
Abstract
Arrhythmogenic cardiomyopathy (ACM) is a genetically determined heart muscle disease characterized pathologically by fibrofatty myocardial replacement of both the right and left ventricle and clinically by ventricular arrhythmias and risk of sudden cardiac death (SCD). Mutations in the genes encoding desmosomal proteins play a key role in the pathogenesis of ACM. These proteins include desmoplakin (DSP), plakophilin 2 (PKP2), desmoglein 2(DSG2), and desmocollin 2(DSC2) [1–3]. Although the classic disease phenotype was characterized by predominant right ventricular (RV) involvement, clinical variants with early and prevalent LV involvement, which may parallel (i.e., biventricular ACM) or even exceed (i.e., left-dominant ACM) the severity of RV involvement, have been increasingly observed [4]. These findings have led over the past few years to use the broader term of “arrhythmogenic cardiomyopathy” (ACM), which encompasses all the phenotypic expressions. This chapter addresses pathogenesis, clinical presentation, diagnosis, and management strategies in patients with ACM.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
Arrhythmogenic cardiomyopathy (ACM) is a genetically determined heart muscle disease characterized pathologically by fibrofatty myocardial replacement of both the right and left ventricle and clinically by ventricular arrhythmias and risk of sudden cardiac death (SCD). Mutations in the genes encoding desmosomal proteins play a key role in the pathogenesis of ACM. These proteins include desmoplakin (DSP), plakophilin 2 (PKP2), desmoglein 2(DSG2), and desmocollin 2(DSC2) [1,2,3]. Although the classic disease phenotype was characterized by predominant right ventricular (RV) involvement, clinical variants with early and prevalent LV involvement, which may parallel (i.e., biventricular ACM) or even exceed (i.e., left-dominant ACM) the severity of RV involvement, have been increasingly observed [4]. These findings have led over the past few years to use the broader term of “arrhythmogenic cardiomyopathy” (ACM), which encompasses all the phenotypic expressions. This chapter addresses pathogenesis, clinical presentation, diagnosis, and management strategies in patients with ACM.
Pathogenesis
Histopathological Features
The hallmark lesion of ACM is the replacement of the ventricular myocardium by fibrofatty tissue [5, 6]. Patchy inflammatory infiltrates (mainly T lymphocytes) are often observed in association with dying myocytes, both in animal models and human patients, suggesting that the pathologic process may be immunologically mediated [7, 8]. The fibrofatty scar tissue progresses from the epicardium toward the endocardium and predominantly involves the right ventricular free wall, resulting in wall thinning and aneurysmal dilatation, which are typically localized in the so-called triangle of dysplasia, that includes the inflow tract (sub-tricuspid region), the outflow tract (infundibular region), and the apex of the RV [5,6,7]. Up to 76% of the ACM hearts studied at postmortem also disclosed involvement of the LV, which is usually limited to the subepicardial or mid-mural layers of the free wall or septum [8].
Molecular Genetic Features and Pathogenesis
The disease process is caused by the progressive loss of myocytes because of genetically defective desmosomes, which are cell-to-cell adhesive structures, with subsequent fibrofatty replacement of the ventricular myocardium of both ventricles. The desmosome contains three major components: desmoplakin, which binds to intermediate filaments (i.e., cardiac desmin); transmembrane proteins (i.e., desmosomal cadherins), including desmocollin 2 and desmoglein 2; and linker proteins (i.e., proteins of the armadillo family), including plakoglobin and plakophilin 2, which mediate interactions between the desmosomal cadherin tails and desmoplakin (Fig. 32.1). That ACM is a cell adhesion disorder was first suggested by molecular genetic study involving patients with Naxos disease, which is an autosomal recessive cardiocutaneous syndrome, characterized by co-segregation of abnormalities of the heart (ACM), skin (palmoplantar keratosis), and hair (woolly hair), and caused by mutations in the gene encoding plakoglobin (JUP) [9]. The discovery that ACM is a cell-to-cell adhesion disease led to research for other related genes. The next clue on the role of desmosome mutations in ACM arose from South America, where Luis Carvajal-Huerta recognized a similar constellation of keratoderma, woolly hair, and ACM with LV predominance. Thus, mutations in genes encoding other desmosomal proteins were subsequently shown to cause the more common (non-syndromic) autosomal dominant form of ACM [10]. Desmoplakin (DSP) was the first defective gene to be associated with autosomal dominant non-syndromic ACM [11]. Because DSP and JUP are both components of the cardiac desmosome, Gerull et al. extended this research to another component of the cardiac desmosome and identified heterozygous mutations in plakophilin-2 (PKP2) in 32 of 120 probands (27%) [12]. After recognition that PKP2 mutations are a common cause of ACM, several additional candidate gene analyses facilitated reports of mutations in other components of the cardiac desmosome. Two groups reported several families with DSG2 mutations causing classic ACM without abnormalities of the skin or hair [13, 14]. Next, DSC2 mutations were recognized to cause ACM. Predictably, heterozygous mutations in JUP also cause non-syndromic ACM [15]. Although the distribution of mutations among these five genes (PKP2, DSG2, DSP, DSC2, and JUP) varies among separate cohorts with ACM, large studies consistently demonstrated that desmosome mutations are the most common cause of ACM. Non-desmosomal genes have been reported to cause arrhythmogenic cardiomyopathy. They include genes encoding for the following: (1) the area composita, a mixed type of junctional structure composed of both desmosomal and adherens junctional proteins, such as alpha-T-catenin (CTTNA3) [16] and cadherin C (CDH2) [17, 18]; (2) proteins for other subcellular structures such as laminin A/C (LMNA) [19, 20]; (3) transmembrane protein 43 (TMEM43) [21, 22]; (4) desmin (DES) [23]; (5) phospholamban (PLN) [24]; (6) transforming growth factor-3 (TFGB3); and (7) sodium voltage-gated channel alpha subunit 5 (SC5NA).

