
Abstract
Arrhythmogenic cardiomyopathy (ACM) is an inherited heart muscle disorder, predisposing to sudden cardiac death (SCD), particularly in young patients and athletes. Pathological features include loss of myocytes and fibrofatty replacement of right ventricular myocardium; biventricular involvement is often observed. It is a cell-to-cell junction cardiomyopathy caused by genetically determined abnormalities of cardiac desmosomes that predisposes to detachment and death of myocytes, mostly under conditions that increase the ventricular wall stress such as sports activity. The diagnosis of ACM does not rely on a single gold standard test but is achieved using a scoring system, which encompasses familial and genetic factors, ECG abnormalities, arrhythmias, and structural/functional ventricular alterations. The main goal of treatment is the prevention of SCD. Life-style changes with restriction from sports activity are recommended not only to patients with overt ACM, but also to asymptomatic patients and healthy gene carriers with the aim to prevent the occurrence and/or progression of the disease phenotype and to reduce the risk of lethal arrhythmias. Implantable cardioverter defibrillator is the only proven lifesaving therapy in high risk patients; selection of patients who most benefit from device implantation is one of the most challenging issues in the disease clinical management.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Athletes
- Primary prevention
- Screening
- Sudden death
- Arrythmogenic cardiomyopathy
- Ventricular tachycardia
- Ventricular fibrillation
-
1.
Understand the pathophysiology of arrhythmogenic cardiomyopathy and its role as a cause of sudden death in the young.
-
2.
Learn how to diagnose arrhythmogenic cardiomyopathy and to recognize the different phenotypes.
-
3.
Differentiate arrhythmogenic cardiomyopathy from athlete’s heart.
-
4.
Become familiar with risk stratification and management of arrhythmogenic cardiomyopathy.
-
5.
Be able to understand the implications for sports eligibility and exercise prescription of patients with arrhythmogenic cardiomyopathy.
1 Introduction
Arrhythmogenic cardiomyopathy (ACM) is an inherited heart muscle disease characterized pathologically by fibrofatty myocardial replacement and clinically by prominent ventricular arrhythmias and impairment of ventricular systolic function [1]. Although the original disease phenotype was characterized by predominant right ventricular (RV) involvement, with minor and late left ventricle (LV) disease, clinical variants characterized by early and greater LV involvement, which may parallel (i.e. biventricular ACM) or exceed (i.e. left-dominant ACM) the severity of RV involvement, have been increasingly reported. These findings have led over the past few years to the increasing use of the broader term of “arrhythmogenic cardiomyopathy,” which encompasses all the phenotypic expressions [2].
The fibro-fatty myocardial scar acts as a substrate of ventricular electrical instability which may lead to arrhythmic cardiac arrest, mostly in young people [3, 4]. Life-threatening ventricular arrhythmias are triggered by physical exercise, and participation in competitive athletics has been associated with an increased risk for sudden cardiac death (SCD) (Fig. 14.1) [5,6,7]. In addition, sport activity has been implicated as a factor promoting acceleration of disease progression [8,9,10]. This chapter will examine the pathophysiology, natural history, diagnosis, prognosis and clinical management of ACM, with particular reference to the athletic population and relevance to risk of SCD during sports.

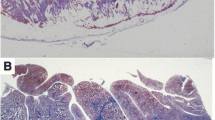
Pathologic features of arrhythmogenic cardiomyopathy. Classical right ventricular (RV) variant: (a) Gross transverse section of the heart that shows anterior and posterior RV wall thinning because of myocardial atrophy and a subtricuspidal aneurysm. Full-thickness histology of the posterior (b) and anterior (c) RV free wall that shows fibrofatty tissue replacement. There is thinning and residual myocardium confined to the endocardial trabeculae (trichrome stain). Biventricular variant: (d) Gross examination of a transverse section of the heart. Note the transmural RV free wall involvement as compared with the subepicardial midmural left ventricular (LV) free wall involvement. (e) Histology of the RV free wall confirms the transmural myocardial atrophy with fibrofatty replacement. (f) The histology of the LV free wall shows replacement-type fibrosis of the outer layer with preserved wall thickness (trichrome stain). (Reproduced with permission from Corrado D et al. [2])
2 Disease Mechanism and Natural History
The prevalence of ACM is estimated to range from 1 case in 2000–5000 persons in the general population. Clinical presentation of ACM becomes clinically apparent between the second and fourth decades of life. The disease is more malignant in men and this has been in part ascribed to the sex-based differences in the amount or intensity of exercise [1, 2].
The disease process is characterized by
-
the progressive loss of myocytes because of a genetically determined defect of the intercellular junction structures (desmosomes)
-
subsequent fibrofatty replacement of the ventricular myocardium of both ventricles.
