER, Long short-term memory, Neural Computation, 9 (1997), pp. 1735–1780.
IBM, Deep blue. https://www.ibm.com/blogs/think/2017/05/deep-blue/.
——–, IBM project debater. https://www.research.ibm.com/artificial-intelligence/project-debater/.
S. ICHI AMARI, Natural gradient works efficiently in learning, Neural Computation, 10 (1988).
S. ICHI AMARI, R. KARAKIDA, AND M. OYIZUMI, Fisher information and natural gradient learning of random deep networks, arXiv preprint: 1808.07172v1, (2018).
G. INC., Google AlphaGo. https://ai.google/research/pubs/pub44806.
C. JIN, R. GE, P. NETRAPALLI, S. M. KAKADE, AND M. I. JORDAN, How to escape saddle points efficiently, arXiv preprint arXiv:1703.00887, (2017).
P. H. JIN, Q. YUAN, F. IANDOLA, AND K. KEUTZER, How to scale distributed deep learning?, arXiv preprint arXiv:1611.04581, (2016).
R. JOHNSON AND T. ZHANG, Accelerating stochastic gradient descent using predictive variance reduction, in Advances in Neural Information Processing Systems, 2013, pp. 315–323.
D. KINGMA AND J. BA, Adam: A method for stochastic optimization, arXiv preprint arXiv:1412.6980, (2014).
S. B. KYLASA, F. R. KHORASANI, M. W. MAHONEY, AND A. Y. GRAMA, GPU accelerated sub-sampled newton’s method for convex classification problems, in Proceedings of the 2019 SIAM International Conference on Data Mining, SIAM, ed., SIAM, 2019, pp. 702–710.
K. LEVENBERG, A method for the solution of certain problems in least squares, Quarterly of Applied Mathematics, 2 (1944), pp. 164–168.
K. Y. LEVY, The power of normalization: Faster evasion of saddle points, arXiv preprint arXiv:1611.04831, (2016).
M. LI, T. ZHANG, Y. CHEN, AND A. J. SMOLA, Efficient mini-batch training for stochastic optimization, in Proceedings of the 20th ACM SIGKDD international conference on Knowledge discovery and data mining, ACM, 2014, pp. 661–670.
C.-J. LIN, R. C. WENG, AND S. S. KEERTHI, Trust region Newton method for logistic regression, The Journal of Machine Learning Research, 9 (2008), pp. 627–650.
D. C. LIU AND J. NOCEDAL, On the limited memory BFGS method for large scale optimization, Mathematical programming, 45 (1989), pp. 503–528.
D. W. MARQUARDT, An algorithm for least-squares estimation of nonlinear parameters, Journal of the Society for Industrial & Applied Mathematics, 11 (1963), pp. 431–441.
J. MARTENS, Deep learning via Hessian-free optimization, in Proceedings of the 27th International Conference on Machine Learning (ICML-10), 2010, pp. 735–742.
J. MARTENS, New insights and perspectives on the natural gradient method, arXiv preprint: arXiv:1412.1193v9, (2017).
J. MARTENS AND R. GROSSE, Optimizing neural networks with Kronecker-factored approximate curvature, arXiv:1503.05671v6, (2016).
G. MONTAVON, G. B. ORR, AND K.-R. MULLER, Neural Networks: Tricks of the Trade, Springer, 2nd ed., September 2012.
W. R. MORROW, Hessian-free methods for checking the second-order sufficient conditions in equality-constrained optimization and equilibrium problems, arXiv preprint arXiv:1106.0898, (2011).
Y. NESTEROV, Introductory lectures on convex optimization, vol. 87, Springer Science & Business Media, 2004.
J. NOCEDAL, Updating quasi-Newton matrices with limited storage, Mathematics of computation, 35 (1980), pp. 773–782.
J. NOCEDAL AND S. WRIGHT, Numerical Optimization, New York: Springer, 1999.
J. NOCEDAL AND S. WRIGHT, Numerical optimization, Springer Science & Business Media, 2006.
B. A. PEARLMUTTER, Fast exact multiplication by the hessian, Neural Computation, (1993).
B. T. POLYAK, Some methods of speeding up the convergence of iteration methods, USSR Computational Mathematics and Mathematical Physics, 4 (1964), pp. 1–17.
S. J. REDDI, J. KONEčNỲ, P. RICHTáRIK, B. PóCZóS, AND A. SMOLA, AIDE: Fast and communication efficient distributed optimization, arXiv preprint arXiv:1608.06879, (2016).