Histopathological features and pathogenesis of arrhythmogenic cardiomyopathy. The hallmark histopathological lesion of ACM is the fibrofatty replacement of the ventricular myocardium. Panel A shows a full-thickness histologic section (azan trichrome stain) of the anterior right ventricular wall in a normal heart; Panel B illustrates an analogous section from the heart of a patient with ACM who died suddenly. With the azan trichrome stain, myocytes appear red, fibrous tissue appears blue, and fatty tissue appears white. ACM is caused by genetically defective desmosomes that are composed by three major components: desmoplakin, which binds to intermediate filaments (i.e., cardiac desmin); transmembrane proteins (i.e., desmosomal cadherins), including desmocollin 2 and desmoglein 2; and linker proteins (i.e., proteins of the armadillo family), including plakoglobin and plakophilin 2, which mediate interactions between the desmosomal cadherin tails and desmoplakin, as shown in Panel C. Abnormal desmosomes predispose over time to disruption of the intercellular junction, as shown in Panel D (double-headed arrow). This process occurs mostly under conditions of increased mechanical stress, such as sports activity. A parallel pathogenic process involves the Wnt–β-catenin signaling pathway. During canonical Wnt–β-catenin signaling, β-catenin forms complexes with members of the TCF–LEF (T-cell factor–lymphocyte-enhancing factor) family of transcription factors in the nucleus to prevent the differentiation of mesodermal precursors into adipocytes and fibrocytes by suppressing the expression of adipogenic and fibrogenic genes (Panel C). Impairment of desmosomal assembly causes the translocation of plakoglobin from the sarcolemma to the nucleus (arrows in Panel D), where it may antagonize the effects of β-catenin, suppressing Wnt–β-catenin signaling and therefore inducing a gene transcriptional switch from myogenesis to adipogenesis and fibrogenesis (Panel D). (From Corrado et al. [57], © 2017 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society)
The traditional explanation for desmosome mutations causing cardiomyopathy relies on loss of adhesion between cardiac myocytes, which predisposes them to detachment and death with replacement by fibrofatty tissue [9]. Physical activity worsens this adhesion defect, with greater consequences for the thinner-walled RV as compared with the LV. Other than being specialized structures that supply mechanical cell attachment, desmosomes are important mediators of intracellular and intercellular signal transduction. The mutant form of the plakoglobin, also known as y-catenin, does not integrate into desmosomes and shifts from intercalated disks to cytosol and nuclear pools, where it causes changes in nuclear signaling and transcriptional activity, through pathways regulated by the protein β-catenin. The inhibition of the established Wnt–β-catenin signaling pathway may increase the expression of adipogenic and fibrogenic genes and contribute to the development of fibrofatty myocardial scarring (Fig. 32.1) [25].
The fibrofatty tissue that replaces myocardium contributes to the development of ventricular arrhythmias in ACM by slowing intraventricular conduction and acting as a substrate for arrhythmias through a scar-related macroreentry mechanism. However, life-threatening ventricular arrhythmias in ACM may also be the result of mechanisms working at the molecular and cellular levels. Desmosomes, sodium channels, and gap-junction proteins interact synergistically to regulate adhesion, excitability, and coupling of myocytes; this coordinated network of proteins found at the intercalated disks has been termed the “connexome” [26]. According to this view, loss of expression of desmosomal proteins may cause (or contribute to) potentially fatal arrhythmias by inducing gap-junction remodeling, with reduction of total content and substantial redistribution of the gap-junction protein connexin 43, and decreasing the amplitude and kinetics of the sodium current [27,28,29]. Moreover, there is some evidence that the Brugada syndrome and ACM may share clinical features and arrhythmic mechanisms because of their common origin from the connexome [30].
Clinical Presentation and Natural History
Epidemiology
The prevalence of ACM has been estimated to range from 1 case in 2000 to 5000 persons in the general population. A familial incidence is present in more than half the ACM cases. The disease is typically transmitted with an autosomal dominant pattern of inheritance, although rare autosomal recessive forms have been described [31]. The disease is more malignant in men than in women, and this finding has been ascribed either by a direct influence of sex hormones on the pathogenic mechanisms involved in the phenotypic expression of the disease [3] or by sex-based difference in the amount or intensity of exercise [32].
Phenotypic Expression
The phenotypic expression of ACM varies considerably, ranging from asymptomatic family members with concealed structural abnormalities and no arrhythmias to symptomatic patients experiencing arrhythmic cardiac arrest or undergoing cardiac transplantation because of refractory heart failure. The disease usually becomes clinically overt within the second to the fourth decades of life and is preceded by a preclinical phase, which is characterized by minimal or no structural abnormalities (concealed disease). Pathobiological and clinical landmarks which characterize the different phases of the disease natural history until clinical diagnosis include the following: (1) the time period between birth and the beginning of the disease process (pre-histologic phase); (2) the development of initial histologic changes of the myocardium (myocyte death and fibrofatty replacement) which are clinically concealed (preclinical phase); (3) the period of clinically detectable electrical (ECG changes and premature ventricular beats) and structural (ventricular remodeling with dilatation and dysfunction, either global or segmental) abnormalities, in the absence of relevant symptoms (presymptomatic phase); and (4) occurrence of symptoms (symptomatic phase). A sizeable proportion of affected patients have a silent clinical course and remain asymptomatic for many years, so delaying the identification of the disease unless they undergo screening programs, such as cascade screening of family members after identification of a proband or universal pre-participation athletic screening including an ECG. SCD may be the first clinical manifestation of the disease. In a study in the Veneto region of Italy, 20% of SCDs in young people and athletes were caused by a previously undiagnosed ACM [6].
