
Abstract
Peptides are short sequences of amino acids. Peptides with biological functionality can be derived from the active domain of proteins or determined from peptide screening experiments. Combined with modern chemical techniques to facilitate peptide synthesis, this leads to peptide modification as an interesting approach to render synthetic biomaterials bioactive. Peptides have been used to functionalize implant surfaces as well as bulk biomaterials, and they can be incorporated within controlled release systems. This chapter considers both osteoinductive peptides and anti-biofilm peptides with the goals to improve bone regeneration and reduce implant-associated infection, respectively.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Peptide synthesis
- Phage display
- Surface modification
- Hydrogels
- Nanofibers
- Titanium
- Controlled release
- Osteoinductive peptides
- Osteogenic differentiation
- Bone morphogenetic protein
- Osteogenic growth peptide
- Hydroxyapatite
- Antimicrobial peptides
- Anti-biofilm
- Bone tissue engineering
- Dentistry
Introduction
Proteins are the functional building blocks of cells and tissues. They are made up of differing combinations of 20 natural amino acids (Table 1), which have an amino group on one end, a carboxylic acid group on the other end, and a variable side chain coming off the central carbon atom. Within a protein or polypeptide, the amino acid residues are joined together by the formation of an amide bond between the carboxylic acid of one amino acid and the amino group of the next. The sequences of natural proteins are determined from the genetic code whereby DNA is first transcribed to messenger RNA and then translated to protein with each three-base-long codon specifying an amino acid. Similar to proteins, peptides are shorter chains of amino acids, also linked by amide bonds, that are typically less than 50 amino acids in length. With advances in chemical and molecular biology techniques, peptides can be chemically synthesized and modified, and de novo sequences can be designed. As a treatment strategy, peptides offer many of the advantages of protein therapeutics while addressing some of their limitations .
This chapter focuses on the use of peptides in biomaterial-based approaches for bone regeneration and anti-biofilm applications. First, a perspective is given on peptide design with two main approaches being the derivation of amino acid sequences from natural proteins and the screening of de novo libraries of peptides for specific functions. Then, relevant peptide synthesis techniques are presented along with chemical methods that allow functionalization of surfaces and bulk materials with peptides. The main focus of the chapter is the introduction of specific peptides from two main classes: osteoinductive peptides and anti-biofilm peptides. In both cases, in vitro assays to assess peptide activity as well as in vivo studies to demonstrate efficacy are presented. To date, however, few studies have combined osteoinductive peptides with antimicrobial peptides to stimulate bone formation while simultaneously reducing infection. The chapter concludes with some future perspectives on the topic.
Peptide Design
Peptides Derived from Proteins
The primary structure of a protein is its chemical composition determined by the specific sequence of amino acids. Protein secondary structure, which includes units such as α-helices and β-sheets, arises from the organization of portions of the protein by weak intramolecular bonds, such as hydrogen bonding and ionic interactions. Hydrogen bonding can occur between atoms on the protein backbone, but in other cases, these interactions are driven by the chemical composition of the side chains of the amino acids. With some exceptions, these side chains can be either positively or negatively charged, polar uncharged, or hydrophobic (Table 1). In de novo protein design, specifying the pattern of hydrophobic and hydrophilic amino acids in the protein sequence is in many cases sufficient to drive protein folding into α-helices [1] or β-sheets [2], as measured by circular dichroism. As with proteins, peptides with specific patterns of amino acids can self-assemble to form these structural units, such as RADA16 [RADARADARADARADA [3]; single-letter amino acid abbreviations used throughout this chapter, see Table 1] and Q11 [QQKFQFQFEQQ [4]] that form β-sheets or LD6 [LIVAGD [5]] that forms α-helical intermediates. These self-assembling peptides have also been used to generate cytocompatible hydrogels for biomedical applications, but further discussion of their use is outside the scope of the current chapter.
