
Abstract
Metastatic brain tumors pose a clinical challenge with a high risk of morbidity and mortality for patients. Standard treatment options include surgery, whole-brain radiotherapy, and stereotactic radiosurgery (SRS). Few biologic agents have shown efficacy for brain metastases. However, immunotherapy drugs are expanding the repertoire of treatment options for advanced stage cancers, including metastatic melanoma within the brain. Most of the progress with immunotherapy for cancer has been achieved with use of checkpoint inhibitors, which are monoclonal antibodies that target regulatory molecules on T cells. Blocking checkpoint signaling lowers the barrier for host T cell activation, including exhausted T cells in the tumor microenvironment, which unleashes anti-tumor immune responses. At the same time, radiation therapy promotes release of immunogenic signals that can promote anti-tumor immunity. This chapter will examine the function of T cells in tumor immunosurveillance, highlight features of CNS immunity and properties of the tumor immune microenvironment, evaluate the emerging role of radiation therapy as an adjuvant treatment with immunotherapy, and examine the rationale of combining modalities for brain metastases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Radiation
- Stereotactic radiation
- Immunotherapy
- CNS
- Brain
- Metastases
- Checkpoint inhibitors
- Tumor Immunology
- Immune tolerance
- PD-1
- CTLA-4
T Cells in Immunity
The immune system encompasses a broad spectrum of cells from the hematopoietic lineage. Each cell type contributes specialized functions which together perform key steps in host immunity: sensing danger stimuli, secreting cytokines that recruit and activate effector cells, displaying peptide fragments, detecting antigens, and engulfing and lysing targets. A guiding hallmark of immune activity was defined by immunologists Burnet and Medawar, who proposed the Self-Nonself Model (SNS), which posited that immune cells cooperate to recognize and attack foreign (non-self) antigens [1]. This framework rationalized how microbes and infected or transformed cells harbor aberrant “Nonself” protein antigens, which are recognized and subjected to immune attack, whereas regular normal “Self” host tissues are spared from immune targeting. SNS helps explain how B and T cell repertoires emerge in the host to respond against certain antigens and ignore others [2]. During development, each T cell clone is individually educated to recognize a peptide: major histocompatibility complex (MHC), but emerging clones that respond to self-antigens are directed to undergo apoptosis. Despite its broad applicability, SNS does not completely explain key phenomena such as tumor immunity. The “Danger Model” is a newer theory developed by Matzinger, and it proposes that immune activation is controlled by the context of innate immune signals in the tissue environment that are generated by perturbation of homeostasis. Viewing immune responses through the lens of “danger” has provided a logical way to interpret fundamental principles of tumor immunology [3]. These include the requirement of co-stimulation signaling to activate naïve T cells and the regulatory role of checkpoint molecules, which attenuate T cell activation and proliferation. In the tumor microenvironment, homeostatic regulatory processes and suppressive signals maintain a balance that suppresses T cell function. These mechanisms create a high threshold for the immune system to activate effective anti-tumor responses.
αβ T cells play a central role in the adaptive immune system and are also the key constituent of anti-tumor immunity. Individual T cell clones express a unique T cell receptor (TCR) dimer on the plasma membrane. The receptors contain an immunoglobulin-like subunit with a unique variable sequence at the surface, which scans MHC:peptide complexes expressed on adjacent antigen-presenting cells (APCs). MHC class I molecules are expressed on nearly all cell types, and these complexes are recognized by CD8+ T cells. MHC class II molecules are primarily presented by professional APCs, which include dendritic cells (DCs) and macrophages, and they are recognized by CD4+ T helper cells. When a TCR is engaged by an MHC:peptide complex with sufficient affinity to bind, activation signals are transmitted from the TCR through downstream signaling cascades that activate T cell effector functions and clonal proliferation. Class I antigens stimulate CD8+ T cells to produce TNF-α and IFN-γ and to secrete cytotoxic granules that release perforin and granzyme, which cause lysis of the targeted cell [4]. Class II antigens stimulate CD4+ T cells, which mediate helper activities including release of supportive cytokines and expression of CD40 ligand, which binds CD40 on adjacent APCs and promotes their activation. Type I helper cells (Th1) can promote anti-tumor activity when activated to secrete IFN-γ, which is a strong paracrine signal that promotes surrounding cells to present class I and class II MHC complexes. For tumor cells, this increases recognition by cytotoxic CD8+ T cells [5]. Additionally, Th1 cells release IL-2, which is a growth factor that promotes survival and proliferation of surrounding T cells. Collectively, CD8+ and CD4+ T cells directly attack tumor cells and produce immunostimulatory signals that promote anti-tumor activity of other immune populations. In the following sections, we will describe some of the mechanisms cancers utilize to escape immune recognition and rejection.
APC Activation of T Cells
T cell clones emerging from the thymus are in a naïve phenotypic state prior to antigen encounter. The context of a T cell’s initial recognition of cognate antigen is critical for its long-term fate in the immune system. When a naïve T cell’s TCR binds MHC:peptide antigen without co-stimulation, the cell is induced to undergo anergy and enters a state of reduced proliferation and diminished IL-2 secretion [6]. The Danger Model predicts that immune cells are activated by alarm signals from pathogens or distressed cells. Toll-like receptors (TLRs) represent a prominent family of innate danger sensors expressed by APCs, and activation through these receptors induces their maturation. When mature, professional APCs, such as dendritic cells, increase surface expression of B7-1 and B7-2; these molecules co-stimulate naïve T cells by binding CD28 [7]. TCR stimulation coupled with CD28 co-stimulation activates a naïve T cell to adopt its mature, effector status; its TCR subunits reorganize at the plasma membrane to respond to future antigen encounters at a lower threshold. The cell also expresses CD25 to enable rapid proliferation in response to IL-2. With these changes, the activated T cell can clonally expand and effectively attack antigenic targets in the periphery. It also spawns effector and memory daughter cells to expand the reach and longevity of cells recognizing the antigen in question.
