
Abstract
Cancer metabolism is a well-known target of cancer therapeutics. Classically, cancer metabolism has been studied in terms of the dependence of cancer cells on crucial metabolites, such as glucose and glutamine. But, the accumulating data show that iron metabolism in tumor microenvironment is also an important factor in preserving the survival of cancer cells. Cancer cells have a distinct phenotype of iron metabolism, which secures the much-needed iron for these metabolically active cells. In order to use this iron efficiently, cancer cells need to increase their iron supply and decrease iron loss. As recent research suggests, this is not only done by modifying the expression of iron-related proteins in cancer cells, but also by interaction of cancer cells with other cells from the tumor milieu. Tumor microenvironment is a dynamic environment characterized with intricate relationship between cancer cells, tumor-associated macrophages, fibroblasts, and other cells. Some of the mechanistic aspects of this relationship have been elucidated, while others are yet to be identified. In any case, identifying the details of the iron phenotype of the cells in tumor microenvironment presents with a new therapeutic opportunity to treat this deadly disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Iron
- Tumor microenvironment
- Transferrin receptor 1
- Divalent metal transporter 1
- Vacuolar ATPase
- Hepcidin
- Ferroportin
- Tumor-associated macrophages
- Cancer-associated fibroblasts
- Cancer stem cells
- Interleukin 6
- Iron chelation
- Ferritinophagy
- Ferroptosis
- Nanomedicine
Tumor cells are highly adaptive cells. They need this plasticity considering the unfavorable conditions that they are exposed to (lactic acidosis, hypoxia, and lack of sufficient nutrients) [1]. The adaptability of cancer cells is partly genetic in nature, but it also depends on the cellular origin of the tumor and its microenvironment [1]. One of the aspects of this adaptability includes specific changes in tumor metabolism; it is well known that cancer cells are able to increase the uptake of glucose and glutamine for their metabolic needs [1]. But, there are many other nutrients that are crucial for cancer cell survival. An increasing amount of research suggests that iron is one of the most fundamental metals needed for cancer proliferation. This finding is based on a wide array of changes found in the iron metabolism of different cancers [2]. What is more, recent evidence suggests that rewiring of the iron metabolism does not include only tumor cells but other cells in the tumor microenvironment, such as cancer stem cells, neighboring normal cells, stromal cells, leukocytes, and even senescent cells [3, 4]. Also, it seems that tumor cells have the ability to affect the iron turnover in tumor microenvironment in order to facilitate their proliferation [2].
The importance of iron for cancer cells should not come as a surprise. It is one of the most abundant metals in human body, while its favorable chemical properties make it an ideal metal to be used by living cells as a transporter of electrons in crucial biochemical reactions [5]. This is why iron is involved in some of the most important biochemical reactions in the cellular milieu, namely, DNA synthesis, cellular respiration, and oxygen metabolism [2]. Experimental evidence shows that manipulating tumor iron supply can have dramatic effects in tumor’s ability to proliferate [6,7,8,9].
Iron dysmetabolism in tumor microenvironment is characterized with differential regulation of cellular iron import and export proteins, changes in the activity of intracellular proteins involved in the regulation of iron metabolism, and an overall propensity towards increased accumulation of cellular iron. These changes seem to be independent from the homeostasis of systemic iron metabolism, which has implications for the treatment of cancer via iron therapeutics.
3.1 Iron homeostasis in physiological conditions
Systemic iron metabolism is mostly controlled through iron absorption in intestines due to lack of a specific excretory mechanism. In duodenum, iron is first reduced and then enters into intestinal cells via divalent metal transporter 1 (DMT1). Its export is mediated via ferroportin (FPN), which is the main target protein of hepcidin, known as the major regulator of systemic iron metabolism [2]. Hepcidin is produced in liver hepatocytes through a series of intricate pathways influenced by iron levels, hypoxia, and inflammation [10]. After being released from intestinal cells, iron is oxidized and is bound to transferrin [2]. This complex is named transferrin-bound iron (TBI). TBI travels in plasma until it binds to its target protein, which is transferrin receptor 1 (TFR1), found in different cells such as macrophages, erythrocyte precursors, and hepatocytes [2]. This binding induces endocytosis and creation of an endosome, in which iron is released from its complex with transferrin and TFR1 via proton pumps such as V-ATPase, and then reduced via metalloreductases [2, 11, 12]. Finally, iron is released in cytoplasm through DMT1 [2]. The fate of the released iron is manifold: it can be transported to ferritin, which serves as a cellular iron depot, it can be part of free cellular iron (also called labile iron pool), or it can be exported out of the cell via FPN [2]. The main regulators of intracellular iron metabolism are iron-responsive element-binding proteins (IRPs). Their activity is influenced by cellular iron availability, which means that in iron-replete conditions, IRPs increase iron import and reduce iron export and vice versa [2].
As it can be seen, most of the time iron is bound to different proteins in our body. Iron sequestration via proteins not only serves as a protective measure against the production of reactive oxygen species, but also prevents the “hijacking” of iron from microorganisms [5]. In physiological conditions, most of the iron is in TBI form, but in some cases (such as in iron overload) a significant amount of iron is in the form of non-TBI (NTBI). This type of iron enters the cells via DMT1 and zinc transporter proteins (Zip) [13].
