
Abstract
Not all aspects of the disruption of iron homeostasis in cancer have been fully elucidated. Iron accumulation in cancer cells is frequent for many solid tumours, and this is often accompanied by the contemporary rise of two key iron regulators, HIF2α and Hepcidin. This scenario is different from what happens under physiological conditions, where Hepcidin parallels systemic iron concentrations while HIF2α levels are inversely associated to Hepcidin. The present review highlights the increasing body of evidence for the pro-tumoral effect of HIF2α and Hepcidin, discusses the possible imbalance in HIF2α, Hepcidin and iron homeostasis during cancer, and explores therapeutic options relying on these pathways as anticancer strategies.
Similar content being viewed by others

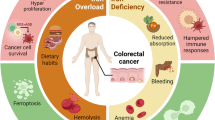
Introduction
In recent years, the relevant role of iron pathways in cancer development and progression has been increasingly recognised [1]. Data exist that demonstrate that iron is capable of enhancing cancer cell proliferation and metastatisation and also, in specific experimental models, of initiating cancer development [2]. In clinical studies, increased circulating and tissue iron concentrations have been associated to the inferior prognosis of cancer patients [3].
Iron is a vital micronutrient with key roles in oxygen distribution and enzymatic redox reactions occurring in many biological processes. Under normal conditions, iron homeostasis is principally regulated by the action of liver Hepcidin and intestinal and renal hypoxia-inducible factor-2-alpha (HIF2α) [4]. Systemic iron levels parallel systemic Hepcidin levels as Hepcidin is the major sensor of iron concentrations, while HIF2α is inversely associated to iron concentrations.
With reduced iron availability, enhanced expression of HIF2α is observed in the duodenum, where it increases intestinal absorption of iron, and in the kidneys, where it stimulates erythropoietin (EPO) synthesis, which ultimately induces erythropoiesis and inhibits liver Hepcidin [5]. Downregulation of Hepcidin is essential to mobilise body iron storage into circulation. In the case of iron abundance, inhibition of HIF2α and upregulation of Hepcidin are both induced. This favours the influx of iron into cells for storage or metabolic use since Hepcidin inhibits the only known mammalian iron cellular exporter, ferroportin (FPN) [6] (Fig. 1).

When transferrin saturation by circulating iron and/or high intracellular iron in the liver occurs, BMP6 are secreted from liver sinusoidal endothelial cells. These proteins, by binding its receptors and co-receptors (i.e., Hemojuvelin and Neogenin) on hepatocytes' surface, promote SMAD complex activation. Once activated, SMAD complex translocates to the nucleus for the synthesis of Hepcidin, encoded by the gene HAMP. HIF2α, synthesised under hypoxic conditions, is able to suppress Hepcidin synthesis directly or by means of renal EPO and erythroid ERFE. Inflammation-derived IL-6 is able to trigger the BMP-SMAD pathway, further increasing Hepcidin levels. Once released, Hepcidin inhibits iron export through ferroportin from macrophages, enterocytes and hepatocytes. BMP bone morphogenetic protein, HAMP hepcidin antimicrobial peptide gene, hep Hepcidin, HJV hemojuvelin, N Neogenin, HIF2α hypoxia-inducible factor-2-alpha, IL-6 interleukin-6, SMAD small mothers against decapentaplegic, CV cardiovascular, EPO erythropoietin, ERFE erythroferrone, IL-6R IL-6 receptor, JAK Janus kinase, STAT signal transducer and activator of transcription.
HIF2α and Hepcidin are also associated with cancer progression independently of iron. They both cover a number of physiological and pathological functions; nonetheless, iron homeostasis could represent the common pathway linking the two mediators and the disruption of the HIF2α/Hepcidin-mediated pro-tumoral effect may represent a promising anticancer therapeutic strategy [7].
The present review has the aim of disentangling the tumour-promoting effect of HIF2α and Hepcidin and the crosstalk between the two mediators as a possible mechanism granting growth advantage to cancer cells in both an iron-dependent and iron-independent manner.
Physiology and perturbation of iron homeostasis. Role of HIF2α and hepcidin
Iron is a vital micronutrient for human beings, and controlling mechanisms have been evolutionary selected in order to guarantee iron homeostasis, with HIF2α and Hepcidin being the two mediators at the extremities of the ‘iron chain’.
HIF2α is a key protein of hypoxia-Inducible pathways activated by the reduced oxygen supply in the tissue [8]. Upregulation of two subunits, HIF-α (labile) and HIF-β (more stable), is induced by hypoxia. Three alternative α-subunits exist (HIF1α, HIF2α, and HIF3α) with distinct biological properties [9]. HIF2α and HIFβ heterodimerize to form a direct transcription factor that regulates over 1000 genes with the coordinated aim of adapting cells to poor oxygen availability [10]. This consists of metabolic reprogramming, neoangiogenesis, cell plasticity and migration, all landmark features also of cancer [11]. HIF2α has long been known as an important mediator in cancer development. It is synthesised under the hypoxic condition that occurs during cancer and orchestrates multiple processes such as neoangiogenesis, epithelial-to-mesenchymal transition and metastatic spread. HIF2α has also the function of enhancing intestinal iron absorption from food. This is explained, from an evolutionary point of view, by the need of iron to increase iron-haem and haemoglobin synthesis to counteract hypoxia. Moreover, HIF2a is the key regulator of erythropoietin (EPO) in the kidneys.
In conditions of hypoxia or hypoferremia, HIF2α increment is generated in the duodenum and in the kidney via inhibition of its degrading enzymes prolyl hydroxylases (PHs). PHs require both iron and oxygen to catalyse HIF2α, and decreased intracellular iron content or oxygen tension result in enzymatic inhibition and stabilisation of HIF2α [12].
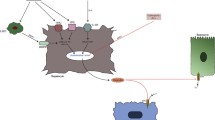
In the duodenum, HIF2α induces the expression of transporters favouring the absorption of iron from food. In particular, HIF2α stabilisation in enterocytes activates the translation of the gene of an apical iron importer, the divalent metal transporter 1 (DMT1, also known as SLC11A2). Once iron is introduced into the enterocytes via DMT1, it is then effluxed into the circulation through the exporter Ferroportin (FPN) expressed in the basolateral membrane (Fig. 2) [13].

Effects on enterocytes are displayed. In physiological conditions, Hepcidin regulates negatively HIF2α with increase of HIF2α when iron and Hepcidin are low (left panel) and decrease when iron and Hepcidin are high (middle panel). In cancer, an opposite regulation exists with HIF2α, which is increased because of the hypoxic tumour microenvironment, inducing an upregulation of cancer-derived Hepcidin that eventually leads to high intracellular iron concentrations essential for nucleotide synthesis. DMT1 divalent metal transporter 1.
In the kidney, HIF2α directly induces the translation of the EPO gene. EPO is a master signal for erythropoiesis expansion which is strictly dependent on iron availability in the bone marrow. It is produced by the interstitial fibroblasts of the medulla of the kidneys (renal EPO-producing cells, REP cells) upon HIF2α upregulation if functional hypoxia is sensed, such as in case of acute blood loss, anaemia, low oxygen tension due to altitude, pulmonary diseases or other conditions [14]. Erythropoietic stimuli need to ultimately result in liver Hepcidin inhibition in order to increase circulating iron availability. Nonetheless, EPO has been found not to directly regulate liver Hepcidin [15]. EPO activates in the erythroblasts of the bone marrow the synthesis of erythroferrone (ERFE), a recently discovered intermediary hormone with major inhibitory effect on liver Hepcidin [16].
Hepcidin is a liver-derived hormone with a fundamental role in systemic iron concentrations and has lately been identified as possible promoter of cancer growth. It mainly acts by inhibiting the iron efflux from the intracellular compartment of iron-storing cells to the plasma via the induction of internalisation and degradation of Ferroportin (FPN), the only known transmembrane iron exporter. FPN is expressed on cells committed to maintaining iron storage in the body, such as hepatocytes and macrophages in the spleen, or on the basolateral membrane of cells at the interface of possible iron-replete external environment such as enterocytes or cells of the distal renal tubules [17]. Iron mobilisation induced by Hepcidin decrement is paramount for haem and haemoglobin production.
Circulating iron participates with a feedback mechanism to Hepcidin regulation. Hepcidin synthesis in the liver is regulated by the bone morphogenetic protein (BMP) pathway, and specifically it is stimulated upon binding of iron-induced BMP2/6 proteins to the BMP receptor expressed on the hepatocyte cell surface [18]. Activated BMP receptor cooperates with two transmembrane co-receptors, Hemojuvelin (HJV) and Neogenin (N), to transmit downstream signalling [19] (Fig. 1).
The so-formed BMP receptor complex leads to intracellular Smad1/5/8 activation and ultimately to Hepcidin synthesis and release from the liver [20]. ERFE has been found to sequester extracellular BMP2/6 produced by sinusoidal endothelial cells thus down-regulating Hepcidin expression [21].
Other two important signals may induce liver Hepcidin production. Iron is carried in the bloodstream by protein transferrin. In the case of circulating iron abundance, iron-saturated transferrin binds to the transferrin receptor (TfR) expressed on hepatocytes and the activated transferrin/TfR complex interacts with the hemochromatosis protein (HFE) that acquires the ability of enhancing the signalling of the BMP complex, thus inducing Hepcidin production [22].
The other Hepcidin-stimulating signal is that driven by inflammatory mediators. A number of substances associated to conditions with enhanced systemic inflammation are able to induce liver Hepcidin synthesis. LPS released during bacterial infection [23], leptin produced in obese subjects [24] and acute-phase proteins released in chronic illnesses such as cancer and cardiovascular diseases [25] were all found to trigger liver Hepcidin synthesis. The main mediator responsible for inflammatory Hepcidin overproduction is IL-6, which is capable to directly induce Hepcidin synthesis in the hepatocytes by binding relevant receptor that converges to the JAK/STAT intracellular transduction signal [26]. JAK/STAT is an activator of downstream Hepcidin gene translation.
In Fig. 1 are summarised the main factors influencing the iron homeostasis in human beings.
HIF2α and Hepcidin are both over-expressed in many cancer types and both have been linked to cancer development and progression. Iron is a potent activator of cell proliferation [27]. Iron increase in cancer cells and the resulting proliferative stimulus have been identified as key oncogenic mechanisms for HIF2α and Hepcidin, however also iron-independent effects have been documented. Role of HIF2α and Hepcidin in cancer pathogenesis will be discussed in the following sections.
HIF2α in cancer
The HIF (hypoxia-inducible factor) is a heterodimer protein constituted of a common stable beta subunit (HIFβ, usually HIF1β also called ARNT) and three possible subtypes of unstable alpha unit (HIF1α, HIF2α, HIF3α) [28]. It is a hallmark of hypoxic state virtually in all tissues and is upregulated following hypoxia-induced inhibition of its degrading enzymes, prolyl hydroxylases (PHs). Under normal oxygen tension, PHs hydroxylate one of two conserved prolyl residues of the HIFα. Hydroxylated HIFα subunits become substrate of pVHL (product of the Von Hippel–Lindau gene) which mediates their proteosomal degradation [29]. HIFs are transcription factors that orchestrate a number of processes to counteract hypoxic stress by directly translating hundreds of target genes. These processes include neoangiogenesis (in particular by activating the VEGF and PDGF pathways) [30,31,32], inflammatory/immune response [33, 34], metabolic reprogramming (such as induction of the glucose or glutamine transporters and lipid accumulation) [35,36,37], iron availability [38] and erythropoiesis (by directly regulating the translation of the erythropoietin gene EPO in the kidneys) [39]. Furthermore, upregulation of growth factors and cell-cycle genes, such as cyclin D1, EGFR, TGFα, has been observed [40]. Hypoxia is a key feature of solid malignancies as the cancer grows and the centre of the tumour invariably suffers of reduced oxygen tension [41]. HIFs upregulation is frequently observed in many cancer types and activation of hypoxia-inducible genes is exploited by the tumour to mediate neoangiogenesis, cell infiltration and metastatic spread [42].
A description of cancer types that are more frequently linked to HIF2α dysregulation follows.