Mature DCs perform key functions necessary for T cell priming. They phagocytose distressed or dead cells, process and present antigens for T cell recognition, and release stimulatory cytokines [8]. In tumors, DCs are activated if they encounter danger-associated molecular patterns (DAMPS) which bind their TLRs. In particular, a subpopulation of BATF-3-dependent CD103+ DCs have been found to efficiently engulf and process tumor cells and vesicles and transport this cargo to tumor draining lymph nodes [9]. Upon arrival, the DCs present MHC class I and II peptide antigens from the tumor and prime anti-tumor T cells. The presence of sufficient DAMPs in the tumor microenvironment is critical for this initial step in generating an anti-tumor response.
Multiple innate regulatory signals are utilized by the immune system to prevent overactive or redundant T cell responses and maintain homeostasis. This includes checkpoint signals, which are transmitted through an array of receptors to control the duration and amplitude of T cell activity. The two most prominent checkpoint targets with a proven efficacy in immunotherapy are CTLA-4 and PD-1. T cells upregulate surface expression of CTLA-4 following stimulation of their TCR. This provides a negative signaling axis, wherein the B7-1 and B7-2 costimulatory molecules can transmit regulatory signals by binding CTLA-4. In mouse models, a germline knockout of CTLA-4 leads to fatal autoimmunity associated with generalized T cell activation, illustrating the regulatory power of this checkpoint molecule in suppressing T cells [10]. A second checkpoint pathway is mediated by the receptor, PD-1. T cells upregulate PD-1 expression following activation, and ligand binding regulates tissue inflammation, which protects against autoimmunity. Its ligands, PD-L1 and PD-L2, are expressed on tumor cells and regulatory immune cells. When PD-1 is bound, the T cell downregulates kinases involved in activation and acquires an “exhausted” phenotype with limited function and potentially apoptosis [11]. Mice with a genetic knockout of PD-1 demonstrate tissue-specific autoimmunity, though less severe than CTLA-4 [12]. Overall, the immune checkpoint molecules maintain homeostasis by dampening immune activation. They also are utilized in the tumor microenvironment to create a barrier to anti-tumor immunity.
Immunity in the Brain
The central nervous system (CNS) has a unique landscape compared to other tissue types with its own resident immune cells, a distinct lymph drainage pathway, and restricted vascular permeability maintained by the blood-brain barrier. Microglia reside exclusively within the CNS, and they perform similar functions to macrophages, including processing and presentation of antigens and expression of MHC class II complexes. At baseline, microglial cells maintain immune homeostasis; they also stimulate and remove various neighboring cells for maintenance of the microenvironment. Innate immune signals can activate microglia and turn on their antigen-presenting and immune-priming functions. Their persistent activation has been associated with destructive inflammation and neurodegenerative diseases [13]. In metastatic and primary brain tumors, microglia function can be subverted to a tolerogenic phenotype, similar to M2 macrophages. Peripheral macrophages and monocytes are often recruited to brain tumors and can function together with altered microglia to release tumor-promoting cytokines and growth factors. In doing so, they help vascularize the tumor tissue and promote tumor cell growth and invasion [14].
The blood-brain barrier stringently regulates passage of substrates and cells into the brain from the vasculature. It is comprised of tight junctions between endothelial cells and support from astrocytes and pericytes. This tight barrier hinders immune cell trafficking, and therefore the brain has been sometimes characterized as an “immune privileged” site due to the limited cross-talk between its tissue epitopes and the APCs and lymphocytes of the immune system, though this interpretation has been challenged. Three sites of immune cell access into the brain have been proposed: choroid plexus, leptomeningeal vessels, and parenchymal vessels [15]. Metastatic tumors exhibit heterogenous vascular regions with selective disruption of the blood-brain barrier, which is due, in part, to their suppression of molecular signaling pathways of CNS endothelial cells [16]. Nevertheless, modeling of the “blood tumor barrier” has found that chemotherapeutic agents are significantly excluded from brain tumors relative to non-CNS tissues [17]. Tumor-directed radiation has been found to disrupt the blood-brain barrier. It is not clear how or whether more extensive alterations of the blood-brain barrier in tumors would impact the systemic anti-tumor immune response.
The origins and extent of tumor immunosurveillance in the CNS are not fully defined. Preclinical evidence has shown that APCs are present in the brain parenchyma and ultimately drain into cervical nodes of the neck, where they can present tumor antigens to circulating T cells to generate a systemic immune response. Recently, discovery of draining lymphatics along the dura has provided more insight into this pathway. Intraparenchymal cerebrospinal fluid (CSF) carrying cells and antigens from the brain tissue flow out to the subarachnoid reservoirs of CSF [18]. Enriched by these substrates, the CSF diffuses into lymphatic vessels, which run in parallel along the dura. The lymph fluid follows this path along the sagittal sinus, which ultimately reaches deep cervical lymph nodes to interface with the peripheral immune system (Fig. 25.1). Overall, the brain has a unique immune microenvironment. Its resident immune cell population, blood-brain barrier, and distinct lymph drainage channels add to the complexity of strategically targeting metastatic CNS tumors with immunotherapy.

