
Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiovascular disorder leading to life-threatening ventricular arrhythmias, progressive biventricular dysfunction, and heart failure. Sudden death can be the unique feature of the disease. Genetic studies indicate that ARVC should be considered a disease of desmosome dysfunction. Diagnosis remains a clinical challenge mainly in its early stages and in patients with minimal imaging structural abnormalities. ARVC shares some common features with other cardiac diseases, such as RV outflow tract ventricular tachycardia, Brugada syndrome, dilated cardiomyopathy, and myocarditis, due to arrhythmic expressivity and biventricular involvement. Diagnosis is based on major and minor criteria listed in the Revised Task Force Criteria.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cardiac Magnetic Resonance
- Late Gadolinium Enhancement
- Arrhythmogenic Right Ventricular Cardiomyopathy
- Brugada Syndrome
- Task Force Criterion
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiovascular disorder characterized by myocyte loss and fibrofatty tissue replacement leading to life-threatening ventricular arrhythmias, progressive ventricular dysfunction of the right (RV) and left ventricle (LV), and heart failure (HF) [1, 2]. The estimated prevalence of ARVC in the general population ranges from 1:2,000 to 1:5,000 individuals; men are more frequently affected than women, with an approximate ratio of 3:1 [3]. A familial history of ARVC is present in 30–50 % of cases, and the disease is usually inherited in an autosomal dominant pattern, with variable penetrance and expressivity, although autosomal recessive forms are also reported (Naxos disease and Carvajal syndrome). ARVC is considered to be a disease of myocyte adhesion caused by defects at the intercellular junctions. Cardiac myocyte-to-myocyte adhesion is maintained by desmosomes, adherens junctions, and gap junctions, which together comprise the intercalated disc.
A genetic defect can be confirmed in approximately 40 % of cases, and 12 different ARVC loci have been reported, among which five genes (DSP, PKP2, DSG2, DSC2, and JUP) encode proteins of cell–cell junctions at the intercalated disc. The role of the other three nondesmosomal genes is well established: transforming growth factor beta-3 (TGF-β3), the ion-channel subunit RYR2, and the transmembrane protein 43 (TMEM43) [4]. Novel variants in the giant sarcomeric protein titin (TTN) are also associated with ARVC [5]. Structural impairment of titin, which probably leads to proteolysis and apoptosis, constitutes to be a novel mechanism underlying myocardial remodeling and sudden death (SD).
2 Clinical Features
ARVC onset usually occurs after childhood, with palpitations and/or syncope. In some cases, severe ventricular arrhythmias are the first presentation of the disease and lead to SD. Since 1995, a recessive form of ARVC has been recognized with a distinct phenotypic expression and cardiocutaneous aspects characterized by palmoplantar keratoderma and woolly hair, well known as Naxos disease. Furthermore, ARVC is the second most frequent cause of SD in young adults and athletes, and cardiac arrest may occur in up to 50 % of index cases [6]. According to Dalal et al. [7], the median age at onset is 29 years. It is rare to manifest clinical signs or symptoms of ARVC before the age of 12 years or after the age of 60 years.
According to the Padua group [8], the natural history of ARVC may be separated into four distinct phases, with progressive development of symptoms and structural abnormalities:
-
1.
Concealed phase: This phase is characterized by minor arrhythmias that usually go unnoticed and subtle or absent structural RV abnormalities. The diagnosis is usually made during family screening in asymptomatic individuals. SD may be the first and unique manifestation of the disease at this initial stage.
-
2.
Overt electrical disorder with palpitations, syncope, and ventricular arrhythmias of RV origin: This manifestation is usually triggered by effort. Arrhythmias range from isolated premature ventricular beats to nonsustained ventricular tachycardia with left bundle-branch block (LBBB) morphology up to ventricular fibrillation leading to cardiac arrest.
-
3.
RV failure due to progressive myocardial fibrofatty replacement: This manifestation leads to RV enlargement and systolic dysfunction with consequent HF.
-
4.
Biventricular failure, which usually develops late in the natural history of the disease: Progressive structural abnormalities involve the LV, with symptoms of overt congestive HF. In such conditions, contractile dysfunction may be so severe as to require cardiac transplantation. Endocavitary mural thrombosis may occur, especially within RV aneurysms or in the atria in the presence of atrial fibrillation (AF). The ultimate phenotype may resemble features of dilated cardiomyopathy (DCM) with biventricular involvement, making differential diagnosis difficult.
The diagnosis of ARVC is often challenging due to heterogeneous clinical presentation, highly variable intra- and interfamily expressivity, and incomplete penetrance. This genotype–phenotype plasticity is as yet largely unexplained.