It has been postulated that the genetically-determined impairment of myocyte cell-to-cell adhesion may lead to tissue and organ fragility that is sufficient to promote myocyte death and subsequent fibrofatty repair, especially under conditions of mechanical stress such as those occurring during physical activity [4, 11]. The resulting alterations in the myocardial structure not only can impair the mechanical function of the ventricles but render affected individuals vulnerable to life-threatening ventricular arrhythmias. Patients may experience scar-related monomorphic ventricular tachycardia, which is caused by a re-entrant circuit related to the underlying fibrofatty ventricular scar [12]. Ventricular fibrillation and SCD may also occur in young patients during the so-called “hot phases” of the disease natural course, as a consequence of myocarditis-mediated bouts of acute myocyte death leading to acute electrical instability [4]. Loss of expression of desmosomal proteins might “per se” induce electrical myocardial instability by secondary sodium ion-channel dysfunction, and current reduction that predisposes to lethal ventricular arrhythmias even prior to the expression of an overt structural abnormality [13].
The natural history of the disease is typically characterized by four different phases:
-
1.
“Concealed”, characterised by the absence of subtle RV structural changes, with or without minor ventricular arrhythmias, during which SCD may occasionally be the first manifestation of the disease, mostly in young people during competitive sports or intense physical exercise.
-
2.
“Overt electrical disorder” in which symptomatic RV arrhythmias possibly leading to sudden cardiac arrest are associated with overt RV functional and structural abnormalities.
-
3.
“RV failure” caused by the progression and extension of RV muscle disease that provoke global RV dysfunction with relatively preserved LV function.
-
4.
“Biventricular pump failure” caused by pronounced LV involvement. At this stage, ACM mimics biventricular dilated cardiomyopathy of other causes leading to congestive heart failure. A sizeable proportion of patients show early and severe LV involvement, which may either parallel (“biventricular”) or exceed (“left dominant”) the severity of RV disease [1, 2, 4, 11].
Exercise has been implicated as the most important environmental factor for promotion and progression of the ACM phenotypic expression: physical activity acutely increases the ventricular stress and accelerates the process of detachment, degeneration and death of genetically defective myocytes by disrupting intercellular junctions [4, 7, 11, 14]. In an animal model of plakoglobin-deficient mice, endurance training worsened the mechanical and electrical pathologic ventricular remodelling and induced RV dilatation, dysfunction and ventricular ectopy, supporting the concept that sport-related chronic ventricular overload might contribute to the development of the ACM phenotype in genetically susceptible individuals [15]. Studies in ACM-gene mutations carriers confirmed that endurance sports and sustained exercise increase age-related penetrance, risk of ventricular tachycardia/ventricular fibrillation and occurrence of heart failure. Moreover, in affected individuals physical activity is a strong pro-arrhythmic factor, because of adrenergic and mechanical stimulation of the diseased myocardium [7,8,9,10, 14].
It has been postulated that intense physical exertion may cause an ACM phenocopy characterized by dilatation and dysfunction of the RV in the absence of a pathological genetic substrate [16]. The theory, which has not been confirmed by other studies on large populations of highly trained athletes, was based on the study observations of a group of endurance athletes showing ventricular arrhythmias of RV origin and clinical features of underlying RV disease. However, the imaging features of the acquired phenocopy differed from those of desmosomal-gene related ACM, particularly due to the lack of RV wall motion abnormalities and fibrosis/late-gadolinium enhancement, which represent the distinctive pathologic abnormalities of ACM [17].
3 Diagnosis
The diagnosis is based on a series of criteria including
-
histopathological manifestations
-
alterations in cardiac structure and function
-
electrocardiographic abnormalities
-
arrhythmic manifestations
-
identification of disease-causing genetic mutations (Figs. 14.2 and 14.3)

Electrocardiographic findings in arrhythmogenic cardiomyopathy. (a) Right precordial repolarization alterations characterized by negative T waves in leads V1 through V3 and (b) depolarization abnormalities consisting of epsilon waves (arrow) and prolongation of QRS complex because of delayed S-wave upstroke leading to a significant terminal activation delay (TAD). (c) Late potentials on signal-averaged ECG. (d) Low QRS voltages (<0.5 mV) in the limb leads. (e) Ventricular tachycardia with a left bundle brunch block and superior axis morphology. (Reproduced with permission from Corrado D et al. [2])

Imaging features of arrhythmogenic cardiomyopathy. Morphofunctional and histologic abnormalities on imaging and endomyocardial biopsy. (a) Two-dimensional echocardiogram (parasternal short-axis view), showing dilatation of the right ventricular outflow tract (PSAX-RVOT = 37 mm). (b) Diastolic frame of 4-chamber view on post-contrast sequences by cardiac magnetic resonance showing biventricular dilatation and wall thinning with evidence of a thrombus in the right ventricular (RV) apex (black arrow), diastolic bulging of the peritricuspid region (white empty arrow), and intramyocardial late gadolinium enhancement because of fibrofatty scar involving the left ventricular free wall (solid white arrows) and septum (solid white arrow). (c) Angiography showing RV dilatation with a bulging of the right ventricular outflow tract (arrows). (d) Endomyocardial biopsy revealing myocyte loss with fibrofatty replacement. Reproduced with permission from Corrado D et al. [2]. AO aorta, LA left atrial, LV left ventricle, RA right atrial
that were elaborated by an International Task Force of experts (Table 14.1) [18]. As no single criterion is accurate enough, the diagnosis requires a combination of criteria (classified as minor or major according to their specificity). Specifically, the diagnosis of “definite” disease is fulfilled in the presence of two major criteria, or, one major and two minor, or, four minor criteria from different categories. The diagnosis is considered “borderline” in the presence of one major and two minor criteria or three minor criteria, and “possible” when two minor criteria are met.