HIF2α in renal cell carcinoma
The role of HIF2α in renal cell carcinoma has been extensively investigated. In clear cell renal cell carcinoma (ccRCC), HIF2α increases because of the genetic alteration of the physiological HIF2α degrading machinery that is led by the von Hippel–Lindau (VHL) protein (pVHL). This may occur in normoxic conditions (sometimes called ‘pseudohypoxic state’). In particular, pVHL deficiency has been observed in up to 90% of renal cell carcinoma [43]. It is mostly due to inactivating gene mutations but also hypermethylation or post-transcriptional deregulation have been documented. The inheritance of inactivating mutation of VHL gives rise to the von Hippel–Lindau syndrome (VHL disease) which is characterised by the development of multiple paragangliomas, ccRCCs, hemangioblastomas and paraneoplastic erythrocytosis [44]. In VHL disease, an acquired alteration also of the normal VHL allele is required for kidney cancer development to take place. When this happens a parallel rise in HIF2α is observed that is a clear risk factor for preneoplastic lesions to transform to overt cancer [45]. A number of lines of evidence have confirmed the importance of HIF2α in renal cancer progression. Renal cancer cells with VHL−/− status hyperexpress HIF2α and this seems to be both necessary and sufficient to maintain the malignant phenotype [46]. Suppression of HIF2α in VHL−/− cells inhibits their ability to form tumours when implanted in nude mice [47, 48]. Moreover, experimental induction of HIF2α overexpression may fully mimic cancer models of VHL deletion [49].
HIF2α acts as a transcription factor by binding the beta subunit hypoxia-inducible factor-1β (also known as aryl hydrocarbon receptor nuclear translocator, ARNT) and activating a number of target genes harbouring ‘hypoxia response elements’ (HRE), collectively called hypoxia response genes that orchestrate tumour progression. In renal cancer, four HRE-carrying genes seem to be crucial for progression: VEGFα, PDGFβ, TGFα and Cyclin D1.
Renal cancer is probably the most VEGFα overexpressing cancer, while other solid tumours may activate pathways alternative to VEGFα [50]. Indeed, this relative VEGFα dependency seems to rely on the privileged transcriptional activation by HIF2α. VEGFα therapeutic inhibition has proven consistently effective in renal cancer treatment [51]. Many anti-VEGFα agents have received regulatory approval and represent standard options in the therapeutic algorithm [52].
PDGFβ supports renal cancer neoangiogensis, since it guarantees the proliferation and differentiation of pericytes that ensure proper functionality of neo-vessels [53].
TGFα has been found to contribute as a direct cancer growth factor in renal cancer, since it binds and activates the growth factor receptor EGFR [54]. However, selective inhibition of EGFR has failed to demonstrate meaningful clinical benefit [55], conceivably because downstream signals, such as c-MET, are also independently enhanced [56].
Cyclin D1 is an important positive regulator of the cell cycle. In renal cancer it is upregulated by HIF2α and this is also accompanied by the downregulation of a natural Cyclin D1 inhibitor, the Dachshund (DACH1) product [57].
Targeted inhibitors have been developed that bind to the HIF1β/HIF2α dimerisation pocket and induce conformational changes. Initial HIF2α inhibitors were proven effective in suppressing hypoxia-inducible genes in ccRCC cultures and mouse xenografts with resulting reduced cancer growth [58]. More recently, inhibitors with superior potency and selectivity have been developed [59]. In a Phase 1 trial, treatment with the new-generation HIF2α inhibitor belzutifan (MK-6482/PT2977) achieved 25% of confirmed objective response rate in patients with ccRCC [60]. Encouraging results have also been obtained with the use of belzutifan in ccRCC patients with inherited VHL disease [61].
Dysregulation of HIF2α and iron is present in ccRCC, with the coexistence of HIF2α overexpression and intracellular iron sequestration. It has been demonstrated that activation of HIF pathways in renal cancer cells results in the expression of iron importers, and this might be coupled with inhibition of iron efflux [62].
The involvement of iron and iron-induced reactive oxygen species in ccRCC tumorigenesis has been documented in previous studies [63]. Higher lifelong exposure to iron such as in cases of occupational pollution, or chronic iron overload observed in cases of thalassaemia, have been associated to ccRCC risk [64, 65]. Moreover, risk factors highly associated to ccRCC, such as tobacco use, have also been found to predispose to iron overload [66]. Last, several groups have demonstrated that administration of ferric compounds in rat models are able to produce subacute renal tubular toxicity and induce renal carcinoma [67, 68]. Greene et al. have investigated differences in iron content and iron-related pathways between renal cancer and normal kidney cell lines [69]. Baseline free iron levels were significantly higher in 4 different ccRCC lines than in normal renal proximal tubule epithelial cells and renal cortical epithelial cells. Moreover, iron depletion using iron chelators (deferasirox and deferoxamine) induced a growth suppression in >90% of cancer cell lines, while growth arrest was modest for normal cell lines, implicating an iron-dependency for cancer cell survival. Intriguingly, it was observed that the anticancer effect of iron chelation was due to HIF2α suppression and induction of apoptosis, thus suggesting also a possible feedback by which iron further sustains HIF2α expression in ccRCC with chelation of iron being able to reduce HIF2α levels [69]. VHL restoration in these cells prevented the anticancer activity of iron chelators. It has also been documented that the therapeutic effect of HIF2α inhibition partly depends on intracellular iron availability since compounds that deplete HIF2α in ccRCC eventually induce apoptosis by means of iron-dependent phospholipid peroxidation, a programmed cell death called ‘ferroptosis' [70].
HIF2α in colorectal cancer
HIF2α fuels colorectal cancer growth in many ways [71]. HIF2α generated under hypoxia favours cancer cell survival and metastatic seeding. It is also implicated in the first steps of colorectal carcinogenesis, and mediates iron influx via the HIF2α-induced iron transporter Divalent Metal Transporter 1 (DMT1), that is vital for cancer cell energetic [72]. HIF2α is more often expressed in KRAS-mutated colon cancer where it regulates cardiolipin-based mitochondrial metabolism, maximises ATP production and minimises reactive oxygen species generation, thus favouring cancer cell survival and replication [73].
In a cohort of 87 curatively resected colorectal cancer patients, histological expression of HIF2α was observed in 30% of cases and was associated with adverse features, such as increased microvessel density, neoangiogenesis and short survival [74]. HIF2α has also been associated to resistance to standard chemotherapy, radiotherapy or other forms of local treatment [75]. For decades fluorouracil has been the mainstay in the chemotherapy armamentarium against colorectal cancer. The drug is catabolized by the enzyme dihydropyrimidine dehydrogenase (DPD). It has recently been found that cancer hypoxia induces the expression of DPD in tumour-associated macrophages (TAMs) via HIF2α, with consequent acquisition of chemoresistance to standard dose fluorouracil [76]. It has been suggested that metronomic (i.e., low dose) chemotherapy, rather than standard-dose chemotherapy, would reduce synthesis of HIF2α and thus be preferable in tumours with HIF2α upregulation [77].
HIF2α-overexpressing colorectal cancer presents molecular vulnerabilities that can be pharmacologically exploited to induce synthetic lethality. In particular, HIF2α seems to sensitise cells to apoptosis using iron to generate toxic lipid peroxides (ferroptosis). In particular, HIF2α-positive CRC cells are highly sensitive to the known ferroptosis inducers Erastin and RSL3 [78, 79].
HIF2α in breast cancer
In breast cancer, HIF2α generated in hypoxic microenvironment has been associated to reprogramming towards a cancer stem cell-like phenotype. Breast cancer stemness is characterised by more aggressive behaviour and chemoresistance. Research by Yan et al. has demonstrated that HIF2α, but not HIF1α, activates an endoplasmic reticulum response that eventually converts chemosensitive breast cancer cells to chemoresistant cells with stem cell-like phenotype [80]. This conversion is abrogated by HIF2α inhibitors. In another experiment by Fu et al., HIF2α was found to mediate also the development of resistance to endocrine therapy in oestrogen receptor-positive breast cancer cells. It has recently been demonstrated that amplification or other forms of overexpression of the transcription regulator Forkhead box A1 (FOXA1) is associated to poor outcome and resistance to hormone therapy in oestrogen receptor-positive breast cancer [81]. Fu et al. have demonstrated that the top target of the FOXA1-mediated transcriptional reprogramming leading to endocrine therapy resistance and aggressive behaviour is HIF2α. FOXA1-induced HIF2α upregulation was associated to the activation of multiple oncogenic pathways including those associated with extracellular matrix remodelling (TNC, SERPINE1, FN1), reduced focal adhesion (CD44, ANXA1, PLAUR) and induced angiogenesis (VEGFA, EPAS1, KLF5) [82]. Inhibition of HIF2α determined a reduction in clonogenicity, migration, and invasion of FOXA1-positive breast cancer cells.
Intracellular iron availability seems to support HIF2α activity in breast cancer cells, since agents known to be iron chelators, such as curcumin, induce a lowering in HIF2α concentration with impaired cell survival [83].
HIF2α in pancreatic cancer
Pancreatic cancer is featured by excessive desmoplastic tissue deposition and consequent low oxygen tension in the interstitium [84]. This pathologic hypoxic milieu is thought to be responsible for many unfavourable peculiarities that are observed in pancreatic cancer, such as reduced drug delivery, treatment resistance, immune escape and infiltrative growth [85, 86].
In particular, HIF2α has been found to mediate the immunosuppressive properties of pancreatic cancer-associated stromal tissue which is mainly produced by cancer-associated fibroblasts (CAFs). In a study by Garcia Garcia et al., selective deletion of HIF2α in CAFs of a mouse model of spontaneous pancreatic cancer was associated with slower cancer growth and improved survival. Interestingly, CAF HIF2α deletion determined only a minor reduction in fibrosis, while it induced a marked reduction in intratumoural M2 macrophages and regulatory T cells which are recognised to have remarkable immunosuppressive functions. Treatment with immunotherapeutic agents targeting PD-1 and CTLA4 in combination with the HIF2α inhibitor PT2399 was associated with enhanced tumour shrinkage [87].
Gemcitabine is a mainstay drug in the cure of pancreatic cancer, however resistance inevitably develops after a few months. It has been shown that HIF2α contributes to ATP and glutathione synthesis in pancreatic cancer cells and this results in tumour-favourable energetic reprogramming and gemcitabine resistance. HIF2α determines overexpression of the SLC1A5 variant, a protein that localises to mitochondria where it functions as glutamine transporter. Enhanced glutamine use in pancreatic cancer cells provides metabolic advantage since it guarantees adequate ATP production. Moreover glutamine is crucial for glutathione synthesis which is a known antidote against gemcitabine [88, 89].
Finally, HIF2α in pancreatic cancer has been found to activate Wnt/β-catenin and E-cadherin signalling which induces epithelial-mesenchymal transition, stroma deposition, neoangiogenesis and a stem cell-like phenotype, all features associated to poor prognosis [90, 91].
HIF2α in hepatocellular carcinoma
HIFs have been strongly associated to HCC development and prognosis. In a recent meta-analysis by Méndez-Blanco et al., immunohistochemistry (IHC) expression of HIF1α and HIF2α in the surgical specimens of radically resected HCC patients was analysed for the association with recurrence and survival outcomes [92]. After selecting 32 publications including nearly 4000 HCC patients, high IHC expression of both HIF1α and HIF2α was found to be associated with shorter overall and disease-free survival. Moreover, it was associated to clinicopathological features of aggressiveness, such as Barcelona Clinic Liver Cancer (BCLC) advanced stage, capsule infiltration and vascular invasion.
Chronic liver diseases characterised by enhanced inflammatory response (e.g., viral hepatitis, non-alcoholic fatty liver disease (NAFLD) and hereditary hemochromatosis) are all invariably associated to increased risk of developing HCC [93, 94]. NAFLD has been associated to HCC both following and independently of cirrhosis [95]. Recently, Foglia et al., by using conditional deletion of hepatocyte genes in mouse models, have unequivocally demonstrated that HIF2α has a crucial role in NAFLD-associated carcinogenesis. Hepatocyte HIF2α−/− mice developed less and smaller HCC lesions in experiments of NAFLD carcinogenesis as compared to HIF2α competent littermates. HIF2α deletion was associated to lower Ki67 proliferation index and downregulation of the Hippo/Yes-associated protein (YAP)/c-Myc oncogenic pathway [96].