3.2 Iron Metabolism in Cancer Cells
Iron import in cancer cells
Iron import in cancer cells is directed by different proteins expressed in cell membranes. Most of the TBI enters through TFR1, while other means of iron entry including NTBI are realized through proteins such as DMT1, Zip proteins, and probably through other as yet unidentified ways [13, 14]. In many cancers, TFR1 is upregulated, which helps cancer cells to increase their iron supply. In some tumors, there is evidence that TFR1 is not only subject of changes in its expression, but also it is differentially distributed inside cells after endocytosis compared to normal cells [15,16,17,18]. Nevertheless, blocking TFR1 action has been shown to be an effective way to suppress tumor growth [15, 16, 18, 19]. On the other hand, blocking DMT1 in colorectal cancer has been shown to have similar effects in terms of cancer progression [20]. This occurs due to DMT1 serving as the main gateway for iron entry in epithelial cells of intestines. But the release of iron inside cancer cells is an important process as well, since in many tumors one finds overexpression of metalloreductases such as six-transmembrane epithelial antigen of prostate (STEAP) proteins. The expression of STEAP proteins is in direct correlation with tumor proliferation [21, 22]. Other proteins involved in intracellular iron release from endosomes also seem to play a role in cancer cells, such as vacuolar ATPase (V-ATPase). The link between V-ATPase inhibition and cellular iron metabolism has been observed in different cancer cells [23]. An unexplained issue remains the role of cellular iron chaperons called poly(C)-binding proteins (PCBPs), which have been shown to be upregulated in different cancers [24,25,26], though the link between PCBPs and iron dyshomeostasis in cancer has not been elucidated. What is more, PCPBs are known to have different physiological functions independent from iron metabolism [24,25,26].
Cellular iron regulators in cancer cells
IRPs are known as major regulators of cellular iron homeostasis [2]. They bind to iron-responsive elements present in different mRNAs of iron-related proteins [2]. In this way, IRPs influence the translation of FPN, TFR1, and DMT1 depending on the cellular iron availability [2].
IRPs have been studied in cancer cells as well, especially IRP2. It has been found upregulated in different cancers, where it is involved in tumor proliferation [27,28,29]. In some tumors, IRP2 expression has been observed as an early sign of iron dyshomeostasis, which correlates with tumor stage [15]. Also, IRP suppression has been used as a therapeutic modality to reduce tumor growth in cultured mediums [15, 27,28,29].
Iron export in cancer cells
FPN and hepcidin are two major players controlling iron export in human cells. FPN is controlled translationally by IRPs, but also by transcriptional factors, and postranslationally through degradation via hepcidin binding [30]. Since FPN is the only protein involved in cellular iron export, it has been the subject of study in different cancer cells. In majority of cancers, FPN is downregulated or does not realize its function properly as a cellular iron exporter [31,32,33,34] (Table 3.1). Furthermore, FPN overactivation has been shown to reduce tumor growth and even metastasis [31,32,33,34]. Similarly to FPN, hepcidin expression has also been related to tumor proliferation. It has been frequently found to be overexpressed in cancer, which is mostly related with its ability to suppress iron export via FPN endocytosis [10]. In some cancers, aggressive behavior of tumors is related not only to local hepcidin expression but also to plasma (systemic) levels of hepcidin [10].
Role of mitochondria in cancer
There is ample evidence through which mitochondria have been “exposed” as promoters of tumor progression [35]. These cellular powerhouses are attractive target organelles in cancer therapy. Mitochondria have their own set of iron-related proteins which regulate the iron flux in mitochondria [36]. Mitochondrial iron homeostasis is important for the proper functioning of mitochondria due to the dependence of crucial mitochondrial enzymes on using iron as a co-factor [36]. Surprisingly, there is not much research relating mitochondrial iron metabolism and cancer. The rationale exists, since some relatively specific mitochondrial iron chelators can inhibit tumor growth by disturbing mitochondrial functional parameters [37]. Some recent data even suggest an impressive potency of specific mitochondrial iron chelators compared to their classical nonspecific counterparts [38]. Still, the exact role of mitochondrial iron homeostasis in cancer awaits confirmation by future studies.
Cause of iron dyshomeostasis in cancer cells
Tumors are heterogenous diseases characterized with a complex pathophysiology and a dynamic microenvironment. Changes in iron metabolism are just one piece of this puzzle, but which require further examination. Culprits for changes in iron metabolism in cancer are heterogeneous in nature. For example, mutations in oncogenes such as C-myc and BRAF are responsible for IRP2 upregulation, while adenoviral oncogene E1A is responsible for increasing labile iron pool [29, 39, 40]. Interestingly, the transcription factor p53 has opposite effects on labile iron pool [8]. p53 gene is known as a tumor suppressor gene which undergoes loss-of-function mutation in many cancers [41]. These observations suggest that the asymmetry between tumor oncogenes and suppressors found in different tumors might be an important instigator of iron dyshomeostasis in cancer cells.
Other molecules are also responsible for changes of iron metabolism in cancer. One such factor is epidermal growth factor receptor (EGFR), which is a growth factor known for its oncogenic properties. It increases cellular iron pool by promoting TFR1 activity [18]. On the other hand, reduction in cellular iron export in cancer has been shown to be mediated via sclerostin domain containing 1 (SOSTDC1), wingless and int (Wnt) pathway, bone morphogenetic protein (BMP) pathway, inflammatory cytokines such as interleukin 6 (IL6), and transcriptional factor zinc-figure protein 217 (ZNF217) [10, 42]. Another level of complexity involved in the control of iron metabolism in cancer cells is realized through epigenetic mechanisms. Epigenetic mechanisms have been shown to exert control in the activity of different iron-related proteins in cancer. In any case, the extent of epigenetic control of iron metabolism in cancer is yet to be revealed [13].
It has to be mentioned that although iron dyshomeostasis is a product of an overall cancer pathophysiology, iron may contribute directly to the initiation of the disease. Iron can cause damage to DNA structure through increases in production of reactive oxygen species (ROS) [7]. This is the reason why repeated intraperitoneal or intramuscular injections with concentrated iron cause cancer in rodents [6, 7, 43]. Many epidemiological data have examined the role of iron intake and cancer prevalence [2]. Results are contradictory and not uniform across different types of cancers. One must not forget that our cells have developed homeostatic control mechanisms to protect themselves from global changes in iron metabolism. This means that only in extreme cases of global iron load our cells will not be able to control their iron depots, in which case cancer rates will increase progressively, especially in older adults [44].