3.1 Classic Right Ventricular Phenotype
The classic phenotype is characterized by prevalent RV involvement with progressive fibro-fatty replacement of the ventricular myocardium, which is the histopathologic hallmark of the disease. Demonstration of fibrous or fibrofatty replacement on endomyocardial biopsy is the most specific diagnostic criteria [19].
The process of progressive myocyte loss causes a gradual dilation and systolic dysfunction of the RV, initially at a regional level and, as the disease progresses, at a global level. The regional distribution of RV wall motion abnormalities (bulging, akinesis or diskinesis) is highly specific for ACM and allows differentiation from other RV conditions (e.g. congenital heart diseases, pulmonary hypertension, or athlete’s heart), characterized by uniform dilation/dysfunction of the entire RV. Hence, imaging diagnosis of ACM requires demonstration of regional wall motion abnormalities [20].
Electrical abnormalities secondary to the fibro-fatty scarring process are also the basis for the typical ECG changes of ACM including depolarization (delayed intraventricular conduction with widening/slurring of the S wave in V1–V3, epsilon waves and late potentials) and/or repolarization (T wave inversion) abnormalities. Such features usually involve the right precordial leads (V1–V3/V4) [21].
Ventricular arrhythmias with a left-bundle branch block morphology (negative QRS complex in V1), suggestive of a RV origin, are another key feature of ACM [12]. However, arrhythmias originating from the RV outflow tract (negative QRS complex in V1 and inferior QRS axis in the limb leads) are less specific for ACM compared to those arising from other RV regions, because in the majority of cases are benign, non-familial and not related to an underlying cardiomyopathy (“idiopathic” RV outflow tract ventricular tachycardia) [22].
Finally, because of the genetic nature of the disease, a positive family history for SCD and/or ACM or the demonstration of a pathogenetic mutation in the genes encoding for desmosomal proteins represent a major diagnostic criterion.
3.2 Left-Dominant Phenotype
The classic ACM phenotype mostly involves the RV while morpho-functional abnormalities of the LV become evident only in the late stages of the disease. However, there are “left dominant” ACM variants characterized by early and predominant LV involvement, often because of specific genetic defects (i.e., mutations of genes encoding for desmoplakin, phospholamban, or filamin C) [1]. The phenotype is the counterpart of the classic variant, with
-
T-wave inversion in the left precordial leads (V4–V6) and
-
LV arrhythmias (right bundle branch block pattern, i.e. positive QRS complex in V1) [11].
A suggestive ECG finding is the presence of low QRS voltages (<0.5 mV) in the limb leads which reflects the reduction of the electrical activity of the LV wall due to fibro-fatty myocardial replacement. In contrast with the classic variant, the diagnostic power of echocardiography is limited because LV dilatation and systolic dysfunction, either regional or global, may be absent in patients with predominantly left-sided disease. The reason is that the fibro-fatty scarring process initially involves the sub-epicardial myocardial layers of the LV wall, which contribute marginally to the development of the contractile power and does not translate into prominent wall motion abnormalities or ejection fraction reduction. Therefore, the left-dominant ACM phenotype in isolation is difficult to diagnose and its incidence is probably underestimated. Contrast-enhanced cardiac magnetic resonance increases the diagnostic sensitivity because it allows identification of non-transmural LV scars (areas of late gadolinium enhancement) at a subepicardial and/or mid-mural level [23, 24].