Histologic evaluation has revealed the presence of lymphatic vessels in the meninges of the brain. They line the dural sinuses and serve as an interface with cerebrospinal fluid carrying cells and soluble particles from the brain parenchyma. The brain lymphatics are a channel for immune cells and fluids to drain to the deep cervical nodes where they can interact with the peripheral immune system
Tumor Immunosurveillance
Tumor immunosurveillance is a model of the dynamic interaction between the immune system and emerging cancers. It postulates that most neoplastic cells are eliminated before they proliferate to form tumors. Newly transformed cells possess genetic or cellular aberrations that are presented in antigen complexes and recognized by circulating T cells. Schreiber et al. defined three main categories of tumor antigens: tumor-associated antigens, cancer germline antigens, and tumor-specific antigens [19]. Tumor-associated antigens (TAAs) are proteins associated with cell function that may be recognized by T cells when expressed at aberrant levels. In melanoma, several substrates involved with pigment synthesis are TAAs, such as MART-1 and GP100. In breast cancer, HER2/neu is a TAA. Germline antigens are proteins normally restricted to the gonads but ectopically expressed by tumor cells. MAGE-A and NY-ESO-1 are well-characterized germline antigens expressed by various cancers. Tumor-specific antigens, also known as “neoantigens,” are proteins expressed from nonsynonymous gene mutations occurring in cancer cells that result in novel peptide epitopes recognized as foreign by lymphocytes. Innovations in bioinformatics are creating new applications to apply whole genome sequencing and mass spectrometry data from tumor samples to predict the presence of neoantigens and identify corresponding reactive lymphocytes from the patient [20]. Tumors that contain a high mutational load, such as in the setting of defective mismatch repair genes, have shown an increased response to immunotherapy. This may be due to an increased abundance of neoantigens susceptible to T cell attack. The ability to analyze tumors and predict antigenic targets may lead to new opportunities in immunotherapy.
Immune Suppression by Tumors
In addition to immunoediting, tumors also activate regulatory processes that suppress host anti-tumor immunity. Histologically, the tumor microenvironment contains supporting and regulatory stromal cells dispersed among the primary cancer cells. They include fibroblasts, myeloid cells, and tumor-associated vascular endothelium. In cancer, these populations converge to create an immunosuppressive network resembling an unhealed wound [21]. They condition the microenvironment by secreting growth factors and chemokines including vascular endothelial growth factor (VEGF), chemokine ligand 2 (CCL2), and granulocyte-macrophage colony-stimulating factor (GM-CSF), which attract myeloid cells from the periphery that differentiate into myeloid-derived suppressor cells (MDSCs) and macrophages; these cells potently suppress APCs and T cells within the tumor [22]. Clinically, high levels of tumor infiltrating polymorphonuclear MDSCs have been associated with disease progression and worse prognosis in cancer patients, which illustrates how local immunosuppression favors tumor persistence and growth [23]. They produce reactive oxygen species which affect CD8+ T cells by reducing levels of the TCR zeta chain and BCL-2, which increases their proclivity to undergo apoptosis [24]. MDSC metabolism also suppresses immune function, by depleting arginine in the tumor microenvironment, disrupting the function of the TCR complex and limiting proliferation of activated T cells [25]. MDSCs also express the enzyme IDO, which catabolizes tryptophan to kynurenines. Low tryptophan concentration sensitizes T cells to apoptosis, and kynurenines induce Treg cell differentiation [26]. Tolerogenic DCs also synthesize IDO and metabolize tryptophan. MDSCs, tumor macrophages, and Tregs all produce IL-10 and transforming growth factor (TGF)-β. IL-10 attenuates DC activation and reduces macrophage expression of both MHC class II complexes and CD86 costimulatory molecules [21]. TGF-β promotes expansion of Tregs and induces differentiation of naïve CD4+ T cells to Foxp3+ Tregs. It also induces apoptosis of activated CD8+ T cells, attenuates DC activation, and directs macrophages toward a suppressor phenotype [27]. In summary, the tumor microenvironment maintains specific populations of cells and produces a profile of cytokines that are potently immunosuppressive, establishing a significant barrier to effective anti-tumor immune responses.
Radiation Therapy in Cancer
Radiation biology dogma has traditionally attributed anti-tumor effects of radiotherapy to cytocidal DNA damage. Measurements of tumor cell sensitivity to radiation, such as survival curves generated from clonogenic assays, have traditionally provided a means to model therapeutic efficacy of various dose and fractionation approaches [28]. This approach interprets radiotherapy through the lens of cell kill. However, more modern data has revealed that radiation also has a substantial effect on the tumor microenvironment that influences systemic processes. In vivo mouse studies have shown that radiation treatment can activate anti-tumor immune responses and synergize with immunotherapeutic agents [29]. Radiation releases cell death substrates that activate innate immune receptors that promote T cell priming [30]. Furthermore, the production of double-stranded DNA breaks and formation of micronuclei turn on the type I interferon pathway [31, 32]. These phenomena, which will be elaborated in greater detail later, are established mechanisms by which radiation stimulates tumor immunity, and they substantiate the beneficial role of radiotherapy as an adjuvant when combined with immunotherapy.
Immunogenic Cell Death
The contribution of radiation to anti-tumor immunity is partly due to how the malignant cells die and the associated signals that are released into the microenvironment. Zitvogel and Kroemer reported that various cell death pathways can produce DAMPS, which are danger signals that activate innate immune receptors and ultimately trigger adaptive T cell activation against antigens from the dying cells. This type of cell death is categorized as “immunogenic cell death” (ICD) [33]. Strategic induction of ICD is an emerging therapeutic strategy to elicit activation of the immune system within the tumor. Three important DAMPs have been conventionally associated with cells undergoing ICD [34]:
-
1.
Calreticulin, an endoplasmic reticulum protein, translocates to the extracellular surface of the plasma membrane. External exposure of calreticulin corresponds to endoplasmic reticulum stress and the molecule signals CD91 on DCs and macrophages, leading to phagocytosis of the dying cell [35].
-
2.
HMGB2 is a chromatin-binding factor that is released from the cell. It signals TLR4 on DCs leading to maturation. Mature DCs upregulate costimulatory molecules such as CD80, efficiently phagocytose dead cells, and cross-present exogenous antigens [36].
-
3.