Role of tumor associated leukocytes in cancer
Tumor microenvironment is abundant with different types of leukocytes, of which macrophages dominate. Their migration into tumor environment is caused by different chemical substances such as interleukins, angiogenic factors like vascular endothelial growth factor A (VEGF-A), or chemokines like C-C motif ligand 2 (CCL2) [45]. Activated tumor-associated macrophages or TAMs are responsible for tumor growth, immunosuppression, and angiogenesis [45]. Furthermore, the density of TAMs is correlated with poor prognosis and high tumor grade [46]. These TAMs are so-called M2 phenotype macrophages compared to M1 phenotype macrophages found in the early stages of cancer, which generally have antitumor properties [45]. M1 macrophages show iron-accumulating properties, while M2 macrophages show iron-releasing properties [47]. This means that iron release from TAM M2 macrophages can potentially exacerbate iron dyshomeostasis in cancer cells. There are different strategies through which M2 macrophages achieve this effect; first by exporting high amounts of iron through FPN and second by increasing the production of iron-related proteins (Fig. 3.1). The candidate for the latter is lipocalin 2 (Lcn2). It is frequently upregulated in cancer locally but also in different body fluids [48]. Data from tumor microenvironment studies suggest that most of Lcn2 comes from tumor stroma (that is, macrophages) and not from tumor cells [49]. In cultured medium, macrophages do not change their iron-releasing phenotype even when FPN is blocked [49]. But, iron-releasing phenotype of tumor macrophages is reversed when Lcn2 is blocked [49]. Furthermore, Lcn2 suppression reverses tumor proliferation in this setting [49]. It is believed that Lcn2 production in M2 macrophages occurs from their interaction with apoptotic tumor cells via sphingosine-1-phosphate/signal transducer and activator of transcription 3 (S1P-STAT3) pathway [50].

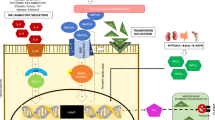
Dysregulation of iron metabolism in tumor microenvironment. Normal cells express different iron-related proteins in the membrane, cytoplasm, and mitochondria as seen in the upper part of this figure. Iron bound with transferrin enters via TFR1 in cells. This is accompanied with endocytosis and creation of an endosome. Iron is released from endosome via DMT1 after being reduced by STEAP metalloreductases. In cytoplasm, iron is bound with PCBs, which transport iron to ferritin, FPN, or transport it to enrich LIP. Some of this iron enters mitochondria via mitochondrial iron import proteins such as MCU and Mfn2. Mitochondria have their own ferritin and their iron export proteins (such as ABCB 8). The major exporter of iron out of cells is FPN which is under negative regulatory control from hepcidin. Many of these proteins are differentially expressed in tumor microenvironment. For example, the iron import protein TFR1 is often upregulated in cancer cells, as well as STEAP proteins. Also the major regulator of cellular iron metabolism (IRP2) is overexpressed in cancer cells, which causes increased iron entry into cells. IRP2 is under regulatory control of different genes involved in tumorigenesis (such as C-myc or BRAF). Another important protein that is overexpressed in cancer is hepcidin. By increasing hepcidin production, cancer cells reduce iron export. Wnt and BMPs are some of the pathways involved in increased hepcidin secretion in cancer cells. But, hepcidin production in cancer cells is increased via IL6 produced by CAFs as well. Another strategy used by cancer cells to secure more iron for their metabolic needs is by using the iron-releasing phenotype of M2 macrophages either through increased iron supply from overactive FPN in M2 macrophages or through increased production of Lcn2. Another important player in the iron metabolism of the tumor microenvironment is CSC. They have voracious appetite for iron, which is secured through increase in TFR1 expression, but also through increased production of transferrin which serves as an iron scavenger. CSCs are important cells in this environment because they secure the replenishment of cancer cells, promote metastasis, and are responsible for resistance to chemotherapy. Abbreviations: ABCB8 ATP-binding cassette subfamily B member 8, BMP7 bone morphogenetic protein 7, CAF cancer-associated fibroblast, CSC cancer stem cell, DMT1 divalent metal transporter 1, Fer ferritin, FPN ferroportin, Hepc hepcidin, IL6 interleukin 6, IRP2 iron-responsive element-binding protein, LIP labile iron pool, Lcn2 lipocalin 2, Lcn2R Lcn2 receptor, Mfn2 mitoferrin 2, MCU mitochondrial calcium uniporter, PCB poly(C)-binding protein 2, STEAP six-transmembrane epithelial antigen of prostate, TFR1 transferrin receptor 1, Wnt wingless and int pathway
Iron metabolism in M2 macrophages is a potential therapeutic possibility in suppressing tumor growth. Iron chelators have already been used in in vitro studies to prevent iron release from M2 macrophages by sequestering iron and by reversing their polarization from an iron-releasing phenotype to an iron-sequestering one [51]. In recent ex vivo experiments, which reflect more faithfully the in vivo tumor microenvironment, iron chelation was confirmed as a therapeutic tool which can reverse the iron-releasing phenotype of M2 macrophages [52]. In addition, iron chelation did not increase the population of M2 macrophages compared to chemotherapeutic agents, which means that iron chelation can be used with traditional chemotherapy to exert additional anticancer effects [52]. Finally, level of iron load in M2 macrophages is an important feature of the tumor microenvironment, since it is correlated with the success of iron chelation therapy in cancer [53]. In conclusion, the iron flux of M2 macrophages is a crucial therapeutic knot, further corroborated by the findings that show TAMs are some of the most abundant cells in tumor microenvironment, which can affect the metabolism of cancer cells.