3.3 Diagnosis in Athletes
The ultimate diagnosis of cardiomyopathy in a young competitive athlete may be problematic due to the presence of physiologic (and reversible) structural and electrical adaptations of the cardiovascular system to long-term athletic training. This condition, known as “athlete’s heart”, is characterized by an increase in ventricular cavity dimension and wall thickness which may overlap with cardiomyopathies. In these circumstances, an accurate diagnosis is crucial because of the potentially adverse outcome associated with cardiomyopathy in an athlete and, conversely, the possibility of an erroneous diagnosis of a pathologic condition leading to unfair disqualifications from sport, with financial and psychological consequences. A sizable proportion of highly trained athletes have increased RV cavity dimensions which raises the question of ACM. Morphologic criteria in favour of a physiologic RV enlargement consist of preserved global and regional ventricular function, without evidence of wall motion abnormalities such as dyskinetic regions and/or diastolic bulging [25, 26]. During the last two decades, the advances in molecular genetics have allowed the identification of a growing number of defective desmosomal genes involved in the pathogenesis of ACM, and nowadays molecular genotyping is clinically available for differential diagnosis between desmosomal-gene related ACM and training-related physiologic RV changes. However, the presence of a large variety of polymorphisms and variants of uncertain significance of ACM-related genes makes it difficult to interpret the results of genetic testing (see also Chap. 12) [1, 27].
4 Prevention of Sudden Cardiac Death
Systematic monitoring and pathologic investigation of sudden death in young people and athletes of the Veneto Region of Italy showed that ACM is the most common pathologic substrate accounting for nearly a quarter of fatalities in young athletes and that the risk of sudden death from ACM is five times higher during competitive sports than during sedentary activity.
-
The incidence of sudden death from ACM in athletes was estimated to be 0.5 per 100,000 persons per year. Sudden death victims with ACM were all males with a mean age of 22.6 ± 4 years [6].
Although ACM has been demonstrated to be the leading cause of SCD in athletes of the Veneto region of Italy, previous studies from other countries showed a higher prevalence of other pathologic substrates such as hypertrophic cardiomyopathy, anomalous coronary arteries and myocarditis [14, 28]. This discrepancy may be explained by several factors, including the experience of pathologists or coroners who perform post-mortem investigation of athletes who die suddenly. ACM is rarely associated with cardiomegaly and usually spares the LV, so that affected hearts may be erroneously diagnosed as normal hearts. Therefore, a number of SCDs in young people and athletes, in which the routine pathologic examination discloses a normal heart, may, in fact, be due to an unrecognised ACM. On the other hand, the high incidence of ACM in the Veneto region may be due to a genetic factor in the population of north-eastern Italy, although ACM can no longer be considered as peculiar “Venetian disease” since there is growing evidence that it is ubiquitous and still largely underdiagnosed both clinically and at post-mortem investigation [29].
-
The risk to die suddenly from ACM has been estimated to be 5.4 times greater during competitive sports than during sedentary activity and early identification of athletes with ACM plays a crucial role in the prevention of SCD during sport [6] (Fig. 14.4).

Incidence and relative risk (RR) of sudden death (SD) for specific cardiovascular causes among athletes and non-athletes. Reproduced with permission from Corrado D et al. [5]. ARVC arrhythmogenic right ventricular cardiomyopathy, CAD coronary artery disease, CCA congenital coronary artery anomaly, MVP mitral valve prolapse
By reviewing clinical and ECG findings of 22 young competitive athletes who died suddenly from ACM proven at autopsy, it has been demonstrated that the majority of SCD victims exhibited ECG changes, ventricular arrhythmias, or both:
-
1.
Right precordial T-waves inversion had been recorded in 88% of athletes.
-
2.
Right precordial QRS duration >110 ms in 76%.
-
3.
Ventricular arrhythmias with a left bundle branch block pattern in the form of isolated/coupled premature ventricular beats or non-sustained ventricular tachycardia in 76%.
-
4.
Limited exercise testing induced ventricular arrhythmias in 50%.
Thus, the majority of young competitive athletes who died suddenly from ACM showed ECG abnormalities that could raise the suspicion of the underlying cardiovascular disease at preparticipation evaluation and could thus lead to further testing for a definitive diagnosis [11].
For more than 20 years a systematic preparticipation screening, based on 12-lead ECG in addition to history and physical examination, has been the practice in Italy. A time-trend analysis of the incidence of SCD in young competitive athletes 12–35 year-old in the Veneto region of Italy between 1979 and 2004 has provided compelling evidence that ECG screening is a lifesaving strategy. The analysis demonstrated a 90% decrease of SCD in athletes after the introduction of the nationwide screening program. By comparison, the incidence of SCD in the unscreened nonathletic population of the same age did not change significantly over that time. Most of the mortality reduction was attributable to fewer deaths from hypertrophic cardiomyopathy and ACM. A parallel analysis of the causes of disqualifications from competitive sports at the Center for Sports Medicine in the Padua country area showed that the proportion of athletes identified and disqualified for cardiomyopathies doubled from the early- to the late-screening period. This indicates that mortality reduction was a reflection of a lower incidence of SCD from cardiomyopathies, as a result of increasing identification over time of affected athletes at ECG pre-participation screening [5].