ATP is secreted by the dying cells, which recruits professional APCs and stimulates IL-1β production by DCs, thus promoting antigen cross-presentation.
Altogether, ICD facilitates anti-tumor immunity by producing an array of DAMPs that promote tumor infiltration and activation of APCs, engulfment of dead and dying tumor cells, and effective cross-presentation and priming of tumor-specific T cells.
The discovery that some cell death pathways promote adaptive immunity has led to evaluation of various anti-neoplastic therapies for their immunogenicity. Among various chemotherapy classes, anthracyclines, cyclophosphamide, and oxaliplatin have been demonstrated to induce ICD in vitro and in vivo [37]. Classic tumor vaccination/re-challenge assays have also shown that radiation induces ICD. Mice injected with irradiated cells fail to grow tumors following a second challenge injection. Importantly, this finding was not recapitulated when the experiment was repeated in immunodeficient mice, bolstering the causal relationship between adaptive immunization and protection against tumor growth. Golden et al. evaluated levels of ICD biomarkers produced in tumor cell cultures and found that tumor cell radiation results in release of ATP and HMGB1 and promotes externalization of plasma membrane calreticulin, all in a dose-dependent fashion [38]. These studies have shown that radiation of tumor cells induces bona fide ICD with production of the hallmark DAMPs.
Radiation Upregulates MHC and IFN-β
Radiation also promotes tumor MHC:peptide antigen presentation. Reits et al. showed that radiation of human melanoma cultures increased the level of tumor MHC class I molecules in a dose-dependent fashion. Radiation was also shown to upregulate MHC expression on normal host tissues in vivo [39]. An orthotopic murine glioma model demonstrated that whole-brain radiation upregulated MHC-I expression on GL261 tumor cells, which improved the efficacy of concomitant vaccination [40]. Radiation also broadens the antigen peptide pool by activating mammalian target of rapamycin (mTOR), which promotes processing of proteins into peptide fragments and increases synthesis of new proteins. Moreover, radiation of different types of human tumor cells demonstrably increased the production of cancer-testis antigens, including MAGE-A1 and NY-ESO-1, which lead to activation of corresponding T cells reactive against these epitopes. These findings taken together show that radiation promotes MHC display with a diverse ensemble of peptide antigens.
Radiation of tumors also stimulates an innate immune pathway that leads to type I interferon production. Specifically, production of double stranded DNA (dsDNA) breaks followed by cell mitosis generates micronuclei that contain chromosome fragments. The cGAS molecule senses these dsDNA fragments and activates downstream STING, which ultimately leads to transcription of type I interferon [41]. Production of IFN-β stimulates maturation of DCs with increased expression of costimulatory molecules and efficient cross-presentation of antigens to T cells, which enhances priming of adaptive immunity. Combinatorial therapy with radiation and checkpoint blockade relies on IFN-β activation of Batf-3-dependent DCs to cross-prime CD8+ T cells and generate effective anti-tumor responses [32]. An in vivo mouse model of breast cancer utilizing combination anti-CTLA-4 and tumor radiotherapy showed that doses greater than 12–15 Gy per fraction attenuated anti-tumor immune responses. Mechanistically, higher doses of radiation induce expression of the nuclease, Trex1, which degrades cytosolic dsDNA and thereby removes the immune signal for activation of the cGAS-STING pathway [31]. This model demonstrated the significance of radiation dose and fractionation for immunotherapy applications.
The immune -activating effects of radiation have provided a basis for models combining tumor radiotherapy with immune targeting drugs. One of the first preclinical models testing this concept utilized Flt-3 ligand, a growth factor for DCs, together with radiation to treat mice challenged with Lewis lung carcinoma. The cohorts that received monotherapy of either agent alone showed limited survival because of lung metastases. However, a combination of radiation with Flt-3 ligand reduced the number of pulmonary metastases and improved overall survival [42]. Subsequently, Demaria and Formenti showed a bona fide abscopal effect with a combination of radiation and immunotherapy. In mice with bilateral flank tumors of mammary carcinoma (67NR), radiation of one tumor and Flt-3 ligand treatment reduced the growth of the contralateral tumor [43]. This effect was abrogated in athymic mice lacking αβ T cells, highlighting a synergy of the two therapies for the adaptive immune response.
Immune Regulation Induced by Radiation
Radiation also activates homeostatic mechanisms of the immune system that play an important role in suppressing immune attack. Irradiated tumors increase HIF1-α expression, TGF-β production, and activation and release of chemokines that recruit Tregs, MDSCs, and macrophages. These phenomena have prompted research into regimens combining radiation with immunomodulatory drugs to “release the brakes” from these regulatory signals. TGF-β is a prominent target for this objective; it diminishes cross-priming by APCs, reduces activation of CD8+ T cells, and increases the prevalence of Tregs. A preclinical model with 4T1 breast cancer evaluated tumor radiation and TGF-β blockade, which showed increased activation of anti-tumor T cells, decreased tumor growth and metastases, and improved survival [44]. This approach was incorporated in a clinical trial for metastatic breast cancer: patients received three fractions of 7.5 Gy to one lesion and either low- or high-dose anti-TGF-β antibody; receipt of the high dose of immunotherapy boosted memory CD8+ T cells and was associated with improved overall survival [45]. Chemokine receptor 2 (CCR) is also a relevant target for combination therapy. Radiation signals through cGas-STING to increase intratumor levels of chemokines that bind CCR2 and attract MDSCs to the tumor microenvironment. Notably, tumor-challenged mice treated with radiation and CCR2 blockade demonstrated enhanced CD8+ T cell-mediated tumor rejection versus cohorts receiving radiation alone [46]. Overall, radiation has both stimulatory and suppressive effects on the immune system. Strategic molecular targeting of potent regulatory pathways together with radiotherapy can successfully elicit anti-tumor immunity.