SCD is caused by an arrhythmic cardiac arrest that may occur unexpectedly in previously asymptomatic individuals, mostly young people and competitive athletes, with a previously undiagnosed (and unsuspected) ACM [9]. Patients with clinically overt disease may experience scar-related monomorphic ventricular tachycardia (VT), which is caused by a reentrant circuit related to the underlying fibrofatty ventricular scar [33]. Ventricular arrhythmias are worsened by adrenergic stimulation and neuroautonomic imbalance occurring during or immediately after exercise [34]. Abrupt ventricular fibrillation (VF) and SCD may also occur in young patients during an active phase of disease progression, the so-called “hot phases”, as a consequence of myocarditis-mediated bouts of acute myocyte death leading to acute electrical instability [9].
While VF is an arrhythmic complication which usually occurs “early” during the natural history of ACM, sustained VT is more commonly observed in advanced disease [35,36,37,38]. A pathobiological explanation of these findings is that VT and VF arise from a myocardial substrate which varies over time in relation to different phases of the natural history of ACM. Episodes of VF most often occur as the first clinical manifestation in young patients because they reflect acute electrical instability of early disease, which progresses through so-called “hot phases” that are recurrent bouts of acute myocyte death with reactive inflammation, most often characterized by a clinically silent course. Older patients with advanced disease more often experience symptomatic VT which is monomorphic (left bundle branch block pattern), hemodynamically well-tolerated and unlikely to degenerate into VF. This VT is caused by a reentry mechanism around a stable fibrofatty myocardial scar as the result of a repair process that occurs in a later stage of the disease course. New insights from molecular biology/genetics studies, genotype-phenotype correlations, and long-term clinical experience have improved our understanding of the natural history of ACM, showing that presentation with lethal arrhythmias may occur “earlier” than previously thought. Loss of expression of desmosomal proteins might “per se” induce electrical myocardial instability, because of the cross talk of genetically defective desmosomal proteins with the voltage-gated sodium channel complex, leading to sodium ion channel dysfunction and reduced sodium current that predisposes to lethal ventricular arrhythmias even prior to the expression of an overt structural abnormality [31, 39,40,41]. However, whether the mechanism leading to VF during the so-called concealed disease is primarily electrical or triggered by initial (clinically undetectable) histologic structural changes remains elusive.
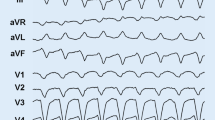
The most common clinical presentation is the classic right ventricular phenotype , characterized by ventricular arrhythmias and related symptoms/events, which include palpitations or effort-induced syncope in an adolescent or young adult, negative T waves in the anterior precordial leads (V1 through V4) on the ECG, ventricular tachycardia with a left bundle branch block morphology, and right ventricular abnormalities on imaging tests (Fig. 32.2).
-
Electrical anomalies secondary to the fibrofatty replacement are the basis for the typical ECG findings, including repolarization (T-wave inversion in the right precordial leads) and/or depolarization (epsilon waves and/or prolongation of QRS complex because of delayed S-wave upstroke) abnormalities. These ECG changes usually involve the right precordial leads (V1 through V3/V4) [42].
-
Ventricular arrhythmias with a left bundle branch block morphology and superior axis are another hallmark of ACM [7,8,9,10, 43,44,45,46]. The spectrum of ventricular arrhythmias ranges from isolated premature ventricular beats to sustained VT, which may degenerate to VF. Ventricular arrhythmias with a left bundle branch block morphology and inferior axis are less specific because in most cases they are benign, non-familial, and not related to an underlying cardiomyopathy (“idiopathic” RV outflow tract ventricular tachycardia) [47].
-
Global or regional dysfunction/structural alterations of the right ventricle on imaging are other typical features of ACM and include: global RV dilatation and dysfunction and regional RV wall motion abnormalities such as diastolic bulging, systolic akinesia, and dyskinesia, typically localized in the “triangle of dysplasia,” namely, subtricuspidal, apical, and infundibular regions. The left ventricle and the septum are usually involved to a lesser extent, if at all [48,49,50]. These structural abnormalities can be detected by imaging techniques such as echocardiography [51], cardiac magnetic resonance (CMR) [52], and angiography [53]. RV angiography has been long regarded as the gold standard imaging test for the diagnosis. Angiographic evidence of systolic akinesia, dyskinesia, and diastolic bulging localized in infundibular, apical, and subtricuspidal regions provides a diagnostic specificity of >90% [20]. Echocardiography represents a first-line approach to evaluate patients with suspected ACM or to screen family members; it is also useful for serial examinations during the follow-up. In recent years, CMR has become the preferred imaging technique, thanks to its capacity to combine the evaluation of morphofunctional abnormalities with myocardial tissue characterization by the gadolinium enhancement, which provides information of the presence and amount of fibrofatty myocardial scarring [21, 54].

Classic right ventricular phenotype. Depolarization (negative T waves in the right precordial leads, a) and repolarization abnormalities (epsilon waves, arrow, and prolongation of QRS complex because of delayed S-wave upstroke, a and b). Ventricular arrhythmia with left bundle branch block and inferior axis morphology is shown in C. Parasternal long-axis view (PLAX) by echocardiography showing dilatation of the right ventricular outflow tract (d). Cardiac MRI illustrating an aneurysm of the RVOT (e, arrowhead) and multiple sacculations of the inferior and apical regions (e, arrows). (From Corrado et al. [57], © 2017 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society)
The clinical presentation of ACM may occasionally simulate an acute myocarditis, characterized by chest pain, transient ST segment and T-wave changes, and increased muscle enzyme levels, with or without ventricular arrhythmias [55].