5 Interpretation of Repolarization Abnormalities in Athletes
The 12-lead ECG is one of the most important tools for the disease diagnosis, follow-up and risk stratification. The inversion of the T-waves in the right precordial leads V1–V3/V4 and/or in the left infero-lateral leads is the most suggestive ECG sign. While inverted T-waves in the inferolateral leads are highly specific for a myocardial disease including left-dominant ACM, persistence of T-wave inversion in right precordial leads (known as persistence of the juvenile pattern of repolarization) may be occasionally observed in young athletes.
-
It is noteworthy that the prevalence of a benign persistence of the juvenile pattern of repolarization declines sharply after puberty, whereas the clinical manifestations of cardiomyopathy usually occur after puberty. Hence, the persistence of right precordial T wave inversion in the post-pubertal age should raise the suspicion of an underlying ACM and prompt further imaging investigation [30].
Another cause of benign right precordial negative T waves in healthy athletes, especially of Afro-Caribbean descent, is an anterior early repolarization variant characterized by domed ST-segment elevation followed by negative T-wave in anterior leads V1–V4. The differential diagnosis requires careful analysis of the ST-segment morphology preceding the negative T-wave. Athletes exhibit J-point elevation (the hallmark of early repolarization), followed by an up-sloping ST-segment elevation with a domed morphology. On the other hand, ACM patients usually show no J-point elevation and no or minimal ST-segment elevation [31, 32] (Fig. 14.5).

Differential diagnosis between cardiomyopathic negative T-waves and early repolarization of athlete’s heart. Right-precordial leads V2–V3 of a patient with arrhythmogenic right ventricular cardiomyopathy (ACM) showing no J-point elevation and negative T-wave (a). Right-precordial leads V2–V3 of an Afro-Caribbean athlete showing an early repolarization pattern characterized by J-point elevation, dome ST-elevation and negative T-wave (b)
The clinical workup for differential diagnosis between pathological and nonpathological negative T waves traditionally includes electrocardiographic exercise testing. The current perception is that negative T waves usually revert to normal with exercise in healthy subjects whereas they persist in patients with structural heart muscle disease. However, the available data in favor of this concept are limited. In fact, by comparing ACM patients with healthy athletes with right precordial T wave inversion, the prevalence of complete or partial normalization of T wave polarity with exercise is observed in the majority of both groups [33].
6 Clinical Management
Patients with ACM should undergo lifelong clinical follow-up (every 6–24 months depending on the age, symptoms and disease severity) including echocardiography, 24-h Holter monitoring, and exercise testing to periodically evaluate new onset or worsening of symptoms, progression of morphological and/or functional ventricular abnormalities, and reassess the risk of SCD. Due to the age-related penetrance of ACM, healthy gene carriers and family members should also be offered repeat clinical assessment, mostly during adolescence and young adulthood. The four cornerstones of clinical management include:
-
1.
Life-style changes
-
2.
Drug therapy
-
3.
Catheter ablation
-
4.
ICD-Implantation
Life-style changes include restriction from sports, with the only possible exception of low-intensity activities. Not only patients with overt ACM, but also asymptomatic patients and healthy gene carriers should be prudently advised to avoid vigorous exercise, not only for reducing the risk of ventricular arrhythmias and SCD, but also to prevent disease progression (Fig. 14.6).

Impact of sports activity on arrhythmogenic cardiomyopathy. Schematic representation of natural history of arrhythmogenic cardiomyopathy from desmosomal gene mutation to phenotypic expression and life-threatening ventricular tachycardia. Sports activity may influence the disease course by promoting development of phenotypic expression, accelerating disease progression, and triggering malignant ventricular arrhythmias. (Reproduced with permission from Corrado D et al. [2])
Drug therapy may include antiarrhythmic agents, beta-blockers, and heart failure drug therapy. Beta-blocker drugs should be offered to all patients with a definite diagnosis of ACM and evidence of morpho-structural ventricular abnormalities or ventricular arrhythmias because of their proven efficacy to prevent effort-induced arrhythmias, their proven efficacy in heart failure management, and their potential but unproven ability to hinder myocardial disease progression by lowering the ventricular wall stress. Adjunctive anti-arrhythmic drug therapy is indicated to reduce the arrhythmia burden in symptomatic patients with frequent premature ventricular beats and/or complex ventricular arrhythmia. For patients who developed heart failure, standard pharmacologic treatment with angiotensin-converting-enzyme inhibitors, angiotensin II receptor blockers, beta-blockers, and diuretics was prescribed as appropriate.
Catheter ablation is a therapeutic option for ACM patients who have recurrent ventricular tachycardia despite antiarrhythmic drug therapy. However, catheter ablation has not been proven to prevent SCD and should not be considered as an alternative to ICD therapy. Also, because of the progressive nature of the disease, repeated ablation procedures may be required to provide clinical control of ventricular arrhythmias.
Implantation of an implantable cardioverter defibrillator (ICD) is the most logical therapeutic strategy for patients with ACM, whose natural history is primarily characterised by the risk of arrhythmic cardiac arrest.