YTH domain family 2A (YTHDF2) is a key regulator of transcription in hepatocytes. It is an important player in cell epigenetic adaptation, since it works as a reader of N6-methyladenosine (m6A) sites in mRNAs and distributes m6A-containing mRNAs to degrading machineries. HIF2α has been found to strongly downregulate YTHDF2 in HCC thus increasing at the post-transcriptional level the stabilisation of a number of tumour-promoting mRNAs. In particular mRNAs with potent effects on inflammation, cancer cell survival and vascular formation, such as IL-11 and Serpin E2 mRNAs, are over-stabilised [97]. Inhibitors of HIF2α restored YTHDF2 activity, repressed IL-11 and Serpin E2 and eventually lead to liver cancer inhibition. HIF2α was also found to favour resistance to commonly used anticancer agents in HCC [98].
Hepcidin in cancer
Hepcidin, encoded by the Hepcidin antimicrobial peptide (HAMP) gene located on chromosome 13, is a 25-amino acid protein initially identified as antimicrobial peptide [99]. It was soon found crucial for iron homeostasis and is now considered the master iron regulator [100]. Hepcidin binds to the only known iron exporter, ferroportin, and induces its degradation in the cytoplasmic side of iron-storing cells thus preventing iron mobilisation into the circulation.
Hepcidin was initially thought to be produced only in the liver, but subsequent studies have shown that it is also synthesised in a number of extra-hepatic tissues, such as the kidney, heart and adipose tissue [101].
Iron and iron-regulating proteins have recently been recognised as crucial tumour promoters [102]. Tumour cells can themselves synthesise hepcidin, and this is associated with increased intracellular iron trapping and activation of iron-based energetic and metabolic pathways that favour a malignant phenotype and cell proliferation [103]. It is possible that specific subtypes of cancer are particular dependent on iron-based axes for their survival. Some tumours seem to use the degradation of the iron-storing protein ferritin (ferritinophagy) to mobilise iron for cellular functions and induce synthesis of iron–sulphur cluster proteins that are needed for mitochondrial activities [104]. Disrupting the cancer iron biology might represent a potential therapeutic approach.
In the following section, we will be discussing the evidence on the possible role of Hepcidin across different tumour types [105].
Hepcidin in colorectal cancer
It has long been known that colorectal cancer consistently sequesters massive iron quantity. Conditions characterised by systemic iron overloads such as hemochromatosis and excessive dietary intake of red meat are associated with high risk of developing colorectal cancer [106]. Conversely, experimental models where iron uptake into colonic cells is inhibited are resistant to tumorigenesis [107].
Brookes et al. have demonstrated that the iron exporter ferroportin can be over-expressed in colon cancer cell lines [108]. Its localisation in cancer cells, though, is chiefly in the cytoplasm where it is not operational and thus not determining the intracellular iron-lowering effect that is obtained when ferroportin is expressed on cell surface.
Schwartz et al. have recently demonstrated that the main mechanism by which ferroportin is internalised in colorectal cancer is the intestinal ectopic production of hepcidin [109]. In patient-derived three-dimensional colorectal enteroid models, it was demonstrated that colorectal cancer cells produced hepcidin that, in turn, determined an increase in inner iron storage by localising ferroportin in the intracellular compartment. In a mouse model of colorectal cancer, iron was fundamental for cell energetic homeostasis at the mitochondrial level, nucleotide synthesis and DNA replication. Genetic deletion of hepcidin reduced the iron content with consequent reduced cell proliferation and tumour growth. The antitumor effect of iron depletion was rescued by exogenous nucleoside implementation, thus demonstrating that the main metabolic role of iron in these cells is to contribute to nucleotide synthesis.
Hepcidin in breast cancer
Pinnix et al. have demonstrated that ferroportin downregulation is a peculiar feature of malignant cells as compared to the normal counterpart in breast tissue [110]. This seems to be modulated by an increase in Hepcidin mRNA expression in breast cancer cells [111].
Systemic dysregulation of Hepcidin and ferritin has been suggested as a hallmark of breast cancer development, and the plasmatic increase of the two proteins has been proposed as a non-invasive tool for the early diagnosis of breast cancer [112].
Studies with experimental breast cancer spheroids demonstrated that overproduction of tumour-derived Hepcidin directly correlated with higher tumour stage. In later stages, synthesis of tumour Hepcidin seems to be sustained by fibroblast-derived Interleukin-6 (IL-6) produced in the microenvironment [113]. IL-6 and other key systemic inflammatory mediators are known to stimulate Hepcidin even under normal or low iron conditions. This constitutes one of the mechanisms responsible of the anaemia of inflammation frequently observed in metastatic breast cancer patients [114].
Hepcidin in prostate cancer
Also in prostate cancer an autocrine loop of Hepcidin/Ferroportin has been demonstrated, with increased intracellular iron retention being essential for cancer survival.
In in vitro studies, prostate cancer-derived Hepcidin was able to increase intracellular iron concentrations in three different prostate cancer cell lines (LNCap, PC3 and DU145) and this was associated to enhanced cell proliferation, migratory capacities and anti-apoptotic properties [115].
In prostate cancer a peculiar mechanism of Hepcidin upregulation has been found that implicates the hypermethylation and silencing of the gene encoding for a natural BMP antagonist, the sclerostin domain containing 1 protein (SOSTDC1) [116]. SOSTDC1 silencing was associated to rapid progression in patients with prostate cancer.
Patients with bone metastasis from prostate cancer have particularly increased circulating levels of Hepcidin as compared to non-metastatic patients. This increase seems to be related to a switch in cytokine synthesis towards IL-6 production [117].
Hepcidin in kidney cancer
Given the importance of intracellular iron sequestration for cancer cell proliferation and metastatic spread and that cancer cell-derived Hepcidin might work as autocrine loop to guarantee high iron content also in the kidney and the genitourinary tract [103], Wang et al. have interrogated The Cancer Genome Atlas (TCGA) and Clinical Proteomic Tumour Analysis Consortium (CPTAC) databases for the expression and correlative analyses of Hepcidin in clear cell renal cell carcinoma (ccRCC) [118]. Hepcidin mRNA was significantly higher in ccRCC tissue than in the normal tissue counterpart and there was also a direct correlation with increasing tumour stage (higher Hepcidin mRNA in Stage IV vs Stage I tumours). Moreover, high Hepcidin was associated with worse survival outcomes (hazard ratio 1.52 and 1.44, for overall and progression-free survival, respectively, both P < 0.001), and with increased expression of immune-checkpoint markers such as PD-L1 and CTLA4, which suggests a possible contribution of Hepcidin to immune escape mechanisms.
Hepcidin in lung cancer
Hepcidin expression is significantly higher in lung cancer tissue as compared to the normal counterpart. Moreover, it is associated with the presence of metastasis and shorter survival in lung cancer patients [119]. Hepcidin upregulation in lung cancer seems to be associated with enhanced immune infiltrates in the tumour microenvironment, indicating a possible mechanism of immune escape, which is being evaluated as predictive factor of response in clinical trials of immunotherapeutic agents.
Dysfunctional immune response has been observed in lungs with excessive iron retention. It has been demonstrated that high Hepcidin and IL-6 levels are present in lung tissue of chronic obstructive pulmonary disease (COPD) patients and this determines the accumulation of iron-rich macrophages. Iron-loaded macrophages are less capable of clearing lung infections and other pathogens with a resulting immune response dysfunction [120]. Subjects with increased Hepcidin blood concentrations are less responsive to vaccination against lung pathogens [121]. Dysregulation of Hepcidin and iron metabolism has been put forward to explain the development of cancer in COPD patients in an observational study including more than 100 subjects [122].
Hepcidin in HCC
Iron metabolism strongly influences most liver diseases, including hepatocellular carcinoma (HCC). Liver is the major source of hepcidin and it has been demonstrated that HCC is typically featured by dysregulated iron sensing mechanisms [123].
Hepatocellular carcinoma appears distinct from other malignancies in that low levels of serum Hepcidin have been observed in HCC patients. In humans, reduced hepcidin has also been found in HCC tissues as compared to adjacent non-cancerous liver tissues. Moreover, animal models of HCC and HCC cell lines are characterised by low hepcidin [124]. Kijima et al., by analysing multiple HCC tumours, reported that Hepcidin expression was low independently of the grade of histological differentiation, number of cancer foci or vessel invasion [125]. Low Hepcidin concentrations can be also found in many hepatic diseases, including HCV- and HBV-related hepatitis, hereditary hemochromatosis, and non-alcoholic steatohepatitis/fatty liver disease (NASH/NAFLD), that are all potential precursors of HCC [126].
Several mechanisms seem to underlie the peculiarity of low Hepcidin expression observed in HCC. When plasma iron concentration is high, iron binds to the iron transporter transferrin (TF) that then binds to its receptor TFR2 on the hepatocyte surface, thus triggering the liver synthesis and release of Hepcidin [127]. Liver-derived Hepcidin downregulates ferroportin protein expression on enterocyte and macrophage surfaces, stopping the iron efflux from cells into plasma. A lower expression of TFR2 has been observed in HCC as compared to normal tissue [128], resulting in ‘transferrin-resistance’ and persistently low levels of Hepcidin despite iron overload [129]. Another mechanism of high iron and low Hepcidin observed in HCC involves the bone morphogenetic protein 6—small mother against decapentaplegic (BMP6-SMAD) pathway, that is the signal physiologically responsible for hepatic Hepcidin synthesis in response to iron excess [130]. As part of this pathway, BMP6 ligand, whose activity is mediated by the transcription factor RUNX3, is the major contributor of Hepcidin release by binding its receptors and co-receptors (i.e., Hemojuvelin) on hepatocytes surface [131]. In HCC, it has been demonstrated that the BMP-SMAD pathway could be suppressed because of gene hypermethylation of BMP6 and/or RUNX3 as well as because of reduced expression on hepatocyte surface of Hemojuvelin, thus determining unresponsiveness to iron excess [132, 133].
The HAMP gene, encoding hepcidin, can itself be silenced by the methylation of its promoter in some HCC models. Moreover, HAMP transcription can be induced by p53. Inactivating TP53 mutations, frequently found in HCC, could be responsible of Hepcidin decrease in some HCCs [134].
Abnormal low Hepcidin levels and resulting high circulating iron concentrations cause hepatocellular oxidative stress and increase in the profibrogenic transforming growth factor β1 (TGFβ1) from hepatic stellate cells [135]. Hepatic stellate cell-driven fibrogenesis seems to be a key step in HCC carcinogenesis [136].
Hepcidin downregulation in HCC is associated with increased cancer proliferation and a more aggressive cancer behaviour, and this renders Hepcidin downregulation a predictor of poor prognosis. Shen et al., by revising more than 350 liver tumour tissue samples, confirmed that HAMP expression is significantly suppressed in HCC as compared to normal tissue and that Hepcidin reduction paralleled the extent of metastasis. Moreover, low HAMP expression was demonstrated to affect both in vivo and in vitro cell-cycle checkpoints by increasing the proportion of cells in the S phase of the cell cycle, via activation of the cyclin-dependent kinases 1/signal transducer and activator of transcription 3 (CDK1/STAT3) pathway, thus enhancing HCC proliferation and aggressiveness [137]. In another study by Abd Elmonem et al. liver biopsies were obtained from 36 HCC patients, 30 patients with chronic hepatitis C and 20 healthy donors. Hepcidin mRNA expression was the lowest in HCC, intermediate in chronic hepatitis C and the highest in healthy liver [138]. Moreover, Hepcidin levels were significantly lower in patients with many tumour lesions than in patients with few lesions. Hepcidin mRNA content was also inversely correlated with serum ferritin concentrations and grade of liver fibrosis.
Hepcidin in brain tumours
Blood–brain barrier constitutes a filter for excessive iron entry in the central nervous system (CNS) [139]. Due to its protected anatomical condition, brain regulates all the genes involved in iron metabolism, including the Hepcidin gene, at a local level, and this has been proven in murine models [140] as well as in human specimens with the cortex and the thalamus being the main sites of regulation of iron pathway-related genes [141].