Immune Checkpoint Inhibitors
Clinical trials have demonstrated the efficacy of checkpoint inhibitors for several tumor types and thus established immunotherapy as a mainstream modality in oncology. New applications continue to emerge, and at present, most are focused on metastatic or locally advanced disease. Allison and colleagues originally elucidated the T cell regulatory molecule, CTLA-4, and demonstrated that antibody blockade (anti-CLTA-4) unleashed anti-tumor immunity. Mice that were challenged with characteristically immunogenic tumors showed pronounced rejection of the tumors after receipt of anti-CTLA-4 antibody [47]. An in vivo study with melanoma demonstrated that anti-CTLA-4 therapy contributed to tumor immunity by amplifying effector T cell function and minimizing Treg cell activity [48]. Notably, a subsequent study using anti-CTLA-4 to treat the poorly immunogenic melanoma, B16-BL6, showed minimal ability to inhibit tumor growth. It was only when mice received anti-CTLA-4 therapy combined with a vaccination injection of irradiated B16-BL6 cells modified to express GM-CSF that elimination of tumor could be achieved in vivo [49]. These results highlighted that most tumors may require multiple sources of immunogenic stimuli for a therapeutic response. The preclinical work characterizing anti-CTLA-4 ultimately translated to clinical applications with ipilimumab. In the first major phase III trial with a checkpoint inhibitor, the drug showed improved overall survival for metastatic melanoma, which set the stage for further development of checkpoint inhibitors in oncology [50].
The PD-1 signaling axis is the second T cell checkpoint pathway that has been successfully incorporated for tumor immunotherapy. Several established human tumors such as lung, ovary, colon, and melanoma increase expression of PD-L1 to suppress T cell activity in their microenvironment [51]. Immune cells recruited by tumors, including MDSCs, can also express PD-L1 [52]. When surface PD-1 is engaged by the ligand, T cells adopt an exhausted phenotype and display diminished activity. Anti-PD-1 antibodies block this signal and help revive tumor infiltrating T cells, thus facilitating adaptive anti-tumor responses. PD-1 checkpoint inhibitors have demonstrated success and are approved for use in an increasing number of malignancies, including advanced stage melanoma, non-small cell lung cancer (NSCLC) urothelial carcinoma, Hodgkin’s disease, and head and neck squamous cell cancer, as well as microsatellite instability-high cancers [53]. Though cohorts of cancer patients receiving checkpoint inhibition have improved clinical outcomes as a group, most patients do not achieve a significant response to treatment. New approaches to increase the proportion of responders are needed, and radiotherapy is being investigated for this purpose.
Recent trials have assessed whether combined checkpoint inhibition may synergistically enhance clinical anti-tumor responses. Checkmate 067 was a phase III clinical trial evaluating monotherapy checkpoint inhibition versus a combination of ipilimumab and nivolumab administration for patients with metastatic melanoma [54]. The cohort receiving combined therapy had a longer progression-free survival (PFS) and higher objective response rate compared to the cohort receiving ipilimumab alone, albeit at the price of increased toxicity. Notably, most of the trials utilizing immunotherapy for advanced stage cancer have excluded patients with brain metastases. However, Margolin and colleagues reported a phase II study of dual checkpoint inhibition with nivolumab and ipilimumab for patients with melanoma brain metastases. Combined therapy resulted in a high response rate of 56%. Complete response was seen in 26% of patients [55]. These impressive results provide a foundation for exploring checkpoint inhibition for different types of brain metastases.
Combination of Checkpoint Inhibitors with Radiation
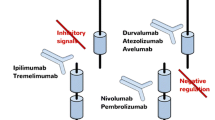
Radiation of tumors associated with off-target responses (abscopal effect) has been described in a small number of case reports dating back several decades. This includes patients with a wide variety of tumor types such as melanoma, renal cell carcinoma, and lymphoma [56,57,58]. The impact of radiation on systemic tumor responses may be related to anti-tumor immunity. As previously described, radiation induces stimulatory immune danger signals that create an in situ vaccine effect in the tumor microenvironment, which helps prime adaptive T cell responses. Potential synergy of these effects with checkpoint inhibition has been extensively explored in preclinical studies [59]. Formenti and Demaria showed that mice challenged with 4T1 breast carcinoma derived minimal benefit from treatment with radiation or anti-CTLA-4 monotherapy. Yet, combined treatment with both agents significantly reduced the number of lung metastases in recipients and improved survival [60]. This approach has also demonstrated efficacy in an orthotopic glioma mouse model: combinatorial therapy with anti-PD-1 and a single fraction of 10Gy to the tumor resulted in a significant improvement in survival over either treatment alone [61]. Minn et al. showed that dual checkpoint therapy with anti-PD-1 and anti-CTLA-4 in addition to tumor radiation provided complementary, non-redundant immune activation signals. The anti-tumor TCR repertoire was expanded by radiation. PD-L1 blockade revived exhausted CD8+ T cells, and CTLA-4 blockade decreased Tregs. Thus, dual checkpoint blockade increased the ratio of CD8/Treg cells [62]. Rudqviist et al. also found that CTLA-4 blockade and radiation therapy for tumor-challenged mice synergized to expand the TCR repertoire within tumor-infiltrating lymphocytes (TIL). Their evaluation identified an increased diversity and number of CDR3 motifs among the population of receptors [63]. The evidence from these and several other preclinical models have provided a compelling rationale to explore combinatorial strategies with radiation and checkpoint inhibitors (Fig. 25.2).