-
There is general agreement that patients who survived an episode of ventricular fibrillation or sustained ventricular tachycardia most benefit from ICD implantation because of their high incidence of malignant arrhythmia recurrences.
Other risk factors identified by studies on ACM patients include
-
unexplained syncope
-
non-sustained VT on 24-h Holter monitoring
-
systolic dysfunction of RV, LV, or both
-
male gender
-
compound and digenic heterozygosity of desmosomal-gene mutations
-
young age at the time of diagnosis
-
proband status
-
inducibility at programmed ventricular stimulation
-
amount of electroanatomic scar and electroanatomic scar-related fractionated electrograms
-
extent of T-wave inversion across precordial and inferior leads
-
low QRS amplitude and QRS fragmentation.
In patients with one of more of these risk factors , the decision to implant an ICD should be made on an individual basis, by assessing the overall clinical profile, the age, the strength of the risk factor identified, the level of SCD risk that is acceptable to the patient, and the potential risk of inappropriate interventions and complications. Finally, asymptomatic patients with no risk factors have a favourable long-term outcome regardless of familial history of SCD and electrophysiologic study findings. These results are particularly relevant for clinical management of the growing cohort of asymptomatic ACM patients or desmosomal gene mutation carriers [34] (Fig. 14.7).

Pyramid of risk stratification in Arrhythmogenic Cardiomyopathy. Pyramid of risk and indications for implantable cardioverter defibrillator (ICD) therapy in arrhythmogenic cardiomyopathy. According to the available data on annual mortality rates associated to previous events and specific risk factors, the estimated risk of major arrhythmic events in the high-risk category (apex of pyramid) is >10% per year, in the intermediate-risk category (mid of pyramid) ranges from 1% to 10% per year and in the low-risk category (base of pyramid) is <1% per year. The recommendations for ICD implantation for different categories of arrhythmic risk are based on the 2015 International Task Force consensus document on treatment of arrhythmogenic right ventricular cardiomyopathy (ACM). Reproduced with permission from Corrado D et al. [2]. LV left ventricle, PVB premature ventricular beats, RV right ventricle, VF ventricular fibrillation, VT ventricular tachycardia
6.1 Eligibility to Sport Activity
According to current recommendations for sports eligibility, athletes with clinical diagnosis of ACM should be excluded from all competitive sports [34]. This recommendation is independent of age, gender, phenotype expression, symptoms, drug therapy, or interventions with surgery, catheter ablation, or implantable defibrillator. The presence of a free-standing automated external defibrillator at sporting events should not be considered absolute protection against sudden death, nor a justification for participation in competitive sports in athletes with ACM.
Patients with ACM may wish to participate in recreational and leisure-time exercise activity, given the recognized beneficial effects of a physically active lifestyle. It has been suggested that the absolute risk of ventricular tachyarrhythmias/death in patients practicing recreational sports does not significantly differ from that of physically inactive patients [10]. However, the conclusion that the recreational sports activity does not increase the risk of SCD or disease progression was not supported by adequate statistical power because of the small sample size.
-
Thus, at the present time patients with a definitive diagnosis of ACM should be prudently restricted from participation in athletic activities, with the possible exception of recreational low intensity sports.
6.2 Implantable Cardioverter Defibrillator and Sports
The accurate prediction of the performance of ICD in athletes remains a challenging subject. Successful ICD therapy depends on a well-functioning device that is capable of both appropriate sensing of the malignant arrhythmia and delivering an adequate amount of energy to depolarize a critical myocardial mass to overcome the ongoing arrhythmic state. Sports may hinder the success of ICD therapy in many aspects:
-
1.
Sinus tachycardia and other supraventricular tachyarrhythmias that are often present during sports activity represent an obstacle for appropriate differentiation of malignant ventricular arrhythmias.
-
2.
Physical trauma whether due to direct or indirect contact may lead to device damage and malfunction.
-
3.
Physiological changes associated with exertion such as high catecholamine levels, electrolyte imbalance, metabolic acidosis, and cardiac loading alterations can lead to persistent arrhythmogenic states in which defibrillation may not be successful (electrical storms) or—even worse—to SCD due to electromechanical dissociation where defibrillation will not be of any benefit [35].
The most compelling evidence on ICD efficacy and safety comes from a prospective, multinational Registry which recruited 372 athletes with an ICD from USA and Europe [36]. Over a median follow-up of 31 months, though ICD shocks occurred during and after sports participation, there were no arrhythmic deaths, resuscitated cardiac arrests, or shock-related injuries. The authors concluded that these data were similar to previously published data on non-athletic ICD patients and therefore do not support competitive sports restriction for all athletes with ICDs [36]. More recently, the same Authors confirmed these outcomes in the athletes enrolled in the same Registry over a long-term follow-up (44 months) [37]. The pro-arrhythmic effect of sports activity accounted for the occurrence of appropriate shocks respectively in 11% of participants during exercise and in 6% at rest. An underlying ARVC was the only variable associated with exercise-induced ICD shock.