The diagnosis of biventricular or predominant left ACM may be missed at onset of symptoms in some patients who present years later with heart failure, with or without ventricular arrhythmias, and are incorrectly diagnosed as having idiopathic dilated cardiomyopathy [15, 56]. Left-dominant ACM variants are characterized by early and predominant LV involvement, often because of specific genetic mutations (i.e., mutations in genes encoding for desmoplakin, phospholamban, or filamin C [36, 57]). The phenotype is the counterpart of the classic variant:
-
T-wave inversion in the left precordial leads (V4–V6) and low QRS voltages (<0.5 mV) in the limb leads, which reflect the fibrofatty replacement of the LV wall.
-
Ventricular arrhythmias with a right bundle branch block morphology [58].
-
Left ventricular dilatation and dysfunction and late gadolinium enhancement of the LV wall with a subepicardial or midmyocardial distribution. In contrast with the classic variant, the diagnostic power of echocardiography is limited because LV dilatation and systolic dysfunction, either regional or global, may be absent in patients with predominantly left-sided disease. This is because the fibrofatty scarring process initially involves the subepicardial myocardial layers of the LV wall, which contributes marginally to the development of the contractile power and does not translate into prominent wall motion abnormalities or ejection fraction reduction. Therefore, the left-dominant ACM phenotype in isolation is difficult to diagnose and its incidence is probably underestimated. Contrast-enhanced CMR increases the diagnostic sensitivity because it allows identification of non-transmural LV scars (areas of late gadolinium enhancement) at a subepicardial and/or mid-mural level [59, 60]. The use of CMR has moved the concept of “triangle of dysplasia” to the concept of “quadrangle of dysplasia”: there is another important disease-specific site of cardiac involvement that is the left ventricle inferolateral wall in terms of isolated subepicardial scar. Accordingly, CMR findings of tissue characterization for the detection of fibrofatty myocardial replacement need to be included in the future revision of ITF diagnostic criteria for ACM.
Differential Diagnosis
Diseases that enter differential diagnosis of ACM include either primary arrhythmic conditions (i.e., RV outflow tract ventricular tachycardia, Brugada syndrome) or structural heart muscle disorders affecting the RV, the LV, or both (cardiac sarcoidosis, myocarditis, neuromuscular diseases). Particularly differential diagnosis between ACM and cardiac sarcoidosis is challenging because of the overlapping phenotype, including ECG abnormalities (right precordial T-wave inversion and epsilon waves) and imaging features such as nonischemic right and left ventricular scar [61].
Prognosis
The prognosis of ACM is related to either ventricular electric instability, which may lead to arrhythmic SCD, and progression of ventricular muscle disease resulting in RV or biventricular systolic dysfunction. The overall mortality rate varies among different studies, ranging from 0.08% to 3.6% per year [10, 62]. This high variability depends on the different populations considered in the studies, and reflects the wide spectrum of ACM clinical presentations and the presence of subgroups with variable degrees of phenotypic expression and different SCD risk. The adverse prognosis of ACM patients was initially overestimated by early reports of patients with severe clinical manifestations. Subsequent studies of a broader ACM patient population, including asymptomatic family members and genetically affected individuals with no phenotypic manifestations, have reported significantly lower SCD rates [17].
Clinical Diagnosis
To standardize the clinical diagnosis , in 1994 the International Task Force (ITF) proposed guidelines in the form of a qualitative scoring system with major and minor criteria [63]. These criteria were designed to diagnose the original right ventricular phenotype and based on the combination of multiple sources of information obtained by a multiparametric approach including electrocardiographic, arrhythmic, morphofunctional, and histopathologic findings. Based on their diagnostic accuracy, the criteria were classified as major and minor, and the diagnosis of ARVC was fulfilled in the presence of two major criteria, one major plus two minor, or four minor criteria from different categories.
Although the ITF criteria have been useful for differentiating ARVC from dilated cardiomyopathy or idiopathic right ventricular outflow tract tachycardia, they have shown a lack of sensitivity for the identification of early/minor phenotypes, particularly in the setting of familial screening. In 2010, a revision of diagnostic guidelines was proposed with the aim to improve the accuracy of the diagnosis in probands and first-degree relatives by providing quantitative criteria for diagnosing right ventricular abnormalities and adding molecular genetic criteria [64]. In this regard, molecular genetic information has been added to family history criteria, and the presence of a disease-causing gene mutation has become a major criterion for the diagnosis of ACM. The diagnosis is based on a series of criteria including histopathological manifestations, alterations in cardiac structure and function, electrocardiographic abnormalities, arrhythmic manifestations, and the identification of disease-causing genetic mutations that were elaborated by the International Task Force of experts. As no single criterion is accurate enough, the diagnosis requires a combination of criteria (classified as minor or major according to their specificity). Specifically, the diagnosis is considered to be “definite” in the presence of two major criteria or one major and two minors or four minor criteria from different categories. The diagnosis is classified as “borderline,” if the patient meets one major and two minor criteria or three minor criteria from different categories, and “possible” when one major or two minor criteria are met.