H. ROBBINS AND S. MONRO, A stochastic approximation method, The annals of mathematical statistics, (1951), pp. 400–407.
F. ROOSTA-KHORASANI AND M. W. MAHONEY, Sub-sampled Newton methods I: globally convergent algorithms, arXiv preprint arXiv:1601.04737, (2016).
——–, Sub-sampled Newton methods II: Local convergence rates, arXiv preprint arXiv:1601.04738, (2016).
S. SHALEV-SHWARTZ AND S. BEN-DAVID, Understanding machine learning: From theory to algorithms, Cambridge university press, 2014.
S. SRA, S. NOWOZIN, AND S. J. WRIGHT, Optimization for machine learning, MIT Press, 2012.
I. SUTSKEVER, J. MARTENS, G. DAHL, AND G. HINTON, On the importance of initialization and momentum in deep learning, in International conference on machine learning, 2013, pp. 1139–1147.
T. TIELEMAN AND G. HINTON, Lecture 6.5-rmsprop: Divide the gradient by a running average of its recent magnitude, COURSERA: Neural Networks for Machine Learning, 4 (2012).
S. WANG, F. ROOSTA-KHORASANI, P. XU, AND M. W. MAHONEY, GIANT: Globally Improved Approximate Newton Method for Distributed Optimization, arXiv preprint arXiv:1709.03528, (2017).
P. XU, F. ROOSTA-KHORASANI, AND M. W. MAHONEY, Newton-Type Methods for Non-Convex Optimization Under Inexact Hessian Information, arXiv preprint arXiv:1708.07164, (2017).
——–, Second-Order Optimization for Non-Convex Machine Learning: An Empirical Study, arXiv preprint arXiv:1708.07827, (2017).
P. XU, J. YANG, F. ROOSTA-KHORASANI, C. Ré, AND M. W. MAHONEY, Sub-sampled newton methods with non-uniform sampling, in Advances in Neural Information Processing Systems, 2016, pp. 3000–3008.
Y. XU, J. RONG, AND T. YANG, First-order stochastic algorithms for escaping from saddle points in almost linear time., in Advances in Neural Information Processing Systems, 2018.
Z. XU, M. A. FIGUEIREDO, X. YUAN, C. STUDER, AND T. GOLDSTEIN, Adaptive relaxed admm: Convergence theory and practical implementation, in 2017 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), IEEE, 2017, pp. 7234–7243.
Z. XU, G. TAYLOR, H. LI, M. FIGUEIREDO, X. YUAN, AND T. GOLDSTEIN, Adaptive consensus admm for distributed optimization, arXiv preprint arXiv:1706.02869, (2017).
H. H. YANG AND S. ICHI AMARI, The efficiency and the robustness of natural gradient descent learning rule, in Neural Information Processing Systems, 1997, pp. 385–391.
Z. YAO, P. XU, F. ROOSTA-KHORASANI, AND M. W. MAHONEY, Inexact non-convex Newton-type methods, arXiv preprint arXiv:1802.06925, (2018).
J. YU, S. VISHWANATHAN, S. GüNTER, AND N. N. SCHRAUDOLPH, A quasi-Newton approach to nonsmooth convex optimization problems in machine learning, The Journal of Machine Learning Research, 11 (2010), pp. 1145–1200.
Y. YU, P. XU, AND Q. GU, Third-order smoothness helps: Even faster stochastic optimization algorithms for finding local minima, arXiv:1712.06585v1, (2017).
Y. YU, D. ZOU, AND Q. GU, Saving gradient and negative curvature computations: Finding local minima more efficiently, arXiv:1712.03950v1, (2017).
M. D. ZEILER, Adadelta: an adaptive learning rate method, arXiv preprint arXiv:1212.5701, (2012).
H. ZHANG, C. XIONG, J. BRADBURY, AND R. SOCHER, Block-diagonal Hessian-free optimization for training neural networks, arXiv preprint: arXiv:1712.07296v1, (2017).
Y. ZHANG AND X. LIN, DiSCO: Distributed optimization for self-concordant empirical loss, in International conference on machine learning, 2015, pp. 362–370.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kylasa, S., Fang, CH., Roosta, F., Grama, A. (2020). Parallel Optimization Techniques for Machine Learning. In: Grama, A., Sameh, A. (eds) Parallel Algorithms in Computational Science and Engineering. Modeling and Simulation in Science, Engineering and Technology. Birkhäuser, Cham. https://doi.org/10.1007/978-3-030-43736-7_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-43736-7_13
Published:
Publisher Name: Birkhäuser, Cham
Print ISBN: 978-3-030-43735-0
Online ISBN: 978-3-030-43736-7
eBook Packages: Mathematics and StatisticsMathematics and Statistics (R0)