Despite these reassuring data, it must be emphasized that the reasons for implementing restrictions from competition in sports in young athletes with ICD go beyond the increased risk of inappropriate interventions, injury to the patient, and damage of the system. Sports participation plays a major role in the disease progression, worsening of the substrate and adverse outcome. Although evidence has emerged in support of the safety of competitive sports in selected individuals carrying ICD, it is prudent to restrict competitive sports participation in ACM patients.
Clinical Pearls
-
The natural history of arrhythmogenic cardiomyopathy progresses from a concealed phase to an overt disease characterized by ventricular arrhythmias and ventricular wall motion abnormalities. Exercise has been implicated as the most important environmental factor for progression of the disease.
-
The diagnosis of arrhythmogenic cardiomyopathy is multiparametric and requires a combination of family history, electrocardiographic abnormalities, ventricular arrhythmias, regional right ventricular dilation/dysfunction associated with wall motion abnormalities and/or positive endomyocardial biopsy. No single criterion is accurate enough to support the diagnosis if present in isolation.
-
The available scientific evidence suggests that patients with a definitive diagnosis of arrhythmogenic cardiomyopathy should be prudently restricted from participation in athletic activities, irrespective from ICD implantation, with the possible exception of recreational low intensity sports.
References
Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72.
Corrado D, Basso C, Judge DP. Arrhythmogenic cardiomyopathy. Circ Res. 2017;121(7):784–802.
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med. 1988;318(3):129–33.
Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2012;9(4):223–33.
Corrado D, Basso C, Pavei A, Michieli P, Schiavon M, Thiene G. Trends in sudden cardiovascular death in young competitive athletes after implementation of a preparticipation screening program. JAMA. 2006;296(13):1593–601.
Corrado D, Basso C, Rizzoli G, Schiavon M, Thiene G. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42(11):1959–63.
Corrado D, Zorzi A. Arrhythmogenic right ventricular cardiomyopathy and sports activity. Eur Heart J. 2015;36(27):1708–10.
James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(14):1290–7.
Saberniak J, Hasselberg NE, Borgquist R, Platonov PG, Sarvari SI, Smith HJ, et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail. 2014;16:1337.
Ruwald AC, Marcus F, Estes NA III, Link M, McNitt S, Polonsky B, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36:1735.
Basso C, Corrado D, Marcus F, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–300.
Lemola K, Brunckhorst C, Helfenstein U, Oechslin E, Jenni R, Duru F. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91(9):1167–72.
Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation. 2014;129(10):1092–103.
Zorzi A, Pelliccia A, Corrado D. Inherited cardiomyopathies and sports participation. Neth Heart J. 2018;26(3):154–65.
Kirchhof P, Fabritz L, Zwiener M, Witt H, Schafers M, Zellerhoff S, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114(17):1799–806.
La Gerche A, Burns AT, Mooney DJ, Inder WJ, Taylor AJ, Bogaert J, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J. 2012;33(8):998–1006.
La Gerche A, Robberecht C, Kuiperi C, Nuyens D, Willems R, de Ravel T, et al. Lower than expected desmosomal gene mutation prevalence in endurance athletes with complex ventricular arrhythmias of right ventricular origin. Heart. 2010;96(16):1268–74.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121(13):1533–41.
Basso C, Ronco F, Marcus F, Abudureheman A, Rizzo S, Frigo AC, et al. Quantitative assessment of endomyocardial biopsy in arrhythmogenic right ventricular cardiomyopathy/dysplasia: an in vitro validation of diagnostic criteria. Eur Heart J. 2008;29(22):2760–71.
Yoerger DM, Marcus F, Sherrill D, Calkins H, Towbin JA, Zareba W, et al. Echocardiographic findings in patients meeting task force criteria for arrhythmogenic right ventricular dysplasia: new insights from the multidisciplinary study of right ventricular dysplasia. J Am Coll Cardiol. 2005;45(6):860–5.
Steriotis AK, Bauce B, Daliento L, Rigato I, Mazzotti E, Folino AF, et al. Electrocardiographic pattern in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2009;103(9):1302–8.
O’Donnell D, Cox D, Bourke J, Mitchell L, Furniss S. Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract tachycardia. Eur Heart J. 2003;24(9):801–10.
Zorzi A, Perazzolo Marra M, Rigato I, De Lazzari M, Susana A, Niero A, et al. Nonischemic left ventricular scar as a substrate of life-threatening ventricular arrhythmias and sudden cardiac death in competitive athletes. Circ Arrhythm Electrophysiol. 2016;9(7):e004229.
Sen-Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175–87.
Bauce B, Frigo G, Benini G, Michieli P, Basso C, Folino AF, et al. Differences and similarities between arrhythmogenic right ventricular cardiomyopathy and athlete’s heart adaptations. Br J Sports Med. 2010;44(2):148–54.