Recently, Dong et al. have explored the role of Hepcidin in primary brain tumours, specifically in glioblastoma mutitiforme, by using the CGGA, TCGA, Rembrandt and Gravendeel glioma databases [142]. Hepcidin was demonstrated to be over-expressed in glioma cells as compared to normal brain tissue. Moreover, tumour Hepcidin levels were independently associated to shorter overall survival. Furthermore, Hepcidin was found to influence tumour immune microenvironment, with increased tumour infiltration of B cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells. A positive relationship was also demonstrated between Hepcidin and mutational tumour burden and expression of immune-checkpoint markers such as PD-1, PD-L1 and CTLA4. This was associated to reduced sensitivity to anticancer drugs such as temozolomide, nevertheless, it could represent a potential target for immunotherapeutic agents.
Hepcidin in haematologic cancers
In haematologic cancers, Hepcidin was found to play a major role in the pathogenesis of anaemia typically observed in leukaemias, lymphomas and myelomas. In particular in Hodgkin’s lymphoma (HL) patients, significantly higher plasma Hepcidin concentrations, lower levels of iron and lower total iron-binding capacity were found in anaemic patients as compared to non-anaemic patients [143].
In Hodgkin’s lymphoma, diffuse large B-cell non-Hodgkin lymphoma and multiple myeloma (MM) patients, high Hepcidin levels have been correlated to upregulation of IL-6 and BMP2, both representative of background inflammation and acute-phase reaction [143,144,145]. In preclinical models, targeted inhibition of BMP2 demonstrated potential efficacy against Hepcidin-driven anaemia [146].
On the other hand, in adults and children with myeloid leukaemia, high Hepcidin levels induced by repeated blood transfusions, given as part of the therapeutic management, were found to be protective against iron overload during allogenic transplant [147]. Accordingly, in the post-transplant phase, a reduction in Hepcidin was observed thus restoring efficient erythropoiesis [148].
Hepcidin was found to correlate with more aggressive disease in Hodginkin’s lymphoma. Plasma Hepcidin concentrations were higher in stage IV disease, in lymphomas with B symptoms and lymphomas with poor prognostic scores [143]. In MM patients, Hepcidin was inversely correlated with treatment response rate and duration of response [149].
Hepcidin in other cancers
Evidence exists indicating that Hepcidin might play a role also in pancreatic, gastric and thyroid cancer. SNPs of genes involved in HAMP regulation (such as BMP2, BMP6, Hemojuvelin and transferrin receptor genes) have been correlated to pancreatic cancer risk [150], and tumour expression of Hepcidin was associated with shorter overall survival in pancreatic cancer patients [151]. In a study by Zuo et al., Hepcidin was over-expressed in gastric cancer tissue as compared to the adjacent normal tissue, and this correlated with enhancement in JAK/STAT3 signalling and STAT3 affinity to the promoter of Hepcidin gene. In thyroid cancer, Hepcidin upregulation was found to be essential for cell proliferation and regulated by the protein E4BP4 [152].
Local tumour synthesis vs systemic increase of hepcidin in cancer patients
Initial investigations on Hepcidin in cancer were focused on the circulating increment of the liver-derived hormone associated with enhanced systemic inflammation and iron-restricted anaemia usually accompanying tumour progression.
More recently, Hepcidin synthesis has been documented in cancer cells of different origin and a number of local modulating effects have been suggested.
An increase in serum Hepcidin levels has been consistently detected across different tumour types. The increase seems to be chiefly correlated with the enhanced tumour production of the inflammatory cytokine IL-6, which is a recognised activator of Hepcidin synthesis in the liver [153, 154]. The clinical effect is the so-called anaemia of inflammation with hypoferremia, restricted iron availability in the active erythropoietic tissue and iron sequestration in the reticuloendothelial system of liver and spleen. High serum Hepcidin and cancer-related anaemia are poor prognostic factors but are more felt as indicator of higher tumour burden than bioactive inducers of cancer progression. In many studies no clear correlation was found between circulating Hepcidin and Hepcidin expression in cancer tissue, thus reinforcing that the main source of circulating Hepcidin in cancer patients is not the tumour itself [155]. In solid cancers where obesity may play a mechanistic role in disease development and progression, adipose tissue was found to be another potential source of circulating Hepcidin [156, 157].
On the other hand, a clear implication in cancerogenesis and acquisition of malignant phenotype was demonstrated for local iron dysregulation and local synthesis of Hepcidin in cancer cells. HAMP, the gene encoding for Hepcidin, has been found over-expressed in many cancer cell lines, including renal cell carcinoma, breast cancer, prostate cancer and colorectal cancer [109, 116, 158, 159]. In studies of human tumour biopsies its overexpression correlates with increasing tumour stage, poor histological features and lymph node metastasis, with little or no expression in the normal cell counterpart [115, 159,160,161,162,163].The increased gene translation seems to be related to the hypomethilation of its promoter [164] and it is further enhanced upon local signalling of IL-6 released by fibroblasts of the tumour microenvironment [113]. From a mechanistic point of view, local Hepcidin upregulation guarantees abundance of intracellular iron in cancer cells which is paramount for cell proliferation [165]. Moreover, it was found that paracrine Hepcidin modulates the expression of many immunosuppressive markers in tumour infiltrating lymphocytes (TILs) thus favouring immune escape [118, 119, 166].
Clinical studies of serum hepcidin levels in cancer patients
Harmonised tests for the measurement of plasma Hepcidin concentration have lately been developed and a number of clinical studies on the diagnostic and prognostic value of circulating Hepcidin levels have been conducted [167]. In general, high Hepcidin levels indicate enhanced cancer-induced systemic inflammation which is associated with poor prognosis especially when other risk factors are present. In a study by Jerkaz et al., plasma Hepcidin was measured with a validated ELISA test in 518 women with early breast cancer enrolled in a prospective trial investigating diet and lifestyle factors after surgery of the primary tumour [168]. Blood samples were drawn 4 to 12 weeks after primary removal and before any adjuvant treatment start. Patients’ characteristics were in line with the expected features of radically resected breast cancer population: half of the patients in pre-menopause, about 70% of patients with hormone-positive breast cancer and about 30% node-positive [156].
Hepcidin levels ranged from 5 to 191 ng/mL, with the median being 16 ng/mL, and the 75th percentile being 28 ng/mL (non-normal distribution). Association of Hepcidin with reduced relapse-free or overall survival was observed in the primary analysis, but this was not statistically significant. However, when the analysis was conducted in obese patients (BMI > 30), the association became more remarkable and statistically significant (Hazard Ratio of about 1.8–1.9 for the highest vs lowest quartile of Hepcidin), and it was maintained in a multivariable model, thus indicating the particularly adverse effect for the concomitant exposure to high systemic Hepcidin levels and high BMI.
Also in another study by Traeger et al., an association between circulating levels of Hepcidin and cancer relapse and metastasis was demonstrated for 38 patients with upper urinary tract urothelial carcinoma and 94 patients with renal cell carcinoma [169].
Another field of research is the diagnostic role of plasma Hepcidin in distinguishing between benign and malignant lesions. Orlandi et al. utilised comprehensive plasma proteomic profiling by means of mass spectrometry to identify proteomic peaks differentially displayed between breast cancer patients, patients with benign breast lesions and healthy volunteers. The peak corresponding to Hepcidin was found to be the one more differentially expressed, together with Ferritin light chain, in 65 breast cancer patients as compared to 88 individuals with benign lesions and 121 healthy controls [112]. The diagnostic power was estimated to be >80% as measured by the Area Under the Curve (AUC). Among breast cancer patients, Hepcidin was particularly high in patients having HER2-positive tumour.
Similar results were found in another series by El-Mahdy et al. including 57 patients with early-stage breast cancer, 16 individuals with benign disease and 30 healthy controls [170].
Circulating Hepcidin levels can be also of potential utility in predicting the risk of developing anaemia with standard oncological treatments, such as chemotherapy or surgery [155, 171, 172].
HIF2α and hepcidin crosstalk
HIF2α and Hepcidin crosstalk is well-known in physiological processes aiming at maintaining iron homeostasis, however, they also cooperate in a number of pathological conditions, including cancer. There are four important processes where the link between HIF2α and Hepcidin has been documented:
-
1.
Hepatic Hepcidin has an inhibitory effect on intestinal HIF2α via a so-called ‘hetero-tissue’ crosstalk.
-
2.
Renal HIF2α increases during hypoxia and anaemia and stimulates renal EPO that ultimately inhibits liver Hepcidin. Another stimulus that increases renal HIF2α to eventually mobilise iron is the thermogenesis that takes place in the adipose tissue. Experimental models characterised by accumulation of renal HIF2alpha, such as IRP1 knockout mice, display pathologically high EPO levels and low Hepcidin levels.
-
3.
Also the liver can generate HIF2α. Liver HIF2α may directly suppress liver Hepcidin in an autocrine loop. Selective deletion in the liver of the VHL gene, the protein that drives the degradation of HIF2α, induces HAMP promoter suppression in hepatocytes. HCV-infected hepatocytes are characterised by increase of intracellular HIF2α due to HCV-induced ROS (reactive oxygen species, known HIF2α stabilisers). High liver HIF2α during HCV infection causes Hepcidin suppression and the iron overload observed in this disease.
-
4.
In cancer, the contemporary increase of Hepcidin and HIF2α is often observed. It is possible that a shift from a negative to a positive control of HIF2α on Hepcidin expression comes along with malignant transformation.
Schwartz et al. have demonstrated that hepatic Hepcidin inhibits intestinal HIF2α [173]. The direct control of hepatic Hepcidin on intestinal HIF2α was unknown in the past, since it was thought that intestinal HIF2α only responded to systemic iron levels and hypoxia [174]. Physiologically, liver responds to iron overload by synthesising hepcidin, which is required to degrade the iron exporter ferroportin in iron-storing cells, thus limiting iron mobilisation into the circulation [175]. Liver Hepcidin acts as an inhibitory regulator also of intestinal HIF2α. It has been demonstrated that Hepcidin-induced ferroportin internalisation in intestinal cells is a regulatory mechanism that enhances HIF2α catabolism thus leading to HIF2α decrement [173]. The suppression of ferroportin in intestinal cells causes intracellular iron accumulation which activates the iron-sensitive prolyl hydroxylase domain of the PH enzymes. The enzymes are responsible of HIF2α degradation. This mechanism prevents further intestinal absorption of iron. Conversely, deletion of the liver hepcidin gene in mouse models induced a stabilisation of ferroportin on the basolateral membrane of enterocytes and iron efflux from intestinal cells into circulation with a drop in intracellular iron concentrations. This leads to PH inhibition and consequent HIF2α increase in enterocytes. Increased HIF2α mechanistically favours iron absorption from the intestinal lumen by activating a number of iron apical transporters, such as the DMT1 [176]. In the experimental recapitulation of human hemochromatosis, such as mice knockout for Hepcidin, where Hepcidin secretion is suppressed and iron overload is present, the selective deletion of HIF2α at the intestinal level is associated with an attenuation of iron hyperabsorption and tissue iron intoxication with improved hemochromatosis [177, 178]. HIF1α does not have a role in the intestinal absorption of iron [179, 180]. Also in certain types of anaemia, suppression of intestinal HIF2α has been demonstrated with reduced absorption of iron from the intestine. In this setting, pharmacological interventions aimed at increasing HIF2α availability, such as inhibitors of the PH degrading enzymes, are able to revert anaemia by increasing dietary iron intake [181].