3 Diagnosis
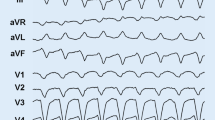
There is no single gold-standard diagnostic test for ARVC. Therefore, diagnosis relies on a scoring system, with major and minor criteria based on demonstration of a combination of abnormalities in RV morphology and function, typical depolarization/repolarization electrocardiographic (ECG) changes (Fig. 14.1), peculiar histological findings, ventricular arrhythmias, family history, and results of genetic testing. Definitive diagnosis, based on the Revised 2010 Task Force Criteria [9] (Table 14.1), requires two major criteria, one major plus two minor criteria, or four minor criteria from different categories. Therefore, initial evaluation of all patients suspected of having ARVC should include physical examination; clinical history; family history of ARVC or arrhythmias or SD; ECG; signal-averaged ECG; 24-h Holter monitoring; and comprehensive nonechocardiography focused on both ventricles. This imaging technique can reveal RV structural abnormalities such as RV dilation, aneurysm formation, and functional abnormalities, including hypo-a-dyskinetic RV regions, RV systolic dysfunction, paradoxical septal motion, and tricuspid regurgitation [10]. In the late stages, LV involvement with biventricular failure is observed. New tools for improving diagnostic accuracy are now available in clinical practice. Among noninvasive investigations, cardiac magnetic resonance (CMR) with late gadolinium enhancement (LGE) can detect myocardial fibrosis and intramyocardial fatty infiltration. CMR allows the clearest visualization of the RV (for dilatation, dysfunction, regional wall motion abnormalities, and aneurysm formation) [11].

Typical electrocardiogram (ECG) in advanced arrhythmogenic right ventricular cardiomyopathy (ARVC). Right atrial enlargement, low QRS voltages, epsilon waves, and negative T waves in anterior precordial leads are present
Diagnosis of ARVC remains a clinical challenge, particularly in its early stages and in patients with minimal echocardiographic RV abnormalities and especially in the absence of structural changes in the typical triangle of dysplasia [subtricuspid region, RV outflow tract (OT), and RV inferoapical region] [12]. Positive endomyocardial biopsy (EMB) of the RV is a recognized gold standard, but it often yields a false-negative result (sensitivity ~67 %) because of the frequently localized fibroadipose infiltration. Consequently, the best approach in making a diagnosis of ARVC is by combining different diagnostic tests [9, 13]. The histological hallmark of the disease is fibrofatty infiltration of the RV myocardium with areas of surviving myocytes (Fig. 14.2) and sometimes inflammatory infiltration. Pathologic abnormalities can progress with time, typically starting from the epicardium and eventually extending down to reach the subendocardium and becoming transmural. This implies a weakness and thinning of the free wall, resulting in RV dilatation and aneurysm formation, bulges, and sacculations, which constitute the typical diagnostic findings on noninvasive imaging tests [14].

Histologic specimen (Azan Mallory, ×20) at the right ventricular level in a case of arrhythmogenic right ventricular cardiomyopathy (ARVC). Severe fatty infiltration associated with patchy and interstitial fibrosis (blue) is present
LV involvement, typically affecting the posterior and lateral walls, is present in more than half of ARVC cases [15, 16]. The frequent, and sometimes predominant, LV involvement suggests that ARVC is not a unique entity but a complex disease, with three possible patterns of expression: classic right-dominant (39 % of cases), left-dominant arrhythmogenic cardiomyopathy (LDAC) (5 %), and biventricular (56 %) forms [17, 18]. Interestingly, recent data showed that the LV may be affected not only in the late stage of the disease but may also occur in absence of alterations in RV systolic dysfunction, characterizing the LDAC form of the disease [16]. This left-dominant pattern is characterized by predominant LV involvement (dilation, systolic impairment, LGE) exceeding that of the RV or in the presence of preserved RV function [19]. Other features of this pattern are the LV origin of arrhythmias (RBBB morphology), inferolateral T-wave inversion on ECG (Fig. 14.3), and family history of LDAC.

Electrocardiogram (ECG) of a patient with arrhythmogenic right ventricular cardiomyopathy with biventricular involvement. Sinus rhythm; epsilon waves in V1; inverted T waves in right precordial leads; negative T waves from V4 to V6 and inferior leads; deep Q waves in inferolateral leads
3.1 Differential Diagnosis
Diagnosis of ARVC should be considered in any patient who does not have known heart disease and who presents with frequent premature ventricular contractions or symptomatic ventricular tachycardia. The main differential diagnoses include the following conditions:
-
1.
Idiopathic RV outflow tract/ventricular tachycardia is a mostly benign condition that is not associated with structural heart disease. In its early stage, ARVC can be difficult to distinguish from this idiopathic type of ventricular arrhythmia in the absence of structural changes. Differential diagnosis is based on the fact that this arrhythmia is nonfamilial, and patients do not have the characteristic ECG/signal average ECG abnormalities of ARVC (inversion T waves in V1–V3, epsilon waves, QRS duration >110 ms) (Table 14.2) [20].