The real experience with the 2010 diagnostic criteria revealed a high risk of disease misdiagnosis due to both the inherent limitation and the inappropriate use of the current criteria. This is mostly related to the limited understanding of the genetic background of the disease, characterized by a large pool of genetic variants of uncertain significance, and the subsequent risk of misinterpreting non-pathogenetic DNA variants as pathogenic mutations, which can lead to misdiagnosis of the disease in probands not fulfilling the phenotypic criteria for diagnosis. Therefore, future ITF diagnostic criteria should be based on phenotypic features of the proband and genotyping used for cascade family screening and preclinical genetic diagnosis of family members. An additional limit of the 2010 criteria is that they were developed to diagnose the classic right ventricular phenotype and did not include criteria for the diagnosis of left-dominant phenotype. Contrast-enhanced CMR has emerged as the “gold standard” imaging test, because it increases diagnostic accuracy by identification of nonischemic LV scars/LGE which are the most typical features of the left ventricular phenotype.
Prognosis and Treatment
Risk Stratification
The clinical course of ACM is characterized by the occurrence of arrhythmic events, which can cause SCD, and the impairment of biventricular systolic function, which can lead to death from heart failure. The estimated overall mortality varies among studies, ranging from 0.08 to 3.6% per year [65]. Risk stratification depends largely on the evaluation of the severity of ACM phenotypic expression in terms of severity of arrhythmic manifestations and amount of fibrofatty myocardial replacement as evaluated by electrocardiographic tests and imaging. History of cardiac arrest because of VF or sustained VT is the most important predictor of major arrhythmic events during follow-up. Unexplained syncope, nonsustained VT, and severe systolic dysfunction of the right ventricle, left ventricle, or both are considered major risk factors. Several minor risk factors have been identified (proband status, male sex, multiple gene mutations [3, 39], greater extent of negative T waves [66], VT/VF inducibility by programmed ventricular stimulation [38, 60]), but their association with an unfavorable outcome is based on either limited scientific evidence or conflicting data (Fig. 32.3) [67].

Prognostic stratification of patients with ARVC based on the clinical presentation. The risk subgroups are defined on the estimated probability of a major arrhythmic event during follow-up. The high-risk group is characterized by an estimated annual risk of more than 10%, the intermediate-risk group by a risk between 1% and 10%, and the low-risk group by a risk below 1%. (From Corrado et al. [57], © 2017 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society)
Therapy
The aims of clinical management of ACM are as follows: (1) to reduce the risk of sudden cardiac death, (2) to prevent the disease progression, and (3) to improve the quality of life by alleviating arrhythmic and heart failure symptoms. Current therapeutic options include lifestyle changes, drug therapy, catheter ablation, ICD, and heart transplantation.
Lifestyle Changes
Physical exercise has been implicated as the most important environmental factor for the promotion and progression of the ACM phenotypic expression and for triggering life-threatening ventricular arrhythmias in ACM [36, 68,69,70,71,72,73]. Accordingly, current guidelines recommend that not only clinically affected individuals but also healthy gene carriers do not participate in competitive or endurance sport activity [67].
Drug Therapy
Drug therapy of patients with ACM includes beta-blockers, anti-arrhythmic agents, and heart failure drug therapy. Adrenergic stimulation promotes ventricular arrhythmias and cardiac arrest that typically occur during or after physical effort. Beta-blocker therapy should be offered to all patients with a definite diagnosis of ACM, regardless of symptoms and arrhythmic manifestations. Beta-blocker therapy has proven efficacy in heart failure management and can prevent effort-induced ventricular arrhythmias; moreover, it offers the potential to lower the myocardial disease progression by lowering the ventricular wall stress. Instead, in genotype-positive but phenotype-negative individuals, prophylactic use of drugs does not seem justified [67]. Adjunctive anti-arrhythmic drug therapy is indicated to reduce the arrhythmia burden in symptomatic patients with frequent premature ventricular beats and/or complex ventricular arrhythmias [67]. Sotalol and amiodarone (alone or in combination with beta-blockers) are the most effective drugs with a relatively low proarrhythmic risk. The standard pharmacological treatment (angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and diuretics) is used in patients who develop right, left, or biventricular heart failure. Oral antithrombotic therapy should be reserved to patients with atrial fibrillation and/or thromboembolic events.
Catheter Ablation
Catheter ablation is a therapeutic option for patients with sustained monomorphic ventricular tachycardia. The initial experience with VT catheter ablation resulted in a high acute success rate followed by a high rate of recurrence [74,75,76]. The poor long-term outcome has been attributed to the progressive nature of the disease, which leads to the development of multiple arrhythmogenic foci over time. In addition, because the lesion wave-front characteristically originates and progresses from the epicardium to the endocardium, VT reentry circuits appear mostly located in the subepicardial layer of the RV wall and are unapproachable from the traditional endocardial approach of catheter ablation. Several studies have shown the feasibility and efficacy of epicardial (through the pericardial space) catheter ablation for patients in whom one or more endocardial procedures have been unsuccessful (Fig. 32.4) [77,78,79,80,81].