Iron and cancer stem cells (CSCs)
CSCs are a small fraction of cells in the tumor milieu, but their renewal ability makes them ideal cells to promote malignancy, metastasis, tumor recurrence, and resistance to chemotherapy in different cancers [54]. These highly active cells need to “fuel” their metabolism by securing the necessary nutrients. In recent years, studies have revealed that iron is important for the proper survival of CSCs [54]. This means that similar to cancer cells, CSCs interact dynamically with their microenvironment. In this respect, CSCs have evolved mechanisms to secure the proper amount of iron for their metabolic needs. Indeed, studies have shown that CSCs are much more efficient in increasing their iron uptake compared to non-CSCs and even compared to macrophages [55,56,57]. Strategies used by CSCs to secure the much-needed iron are manifold: they are able to secrete high amounts of TF (through which they scavenge free iron), they increase TFR expression (to increase iron import), increase ferritin depots, downregulate FPN (to decrease iron export) [55,56,57]. The dependence of CSCs on iron is observed when these cells are exposed to iron chelation therapy, but also when their ferritin depots are depleted, or when iron is forced out of cells; in these cases, proliferation rate and metastatic potential of CSCs drop significantly [55,56,57]. Increased intracellular iron in CSCs is linked with an overactive IL6/STAT3 pathway which promotes the invasive potential of CSCs [55, 57]. It is interesting to notice that iron chelation inhibits the expression of stemness markers in CSCs, which effect has not been observed with standard chemotherapy [58]. This is an important finding with therapeutic implications considering that stemness markers are related directly with malignant potential of CSCs [54]. Another interesting phenomenon has been observed in CSCs of breast and prostate cancer lines. IRP2 in these cells seems to be under the regulatory feedback of cellular iron depots, which is not the case with cancer cells [59]. This finding needs to be confirmed by other studies in order to evaluate its importance in tumor pathophysiology.
The activity of CSCs in cancer is one part of the complex picture that unfolds during one of the most important processes which occurs in cancer, that is, epithelial-to-mesenchymal (EMT) transition. EMT is a process which helps tumor mass to expand, proliferate, and metastasize, but also helps tumors to resist chemotherapeutic treatment [54]. Besides being responsible for transformation of CSCs, EMT enables phenotypic transformations of tumor cells, which increases their mobility and eventual metastasis [54]. This transformation is realized by making tumor cells more mesenchymal-like cells. It is interesting that many biochemical pathways involved in regulation of EMT are also involved in regulation of cellular iron metabolism such as transforming growth factor beta (TGF-β) and Wnt pathway [60, 61]. One of the most known inhibitors of EMT is N-myc downstream-regulated gene 1 (NDRG1) [60]. Its activity is dependent on iron availability, which has been used in experimental setting to reverse EMT via iron chelation [60]. NDRG1 is able to suppress Wnt pathway, which is known to be hyperactive in different tumors [62]. Wnt pathway inhibition has been shown to reduce cancer cell iron pool by reducing hepcidin production [10]. These results are in line with the observation that suppression of FPN (main target of hepcidin) via activation of TGF-β enhances markers of EMT [63].
Iron and cancer-associated fibroblasts (CAFs) in tumor microenvironment
The complex tumor environment is exposed to different modes of signaling from different cells. Along with macrophages, fibroblasts are some of the most abundant cells in tumor milieu [64]. Their heterogeneous phenotype makes them a difficult cellular target for cancer therapy. Our current understanding of CAFs reveals at least three distinct populations: one group promotes tumor growth, another one retards tumor growth, while other populations are neutral in terms of their effect on tumor growth [64]. Tumor-promoting CAFs have multiple roles in promoting tumor growth. CAFs produce different chemical signals through which they affect the stemness of CSCs, transformation of TAMs into M2 macrophages, migration of tumor cells, extracellular matrix remodeling, formation of new blood vessels, and immunosuppression [64]. CAFs which express tumor-promoting markers are present in high numbers in tumor with high density of stroma and in invasive margins of the tumor [64, 65]. Furthermore, CAFs can “donate” their mitochondria for metabolic needs of cancer cells, while their secretome can promote cancer cell metabolism [64, 66]. CAFs have been shown to promote tumor growth via pathways which are known to be involved in iron dyshomeostasis in tumor microenvironment. Although the importance of iron metabolism in the activity of CAFs and tumor growth has just recently been studied, it has unveiled some interesting results. For example, in breast cancer, IL6 produced by CAFs has been shown to increase hepcidin production in cancer cells, which can be reduced significantly with anti-IL6 antibodies [67]. The elegant study done by Blanchette-Farra et al. shows that hepcidin production is affected by the spatial organization of the tumor cell milieu, which has been shown by comparing 2D with 3D models of cultured cells [67].
Senescent cells in tumor microenvironment
Cellular senescence is a process through which cells suffer cell cycle arrest which cannot be reversed with mitogens [68]. The state of senescence can be induced by mutations, chemotherapy, and oxidative stress [68]. These cells are believed to contribute to the tumor microenvironment by their senescence-associated secretory phenotype (SASP) and by their ability for neoplastic transformation during chronic senescence [68]. But, senescent cells exhibit a continuum of phenotypes, from the initial tumor-suppressing one to a tumor-promoting one, which should be taken into account when trying to eliminate senescent cells as part of an antitumor strategy [68]. This means that a two-hit strategy might be the best option for dealing with these cells: the first hit turns cancer cells into senescent cells, and the second hit is used to destroy the senescent cells so they would not be able to undergo neoplastic retransformation. In fact, low dose curcumin in combination with knockdown of specific long noncoding RNAs has already been used to induce senescence and then apoptosis [69]. This is important since curcumin is known for its iron-chelating properties and its effect on iron metabolism [70]. Iron dyshomeostasis has been observed recently in senescent cells as well. It is characterized by increased cellular iron flux [4]. This occurs in response to impaired ferritinophagy, which disrupts the ability of senescent cells to detect true levels of cellular iron [4]. Iron dyshomeostasis in senescent cells can be reversed by promoting ferritinophagy [4]. On the other hand, FPN overactivation in prostate cancer is accompanied with SASP perturbations [32]. Still, the role of iron metabolism in senescent cells during cancer has not been elucidated properly and more studies are needed to address this issue.