Immunotherapy with anti-CTLA-4 and anti-PD-1 monoclonal antibodies activates non-redundant mechanisms that promote clonal expansion of T cells and revive exhausted effector cells. Tumor radiation enhances MHC antigen presentation and increases the diversity of the anti-tumor T cell repertoire. Clinical trials are exploring paradigms for combining immunotherapy with tumor radiation to synergistically activate and expand anti-tumor T cells that mediate systemic tumor rejection
Results from preclinical data have influenced new oncology trials for patients incorporating synchronous immunotherapy and radiation. Most of the findings are limited to small cohort studies or anecdotal case reports. For example, a melanoma patient who reportedly progressed after receipt of ipilimumab received palliative radiation in three fractions for a spinal metastasis. Within 3 months, distant hilar metastases, and splenic lesions responded, representing nearly a complete disease regression [64]. Also a phase II study treating Merkel cell carcinoma with pembrolizumab reported that two patients who received palliative radiation following disease progression had subsequent off-target tumor response [65]. Formenti and colleagues recently reported the results of a trial for patients with NSCLC who, after failing chemotherapy, went on to receive radiation therapy to a single metastasis and concurrent ipilimumab. Notably, two previous prospective randomized trials of CTLA-4 blockade with chemotherapy failed to demonstrate significant activity in advanced NSCLC [66, 67]. However, in Formenti’s trial combining ipilimumab with focal radiotherapy, 31% of the patients achieved disease control, and 18% demonstrated an objective response [68]. One patient who achieved a complete response after originally presenting with synchronous lung cancer and brain metastases was found to have a clonal expansion of T cells recognizing a mutation within his tumor. This result demonstrated translational success of radiotherapy in inducing neo-antigens and converting the tumor into an in-situ vaccine. As ongoing combinatorial trials continue to mature, more sophisticated conclusions can be reached regarding the efficacy of combining tumor radiation with immune checkpoint inhibitors.
Optimal Radiation Parameters for Immunotherapy
The optimal dose and fractionation of radiotherapy in combinatorial regimens with checkpoint inhibitors are yet to be determined. Several cases reported in the literature utilized a hypofractionated course, though no standard prescription has emerged. One core question is the comparative efficacy of different doses per fraction of radiotherapy. In preclinical work with B16 melanoma, a single fraction of 20Gy activated anti-tumor CD8+ T cells in mice, whereas this response was not seen in a comparison cohort treated with 5 Gy × 4 fractions [69]. On the other hand, Vanpouille-Box treated mice bearing two subcutaneous TSA breast carcinomas with anti-CTLA-4 and various radiation regimens directed only to one tumor. Cohorts that received 8 Gy × 3 demonstrated abscopal tumor response (measured in the non-irradiated tumor) and increased survival compared to those that received a single fraction of 20 Gy. In this model, the abscopal response from radiation diminished as doses were escalated above 12 Gy per fraction [31]. This trend paralleled dose-dependent induction of Trex-1, an exonuclease that digests cytoplasmic dsDNA and removes the substrate for cGAS/Sting, which attenuates induction of type I interferon.
With no consensus dose established for immunotherapy applications, clinical trials are utilizing a variety of radiation prescriptions. Chmura et al. conducted a phase I clinical trial treating metastatic solid tumors with pembrolizumab and SBRT doses from 30 to 50 Gy. They reported a favorable toxicity profile, but the objective response was only 13.2%, which was similar to the outcome of pembrolizumab alone in an unselected cohort of patients with metastatic disease. The median PFS was 3.1 months [70]. In comparison, the Netherlands Cancer Institute reported preliminary phase II results from NSCLC patients, who were randomized to pembrolizumab alone versus pembrolizumab with a sub-ablative radiation dose of 8 Gy × 3. The pembrolizumab alone cohort achieve a 19% response rate, while the cohort receiving combination therapy had a 41% objective response. Also, the median PFS was 1.8 versus 6.4 months, respectively [71]. These preliminary findings suggest that a dose/fraction effect may govern the immune activating potential of radiotherapy. Further investigation is needed to validate this phenomenon and, if confirmed, determine whether this is due to Trex-1 induction or other signals.
Modern clinical trials have not yet reported high-level data for combinatorial regimens with checkpoint inhibitors and radiation of brain metastases. Standard whole-brain radiation prescriptions include 30 Gy in 10 fractions and 20 Gy in 5 fractions as palliative options for extensive disease. Stereotactic radiosurgery (SRS) using a single-fraction ablative dose has demonstrated excellent local control for patients with a limited number and size of brain metastases. SRS also has superior preservation of long-term cognition compared to whole-brain radiation. Furthermore, Knisely and colleagues reported findings that bolstered the prospect of combination SRS and checkpoint inhibition. In a retrospective analysis of cases of melanoma brain metastases, they showed that the cohort of patients who received ipilimumab in addition to SRS had an overall survival of 21.4 months versus 4.9 months for patients who received SRS alone [72], a significant difference even if the retrospective nature of the study likely reflects patient selection. Additionally, hypofractionated regimens may have comparable efficacy to SRS for larger brain tumors >2 cm. A meta-analysis of 24 trials showed similar 1-year local control for patients receiving SRS versus multi-fraction RT. The most common multi-fractionation regimen utilized was 27 Gy in three fractions [73], a prescription that aligns well with the preclinical data from Vanpouille-Box modeling optimal immunogenic doses to induce tumor production of type I interferon.