Catheter ablation of ventricular tachycardia in patients with ARVC. Catheter ablation uses an endocardial or an epicardial approach depending on the site of the arrhythmia substrate. For endocardial ablation (Panel A), the catheter is advanced into the right ventricular cavity through the venous system; the black circular arrow shows a reentry circuit of ventricular tachycardia, and the red X indicates its interruption by endocardial catheter ablation. Epicardial ablation (Panel B) is based on the introduction of the catheter into the pericardial space by means of pericardial puncture; the black circular arrow shows a reentry circuit, and the red X indicates its interruption by epicardial catheter ablation. Target sites for catheter ablation can be identified with the use of three-dimensional electroanatomical voltage mapping (Panel C) to reconstruct regions of right ventricular scarring (i.e., either endocardial or epicardial low-voltage areas showing bipolar signal amplitude of <0.5 mV, indicated by red color coding), which represent the substrate for the reentry mechanism of ventricular tachycardia. The reentry circuit is interrupted by delivering radiofrequency energy through the ablation catheter to create point-by-point linear lesions (red circles), eliminating intrascar or interscar conducting pathways. On an electrophysiological recording obtained during catheter ablation (Panel D), ventricular tachycardia is interrupted abruptly after radiofrequency energy application (red arrow). Standard electrocardiographic leads I, III, V1, and V6 are shown, as well as recordings from the ablation catheter itself at proximal (ABL p) and distal (ABL d) sites. LAD denotes left-axis deviation, LBBB left bundle branch block, and RF radiofrequency. (From Corrado et al. [57], © 2017 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society)
ICD Therapy
Although randomized trials to guide ICD therapy have not been performed, data collected from observational studies consistently showed the safety and efficacy of ICD therapy [37, 48, 82,83,84,85]. The indication for ICD therapy should be the result of a balanced evaluation of the arrhythmic patient’s profile and potential risk of device-related complication. Patients who benefit most from ICD therapy are those who had experienced an episode of VF or sustained VT (high-risk category, estimated event rate >10% per year [64]). It remains uncertain whether defibrillator therapy is appropriate for primary prevention of SCD among patients with one or more risk factors and no prior major arrhythmic events (intermediate-risk category, estimated event rate ranging from 1% to 10% per year [88]). Prophylactic defibrillator implantation is not recommended in asymptomatic patients with no risk factors and in healthy gene carriers (low-risk category, estimated event rate <1% per year) because of the low risk of arrhythmias and the concomitant significant risk of device-related complications over time (Fig. 32.5). Subcutaneous ICD has emerged as a valid alternative to transvenous device because it reduces or eliminates morbidity and potentially life-threatening lead-related complications over the time, mostly in the young population with a long life expectancy. Preliminary data on the use of S-ICD in patients with ACM demonstrate that the device successfully treats VT/VF in the absence of electrode and device-related complications with a rate of inappropriate shock comparable to that of transvenous ICD. At present time, S-ICD is mainly indicated for the primary prevention of SCD in young patients with ACM and a high-risk profile [86,87,88,89,90].

Flowchart of risk stratification and indications to ICD implantation in ACM. Based on the available data on the annual mortality rates associated to specific risk factors, the estimated risk of major arrhythmic events in the high-risk category is >10%/year, in the intermediate-risk category ranges from 1% to 10%/year, and in the low-risk category is <1%/year. The recommendations for ICD implantation for different categories of arrhythmic risk were determined by consensus considering not only the statistical risk but also the general health, the socioeconomic factors, the psychological impact, and the adverse effects of the device. SCD, sudden cardiac death; VF, ventricular fibrillation; VT, ventricular tachycardia; RV, right ventricle; and LV, left ventricle. (From Corrado et al. [65]. Reprinted with permission from Wolters Kluwer Health, Inc.).*See the text for distinction between major and minor risk factors
Heart Transplant
Heart transplant is the final treatment option for patients with debilitating heart failure refractory to pharmacological and non-pharmacological therapy or potentially lethal arrhythmias that are uncontrollable with anti-arrhythmic therapy or catheter ablation. Posttransplant survival rates are reported of 94% and 88% at 1 and 6 years, respectively [91].
Competing Interest Declaration
None to declare.
References
Corrado D, Basso C, Pilichou K, Thiene G. Molecular biology and clinical management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart. 2011;97:530–9.
Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113:1634–7.
Rigato I, Bauce B, Rampazzo A, Zorzi A, Pilichou K, Mazzotti E, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6:533–42.
Norman M, Simpson M, Mogensen J, Shaw A, Hughes S, Syrris P, et al. Novel mutation in desmoplakin causes arrhythmogenic left ventricular cardiomyopathy. Circulation. 2005;112:636–42.
Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65:384–98.
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318:129–33.
Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94:983–91.
Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicentre study. J Am Coll Cardiol. 1997;30:1512–20.
McKoy G, Protonotarios N, Crosby A, Tsatsopoulou A, Anastasakis A, Coonar A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355:2119–24.
Carvajal-Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol. 1998;39:418–21.
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71:1200–6.
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–4.
Awad MM, Dalal D, Cho E, Amat-Alarcon N, James C, Tichnell C, et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79:136–42.
Pilichou K, Nava A, Basso C, Beffagna G, Bauce B, Lorenzon A, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–9.
Syrris P, Ward D, Evans A, Asimaki A, Gandjbakhch E, Sen-Chowdhry S, McKenna WJ. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79:978–84.
Van Hengel J, Calore M, Bauce B, Dazzo E, Mazzotti E, De Bortoli M, et al. Mutations in the area composita protein αT-catenin are associated with arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2013;34:201–10.
Mayosi BM, Fish M, Shaboodien G, Mastantuono E, Kraus S, Wieland T, et al. Identification of cadherin 2 (CDH2) mutations in arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2017;10:001605.
Turkowski KL, Tester DJ, Bos JM, Haugaa KH, Ackerman MJ. Whole exome sequencing with genomic triangulation implicates CDH2-encoded N-cadherin as a novel pathogenic substrate for arrhythmogenic cardiomyopathy. Congenit Heart Dis. 2017;12:226–35.
Young SG, Jung HJ, Lee JM, Fong LG. Nuclear lamins and neurobiology. Mol Cell Biol. 2014;34:2776–85.