It has recently been clarified that the main mechanism by which hypoxia induces Hepcidin suppression is not via a direct HIF-mediated inhibition, but by means of the induction of renal EPO and erythropoiesis mainly driven by an increase in HIF2α in the kidney [182]. In other physiological processes, evidence exists that renal HIF2α induces Hepcidin suppression in the liver. In experiments of adaptive thermogenesis where iron is necessary to perform thermogenesis in adipose tissue, the activation of thermogenic fat with the use of adrenoreceptor agonists determined a rapid increase of renal HIF2α which, in turn, induced erythropoietin synthesis in kidney, inhibition of hepatic hepcidin and, eventually, iron mobilisation in the circulation for adipose tissue thermogenesis [183]. In a research by Wilkinson et al., a mouse model of polycythemia with constitutive overexpression of renal HIF2α was generated by deleting IRP1 gene (IRP1–/– mice). Under normal conditions, IRP1 represses HIF2α mRNA, and its deletion determines HIF2α upregulation. Renal HIF2α was the major source of HIF2α overexpression in IRP1−/− mice, and this was associated with significant increase in circulating erythropoietin concentrations, reticulocytosis, polycythemia, and inhibition of Hepcidin mRNA in the liver [184]. Similarly, knockout models of the enzymes degrading HIF2α, PHs, are associated with increased levels of renal HIF2α and erythropoietin and reduced levels of Hepcidin [185]. Also in human settings, inherited conditions with constitutive elevation of HIF1α and HIF2α, such as individuals homozygotes for the loss-of-function VHL(R200W) mutation, an upregulation of renal HIF2α and erythropoietin, increase in red cell mass and haemoglobin and a downregulation of liver Hepcidin have been documented [186]. Pharmacological modulation of HIF2α is now possible, with a number of PH inhibitors, also known as HIF stabilisers, such as daprodustat, vadadustat and roxadustat [187,188,189]. PH inhibitors exploit the adaptive response to hypoxia driven by the upsurge of HIF1α and HIF2α. PH inhibitors induce HIFs upregulation primarily in the kidneys, where they determine an increase in the endogenous production of Erythropoietin, downregulation of liver Hepcidin and improvement in iron absorption. Beneficial effects in terms also of renal and myocardial protection and resolution of chronic disease-associated anaemia have been demonstrated with these drugs [190,191,192]. Conversely, artificial induction of renal damage, such as that induced by cisplatin, is associated with a decrease in renal HIF2α and erythropoietin and an increase in Hepcidin [193].
Liver HIF2α acts as a negative regulator of Hepcidin in HCV infection. Patients with chronic HCV have enhanced iron accumulation in the liver which contributes to the toxic effect of HCV infection. Iron accumulation is guided by HCV-induced upregulation of liver HIF1α and HIF2α. These two mediators directly downregulate Hepcidin expression in the liver [194]. Low Hepcidin expression occurring despite iron overload is a condition that determines iron intoxication similar to that seen in hemochromatosis. This happens when liver remains unresponsive to iron increase because of persistent inhibitory signalling to Hepcidin secretion.
In cancer, as well as in other diseases that imply enhanced systemic inflammation, both hypoxic signals leading to a HIF2α increase, and inflammatory cytokines such as IL-6 that simulate Hepcidin synthesis, coexist. It is therefore not infrequent to observe the contemporary increase in HIF2α and Hepcidin in many solid cancers. Furthermore, a control of HIF2α on hepcidin with direction opposite to what happens in normal processes has been recently documented in cancer at a local level. A peripheral autocrine loop of cancer-derived Hepcidin as tumour-promoting pathway has been correlated to HIF2α expression in colorectal cancer models. Opposite to what is observed for liver-derived hepcidin, tumour-derived Hepcidin is upregulated by local HIF2α, since deletion of HIF2α gene causes a marked reduction of hepcidin expression in cancer cells [109]. Tumour Hepcidin induces an abundance of intracellular iron levels which is required for active cell proliferation. HIF2α, however, seemed to be necessary but not sufficient to drive Hepcidin expression in this model, since activators of HIF2α alone did not trigger hepcidin overexpression [195]. Further, necessary clues of Hepcidin upregulation in this model are currently unknown (Fig. 2). It is possible that tumour-generated HIF2α may have also systemic effect with induction of renal EPO and polycythemia. If an inflammation-induced liver Hepcidin upregulation is present, an anaemia refractory to treatment with exogenous EPO can occur. This has been observed in a breast cancer model [196]. Hepatocellular carcinoma seems to represent a unique condition, in this regard, since it is the only known solid cancer where HIF2α increase is accompanied by a decrease in Hepcidin with mechanisms similar to those observed in hemochromatosis. In HCC, systemic iron intoxication and iron-derived reactive oxygen species seems to play a key role in carcinogenesis and little is known about the influence of intracellular iron levels in hepatocytes. How the difference between HCC and the other solid cancers can be exploited from a therapeutic point of view is still to be investigated.
Table 1 reports the evidence of HIF2α and Hepcidin crosstalk and dynamics of key iron regulators in normal and pathological conditions.
Remarkable evidence now exists that HIF2α and Hepcidin are tightly interconnected for iron regulation in humans and that this might be key for the growth of certain cancers. Overall, HIF2α induces the expression cellular iron importers while Hepcidin suppresses the iron exporter FPN. The contemporary upregulation of HIF2α and Hepcidin determines a powerful synergism whereby iron is trapped into the cell and this sustains cancer cell proliferation. Since pharmacological modulators of both HIF2α and Hepcidin have been developed, there would be a strong rationale to combine them in clinical trials of patients with ‘HIF2α/Hepcidin/iron-dependent’ cancer, and also the use of iron chelators can be taken into consideration. Moreover, given the effect that this network can have on anti-tumour immune response, also combination regimens with immunotherapeutic agents can be considered.
Clinical applications of HIF2α and Hepcidin modulators
Agonists and antagonists of HIF2α and Hepcidin have been developed with potential utility in the treatment of cancer patients. Moreover, iron chelators have long been tested for cancer treatment with alternate results.
Hepcidin antagonists may have a role in treating anaemia of inflammation observed in cancer patients. LY2787106 is a monoclonal antibody binding hepcidin and preventing its interaction with ferroportin. In a Phase I study, 33 cancer patients (mainly MM patients) with hepcidin levels ≥5 ng/mL and an overall mean Haemoglobin of 9 g/dL were treated with LY2787106. All patients experienced an increase in iron concentrations and transferrin saturation, and 4 out of 33 patients experienced a ≥ 0.5 g/dL increase in haemoglobin at week 12 [197].
The direct antitumor effect of inhibiting the local activity of tumour-derived hepcidin is yet to be investigated in human subjects. However, preclinical evidence in colorectal cancer suggests that hepcidin inhibition may reduce cancer growth [109].
Cancers that are crucially driven by HIF2α, such as those associated to the VHL disease, renal cell carcinomas, paragangliomas and neuroendocrine tumours, have recently been found to be highly responsive to specific HIF2α inhibitors.
Belzutifan (also known PT2977 or MK-6482), a first-in-class HIF2α inhibitor, has demonstrated a clinical benefit rate (tumour response plus disease stabilisation) as high as 95% in early phase studies of patients with VHL disease [61]. As expected, anaemia was a side effect occurring in a high percentage of treated patients, almost 90%, and it was usually reversed by erythropoietin (EPO) administration. Future studies are including pretreatment with EPO before HIF2α commencement. Interestingly, the contribution of iron deficiency following reduced iron absorption due to HIF2α inhibition in the small intestine has not been investigated in these patients.
In sporadic ccRCC, early HIF2α inhibitors demonstrated a 14% response rate [198]. Their pharmacokinetics, however, was suboptimal as compared to the next-generation drug belzutifan. In the Phase I trial of belzutifan, a 25% response rate was demonstrated [60], and in a Phase II trial of belzutifan in combination with the multi-tyrosin kinase inhibitor cabozantinib, 31% of response was observed [199].
A report has been published on the use of belzutifan in a heavily pretreated 43-year-old woman with metastatic pancreatic neuroendocrine cancer and VHL disease. The patient experienced a radiographic partial response within 1 month of belzutifan commencement [200].
In another genetic disease characterised by hyper-activation of HIF2α and multiple paragangliomas, the Pacak-Zhuang syndrome, belzutifan yielded rapid and prolonged tumour response in most of the visible lesions [201].
Belzutifan has now obtained FDA approval for the treatment of von Hippel–Lindau disease-associated tumours [202] and is also being tested in association with the antiangiogenic tyrosin kinase inhibitor lenvatinib in Phase III trials of ccRCC patients after failure of immunotherapy, the LITESPARK-011 trial (NCT04586231) [203]. Moreover, Phase III trials are ongoing in association with immunotherapy (pembrolizumab), both in the first-line setting (NCT04736706) and in the adjuvant setting after nephrectomy (NCT05239728).
DFF332 is another HIF2α inhibitor that is being tested in Phase I trials in combination with immunotherapy, namely the immune-checkpoint inhibitor anti-PD-1 spartalizumab (NCT04895748).
Iron chelators have a long history of investigation in cancer trials, however results have been largely disappointing [204]. Novel chelators, such as triapine, have improved pharmacokinetics and produced encouraging results in Phase II trials [205].
There is a strong rationale for combining anti-HIF2α and anti-hepcidin agents in disease settings, such as colorectal cancer, where HIF2α and Hepcidin may play a synergistic pathogenetic role. Also inclusion of iron chelators can be taken into consideration.
HIF2α inducers have been developed (PH inhibitors). A number of studies in patients with kidney disease and anaemia due to suppression of renal HIF2α and EPO have demonstrated the efficacy of PH inhibitors to resolve anaemia. It is possible that some cancer patients will suffer from anaemia of inflammation due to inflammatory induction of hepcidin and suppression of intestinal HIF2α. In this case stabilisation of HIF2alpha with PH inhibitors might be of some benefit, however, the detrimental effect of the HIF2alpha increase must first be ruled out. It is conceivable that cancer types not dependent on HIF2α-related pathways for their growth and with anaemia of inflammation might be candidates to trials with these agents.
Discussion and conclusion
Perturbations of iron homeostasis may consist of deleterious Hepcidin inactivation and intestinal HIF2α induction in the presence of iron load, such as in hemochromatosis, or of inappropriate renal HIF2α suppression and Hepcidin overexpression followed by reduced iron availability and anaemia, such as that observed during kidney disease. A third possible perturbation exists, with an increased inflammatory response that determines a contemporary rise in both Hepcidin and HIF2α and reduced iron availability. This scenario is typical of some solid tumours (Fig. 2).
The latter case is also known as anaemia due to systemic inflammation (anaemia of inflammation) and positive regulators other than iron are able to increase Hepcidin secretion despite low systemic iron levels. In the meanwhile, inflammation and cancer growth are also associated with hypoxia and increase in HIF mediators; thus a contemporary increase of Hepcidin and HIF2α might coexist. Furthermore, in some cancers, such as colorectal cancer, an unexpected direct induction of ectopic hepcidin synthesis driven by HIF2α has been described.
In the present article, we attempted to review the available evidence on the role of HIF2α and Hepcidin in cancer progression and possible evidence for their interaction. There are consistent data demonstrating that HIF2α and Hepcidin are tightly linked in order to guarantee iron homeostasis with a prominent hetero-tissue crosstalk between the liver and intestine. However, the reciprocal regulation of the two mediators seems to work in an opposite fashion during cancer (Fig. 2). Since they have been both separately proven as tumour-promoting factors, and since pharmacological agents acting on their pathways are now available, it would be interesting to study the degree of synergism between the two signals in specific cancer settings and the potential for treatment combinations targeting both mediators. Apart from iron homeostasis, which is known to be necessary for cancer cell survival, other mechanisms, such induction of immunosuppressive environment, neoangiogenesis stimulation, and desmoplastic reaction might be involved in cancer patients with dysregulated HIF2α and Hepcidin signalling. Studies focused on the integrated analysis of the two pathways are highly warranted in oncology.
Data availability
The present review has been written using as source articles that are all publicly available.
References
Morales M, Xue X. Targeting iron metabolism in cancer therapy. Theranostics. 2021;11:8412–29.
Hsu MY, Mina E, Roetto A, Porporato PE. Iron: an essential element of cancer metabolism. Cells. 2020;9:2591.
Guo Q, Li L, Hou S, Yuan Z, Li C, Zhang W, et al. The role of iron in cancer progression. Front Oncol. 2021;11:778492.
Doguer C, Ha JH, Collins JF. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr Physiol. 2018;8:1433–61.
Xu MM, Wang J, Xie JX. Regulation of iron metabolism by hypoxia-inducible factors. Sheng Li Xue Bao. 2017;69:598–610.
Ganz T, Nemeth E. The hepcidin-ferroportin system as a therapeutic target in anemias and iron overload disorders. Hematol Am Soc Hematol Educ Program. 2011;2011:538–42.
Jung M, Mertens C, Tomat E, Brüne B. Iron as a central player and promising target in cancer progression. Int J Mol Sci. 2019;20:273.
Muz B, de la Puente P, Azab F, Azab AK. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia. 2015;3:83–92.
Prabhakar NR, Semenza GL. Oxygen sensing and homeostasis. Physiology. 2015;30:340–8.