Table 14.2 Clinical expressions of RVOT VT and ARVC -
2.
Brugada syndrome is an inherited cardiac condition that, similarly to ARVC, is transmitted with an autosomal dominant pattern, which can lead to SD from malignant ventricular arrhythmias. Conversely, it is also characterized by a distinct typical ECG pattern, with J wave in precordial leads (Fig. 14.4) and by the absence of morphological echocardiographic features.
Fig. 14.4 
Electrocardiogram (ECG) of a patient with Brugada syndrome: sinus rhythm. In precordial leads (V1–3), a typical type 1 pattern is present. “Coved-type” ST elevation with at least 2 mm (0.2 mV) J-point elevation, gradually descending ST segment, followed by a negative T wave
-
3.
Dilated cardiomyopathy may be difficult to distinguish from ARVC, especially in its advanced stage with severe biventricular involvement. In this late phase, signs and symptoms of RV and/or LV failure can be present; finally, severe biventricular congestive HF can occur. In the absence of classic ARVC hallmarks (RV aneurysms, bulging), clinical distinction between these two CMP can be extremely difficult or impossible. Table 14.3 shows some differences between these two pathologies that are useful in differential diagnosis.
Table 14.3 Differential diagnosis between ARVC with biventricular involvement and DCM -
4.
Myocarditis can mimic ARVC, especially when the RV is involved. Myocarditis can cause structural abnormalities, including microaneurysms, as well as the arrhythmic manifestations considered typical of ARVC. Moreover, myocardial inflammatory infiltrates, myocyte necrosis, and replacement fibrosis may lead to functional and structural changes in the RV myocardium, resembling those produced by ARVC fibrofatty replacement. New tools, such as 3D electroanatomic mapping, applied to the standard EMB, have been introduced to improve diagnostic accuracy in clinical practice. In a provocative study, Pieroni et al. [21] found that 50 % of patients with a diagnosis of noninvasive ARVC fulfilled Dallas histological criteria of active myocarditis. These data require confirmation in large patient populations.
-
5.
Sarcoidosis with cardiac involvement can mimic ARVC, making accurate differential diagnosis more challenging. It must be considered if conduction defects with a high-grade atrioventricular block, respiratory, or systemic symptoms are present. Global RV hypokinesis or some regional wall motion abnormalities can be present due to the patchy nature of the granulomatous infiltration. Both sarcoidosis and ARVC can be progressive pathologies, and the accuracy of CMR could vary depending on the stage of the disease at which CMR data are acquired. The absence of myocardial fat infiltrates at CMR could be a useful distinguishing feature by which to suspect sarcoidosis [22].
-
6.
Other pathologies:
-
(a)
Coronary artery disease and myocardial infarction can involve both ventricles and mimic aspects of ARVC.
-
(b)
Pulmonary hypertension (RV-pressure overload) and tricuspid regurgitation (RV-volume overload secondary to increasing stroke volume) can cause RV dilation and dysfunction.
-
(c)
Congenital heart diseases, such as Uhl anomaly (a rare congenital heart disease with a partial or total loss of the RV myocardial muscle) [22] and repaired tetralogy of Fallot must be considered, especially for their prevalent RV involvement.
-
(d)
Intracardiac shunts (e.g., atrial septal defects and anomalous pulmonary venous drainage) may cause RV volume overload. The diagnosis can be missed on standard echocardiogram, and in this cases, transesophageal echocardiography (TEE) and/or CMR (which have excellent correlation with RV angiography) can improve diagnostic accuracy.
-
(a)

4 Conclusions
ARVC is a frequently progressive disease with risk of life-threatening complications and which constitutes a clinical diagnostic challenge for physicians given the different genotypic and phenotypic variations and the wide ranges of clinical manifestations. Genetic studies indicate that ARVC should be considered a disease of desmosome dysfunction. Its diagnosis is based on the modified Task Force Criteria for ARVC [9] and should be approached with great caution. The main challenge is to improve risk stratification in relation to SD and HF and identify patients who will most benefit from early intervention involving lifestyle changes, restriction of physical sport activity, antiarrhythmic drugs, and/or ICD placement.