In addition to dose and fractionation, the optimal sequencing of radiation and immunotherapy continues to be evaluated. Preclinical work comparing different sequences showed that upfront checkpoint blockade with anti-CTLA-4 followed by radiotherapy achieved the greatest tumor treatment efficacy. The study concluded that early depletion of Tregs facilitated immune priming of CD8+ T cells when tumors were irradiated [74]. Limited results from currently available trials suggest that overlapping or close sequencing of checkpoint blockade with radiotherapy is likely to be the most effective approach. For melanoma brain metastases, a retrospective analysis showed that patients receiving anti-PD-L1 and anti-CTLA-4 therapy followed by stereotactic radiosurgery within 4 weeks of checkpoint blockade demonstrated a greater median reduction in lesion volume compared to patients with a longer separation of treatments. However, this result could be attributable to patient selection since progression through ipilumimab may correspond to more aggressive metastatic disease [75]. An unplanned analysis of the Pacific Trial for NSCLC found that patients who received durvalumab (anti-PD-1) after responding to platinum-based chemo-radiation had improved PFS. The finding was particularly significant if checkpoint blockade was administered within 2 weeks from completion of chemoradiation [76]. Also, Chiang and colleagues reported retrospective data of melanoma patients with brain metastases who were treated with SRS and immune checkpoint inhibition. Administration of immune checkpoint therapy within 4 weeks of SRS resulted in greater reduction in tumor size compared with patients who received treatment that was not concurrent [77]. Going forward, results from clinical trials that are currently underway will provide a clearer understanding of the significance of dose/fractionation and sequencing to the overall success of therapy.
Lymph Nodes as an Organ at Risk (OAR)
Utilization of radiotherapy for tumor immune activation will elevate the importance of lymphocytes and lymph nodes as organs at risk for treatment planning. Functional lymph nodes provide an interface for T cells and APCs draining from tumors to interact and receive priming signals for activation and proliferation. Marciscano and colleagues examined the impact of radiation target fields that included tumor-draining lymph nodes in a preclinical model. Mice were challenged with flank tumors and treated with checkpoint blockade and a single fraction of 12 Gy that either included or omitted the regional draining lymph nodes. The cohort that received radiation with a field encompassing their draining lymph nodes had a diminished tumor infiltrating lymphocytes population and worse survival compared to the cohort where draining lymph nodes were avoided [78]. A second area of consideration is the impact of fractionated radiation on lymphocytes in the peripheral blood. Ford and colleagues modeled the radiation dose to the circulating pool of lymphocytes. In their calculation, a single fraction of 2 Gy would deliver 0.5 Gy to 5% of circulating cells. Notably, a 30-fraction course would result in ≥0.5 Gy to 99% of circulating blood cells. These studies support a strategy of lymph node sparing and the utilization of hypofractionated courses of radiation to best protect the host T cell pool if attempting to stimulate an anti-tumor immune response. For radiation of the brain, an additional consideration could be the anatomic avoidance of the previously described lymphoid drainage network that traces along the sinuses to the cervical nodes. Louvea and colleagues showed that ablation of meningeal lymphatics reduces T cells and inflammatory responses in the brain in a model of multiple sclerosis [79]. It has not been determined how treatment such as whole-brain radiation impacts the integrity of the brain lymphatic channels and, furthermore, what impact this has on anti-tumor T cell responses within the brain. Future preclinical studies may be needed to explore how brain radiation specifically impacts all of these variables.
Summary
Immunotherapy is transforming the practice of oncology and rapidly integrating into mainstream treatment paradigms. The utility of radiotherapy as an adjuvant with immunotherapy is well established by preclinical data showing how tumor radiation releases danger signals that may convert the irradiated tumor into an in situ vaccine. The rarity of abscopal effects confirms the evidence of the robust immunosuppressive microenvironment of established tumors. Tipping the balance by adding immunomodulators to local radiotherapy, such as checkpoint inhibitors, can create a synergistic effect that promotes therapeutic anti-tumor T cell responses. Brain metastases present a unique challenge because the brain has a distinct immune profile. Furthermore, many clinical trials with checkpoint inhibitors have excluded such patients. Additional data regarding optimal dose, timing, and targeting with radiation is rapidly emerging. This data should be incorporated into new clinical trials for brain metastases to ultimately develop the most effective combinations of stereotactic radiation and immunotherapy.
References
Ribatti D. Peter Brian Medawar and the discovery of acquired immunological tolerance. Immunol Lett. 2015;167(2):63–6.
Medzhitov R, Janeway CA Jr. Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296(5566):298–300.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301–5.
Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–8.
Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18:635.
Appleman LJ, Boussiotis VA. T cell anergy and costimulation. Immunol Rev. 2003;192:161–80.
Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290(5489):92–7.
Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2(8):675–80.
Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol. 2017;28(suppl_12):xii74.
Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–8.
Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–77.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–42.
Wu SY, Watabe K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front Biosci (Landmark Ed). 2017;22:1805–29.
Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120(5):1368–79.
Tiwary S, Morales JE, Kwiatkowski SC, Lang FF, Rao G, McCarty JH. Metastatic brain tumors disrupt the blood-brain barrier and alter lipid metabolism by inhibiting expression of the endothelial cell fatty acid transporter Mfsd2a. Sci Rep. 2018;8(1):8267.
Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16(23):5664–78.
Da Mesquita S, Fu Z, Kipnis J. The meningeal lymphatic system: a new player in neurophysiology. Neuron. 2018;100(2):375–88.
Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest. 2015;125(9):3413–21.
Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, et al. Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res. 2016;4(9):734–43.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–20.
Weide B, Martens A, Zelba H, Stutz C, Derhovanessian E, Di Giacomo AM, et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin Cancer Res. 2014;20(6):1601–9.
Ezernitchi AV, Vaknin I, Cohen-Daniel L, Levy O, Manaster E, Halabi A, et al. TCR zeta down-regulation under chronic inflammation is mediated by myeloid suppressor cells differentially distributed between various lymphatic organs. J Immunol. 2006;177(7):4763–72.
Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91.
Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–64.
Travis MA, Sheppard D. TGF-beta activation and function in immunity. Annu Rev Immunol. 2014;32:51–82.
Buch K, Peters T, Nawroth T, Sanger M, Schmidberger H, Langguth P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT assay – a comparative study. Radiat Oncol. 2012;7:1.
Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. 2016;2(6):286–94.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25(1):11–7.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618.
Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–52.
Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3. United States:e955691.