Davis L, Recktenwald M, Hutt E, Fuller S, Briggs M, Goel A, et al. Targeting HIF-2α in the tumor microenvironment: redefining the role of HIF-2α for solid cancer therapy. Cancers. 2022;14:1259.
Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9:10–7.
Mole DR. Iron homeostasis and its interaction with prolyl hydroxylases. Antioxid Redox Signal. 2010;12:445–58. https://doi.org/10.1089/ars.2009.2790.
Mackenzie B, Garrick MD. Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G981–6. https://doi.org/10.1152/ajpgi.00363.2005.
Suzuki N, Yamamoto M. Roles of renal erythropoietin-producing (REP) cells in the maintenance of systemic oxygen homeostasis. Pflug Arch. 2016;468:3–12. https://doi.org/10.1007/s00424-015-1740-2.
Gammella E, Diaz V, Recalcati S, Buratti P, Samaja M, Dey S, et al. Erythropoietin’s inhibiting impact on hepcidin expression occurs indirectly. Am J Physiol Regul Integr Comp Physiol. 2015;308:R330–5. https://doi.org/10.1152/ajpregu.00410.2014.
Srole DN, Ganz T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J Cell Physiol. 2021;236:4888–901. https://doi.org/10.1002/jcp.30247.
Drakesmith H, Nemeth E, Ganz T. Ironing out ferroportin. Cell Metab. 2015;22:777–87.
Fisher AL, Babitt JL. Coordination of iron homeostasis by bone morphogenetic proteins: current understanding and unanswered questions. Dev Dyn. 2022;251:26–46. https://doi.org/10.1002/dvdy.372.
Lee DH, Zhou LJ, Zhou Z, Xie JX, Jung JU, Liu Y, et al. Neogenin inhibits HJV secretion and regulates BMP-induced hepcidin expression and iron homeostasis. Blood. 2010;115:3136–45. https://doi.org/10.1182/blood-2009-11-251199.
Nemeth E, Ganz T. Hepcidin and iron in health and disease. Annu Rev Med. 2023;74:261–77. https://doi.org/10.1146/annurev-med-043021-032816.
Arezes J, Foy N, McHugh K, Sawant A, Quinkert D, Terraube V, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132:1473–7. https://doi.org/10.1182/blood-2018-06-857995.
Vela D, Vela-Gaxha Z. Differential regulation of hepcidin in cancer and non-cancer tissues and its clinical implications. Exp Mol Med. 2018;50:e436. https://doi.org/10.1038/emm.2017.273.
Nemeth E, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–6.
Yamamoto K, et al. Interplay of adipocyte and hepatocyte: leptin upregulates hepcidin. Biochem Biophys Res Commun. 2018;495:1548–54.
Savarese G, von Haehling S, Butler J, Cleland JGF, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Eur Heart J. 2023;44:14–27. https://doi.org/10.1093/eurheartj/ehac569.
Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93:1721–41. https://doi.org/10.1152/physrev.00008.2013.
Richardson DR, Kalinowski DS, Lau S, Jansson PJ, Lovejoy DB. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim Biophys Acta. 2009;1790:702–17. https://doi.org/10.1016/j.bbagen.2008.04.003.
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8.
Branco-Price C, Zhang N, Schnelle M, Evans C, Katschinski DM, Liao D, et al. Endothelial cell HIF-1α and HIF-2α differentially regulate metastatic success. Cancer Cell. 2012;21:52–65.
Skuli N, Liu L, Runge A, Wang T, Yuan L, Patel S, et al. Endothelial deletion of hypoxia-inducible factor-2alpha (HIF-2alpha) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–77.
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22.
Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von Hippel-Lindau gene mutation status. Eur Urol. 2016;70:623–32.
Palazon A, Tyrakis PA, Macias D, Veliça P, Rundqvist H, Fitzpatrick S, et al. An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell. 2017;32:669–83.e5.
Xie C, Yagai T, Luo Y, Liang X, Chen T, Wang Q, et al. Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat Med. 2017;23:1298–308.
Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015;5:652–67.
Walter KM, Schönenberger MJ, Trötzmüller M, Horn M, Elsässer HP, Moser AB, et al. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014;20:882–97.
Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–27.
Dengler VL, Galbraith M, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2014;49:1–15.
Bindra RS, Vasselli JR, Stearman R, Linehan WM, Klausner RD. VHL-mediated hypoxia regulation of cyclin D1 in renal carcinoma cells. Cancer Res. 2002;62:3014–9.
Franovic A, Holterman CE, Payette J, Lee S. Human cancers converge at the HIF-2alpha oncogenic axis. Proc Natl Acad Sci USA. 2009;106:21306–11.
Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420–6.
Linehan WM, Ricketts CJ. The cancer genome atlas of renal cell carcinoma: findings and clinical implications. Nat Rev Urol. 2019;16:539–52.
Maher ER, Kaelin WG Jr. von Hippel-Lindau disease. Medicine. 1997;76:381–91.
Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cells. 2002;1:459–68.
Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIFalpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cells. 2008;14:435–46.
Kondo K, Kim WY, Lechpammer M, Kaelin, Jr WG. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:439–44.
Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxiainducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res. 2004;2:89–95.
Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cells. 2002;1:237–46.
Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. 2019;12:27.
Choueiri TK, Kaelin WG Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat Med. 2020;26:1519–30.
Navani V, Heng DYC. Treatment selection in first-line metastatic renal cell carcinoma-the contemporary treatment paradigm in the age of combination therapy: a review. JAMA Oncol. 2022;8:292–9.
Ferician AM, Ferician OC, Cumpanas AD, Berzava PL, Nesiu A, Barmayoun A, et al. Heterogeneity of platelet derived growth factor pathway gene expression profile defines three distinct subgroups of renal cell carcinomas. Cancer Genom Proteom. 2022;19:477–89.
Uhlman DL, Nguyen P, Manivel JC, Zhang G, Hagen K, Fraley E, et al. Epidermal growth factor receptor and transforming growth factor alpha expression in papillary and nonpapillary renal cell carcinoma: correlation with metastatic behavior and prognosis. Clin Cancer Res. 1995;1:913–20.
Dawson NA, Guo C, Zak R, Dorsey B, Smoot J, Wong J, et al. A phase II trial of gefitinib (Iressa, ZD1839) in stage IV and recurrent renal cell carcinoma. Clin Cancer Res. 2004;10:7812–9.
Nakaigawa N, Yao M, Baba M, Kato S, Kishida T, Hattori K, et al. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006;66:3699–705.
Chu Q, Han N, Yuan X, Nie X, Wu H, Chen Y, et al. DACH1 inhibits cyclin D1 expression, cellular proliferation and tumor growth of renal cancer cells. J Hematol Oncol. 2014;7:73.
Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, et al. A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 2016;76:5491–500.
Xu R, Wang K, Rizzi JP, Huang H, Grina JA, Schlachter ST, et al. 3-[(1 S,2 S,3 R)-2,3-difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzo nitrile (PT2977), a hypoxia-inducible factor 2α (HIF-2α) inhibitor for the treatment of clear cell renal cell carcinoma. J. Med. Chem. 2019;62:6876–93.
Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med. 2021;27:802–5.
Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for Renal cell carcinoma in von Hippel-Lindau disease. N Engl J Med. 2021;385:2036–46.
Wei H, Ke HL, Lin J, Shete S, Wood CG, Hildebrandt MA. MicroRNA target site polymorphisms in the VHL-HIF1α pathway predict renal cell carcinoma risk. Mol Carcinog. 2014;53:1–7. https://doi.org/10.1002/mc.21917.
Greene CJ, Attwood K, Sharma NJ, Gross KW, Smith GJ, Xu B, et al. Transferrin receptor 1 upregulation in primary tumor and downregulation in benign kidney is associated with progression and mortality in renal cell carcinoma patients. Oncotarget. 2017;8:107052–75.
Ricchi P, Ammirabile M, Spasiano A, Costantini S, Di Matola T, Cartenì G, et al. Renal cell carcinoma in adult patients with thalassaemia major: a description of three cases. Br J Haematol. 2014;165:887–8.
Partanen T, Heikkilä P, Hernberg S, Kauppinen T, Moneta G, Ojajärvi A. Renal cell cancer and occupational exposure to chemical agents. Scand J Work Environ Health. 1991;17:231–9.
Weinberg ED. Tobacco smoke iron: an initiator/promoter of multiple diseases. Biometals. 2009;22:207–10.
Li JL, Okada S, Hamazaki S, Ebina Y, Midorikawa O. Subacute nephrotoxicity and induction of renal cell carcinoma in mice treated with ferric nitrilotriacetate. Cancer Res. 1987;47:1867–9.
Ebina Y, Okada S, Hamazaki S, Ogino F, Li JL, Midorikawa O. Nephrotoxicity and renal cell carcinoma after use of iron- and aluminum-nitrilotriacetate complexes in rats. J Natl Cancer Inst. 1986;76:107–13.
Greene CJ, Sharma NJ, Fiorica PN, Forrester E, Smith GJ, Gross KW, et al. Suppressive effects of iron chelation in clear cell renal cell carcinoma and their dependency on VHL inactivation. Free Radic Biol Med. 2019;133:295–309.
Green YS, Ferreira Dos Santos MC, Fuja DG, Reichert EC, Campos AR, Cowman SJ, et al. ISCA2 inhibition decreases HIF and induces ferroptosis in clear cell renal carcinoma. Oncogene. 2022. https://doi.org/10.1038/s41388-022-02460-1.
Ramakrishnan SK, Shah YM. Role of intestinal HIF-2α in health and disease. Annu Rev Physiol. 2016;78:301–25.
Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, et al. Hypoxia-inducible factor-2α activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 2012;72:2285–93.
Chun SY, Johnson C, Washburn JG, Cruz-Correa MR, Dang DT, Dang LH. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol Cancer. 2010;9:293.
Yoshimura H, Dhar DK, Kohno H, Kubota H, Fujii T, Ueda S, et al. Prognostic impact of hypoxia-inducible factors 1alpha and 2alpha in colorectal cancer patients: correlation with tumor angiogenesis and cyclooxygenase-2 expression. Clin Cancer Res. 2004;10:8554–60.
Nijkamp MW, van der Bilt JD, de Bruijn MT, Molenaar IQ, Voest EE, van Diest PJ, et al. Accelerated perinecrotic outgrowth of colorectal liver metastases following radiofrequency ablation is a hypoxia-driven phenomenon. Ann Surg. 2009;249:814–23.
Malier M, Gharzeddine K, Laverriere MH, Marsili S, Thomas F, Decaens T, et al. Hypoxia drives dihydropyrimidine dehydrogenase expression in macrophages and confers chemoresistance in colorectal cancer. Cancer Res. 2021;81:5963–76.
Schito L, Rey S, Xu P, Man S, Cruz-Muñoz W, Kerbel RS. Metronomic chemotherapy offsets HIFα induction upon maximum-tolerated dose in metastatic cancers. EMBO Mol Med. 2020;12:e11416.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Singhal R, Mitta SR, Das NK, Kerk SA, Sajjakulnukit P, Solanki S, et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest. 2021;131:e143691.
Yan Y, He M, Zhao L, Wu H, Zhao Y, Han L, et al. A novel HIF-2α targeted inhibitor suppresses hypoxia-induced breast cancer stemness via SOD2-mtROS-PDI/GPR78-UPRER axis. Cell Death Differ. 2022. https://doi.org/10.1038/s41418-022-00963-8.
Fu X, Jeselsohn R, Pereira R, Hollingsworth EF, Creighton CJ, Li F, et al. FOXA1 overexpression mediates endocrine resistance by altering the ER transcriptome and IL-8 expression in ER-positive breast cancer. Proc Natl Acad Sci USA. 2016;113:E6600–9.
Fu X, Pereira R, De Angelis C, Veeraraghavan J, Nanda S, Qin L, et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc Natl Acad Sci USA. 2019;116:26823–34.
Ströfer M, Jelkmann W, Depping R. Curcumin decreases survival of Hep3B liver and MCF-7 breast cancer cells: the role of HIF. Strahlenther Onkol. 2011;187:393–400.
Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48:919–22.
Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32:71.e7.
Colbert LE, Fisher SB, Balci S, Saka B, Chen Z, Kim S, et al. High nuclear hypoxia-inducible factor 1 alpha expression is a predictor of distant recurrence in patients with resected pancreatic adenocarcinoma. Int J Radiat Oncol Biol Phys. 2015;91:631–9.