Quarta G, Syrris P, Ashworth M, Jenkins S, Zuborne Alapi K, Morgan J, et al. Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;33:1128–36.
Merner ND, Hodgkinson KA, Haywood AF, Connors S, French VM, Drenckhahn JD, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82:809–21.
Baskin B, Skinner JR, Sanatani S, Terespolsky D, Krahn AD, Ray PN, et al. TMEM43 mutations associated with arrhythmogenic right ventricular cardiomyopathy in non-Newfoundland populations. Hum Genet. 2013;132:1245–52.
Greenberg SA, Salajegheh M, Judge DP, Feldman MW, Kuncl RW, Waldon Z, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol. 2012;71:141–5.
Van der Zwaag PA, van den Berg MP, Cox MG, et al. One mutation fits all: phospholamban R14del causes both dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2010;122:A17663.
Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, et al. Suppression of canonical Wnt/ beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–21.
Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107:700–14.
Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004;1:3–11.
Sato PY, Musa H, Coombs W, Guerrero-Serna G, Patiño GA, Taffet SM, et al. Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. 2009;105:523–6.
Rizzo S, Lodder EM, Verkerk AO, Wolswinkel R, Beekman L, Pilichou K, et al. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95:409–18.
Corrado D, Zorzi A, Cerrone M, Rigato I, Mongillo M, Bauce B, et al. Relationship between arrhythmogenic right ventricular cardiomyopathy and Brugada syndrome: new insights from molecular biology and clinical implications. Circ Arrhythm Electrophysiol. 2016;9(4):e003631.
Marcus FI, Edson S, Tobwin JA. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide fot physicians. J Am Coll Cardiol. 2013;61:1945–8.
James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:129–7.
Lemola K, Brunckhorst C, Helfenstein U, Oechslin E, Jenni R, Duru F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91(9):1167–72.
Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42:1959–63.
Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–91.
Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JD, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–55.
Bhonsale A, te Riele ASJM, Sawant AC. Cardiac phenotype and long term prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia patients with late presentation. Heart Rhythm. 2017;14(6):883–91.
Corrado D, Zorzi A. Natural history of arrhythmogenic cardiomyopathy: redefining the age range of clinical presentation. Heart Rhythm. 2017;14(6):892–3.
Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129(10):1092–103.
Cerrone M, Noorman M, Lin X, Chkourko H, Liang FX, van der Nagel R, et al. Sodium current deficit and arrhythmogenesis in a murine model of plakophilin-2 haploinsufficiency. Cardiovasc Res. 2012;95:460–8.
Sato PY, Coombs W, Lin X, Nekrasova O, Green KJ, Isom LL, et al. Interactions between ankyrin-G, Plakophilin-2, and Connexin43 at the cardiac intercalated disc. Circ Res. 2011;109:193–201.
Steriotis AK, Bauce B, Daliento L, Rigato I, Mazzotti E, Folino AF, et al. Electrocardiographic pattern in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2009;103(9):1302–8.
Marcus FI, Zareba W, Calkins H, Towbin JA, Basso C, Bluemke DA, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia clinical presentation and diagnostic evaluation: results from the North American Multidisciplinary Study. Heart Rhythm. 2009;6:984–92.
Zorzi A, Rigato I, Pilichou K, Perazzolo Marra M, Migliore F, Mazzotti E, et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18:1086–94.
Protonotarios A, Anastasakis A, Panagiotakos DB, Antoniades L, Syrris P, Vouliotis A, et al. Arrhythmic risk assessment in genotyped families with arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18:610–6.
Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8:437–46.
O'Donnell D, Cox D, Bourke J, Mitchell L, Furniss S. Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract tachycardia. Eur Heart J. 2003;24(9):801–10.
Yoerger DM, Marcus F, Sherril D, Calkins H, Towbin JA, Zareba W, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the Multidisciplinary Study of Right Ventricular Dysplasia. J Am Coll Cardiol. 2005;45:860–5.
Indik JH, Wichter T, Gear K, Dallas WJ, Marcus FI. Quantitative assessment of angiographic right ventricular wall motion in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC/D). J Cardiovasc Electrophysiol. 2008;19:39–45.
Sen-Chowdhry S, Prasad SK, Syrris P, Wage R, Ward D, Merrifield R, et al. Cardiovascular magnetic resonance in arrhythmogenic right ventricular cardiomiopathy revisited: comparison with Task Force Criteria and genotype. J Am Coll Cardiol. 2006;48(10):2132–40.
Blomstrom-Lunqqvist C, Beckman-Suurkula M, Wallentin I, Jonsson R, Olsson SB. Ventricular dimensions and wall motion assessed by echocardiography in patients with arrhythmogenic right ventricular dysplasia. Eur Heart J. 1998;9:1291–302.
Meneghetti L, Basso C, Nava A, Angelini A, Thiene G. Spinecho nuclear magnetic resonance for tissue characterization in arrhythmogenic right ventricular cardiomyopathy. Heart. 1996;76:467–70.
Daliento L, Rizzoli G, Thiene G, Nava A, Rinuncini M, Chioin R, et al. Diagnostic accuracy of right ventriculography in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 1990;66:741–5.
Etoom Y, Govindapillai S, Hamilton R, Manlhiot C, Yoo SJ, Farhan M, et al. Importance of CMR within the Task Force Criteria for the diagnosis of ARVC in children and adolescents. J Am Coll Cardiol. 2015;65:987–95.
Thiene G, Corrado D, Nava A, Rossi L, Poletti A, Boffa GM, et al. Right ventricular cardiomyopathy: is there evidence of an inflammatory aetiology? Eur Heart J. 1991;12(suppl D):22–5.
Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110:1879–84.
Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72.
Basso C, Corrado D, Marcus F, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–300.
Zorzi A, Perazzolo Marra M, Rigato I, De Lazzari M, Susana A, Niero A, et al. Nonischemic left ventricular scar as a substrate of life-threatening ventricular arrhythmias and sudden cardiac death in competitive athletes. Circ Arrhythm Electrophysiol. 2016;9(7):e004229–e004229.
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–87.
Corrado D, Van Tintelen JP, McKenna WJ, Hauer RN, Anastasakis A, Asimaki A, et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur Heart J. 2019;1–16 in press.; https://doi.org/10.1093/eurheartj/ehz669.
Nava A, Bauce B, Basso C, Muriago M, Rampazzo A, Villanova C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36:2226–33.
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom-Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Br Heart J. 1994;71:215–8.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–41.
Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an International Task Force consensus statement. Circulation. 2015;132:441–53.
Zorzi A, Migliore F, Elmaghawry M, Silvano M, Marra MP, Niero A, et al. Electrocardiographic predictors of electroanatomic scar size in arrhythmogenic right ventricular cardiomyopathy: implications for arrhythmic risk stratification. J Cardiovasc Electrophysiol. 2013;24:1321–7.
Priori SG, Blomström-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the Task Force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Europace. 2015;17:1601–87.
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65:366–73.
Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65:1324–36.
Corrado D, Thiene G, Nava A, Rossi L, Pennelli N. Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med. 1990;89:588–96.
Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296:1593–601.
Kirchhof P, Fabritz L, Zwiener M, Witt H, Schäfers M, Zellerhoff S, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin- deficient mice. Circulation. 2006;114:1799–806.
Cruz FM, Sanz-Rosa D, Roche-Molina M, García-Prieto J, García-Ruiz JM, Pizarro G, et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J Am Coll Cardiol. 2015;65:1438–50.
Marchlinski FE, Zado E, Dixit S, Gerstenfeld E, Callans DJ, Hsia H, et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation. 2004;110:2293–8.
Verma A, Kilicaslan F, Schweikert RA, Tomassoni G, Rossillo A, Marrouche NF, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation. 2005;111:3209–16.
Dalal D, Jain R, Tandri H, Dong J, Eid SM, Prakasa K, et al. Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/ cardiomyopathy. J Am Coll Cardiol. 2007;50:432–40.
Garcia FC, Bazan V, Zado ES, Ren JF, Marchlinski FE. Epicardial substrate and outcome with epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2009;120:366–75.
Philips B, Madhavan S, James C, Tichnell C, Murray B, Dalal D, et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:499–505.
Philips B, te Riele AS, Sawant A, Kareddy V, James CA, Murray B, et al. Outcomes and ventricular tachycardia recurrence characteristics after epicardial ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/ cardiomyopathy. Heart Rhythm. 2015;12:716–25.
Santangeli P, Zado ES, Supple GE, Haqqani HM, Garcia FC, Tschabrunn CM, et al. Long-term outcome with catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8:1413–21.
Della Bella P, Brugada J, Zeppenfeld K, Merino J, Neuzil P, Maury P, et al. Epicardial ablation for ventricular tachycardia: a European multicenter study. Circ Arrhythm Electrophysiol. 2011;4:653–9.
Wichter T, Paul M, Wollmann C, Acil T, Gerdes P, Ashraf O, et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004;109:1503–8.
Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010;122:1144–52.
Bhonsale A, James CA, Tichnell C, Murray B, Gagarin D, Philips B, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter- defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58:1485–96.
Link MS, Laidlaw D, Polonsky B, Zareba W, McNitt S, Gear K, et al. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, characteristics, and treatment. J Am Coll Cardiol. 2014;64:119–25.
Orgeron GM, Bhonsale A, Migliore F, James CA, Tichnell C, Murray B, et al. Subcutaneous implantable cardioverter-defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia: a transatlantic experience. J Am Heart Assoc. 2018;7(21):e008782.
Migliore F, Mattesi G, De Franceschi P, Allocca G, Crosato M, Calzolari V, et al. Multicentre experience with the second-generation subcutaneous implantable cardioverter defibrillator and the intermuscular two-incision implantation technique. J Cardiovasc Electrophysiol. 2019;3(6):854–64. https://doi.org/10.1111/jce.13894.
Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rythm Society. Circulation. 2018;138:e272–391.
Basu-Ray I, Liu J, Jia X, Gold M, Ellenbogen K, DiNicolantonio J, et al. Subcutaneous versus transvenous implantable defibrillator therapy: a meta-analysis of case-control studies. JACC Clin Electrophysiol. 2017;3(13):1475–83.
Boersma L, Barr C, Knops R, Theuns D, Eckardt L, Neuzil P, et al. Implant and midterm outcomes of the subcutaneous implantable cardioverter-defibrillator registry the EFFORTLESS study. J Am Coll Cardiol. 2017;70:830–41.
Tedford RJ, James C, Judge DP, Tichnell C, Murray B, Bhonsale A, et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012;17(59):289–90.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mattesi, G., Cipriani, A., Zorzi, A., Corrado, D. (2020). Arrhythmogenic Right Ventricular Cardiomyopathy. In: Yan, GX., Kowey, P., Antzelevitch, C. (eds) Management of Cardiac Arrhythmias. Contemporary Cardiology. Humana, Cham. https://doi.org/10.1007/978-3-030-41967-7_32
Download citation
DOI: https://doi.org/10.1007/978-3-030-41967-7_32
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-41966-0
Online ISBN: 978-3-030-41967-7
eBook Packages: MedicineMedicine (R0)