Pericytes, new players in tumor microenvironment?
Pericytes have been traditionally seen as redundant cells in the tumor environment. But, advances in functional analysis at the cellular level have revealed that pericytes have important features which can be used in cancer therapeutics. This stems from the observation that pericytes do not just act as stabilizers of blood vessels, but also function as stem cells and have immune functions and other functions as well [71]. But pericytes are a heterogeneous group of cells, which means that targeting pericytes in tumor microenvironment is a challenging endeavor [71]. Birbrair et al. have examined the behavior of different pericytes during tumor angiogenesis. They have found that a specific subset of pericytes (type 2 pericytes) is responsible for tumor angiogenesis [72]. Brain pericytes show expression of iron-related proteins (hepcidin, ceruloplasmin, hephaestin) and have been suggested as mediators of iron transport from endothelial cells into brain parenchyma [73, 74]. On the other hand, administration of ferric ammonium citrate (FAC) is known to reduce the extent of angiogenesis in tumor microenvironment, while iron deficiency promotes tumor angiogenesis [75, 76]. Interestingly, administration of FAC does not have an effect in pericyte numbers in tumor microenvironment [75]. This is important since stabile number of pericytes associated with endothelial cells reduces leakage and promotes an organized architecture of blood vessels [77]. The disassociation of pericytes from endothelial cells is a feature of tumor microenvironment. But, what happens with disassociated pericytes is a matter of debate. Some tumor pericytes are able to interact with tumor cells to induce a state of immunosuppression [78], while others can transform to CAFs [79]. Still, the way in which iron metabolism in tumor microenvironment affects the activity of tumor pericytes has still not been examined.
Ferritinophagy, ferroptosis and their role in tumor microenvironment
Ferroptosis is a recently observed form of programmed cellular death with distinct features. It does not involve chromatin margination or caspase activation as seen in apoptosis or non-apoptotic cell death [71]. In order for ferroptosis to occur, there are some conditions which should be met; first, there needs to be a substrate for peroxidation (phospholipids), then iron is needed as the main instigator of peroxidation, and finally these changes should be accompanied with the inability of the cell to eliminate the products of lipid peroxidation [71]. Although we still do not know the role of ferroptosis in human physiology, its importance has been observed in different pathophysiological states, including cancer. The rationale to study ferroptosis in cancer is based on the ability of cancer cells to resist classical apoptosis [71]. Also, different cells of the tumor microenvironment seem to be more sensitive to ferroptosis than normal cells [72]. This includes mesenchymal type cells, detached cells, and CSCs. But, cancer cells need activity; therefore, using iron to induce ferroptosis might be a double-edged sword in fighting cancer. For example, downregulation of TFR reduces the extent of ferroptosis in cancer cells [73]. Similarly, knockdown of FPN accelerates ferroptosis [74]. Still, there are ways to circumvent this scenario. Redistributing iron from cytosol to lysosomes is one possible approach. It causes peroxidation of liposomal membranes and resultant ferroptosis [75]. Another approach would be to induce the process of ferritinophagy in cancer cells, which is accompanied with increased levels of ROS inside the cells. This can induce ferroptosis, as it has been observed in breast cancer cells [76]. It is interesting to note that iron chelators can be used to reverse the process of EMT through induction of ferritinophagy [77].
3.3 Iron therapeutics in cancer
Iron therapy is not a recent idea in the management of cancer. Considering that excessive iron deficiency/overload is detrimental for cancer cell survival, it is expected that stimulating these conditions can affect tumor growth, especially when one takes into account that iron dysmetabolism is a feature of different cells in the tumor microenvironment.
Iron chelators have the ability to bind iron and cause cellular iron deficiency [2]. These agents are able to have synergistic effects when combined with traditional chemotherapy [78]. Thus, they have been used in experimental studies to destroy cancer cells, but they have not shown to be an effective therapeutic strategy for patients with cancer [2, 44]. This probably occurs due to their inefficient accumulation in cancer cells, their ability to cause reactive intracellular iron flux, and also because they can have side effects in normal tissue. Other options used to treat cancer by manipulating iron metabolism of cancer cells involve increasing/decreasing the activity of iron-related proteins. Genetic studies in cancer patients show that low TFR expression and high FPN/low hepcidin combination are markers of favorable outcomes in cancer [31, 79, 80]. Therefore, blocking TFR, increasing FPN activity, and downregulating hepcidin can be used as a useful strategy to prevent tumor growth and metastasis [2, 10] (Table 3.2). Other proteins involved in iron import, iron release, or control of cellular iron metabolism in tumor cells are also potential targets in cancer therapy. Up till now, these targets have been studied mostly in in vitro conditions by using antibodies, small interfering RNAs (siRNAs), or naturally occurring compounds which can modulate iron-related proteins. Hepcidin therapy is being used in clinical trials in the treatment of anemia of cancer, but there is no trial which has considered the effects of hepcidin therapy in tumor growth [10]. The problem with this approach is that blocking systemic hepcidin does not necessary translate into a clinical success in treating cancer. In many tumors, hepcidin is produced locally, while in others, systemic hepcidin also contributes in the hepcidin pool of the tumor milieu [10]. The complicated example of hepcidin shows that in order for the antitumor strategy to work, the treatment regimen should be focused in delivering compounds in specific targets in cancer cells, without affecting normal cells. Using nanotechnology is one of the options to secure this specificity of action. Nanomedicine is based on the principle of the so-called “smart delivery” of drugs [81]. It is a new field in the treatment of patients with cancer that has been evolving progressively in the last decade. Nanomedicine can be used not only to manipulate more efficiently the iron metabolism in tumor microenvironment, but also it can be useful in targeting cancer cells with different antitumor strategies by using the characteristic iron phenotype of the tumor milieu. For example, liposomal nanoparticles have been used to deliver specific genes in pancreatic cancer via TFR binding [82, 83]. Using TFR as an entry point in cancer cells is reasonable, since it is highly expressed in tumor cells compared to normal cells.