D’Ascenzi F, Solari M, Corrado D, Zorzi A, Mondillo S. Diagnostic differentiation between arrhythmogenic cardiomyopathy and athlete’s heart by using imaging. JACC Cardiovasc Imaging. 2018;11(9):1327–39.
Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113(13):1634–7.
Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349(11):1064–75.
Elmaghawry M, Alhashemi M, Zorzi A, Yacoub MH. A global perspective of arrhythmogenic right ventricular cardiomyopathy. Glob Cardiol Sci Pract. 2012;2012(2):81–92.
Migliore F, Zorzi A, Michieli P, Perazzolo Marra M, Siciliano M, Rigato I, et al. Prevalence of cardiomyopathy in Italian asymptomatic children with electrocardiographic T-wave inversion at preparticipation screening. Circulation. 2012;125(3):529–38.
Corrado D, Pelliccia A, Heidbuchel H, Sharma S, Link M, Basso C, et al. Recommendations for interpretation of 12-lead electrocardiogram in the athlete. Eur Heart J. 2010;31(2):243–59.
Calore C, Zorzi A, Sheikh N, Nese A, Facci M, Malhotra A, et al. Electrocardiographic anterior T-wave inversion in athletes of different ethnicities: differential diagnosis between athlete’s heart and cardiomyopathy. Eur Heart J. 2016;37(32):2515–27.
Zorzi A, ElMaghawry M, Rigato I, Cardoso Bianchini F, Crespi Ponta G, Michieli P, et al. Exercise-induced normalization of right precordial negative T waves in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;112(3):411–5.
Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015;132(5):441–53.
Corrado D, Migliore F, Zorzi A. Sport activity in patients with implantable defibrillator: playing with death? Eur J Prev Cardiol. 2019;26:760.
Lampert R, Olshansky B, Heidbuchel H, Lawless C, Saarel E, Ackerman M, et al. Safety of sports for athletes with implantable cardioverter-defibrillators: results of a prospective, multinational registry. Circulation. 2013;127(20):2021–30.
Lampert R, Olshansky B, Heidbuchel H, Lawless C, Saarel E, Ackerman M, et al. Safety of sports for athletes with implantable cardioverter-defibrillators: long-term results of a prospective multinational registry. Circulation. 2017;135(23):2310–2.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Review
Review
1.1 Questions
-
1.
A 19-year-old female dancer with a family history of arrhythmogenic cardiomyopathy was referred for frequent premature ventricular beats with a right-bundle-branch block morphology that increase in number and complexity with increasing workload during exercise testing. Resting electrocardiogram and echocardiography are unremarkable. Should other investigations be prescribed?
-
2.
A 25-year-old black highly-trained competitive asymptomatic athlete shows T-wave inversion preceded by J-point and ST-segment elevation in V1–V3. Echocardiography shows a mildly dilated right ventricle in the absence of regional wall motion abnormalities. Is this pattern suggestive of arrhythmogenic cardiomyopathy?
-
3.
A 36-year-old asymptomatic athlete has been diagnosed with definite arrhythmogenic cardiomyopathy. There is no family history of sudden death, only isolated premature ventricular beats are recorded during exercise test and 24-h Holter monitoring and echocardiography showed only a mild right ventricular dysfunction. Should he/she be allowed to engage in competitive non-professional soccer?
1.2 Answers
-
1.
The presence of premature ventricular beats with a right-bundle-branch block morphology, suggesting the origin from the left ventricular wall, in a young patient with a family history of arrhythmogenic cardiomyopathy may be the sign of an underlying left ventricular involvement. Echocardiography may not be sensitive enough to detect segmental fibrofatty scarring of the left ventricle. Hence, prescription of a contrast-enhanced cardiac magnetic resonance is reasonable.
-
2.
No, the presence of T-wave inversion preceded by J-point/ST-segment elevation is consistent with an early repolarization variant typical of black athletes. Moreover, a mildly dilated right ventricle without regional wall motion abnormalities is a usual finding in highly-trained athletes and does not support the diagnosis of arrhythmogenic cardiomyopathy.
-
3.
There are two reasons for excluding patients with arrhythmogenic cardiomyopathy from all competitive sports. First, the risk of sudden death. Second, the fact that high-intensity exercise has been implicated as the most important environmental factor for progression of the disease.
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Corrado, D., Zorzi, A., Thiene, G. (2020). Specific Cardiovascular Diseases and Competitive Sports Participation: Arrhythmogenic Right Ventricular Cardiomyopathy. In: Pressler, A., Niebauer, J. (eds) Textbook of Sports and Exercise Cardiology. Springer, Cham. https://doi.org/10.1007/978-3-030-35374-2_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-35374-2_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-35373-5
Online ISBN: 978-3-030-35374-2
eBook Packages: MedicineMedicine (R0)