Gebremeskel S, Johnston B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: impact on clinical studies and considerations for combined therapies. Oncotarget. 2015;6(39):41600–19.
Wiersma VR, Michalak M, Abdullah TM, Bremer E, Eggleton P. Mechanisms of translocation of ER chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front Oncol. 2015;5:7.
Pathak SK, Skold AE, Mohanram V, Persson C, Johansson U, Spetz AL. Activated apoptotic cells induce dendritic cell maturation via engagement of Toll-like receptor 4 (TLR4), dendritic cell-specific intercellular adhesion molecule 3 (ICAM-3)-grabbing nonintegrin (DC-SIGN), and beta2 integrins. J Biol Chem. 2012;287(17):13731–42.
Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39(1):74–88.
Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71.
Newcomb EW, Demaria S, Lukyanov Y, Shao Y, Schnee T, Kawashima N, et al. The combination of ionizing radiation and peripheral vaccination produces long-term survival of mice bearing established invasive GL261 gliomas. Clin Cancer Res. 2006;12(15):4730–7.
Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466–70.
Chakravarty PK, Alfieri A, Thomas EK, Beri V, Tanaka KE, Vikram B, et al. Flt3-ligand administration after radiation therapy prolongs survival in a murine model of metastatic lung cancer. Cancer Res. 1999;59(24):6028–32.
Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862–70.
Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75(11):2232–42.
Formenti SC, Lee P, Adams S, Goldberg JD, Li X, Xie MW, et al. Focal irradiation and systemic TGFbeta blockade in metastatic breast cancer. Clin Cancer Res. 2018;24(11):2493–504.
Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8(1):1736.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6.
Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–25.
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800.
Ballbach M, Dannert A, Singh A, Siegmund DM, Handgretinger R, Piali L, et al. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol Lett. 2017;192:1–6.
Tang J, Shalabi A, Hubbard-Lucey VM. Comprehensive analysis of the clinical immuno-oncology landscape. Ann Oncol. 2018;29(1):84–91.
Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345–56.
Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722–30.
Kingsley DP. An interesting case of possible abscopal effect in malignant melanoma. Br J Radiol. 1975;48(574):863–6.
Wersall PJ, Blomgren H, Pisa P, Lax I, Kalkner KM, Svedman C. Regression of non-irradiated metastases after extracranial stereotactic radiotherapy in metastatic renal cell carcinoma. Acta Oncol. 2006;45. England:493–7.
Robin HI, AuBuchon J, Varanasi VR, Weinstein AB. The abscopal effect: demonstration in lymphomatous involvement of kidneys. Med Pediatr Oncol. 1981;9(5):473–6.
Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015;1(9):1325–32.
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2 Pt 1):728–34.
Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–9.
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520(7547):373–7.
Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res. 2018;6(2):139–50.
Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–31.
Xu MJ, Wu S, Daud AI, Yu SS, Yom SS. In-field and abscopal response after short-course radiation therapy in patients with metastatic Merkel cell carcinoma progressing on PD-1 checkpoint blockade: a case series. J Immunother Cancer. 2018;6(1):43.
Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, et al. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non-small-cell lung cancer. J Clin Oncol. 2017;35(30):3449–57.
Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol. 2016;34(31):3740–8.
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C, et al. Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med. 2018;24(12):1845–51.
Lee Y, Auh SL, Wang Y, Burnette B, Meng Y, Beckett M, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114(3):589–95.
Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol. 2018;36(16):1611–8.
Theelen W, Peulen H, Lalezari F, de Vries J, De Langen J, Aerts J, et al., editors. Randomized phase II study of pembrolizumab after stereotactic body radiotherapy (SBRT) versus pembrolizumab alone in patients with advanced non-small cell lung cancer: the PEMBRO-RT study. ASCO Annual Meeting; 2018; Chicago.
Knisely JPS, Yu JB, Flanigan J, Sznol M, Kluger HM, Chiang VLS. Radiosurgery for melanoma brain metastases in the ipilimumab era and the possibility of longer survival. J Neurosurg. 2012;117(2):227–33.
Lehrer EJ, Peterson JL, Zaorsky NG, Brown PD, Sahgal A, Chiang VL, et al. Single versus multifraction stereotactic radiosurgery for large brain metastases: an international meta-analysis of 24 trials. Int J Radiat Oncol Biol Phys. 2019;103(3):618–30.
Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS One. 2016;11(6):e0157164.
Qian JM, Yu JB, Kluger HM, Chiang VL. Timing and type of immune checkpoint therapy affect the early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer. 2016;122(19):3051–8.
Wang Y, Deng W, Li N, Neri S, Sharma A, Jiang W, et al. Combining immunotherapy and radiotherapy for cancer treatment: current challenges and future directions. Front Pharmacol. 2018;9:185.
Qian JM, Yu JB, Kluger HM, Chiang VL. Timing and type of immune checkpoint therapy affects early radiographic response of melanoma brain metastases to stereotactic radiosurgery. Cancer. 2016;122(19):3051–8.
Marciscano AE, Ghasemzadeh A, Nirschl TR, Theodros D, Kochel CM, Francica BJ, et al. Elective nodal irradiation attenuates the combinatorial efficacy of stereotactic radiation therapy and immunotherapy. Clin Cancer Res. 2018;24(20):5058–71.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21(10):1380–91.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Brandmaier, A.G., Ramakrishna, R., Formenti, S.C. (2020). Synergy of Immunotherapy and Radiosurgery. In: Ramakrishna, R., Magge, R., Baaj, A., Knisely, J. (eds) Central Nervous System Metastases. Springer, Cham. https://doi.org/10.1007/978-3-030-42958-4_25
Download citation
DOI: https://doi.org/10.1007/978-3-030-42958-4_25
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-42957-7
Online ISBN: 978-3-030-42958-4
eBook Packages: MedicineMedicine (R0)