3.4 Conclusion and Future Perspectives
It is evident that iron dysmetabolism is prevalent in many tumors. It results from the ability of tumor cells to secure the much-needed iron for their metabolic needs. Iron dysmetabolism can initiate tumorigenesis, enhance tumor growth, promote metastasis, and is even the main instigating factor of ferroptosis, which is a specific form of cellular death. Most of the time iron dysmetabolism is caused by local pathophysiological processes in tumor microenvironment, which means that systemic iron dyshomeostasis is not needed for these processes to occur. This does not mean that global changes of iron metabolism cannot worsen iron dyshomeostasis in tumor milieu. For example, systemic hepcidin can contribute in the total levels of this iron-related peptide in the tumor milieu. Other issues regarding the role of other proteins in cancer iron metabolism include the elucidation of the role of PCBs as intracellular iron chaperons, role of proteins involved in non-TFR iron transport, etc.
Increasing evidence suggests that iron dysmetabolism is related to the dysfunction of some of the most important genes and biochemical pathways involved in tumorigenesis. This means that iron dysmetabolism observed in tumor microenvironment is directly related to the pathophysiology of cancer. This is further corroborated by the existence of iron dysmetabolism in CSCs as well, known also as tumor-initiating cells. On the other hand, the density of CSCs is not uniform in all tumor types, which should be taken into account in the treatment of cancer.
Tumor milieu is a dynamic environment with different types of cells. Some of the most abundant ones are TAMs. A subtype of TAMs known as M2 macrophages are frequently linked with the aggressive phenotype of tumors. M2 macrophages can supply excess iron needed for cancer cells. One way which this occurs is through increased production of Lcn2, though it is still not clear if Lcn2 serves as direct iron supplying molecule for cancer cells. The ability of cancer cells to “manipulate” surrounding TAMs by using their iron flux for their metabolic needs may not be the only way how cancer cells secure increased amount of iron from their surroundings. But, most of the studies relating to TAMs are based on in vitro models which are not ideal replications of the in vivo tumor microenvironment. There is evidence that hijacking mechanisms of cancer cells might be directed towards normal cells localized in the vicinity of tumor cells, as it has been observed in PCa, though the details of this process are not known [44]. Recently, other cells, such as CAFs, have been suggested as important contributors in increasing intracellular iron in cancer cells. But whether CAFs can also be directed by cancer cells to secure more iron is still not known. Furthermore, specific markers of CAFs have still not been elucidated properly. Even less is known about the role of senescent cells in tumor microenvironment, although their SASP includes many substances which are able to modulate cancer cell metabolism.
One recent factor related to iron metabolism in cancer cells is the process of ferroptosis. This type of cellular death occurs in the presence of iron which means that iron can also be detrimental for cancer cell survival. One way to do this is to unleash the sequestered iron from intracellular proteins or from intracellular compartments where iron might be stored. Although ferroptosis is a promising therapeutic target in cancer, it is still a relatively unknown process. In some studies, ferroptosis was known to induce rather than destroy cancer. Therefore, differentiating the mechanistic peculiarities between these two types of ferroptosis is important before considering this form of cellular death as a viable anticancer therapy. On the other hand, the use of iron chelators, antibodies against TFR, or blockers of hepcidin has been met with modest therapeutic success. The problem with this approach is that these compounds are not target specific and often do not have favorable pharmacokinetic properties. The solution to this could be nanomedicine, which is a new branch of medicine with a potential to increase the specificity of anticancer drugs. Still, its actual limitations will have to be resolved before being considered as a therapeutic solution for patients with cancer.
It must be mentioned that the specific iron phenotype of cancer cells and its microenvironment can be used for diagnostic and prognostic purposes in cancer. In many cancers, iron phenotype determines the specificity of the cells of the tumor microenvironment (such as abundance of TFRs in cancer cells or iron-releasing phenotype of M2 macrophages), their aggressiveness (e.g., hepcidin/FPN activity in cancer cells), and even their reaction to chemotherapy. Future studies should evaluate this aspect of iron metabolism in tumor microenvironment in in vivo conditions.
References
DelNero P, Hopkins BD, Cantley LC, Fischbach C (2018) Cancer metabolism gets physical. Sci Transl Med 10:eaaq1011
Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM (2018) Iron and Cancer. Annu Rev Nutr 38:97–125
Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV (2016) Iron and cancer: recent insights. Ann N Y Acad Sci 1368:149–161
Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S et al (2018) Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol 14:100–115
Sánchez M, Sabio L, Gálvez N, Capdevila M, Dominguez-Vera JM (2017) Iron chemistry at the service of life. IUBMB Life 69:382–388
Ebina Y, Okada S, Hamazaki S, Ogino F, Li JL, Midorikawa O (1986) Nephrotoxicity and renal cell carcinoma after use of iron- and aluminum-nitrilotriacetate complexes in rats. J Natl Cancer Inst 76:107–113
Akatsuka S, Yamashita Y, Ohara H, Liu Y-T, Izumiya M, Abe K et al (2012) Fenton reaction induced cancer in wild type rats recapitulates genomic alterations observed in human cancer. PLoS One 7:e43403
Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM (2008) Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem 283:33911–33918