References
Corrado D, Basso C, Thiene G (2000) Arrhythmogenic right ventricular cardiomyopathy: diagnosis, prognosis, and treatment. Heart 83(5):588–595
Pinamonti B, Sinagra G, Camerini F (2000) Clinical relevance of right ventricular dysplasia/cardiomyopathy. Heart 83:9–11
Corrado D, Thiene G (2006) Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 113(13):1634–1637. doi:10.1161/CIRCULATIONAHA.105.616490
Campuzano O, Alcalde M, Allegue C, Iglesias A, Garcia-Pavia P, Partemi S, Oliva A, Pascali VL, Berne P, Sarquella-Brugada G, Brugada J, Brugada P, Brugada R (2013) Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet 50(5):280–289. doi:10.1136/jmedgenet-2013-101523
Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L (2011) Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation 124(8):876–885. doi:10.1161/CIRCULATIONAHA.110.005405
Corrado D, Thiene G, Nava A, Rossi L, Pennelli N (1990) Sudden death in young competitive athletes: clinicopathologic correlations in 22 cases. Am J Med 89(5):588–596
Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H (2005) Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation 112(25):3823–3832. doi:10.1161/CIRCULATIONAHA.105.542266
Basso C, Corrado D, Marcus FI, Nava A, Thiene G (2009) Arrhythmogenic right ventricular cardiomyopathy. Lancet 373(9671):1289–1300. doi:10.1016/S0140-6736(09)60256-7
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 121(13):1533–1541. doi:10.1161/CIRCULATIONAHA.108.840827
Scheinman MM, Crawford MH (2005) Echocardiographic findings and the search for a gold standard in patients with arrhythmogenic right ventricular dysplasia. J Am Coll Cardiol 45(6):866–867. doi:10.1016/j.jacc.2004.12.021
Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA (2005) Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol 45(1):98–103. doi:10.1016/j.jacc.2004.09.053
Basso C, Thiene G (2005) Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc Pathol 14(1):37–41. doi:10.1016/j.carpath.2004.12.001
Angelini A, Basso C, Nava A, Thiene G (1996) Endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy. Am Heart J 132(1 Pt 1):203–206
Fisher NG, Gilbert TJ (2000) Arrhythmogenic right ventricular dysplasia. An illustrated review highlighting developments in the diagnosis and management of this potentially fatal condition. Postgrad Med J 76(897):395–398
Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, Nava A, Silvestri F, Blomstrom-Lundqvist C, Wlodarska EK, Fontaine G, Camerini F (1997) Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol 30(6):1512–1520
Pinamonti B, Sinagra G, Salvi A, Di Lenarda A, Morgera T, Silvestri F, Bussani R, Camerini F (1992) Left ventricular involvement in right ventricular dysplasia. Am Heart J 123(3):711–724
Sen-Chowdhry S, Syrris P, Ward D, Asimaki A, Sevdalis E, McKenna WJ (2007) Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation 115(13):1710–1720. doi:10.1161/CIRCULATIONAHA.106.660241
Sen-Chowdhry S, Lowe MD, Sporton SC, McKenna WJ (2004) Arrhythmogenic right ventricular cardiomyopathy: clinical presentation, diagnosis, and management. Am J Med 117(9):685–695. doi:10.1016/j.amjmed.2004.04.028
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ (2008) Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol 52(25):2175–2187. doi:10.1016/j.jacc.2008.09.019
Pamuru PR, Dokuparthi MV, Remersu S, Calambur N, Nallari P (2010) Comparison of Uhl’s anomaly, right ventricular outflow tract ventricular tachycardia (RVOT VT) & arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) with an insight into genetics of ARVD/C. Indian J Med Res 131:35–45
Pieroni M, Dello Russo A, Marzo F, Pelargonio G, Casella M, Bellocci F, Crea F (2009) High prevalence of myocarditis mimicking arrhythmogenic right ventricular cardiomyopathy differential diagnosis by electroanatomic mapping-guided endomyocardial biopsy. J Am Coll Cardiol 53(8):681–689. doi:10.1016/j.jacc.2008.11.017
Steckman DA, Schneider PM, Schuller JL, Aleong RG, Nguyen DT, Sinagra G, Vitrella G, Brun F, Cova MA, Pagnan L, Mestroni L, Varosy PD, Sauer WH (2012) Utility of cardiac magnetic resonance imaging to differentiate cardiac sarcoidosis from arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 110(4):575–579. doi:10.1016/j.amjcard.2012.04.029
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(MOV 1176 kb)
Clip 14.1b
(MP4 973 kb)
(MOV 1247 kb)
(MOV 1224 kb)
(MOV 2329 kb)
(MOV 1425 kb)
(MOV 1405 kb)
Rights and permissions
Copyright information
© 2014 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Brun, F., Di Nora, C., Merlo, M., Pivetta, A., Mestroni, L., Sinagra, G. (2014). Arrhythmogenic Right Ventricular Cardiomyopathy: Clinical Assessment and Differential Diagnosis. In: Pinamonti, B., Sinagra, G. (eds) Clinical Echocardiography and Other Imaging Techniques in Cardiomyopathies. Springer, Cham. https://doi.org/10.1007/978-3-319-06019-4_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-06019-4_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-06018-7
Online ISBN: 978-3-319-06019-4
eBook Packages: MedicineMedicine (R0)