Garcia Garcia CJ, Huang Y, Fuentes NR, Turner MC, Monberg ME, Lin D, et al. Stromal HIF2 regulates immune suppression in the pancreatic cancer microenvironment. Gastroenterology. 2022;162:2018–31.
Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. 2020;31:267–83.e12.
Chen R, Lai LA, Sullivan Y, Wong M, Wang L, Riddell J, et al. Disrupting glutamine metabolic pathways to sensitize gemcitabine-resistant pancreatic cancer. Sci Rep. 2017;7:7950.
Zhang Q, Lou Y, Zhang J, Fu Q, Wei T, Sun X, et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol Cancer. 2017;16:119.
Yang J, Zhang X, Zhang Y, Zhu D, Zhang L, et al. HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J Exp Clin Cancer Res. 2016;35:26.
Méndez-Blanco C, Fernández-Palanca P, Fondevila F, González-Gallego J, Mauriz JL. Prognostic and clinicopathological significance of hypoxia-inducible factors 1α and 2α in hepatocellular carcinoma: a systematic review with meta-analysis. Ther Adv Med Oncol. 2021;13:1758835920987071.
Llovet JM, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer. 2022;3:386–401.
Cramer T, Vaupel P. Severe hypoxia is a typical characteristic of human hepatocellular carcinoma: scientific fact or fallacy? J Hepatol. 2022;76:975–80.
Younes R, Bugianesi E. Should we undertake surveillance for HCC in patients with NAFLD? J Hepatol. 2018;68:326–34.
Foglia B, Sutti S, Cannito S, Rosso C, Maggiora M, Autelli R, et al. Hepatocyte-specific deletion of HIF2α prevents NASH-related liver carcinogenesis by decreasing cancer cell proliferation. Cell Mol Gastroenterol Hepatol. 2022;13:459–82.
Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. 2019;18:163.
Méndez-Blanco C, Fondevila F, García-Palomo A, González-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50:1–9.
Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–10.
Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768–72.
Kroot JJ, Tjalsma H, Fleming RE, Swinkels DW. Hepcidin in human iron disorders: diagnostic implications. Clin Chem. 2011;57:1650–69.
Mittler R, Darash-Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, et al. NEET proteins: a new link between iron metabolism, reactive oxygen species, and cancer. Antioxid Redox Signal. 2019;30:1083–95.
Sacco A, Battaglia AM, Botta C, Aversa I, Mancuso S, Costanzo F, et al. Iron metabolism in the tumor microenvironment-implications for anti-cancer immune response. Cells. 2021;10:303.
Santana-Codina N, Del Rey MQ, Kapner KS, Zhang H, Gikandi A, Malcolm C, et al. NCOA4-mediated ferritinophagy is a pancreatic cancer dependency via maintenance of iron bioavailability for iron-sulfur cluster proteins. Cancer Discov. 2022;12:2180–97.
Lelièvre P, Sancey L, Coll JL, Deniaud A, Busser B. Iron dysregulation in human cancer: altered metabolism, biomarkers for diagnosis, prognosis, monitoring and rationale for therapy. Cancers. 2020;12:3524.
Le NT, Richardson DR. The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta. 2002;1603:31–46.
Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron uptake via DMT1 integrates cell cycle with JAK-STAT3 signaling to promote colorectal tumorigenesis. Cell Metab. 2016;24:447–61.
Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut. 2006;55:1449–60.
Schwartz AJ, Goyert JW, Solanki S, Kerk SA, Chen B, Castillo C, et al. Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab. 2021;3:969–82.
Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2:43ra56.
Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin–ferroportin signaling promotes breast cancer growth. Cell Signal. 2014;26:2539–50.
Orlandi R, De Bortoli M, Ciniselli CM, Vaghi E, Caccia D, Garrisi V, et al. Hepcidin and ferritin blood level as noninvasive tools for predicting breast cancer. Ann Oncol. 2014;25:352–7.
Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene. 2018;37:4013–32.
Villarroel P, Le Blanc S, Arredondo M. Interleukin-6 and lipopolysaccharide modulate hepcidin mRNA expression by HepG2 cells. Biol Trace Elem Res. 2012;150:496–501.
Zhao B, Li R, Cheng G, Li Z, Zhang Z, Li J, et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol Lett. 2018;15:9953–8.
Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015;75:2254–63.
Tanno T, Rabel A, Alleyne M, Lee YT, Dahut WL, Gulley JL, et al. Hepcidin, anaemia, and prostate cancer. B J U Int. 2011;107:678–9.
Wang X, Shi Q, Gong P, Zhou C, Cao Y. An integrated systematic analysis and the clinical significance of hepcidin in common malignancies of the male genitourinary system. Front Genet. 2022;13:771344.
Fan Y, Liu B, Chen F, Song Z, Han B, Meng Y, et al. hepcidin upregulation in lung cancer: a potential therapeutic target associated with immune infiltration. Front Immunol. 2021;12:612144.
Ho T, Nichols M, Nair G, Radford K, Kjarsgaard M, Huang C, et al. Iron in airway macrophages and infective exacerbations of chronic obstructive pulmonary disease. Respir Res. 2022;23:8.
Frost JN, Tan TK, Abbas M, Wideman SK, Bonadonna M, Stoffel NU, et al. Hepcidin-mediated hypoferremia disrupts immune responses to vaccination and infection. Medicine. 2021;2:164–79.e12.
Brzóska K, Bartłomiejczyk T, Sochanowicz B, Cymerman M, Grudny J, Kołakowski J, et al. Carcinogenesis-related changes in iron metabolism in chronic obstructive pulmonary disease subjects with lung cancer. Oncol Lett. 2018;16:6831–7.
Maegdefrau U, Arndt S, Kivorski G, Hellerbrand C, Bosserhoff AK. Downregulation of hemojuvelin prevents inhibitory effects of bone morphogenetic proteins on iron metabolism in hepatocellular carcinoma. Lab Invest. 2011;91:1615–23.
Udali S, Castagna A, Corbella M, Ruzzenente A, Moruzzi S, Mazzi F, et al. Hepcidin and DNA promoter methylation in hepatocellular carcinoma. Eur J Clin Invest. 2018;48:e12870.
Kijima H, Sawada T, Tomosugi N, Kubota K. Expression of hepcidin mRNA is uniformly suppressed in hepatocellular carcinoma. BMC Cancer. 2008;8:167.
Kew MC. Hepatic iron overload and hepatocellular carcinoma. Liver Cancer. 2014;3:31–40.
Rapisarda C, Puppi J, Hughes RD, Dhawan A, Farnaud S, Evans RW, et al. Transferrin receptor 2 is crucial for iron sensing in human hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2010;299:G778–83.
Tan MG, Kumarasinghe MP, Wang SM, Ooi LL, Aw SE, Hui KM. Modulation of iron-regulatory genes in human hepatocellular carcinoma and its physiological consequences. Exp Biol Med. 2009;234:693–702.
Worthen CA, Enns CA. The role of hepatic transferrin receptor 2 in the regulation of iron homeostasis in the body. Front Pharmacol. 2014;5:34.
Rishi G, Wallace DF, Subramaniam VN. Hepcidin: regulation of the master iron regulator. Biosci Rep. 2015;35:e00192.
Sangkhae V, Nemeth E. Regulation of the iron homeostatic hormone hepcidin. Adv Nutr. 2017;8:126–36.
He Y, Cui Y, Xu B, Gu J, Wang W, Luo X. Hypermethylation leads to bone morphogenetic protein 6 downregulation in hepatocellular carcinoma. PLoS ONE. 2014;9:e87994.
Park WS, Cho YG, Kim CJ, Song JH, Lee YS, Kim SY, et al. Hypermethylation of the RUNX3 gene in hepatocellular carcinoma. Exp Mol Med. 2005;37:276–81.
Weizer-Stern O, Adamsky K, Margalit O, Ashur-Fabian O, Givol D, Amariglio N, et al. Hepcidin, a key regulator of iron metabolism, is transcriptionally activated by p53. Br J Haematol. 2007;138:253–62.
Han CY, Koo JH, Kim SH, Gardenghi S, Rivella S, Strnad P, et al. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat Commun. 2016;7:13817.
Zhang DY, Friedman SL. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology. 2012;56:769–75.
Shen Y, Li X, Su Y, Badshah SA, Zhang B, Xue Y, et al. HAMP downregulation contributes to aggressive hepatocellular carcinoma via mechanism mediated by cyclin4-dependent kinase-1/STAT3 pathway. Diagnostics. 2019;9:48.
Abd Elmonem E, Tharwa EL-S, Farag MA, Fawzy A, El Shinnawy SF, Suliman S. Hepcidin mRNA level as a parameter of disease progression in chronic hepatitis C and hepatocellular carcinoma. J Egypt Natl Canc Inst. 2009;21:333–42.
Burdo JR, Connor JR. Brain iron uptake and homeostatic mechanisms: an overview. Biometals. 2003;16:63–75. https://doi.org/10.1023/a:1020718718550.
Zechel S, Huber-Wittmer K, von Bohlen und Halbach O. Distribution of the iron-regulating protein hepcidin in the murine central nervous system. J Neurosci Res. 2006;84:790–800. https://doi.org/10.1002/jnr.20991.
Hänninen MM, Haapasalo J, Haapasalo H, Fleming RE, Britton RS, Bacon BR, et al. Expression of iron-related genes in human brain and brain tumors. BMC Neurosci. 2009;10:36. https://doi.org/10.1186/1471-2202-10-36.
Dong T, Zhang B, Zhang R, Wang C, Liu X, Wang F, et al. Hepcidin is upregulated and is a potential therapeutic target associated with immunity in glioma. Front Oncol. 2022;12:963096. https://doi.org/10.3389/fonc.2022.963096. Erratum in: Front Oncol. 2022 Dec 06;12:1085757.
Hohaus S, Massini G, Giachelia M, Vannata B, Bozzoli V, Cuccaro A, et al. Anemia in Hodgkin’s lymphoma: the role of interleukin-6 and hepcidin. J Clin Oncol. 2010;28:2538–43. https://doi.org/10.1200/JCO.2009.27.6873.
Tisi MC, Bozzoli V, Giachelia M, Massini G, Ricerca BM, Maiolo E, et al. Anemia in diffuse large B-cell non-Hodgkin lymphoma: the role of interleukin-6, hepcidin and erythropoietin. Leuk Lymphoma. 2014;55:270–5. https://doi.org/10.3109/10428194.2013.802314.
Sharma S, Nemeth E, Chen YH, Goodnough J, Huston A, Roodman GD, et al. Involvement of hepcidin in the anemia of multiple myeloma. Clin Cancer Res. 2008;14:3262–7. https://doi.org/10.1158/1078-0432.CCR-07-4153.
Maes K, Nemeth E, Roodman GD, Huston A, Esteve F, Freytes C, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood. 2010;116:3635–44. https://doi.org/10.1182/blood-2010-03-274571.
Eisfeld AK, Westerman M, Krahl R, Leiblein S, Liebert UG, Hehme M, et al. Highly elevated serum hepcidin in patients with acute myeloid leukemia prior to and after allogeneic hematopoietic cell transplantation: does this protect from excessive parenchymal iron loading? Adv Hematol. 2011;2011:491058. https://doi.org/10.1155/2011/491058.
Słomka A, Łęcka M, Styczyński J. Hepcidin in children and adults with acute leukemia or undergoing hematopoietic cell transplantation: a systematic review. Cancers. 2022;14:4936. https://doi.org/10.3390/cancers14194936.
Katodritou E, Ganz T, Terpos E, Verrou E, Olbina G, Gastari V, et al. Sequential evaluation of serum hepcidin in anemic myeloma patients: study of correlations with myeloma treatment, disease variables, and anemia response. Am J Hematol. 2009;84:524–6. https://doi.org/10.1002/ajh.21448.
Julián-Serrano S, Yuan F, Wheeler W, Benyamin B, Machiela MJ, Arslan AA, et al. Hepcidin-regulating iron metabolism genes and pancreatic ductal adenocarcinoma: a pathway analysis of genome-wide association studies. Am J Clin Nutr. 2021;114:1408–17. https://doi.org/10.1093/ajcn/nqab217.