Chanvorachote P, Luanpitpong S (2016) Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells. Am J Physiol Cell Physiol 310:C728–C739
Vela D, Vela-Gaxha Z (2018) Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp Mol Med 50:e436
Straud S, Zubovych I, de Brabander JK, Roth MG (2010) Inhibition of iron uptake is responsible for differential sensitivity to V-ATPase inhibitors in several cancer cell lines. PLoS One 5:e11629
Miles AL, Burr SP, Grice GL, Nathan JA (2017) The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1α prolyl hydroxylation by regulating cellular iron levels. elife 6:e22693
Wang Y, Yu L, Ding J, Chen Y (2019) Iron metabolism in cancer. Int J Mol Sci 20:95
Ornstein DL, Zacharski LR (2007) Iron stimulates urokinase plasminogen activator expression and activates NF-kappa B in human prostate cancer cells. Nutr Cancer 58:115–126
Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L et al (2014) IRP2 regulates breast tumor growth. Cancer Res 74:497–507
Babu KR, Muckenthaler MU (2019) miR-148a regulates expression of the transferrin receptor 1 in hepatocellular carcinoma. Sci Rep 9:1518
Johnson IRD, Parkinson-Lawrence EJ, Shandala T, Weigert R, Butler LM, Brooks DA (2014) Altered endosome biogenesis in prostate cancer has biomarker potential. Mol Cancer Res 12:1851–1862
Wang B, Zhang J, Song F, Tian M, Shi B, Jiang H et al (2016) EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett 381:331–340
Kovar J, Naumann PW, Stewart BC, Kemp JD (1995) Differing sensitivity of non-hematapoietic human tumors to synergistic anti-transferrin receptor monoclonal antibodies and deferoxamine in vitro. Pathobiology 63:65–70
Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D et al (2016) Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab 24:447–461
Gomes IM, Maia CJ, Santos CR (2012) STEAP proteins: from structure to applications in cancer therapy. Mol Cancer Res 10:573–587
Jin Y, Wang L, Qu S, Sheng X, Kristian A, Mælandsmo GM et al (2015) STAMP 2 increases oxidative stress and is critical for prostate cancer. EMBO Mol Med 7:315–331
Whitton B, Okamoto H, Packham G, Crabb SJ (2018) Vacuolar ATPase as a potential therapeutic target and mediator of treatment resistance in cancer. Cancer Med 7:3800–3811
Zhang X, Hua L, Yan D, Zhao F, Liu J, Zhou H et al (2016) Overexpression of PCBP2 contributes to poor prognosis and enhanced cell growth in human hepatocellular carcinoma. Oncol Rep 36:3456–3464
Li F, Bullough KZ, Vashisht AA, Wohlschlegel JA, Philpott CC (2016) Poly(rC)-binding protein 2 regulates hippo signaling to control growth in breast epithelial cells. Mol Cell Biol 36:2121–2131
Chen C, Lei J, Zheng Q, Tan S, Ding K, Yu C (2018) Poly(rC) binding protein 2 (PCBP2) promotes the viability of human gastric cancer cells by regulating CDK2. FEBS Open Bio 8:764–773
Deng Z, Manz DH, Torti SV, Torti FM (2017) Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget 8:82231–82243
Khiroya H, Moore JS, Ahmad N, Kay J, Woolnough K, Langman G et al (2017) IRP2 as a potential modulator of cell proliferation, apoptosis and prognosis in nonsmall cell lung cancer. Eur Respir J 49:1600711
Horniblow RD, Bedford M, Hollingworth R, Evans S, Sutton E, Lal N et al (2017) BRAF mutations are associated with increased iron regulatory protein-2 expression in colorectal tumorigenesis. Cancer Sci 108:1135–1143
Ward DM, Kaplan J (1823) Ferroportin-mediated iron transport: expression and regulation. Biochim Biophys Acta, Mol Cell Res 2012:1426–1433
Pinnix ZK, Miller LD, Wang W, D’Agostino R, Kute T, Willingham MC et al (2010) Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med 2:43ra56
Deng Z, Manz DH, Torti SV, Torti FM (2019) Effects of ferroportin-mediated iron depletion in cells representative of different histological subtypes of prostate cancer. Antioxid Redox Signal 30:1043–1061
Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y et al (2014) Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal 26:2539–2550
Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H et al (2015) Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res 75:2211–2221
Porporato PE, Filigheddu N, Pedro JMBS, Kroemer G, Galluzzi L (2018) Mitochondrial metabolism and cancer. Cell Res 28:265–280
Vela D (2019) Keeping heart homeostasis in check through the balance of iron metabolism. Acta Physiol 228:e13324. https://doi.org/10.1111/apha.13324
Fryknäs M, Zhang X, Bremberg U, Senkowski W, Olofsson MH, Brandt P et al (2016) Iron chelators target both proliferating and quiescent cancer cells. Sci Rep 6:38343
Sandoval-Acuña C, Tomkova V, Cardenas NT, Neuzil J, Repkova K, Stursa J et al (2018) Mitochondrial iron chelation as a novel anti-cancer strategy. Free Radic Biol Med 120(Supp. 1):S61
Wu KJ, Polack A, Dalla-Favera R (1999) Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 283:676–679
Tsuji Y, Kwak E, Saika T, Torti SV, Torti FM (1993) Preferential repression of the H subunit of ferritin by adenovirus E1A in NIH-3T3 mouse fibroblasts. J Biol Chem 268:7270–7275
Ozaki T, Nakagawara A (2011) Role of p53 in cell death and human cancers. Cancers 3:994–1013
Jiang X, Zhang C, Qi S, Guo S, Chen Y, Du E et al (2016) Elevated expression of ZNF217 promotes prostate cancer growth by restraining ferroportin-conducted iron egress. Oncotarget 7:84893–84906
Li JL, Okada S, Hamazaki S, Ebina Y, Midorikawa O (1987) Subacute nephrotoxicity and induction of renal cell carcinoma in mice treated with ferric nitrilotriacetate. Cancer Res 47:1867–1869
Vela D (2018) Iron metabolism in prostate cancer; from basic science to new therapeutic strategies. Front Oncol 8:547
Lin Y, Xu J, Lan H (2019) Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 12:76
Gollapudi K, Galet C, Grogan T, Zhang H, Said JW, Huang J et al (2013) Association between tumor-associated macrophage infiltration, high grade prostate cancer, and biochemical recurrence after radical prostatectomy. Am J Cancer Res 3:523–529
Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M et al (2010) Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol 40:824–835
Jung M, Mertens C, Bauer R, Rehwald C, Brüne B (2017) Lipocalin-2 and iron trafficking in the tumor microenvironment. Pharmacol Res 120:146–156
Mertens C, Mora J, Ören B, Grein S, Winslow S, Scholich K et al (2018) Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Onco Targets Ther 7:e1408751
Jung M, Ören B, Mora J, Mertens C, Dziumbla S, Popp R et al (2016) Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine-1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal 9:ra64
Mertens C, Akam EA, Rehwald C, Brüne B, Tomat E, Jung M (2016) Intracellular iron chelation modulates the macrophage iron phenotype with consequences on tumor progression. PLoS One 11:e0166164
Prill S, Rebstock J, Tennemann A, Körfer J, Sönnichsen R, Thieme R et al (2019) Tumor-associated macrophages and individual chemo-susceptibility are influenced by iron chelation in human slice cultures of gastric cancer. Oncotarget 10:4731–4742
Leftin A, Ben-Chetrit N, Joyce JA, Koutcher JA (2019) Imaging endogenous macrophage iron deposits reveals a metabolic biomarker of polarized tumor macrophage infiltration and response to CSF1R breast cancer immunotherapy. Sci Rep 9:857
Recalcati S, Gammella E, Cairo G (2019) Dysregulation of iron metabolism in cancer stem cells. Free Radic Biol Med 133:216–220
Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G et al (2017) Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene 36:4089–4099
Kanojia D, Zhou W, Zhang J, Jie C, Lo PK, Wang Q et al (2012) Proteomic profiling of cancer stem cells derived from primary tumors of HER2/Neu transgenic mice. Proteomics 12:3407–3415
Schonberg DL, Miller TE, Wu Q, Flavahan WA, Das NK, Hale JS et al (2015) Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell 28:441–455
Ninomiya T, Ohara T, Noma K, Katsura Y, Katsube R, Kashima H et al (2017) Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget 8:98405–98416
Rychtarcikova Z, Lettlova S, Tomkova V, Korenkova V, Langerova L, Simonova E et al (2017) Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 8:6376–6398
El Hout M, Dos Santos L, Hamaï A, Mehrpour M (2018) A promising new approach to cancer therapy: targeting iron metabolism in cancer stem cells. Semin Cancer Biol 53:125–138
Brabletz T, Kalluri R, Nieto MA, Weinberg RA (2018) EMT in cancer. Nat Rev Cancer 18:128–134
Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK et al (2012) N-myc downstream regulated gene 1 modulates Wnt-β-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol Med 4:93–108
Shan Z, Wei Z, Shaikh ZA (2018) Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol Appl Pharmacol 356:36–43
Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL (2019) Cancer-associated fibroblasts in gastrointestinal cancer. Nat Rev Gastroenterol Hepatol 16:282–295
Sandberg TP, Stuart MPME, Oosting J, Tollenaar RAEM, Sier CFM, Mesker WE (2019) Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 19:284
Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A et al (2019) Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 38:5339–5355
Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P et al (2018) Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene 37:4013–4032
Lee S, Schmitt CA (2019) The dynamic nature of senescence in cancer. Nat Cell Biol 21:94–101
Chen T, Yang P, Wang H, He ZY (2017) Silence of long noncoding RNA PANDAR switches low-dose curcumin-induced senescence to apoptosis in colorectal cancer cells. Onco Targets Ther 10:483–491
Jiao Y, Wilkinson J IV, Christine Pietsch E, Buss JL, Wang W, Planalp R et al (2006) Iron chelation in the biological activity of curcumin. Free Radic Biol Med 40:1152–1160
Hassannia B, Vandenabeele P, Vanden BT (2019) Targeting ferroptosis to iron out cancer. Cancer Cell 35:830–849
Dixon SJ, Stockwell BR (2019) The hallmarks of ferroptosis. Annu Rev Cancer Biol 3:35–54
Ma S, Henson ES, Chen Y, Gibson SB (2016) Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis 7:e2307
Geng N, Shi B-J, Li S-L, Zhong Z-Y, Li Y-C, Xua W-L et al (2018) Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 22:3826–3836
Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J et al (2017) Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem 9:1025–1033
Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D (2019) Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis 10:331. https://doi.org/10.1038/s41419-019-1564-7
Sun Y, Li C, Feng J, Li Y, Zhai X, Zhang L et al (2019) Ferritinophagic flux activation in CT26 cells contributed to EMT inhibition induced by a novel iron chelator, DpdtpA. Oxid Med Cell Longev 2019:1–14
Tury S, Assayag F, Bonin F, Chateau-Joubert S, Servely JL, Vacher S et al (2018) The iron chelator deferasirox synergises with chemotherapy to treat triple-negative breast cancers. J Pathol 246:103–114
Miller LD, Coffman LG, Chou JW, Black MA, Bergh J, D’Agostino R et al (2011) An iron regulatory gene signature predicts outcome in breast cancer. Cancer Res 71:6728–6737
Toshiyama R, Konno M, Eguchi H, Asai A, Noda T, Koseki J et al (2018) Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol Lett 15:8125–8133
Kalaydina RV, Bajwa K, Qorri B, Decarlo A, Szewczuk MR (2018) Recent advances in “smart” delivery systems for extended drug release in cancer therapy. Int J Nanomedicine 13:4727–4745
Camp ER, Wang C, Little EC, Watson PM, Pirollo KF, Rait A et al (2013) Transferrin receptor targeting nanomedicine delivering wild-type p53 gene sensitizes pancreatic cancer to gemcitabine therapy. Cancer Gene Ther 20:222–228
Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M et al (2013) Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. In: Molecular Therapy, vol 21. Nature Publishing Group, New York, pp 1096–1103
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vela, D. (2020). Iron in the Tumor Microenvironment. In: Birbrair, A. (eds) Tumor Microenvironment. Advances in Experimental Medicine and Biology, vol 1259. Springer, Cham. https://doi.org/10.1007/978-3-030-43093-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-43093-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-43092-4
Online ISBN: 978-3-030-43093-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)