Toshiyama R, Konno M, Eguchi H, Asai A, Noda T, Koseki J, et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol Lett. 2018;15:8125–33. https://doi.org/10.3892/ol.2018.8357.
Zhou Q, Chen J, Feng J, Wang J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis. 2018;9:987. https://doi.org/10.1038/s41419-018-1001-3.
Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal. 2014;26:2539–50. https://doi.org/10.1016/j.cellsig.2014.07.029.
Noguchi-Sasaki M. Treatment with anti-IL-6 receptor antibody prevented increase in serum hepcidin levels and improved anemia in mice inoculated with IL-6-producing lung carcinoma cells. BMC Cancer. 2016;16:270. https://doi.org/10.1186/s12885-016-2305-2.
Pan X, Lu Y, Cheng X, Wang J. Hepcidin and ferroportin expression in breast cancer tissue and serum and their relationship with anemia. Curr Oncol. 2016;23:e24–6. https://doi.org/10.3747/co.23.2840.
Jerzak KJ, Lohmann AE, Ennis M, Nemeth E, Ganz T, Goodwin PJ. Prognostic associations of plasma hepcidin in women with early breast cancer. Breast Cancer Res Treat. 2020;184:927–35. https://doi.org/10.1007/s10549-020-05903-z.
Phillips E. A potential role for hepcidin in obesity-driven colorectal tumourigenesis. Oncol Rep. 2018;39:392–400. https://doi.org/10.3892/or.2017.6062.
Tang Y, Ge S, Zheng X, Zheng J. High Hepcidin expression predicts poor prognosis in patients with clear cell renal cell carcinoma. Diagn Pathol. 2022;17:100. https://doi.org/10.1186/s13000-022-01274-9.
Yalovenko TM, Todor IM, Lukianova NY, Chekhun VF. Hepcidin as a possible marker in determination of malignancy degree and sensitivity of breast cancer cells to cytostatic drugs. Exp Oncol. 2016;38:84–8.
Huang J, Liu W, Song S, Li JC, Gan K, Shen C, et al. The iron-modulating hormone hepcidin is upregulated and associated with poor survival outcomes in renal clear cell carcinoma. Front Pharm. 2022;13:1080055. https://doi.org/10.3389/fphar.2022.1080055.
Qiu H, Gu G, Zuo E, Cheng X. Tumoral overexpression of hepcidin is associated with poor prognosis of patients with clear cell renal cell carcinoma. Cancer Invest. 2023;41:84–92. https://doi.org/10.1080/07357907.2022.2133775.
Sornjai W. Iron and hepcidin mediate human colorectal cancer cell growth. Chem Biol Interact. 2020;319:109021. https://doi.org/10.1016/j.cbi.2020.109021.
Xiang-Tao P. Expression of Hepcidin and Neogenin in Colorectal Cancer. Open Med. 2017;12:184–8. https://doi.org/10.1515/med-2017-0027.
Zhou Z, Wu J, Yang Y, Gao P, Wang L, Wu Z. Hepcidin as a prognostic biomarker in clear cell renal cell carcinoma. Am J Cancer Res. 2022;12:4120–39.
Wang F. Hepcidin and iron metabolism in the pathogenesis of prostate cancer. J BUON. 2017;22:1328–32.
Di Grazia A, Di Fusco D, Franzè E, Colella M, Strimpakos G, Salvatori S, et al. Hepcidin upregulation in colorectal cancer associates with accumulation of regulatory macrophages and epithelial-mesenchymal transition and correlates with progression of the disease. Cancers. 2022;14:5294. https://doi.org/10.3390/cancers14215294.
van der Vorm LN, Hendriks JC, Laarakkers CM, Klaver S, Armitage AE, Bamberg A, et al. Toward worldwide hepcidin assay harmonization: identification of a commutable secondary reference material. Clin Chem. 2016;62:993–1001. https://doi.org/10.1373/clinchem.2016.256768.
Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, et al. Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol. 2002;20:42–51. https://doi.org/10.1200/JCO.2002.20.1.42.
Traeger L. Serum Hepcidin and GDF-15 levels as prognostic markers in urothelial carcinoma of the upper urinary tract and renal cell carcinoma. BMC Cancer. 2019;19:74. https://doi.org/10.1186/s12885-019-5278-0.
El-Mahdy RI. Circulating osteocyte-related biomarkers (vitamin D, sclerostin, dickkopf-1), hepcidin, and oxidative stress markers in early breast cancer: their impact in disease progression and outcome. J Steroid Biochem Mol Biol. 2020;204:105773. https://doi.org/10.1016/j.jsbmb.2020.105773.
Durigova A. Anemia and iron biomarkers in patients with early breast cancer. Diagnostic value of hepcidin and soluble transferrin receptor quantification. Clin Chem Lab Med. 2013;51:1833–41. https://doi.org/10.1515/cclm-2013-0031.
Wadowska K. Hepcidin as a diagnostic biomarker in anaemic lung cancer patients. Cancers. 2022;15:224. https://doi.org/10.3390/cancers15010224.
Schwartz AJ, Das NK, Ramakrishnan SK, Jain C, Jurkovic MT, Wu J, et al. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest. 2019;129:336–48.
Lee FS. At the crossroads of oxygen and iron sensing: hepcidin control of HIF-2α. J Clin Invest. 2019;129:72–4.
Hawula ZJ, Wallace DF, Subramaniam VN, Rishi G. Therapeutic advances in regulating the hepcidin/ferroportin axis. Pharmaceuticals. 2019;12:170. https://doi.org/10.3390/ph12040170.
Yeh KY, Yeh M, Polk P, Glass J. Hypoxia-inducible factor-2α and iron absorptive gene expression in Belgrade rat intestine. Am J Physiol Gastrointest Liver Physiol. 2011;301:G82–90. https://doi.org/10.1152/ajpgi.00538.2010.
Mastrogiannaki M, Matak P, Delga S, Deschemin JC, Vaulont S, Peyssonnaux C. Deletion of HIF-2α in the enterocytes decreases the severity of tissue iron loading in hepcidin knockout mice. Blood. 2012;119:587–90.
Mastrogiannaki M, Matak P, Peyssonnaux C. The gut in iron homeostasis: role of HIF-2 under normal and pathological conditions. Blood. 2013;122:885–92.
Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–66.
Simpson RJ, McKie AT. Regulation of intestinal iron absorption: the mucosa takes control? Cell Metab. 2009;10:84–7.
Ogawa C, Tsuchiya K, Maeda K. Hypoxia-inducible factor prolyl hydroxylase inhibitors and iron metabolism. Int J Mol Sci. 2023;24:3037. https://doi.org/10.3390/ijms24033037.
Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122:4635–44. https://doi.org/10.1172/JCI63924.
Yook JS, You M, Kim J, Toney AM, Fan R, Puniya BL, et al. Essential role of systemic iron mobilization and redistribution for adaptive thermogenesis through HIF2-α/hepcidin axis. Proc Natl Acad Sci USA. 2021;118:e2109186118.
Wilkinson N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron homeostasis by controlling HIF2α mRNA translation. Blood. 2013;122:1658–68.
Laitala A, Aro E, Walkinshaw G, Mäki JM, Rossi M, Heikkilä M, et al. Transmembrane prolyl 4-hydroxylase is a fourth prolyl 4-hydroxylase regulating EPO production and erythropoiesis. Blood. 2012;120:3336–44.
Gordeuk VR, Miasnikova GY, Sergueeva AI, Niu X, Nouraie M, Okhotin DJ, et al. Chuvash polycythemia VHLR200W mutation is associated with down-regulation of hepcidin expression. Blood. 2011;118:5278–82.
Singh AK, Carroll K, Perkovic V, Solomon S, Jha V, Johansen KL, et al. Daprodustat for the treatment of anemia in patients undergoing dialysis. N Engl J Med. 2021;385:2325–35.
Eckardt KU, Agarwal R, Aswad A, Awad A, Block GA, Bacci MR, et al. Safety and efficacy of vadadustat for anemia in patients undergoing dialysis. N Engl J Med. 2021;384:1601–12.
Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med. 2019;381:1011–22.
Requena-Ibáñez JA, Santos-Gallego CG, Rodriguez-Cordero A, Zafar MU, Badimon JJ. Prolyl hydroxylase inhibitors: a new opportunity in renal and myocardial protection. Cardiovasc Drugs Ther. 2021. https://doi.org/10.1007/s10557-021-07257-0
Del Balzo U, Signore PE, Walkinshaw G, Seeley TW, Brenner MC, Wang Q, et al. Nonclinical characterization of the hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat, a novel treatment of anemia of chronic kidney disease. J Pharmacol Exp Ther. 2020;374:342–53.
Landau D, London L, Bandach I, Segev Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS ONE. 2018;13:e0196684.
Estrela GR, Freitas-Lima LC, Budu A, Arruda AC, Perilhão MS, Fock RA, et al. Chronic kidney disease induced by cisplatin, folic acid and renal ischemia reperfusion induces anemia and promotes GATA-2 activation in mice. Biomedicines. 2021;9:769.
Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–9.
Ward DG, Roberts K, Brookes MJ, Joy H, Martin A, Ismail T, et al. Increased hepcidin expression in colorectal carcinogenesis. World J Gastroenterol. 2008;14:1339–45.
Bregolat NF, Ruetten M, Da Silva MC, Aboouf MA, Ademi H, Büren NV, et al. Iron- and erythropoietin-resistant anemia in a spontaneous breast cancer mouse model. Haematologica. 2022;107:2454–65. https://doi.org/10.3324/haematol.2022.280732.
Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL Jr, et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol. 2017;10:73. https://doi.org/10.1186/s13045-017-0427-x.
Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. 2018;36:867–74. https://doi.org/10.1200/JCO.2017.74.2627.
McDermott DF, Choueiri TK, Tykodi S, et al. Phase II study of belzutifan plus cabozantinib for previously treated advanced renal cell carcinoma (RCC): update from cohort 2 of LITESPARK-003. Ann Oncol. 2022;33:S1208–9.
Pelle E, Al-Toubah T, Morse B, Strosberg J. Belzutifan in a patient with VHL-associated metastatic pancreatic neuroendocrine tumor. J Natl Compr Canc Netw. 2022;20:1285–7. https://doi.org/10.6004/jnccn.2022.7047.
Kamihara J, Hamilton KV, Pollard JA, Clinton CM, Madden JA, Lin J, et al. Belzutifan, a potent HIF2α inhibitor, in the Pacak-Zhuang syndrome. N Engl J Med. 2021;385:2059–65. https://doi.org/10.1056/NEJMoa2110051.
Fallah J, Brave MH, Weinstock C, Mehta GU, Bradford D, Gittleman H, et al. FDA approval summary: belzutifan for von Hippel-Lindau disease-associated tumors. Clin Cancer Res. 2022;28:4843–8. https://doi.org/10.1158/1078-0432.CCR-22-1054.
Motzer RJ, Schmidinger M, Eto M, Suarez C, Figlin R, Liu Y, et al. LITESPARK-011: belzutifan plus lenvatinib vs cabozantinib in advanced renal cell carcinoma after anti-PD-1/PD-L1 therapy. Future Oncol. 2023. https://doi.org/10.2217/fon-2022-0802.
Ibrahim O, O’Sullivan J. Iron chelators in cancer therapy. Biometals. 2020;33:201–15. https://doi.org/10.1007/s10534-020-00243-3.
Kunos CA, Andrews SJ, Moore KN, Chon HS, Ivy SP. Randomized phase ii trial of triapine-cisplatin-radiotherapy for locally advanced stage uterine cervix or vaginal cancers. Front Oncol. 2019;9:1067. https://doi.org/10.3389/fonc.2019.01067.
Funding
None.
Author information
Authors and Affiliations
Contributions
VF conceived and designed the work that led to the submission, acquired the data, played an important role in interpreting the literature data, drafted and revised the manuscript, approved the final version, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SR, CM and HTA acquired the data, played an important role in interpreting the literature data, revised the manuscript, approved the final version, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SG, FDA, ADG, GDVB, GS, GM and MR played an important role in interpreting the literature data, revised the manuscript, approved the final version, agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Formica, V., Riondino, S., Morelli, C. et al. HIF2α, Hepcidin and their crosstalk as tumour-promoting signalling. Br J Cancer 129, 222–236 (2023). https://doi.org/10.1038/s41416-023-02266-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02266-2
- Springer Nature Limited