Likewise, these interactions can occur intermolecularly, that is, between two proteins, leading to receptor–ligand pairing or enzyme–substrate docking for example. As the amino acid sequences that are involved in these interactions are usually specific, they can serve as a starting point for the design of bioactive peptides that mimic these active domains of the protein. One of the most simple and commonly used peptide mimetics is the cell adhesive RGD sequence. RGD is found in several proteins including fibronectin, laminin, collagen, vitronectin, and others and mediates cell attachment through various integrins on the surface of cells [6]. Cell attachment to RGD is sequence specific, as evidenced by reduced binding to scrambled peptides such as RDG, and exposure to soluble RGD causes cells to detach from their substrate [6]. Functionalization of an otherwise nonadhesive biomaterial with RGD will allow cells to attach to and spread on or within the material. Patterning of RGD can also restrict cell attachment to specific areas, as has been shown by observing encapsulated cell spreading in inert poly(ethylene glycol) (PEG) hydrogels compared to PEG hydrogels with uniform functionalization with RGD and with spatially patterned RGD (Fig. 1) [7].

Effect of the addition of RGD uniformly throughout compared to spatially patterned within PEG hydrogels that were formed by click chemistry on 3 T3 fibroblast spreading. (a) No RGD. (b) Uniform RGD. (c) Patterned RGD. The scale bars are 100 μm. (Reprinted by permission from Springer Nature, Nature Materials, ©2009 [7])
Besides RGD , there are a number of other integrin binding peptides that can be used as cell adhesion ligands , as summarized in Table 2. The use of integrin-binding peptides is a generic strategy in tissue engineering approaches, as most cell types are adherent and thus will attach, spread, proliferate, and migrate on the peptide-functionalized materials. Knowledge of the surface receptor profile of different cells can aid in the selection of which ligand or ligands to choose to be able to bind a specific cell type. These peptides can then be used to functionalize biomaterials in both 2D and 3D. For example, nanofibrous matrices were formed from the self-assembly of peptide amphiphiles functionalized with either YIGSR or VAPG. The materials presenting YIGSR enhanced the attachment of endothelial cells, whereas the matrices containing VAPG promoted greater spreading of smooth muscle cells [8]. Hydrogels formed from photo-cross-linked PEG diacrylate combined with VAPG-functionalized PEG monoacrylate were also shown to preferentially support attachment of smooth muscle cells over fibroblasts, endothelial cells, and platelets [11]. Further, SIKVAV-conjugated chitosan hydrogels were demonstrated to promote angiogenesis and re-epithelialization in vivo [12]. These cell adhesive peptides can potentially be combined with peptides targeting other receptors, and the design of osteoinductive and anti-biofilm peptides that are based on naturally occurring domains within proteins are further described in sections “Osteoinductive Peptides” and “Anti-biofilm Peptides” of this chapter.
Moving beyond the direct use of peptides derived from natural proteins, these mimetics can be varied, often in a systematic way, by changing individual amino acids within the peptide sequence to increase or decrease the activity of the peptide. One good example of this is the use of collagen-derived peptides to explore the sequence specificity of matrix metalloproteinases (MMPs), which are a family of enzymes that are active during normal tissue remodeling processes and also in disease states. Starting from an eight-amino-acid-long MMP substrate sequence found in the α1 chain of type I collagen, GPQGIAGQ, Nagase and Fields systematically varied the amino acids in each position and found that the substitution of A with W led to increases in the Michaelis-Menten parameter kcat/KM and the relative rate of hydrolysis by MMP-1, MMP-8, and MMP-3 [21]. These peptide sequences were then used in innovative enzymatically degradable synthetic hydrogels formed by the Michael-type addition reaction of end-functionalized multi-arm PEG macromers with cross-linker peptides containing the MMP substrate sequences [22, 23]. Indeed, the hydrogels containing the modified GPQGIWGQ substrate degraded faster than the hydrogels containing the collagen-derived GPQGIAGQ sequence, and this allowed for faster remodeling of the matrix and migration of embedded fibroblasts (Fig. 2) [22]. These degradable hydrogels were also found to support bone formation in combination with the osteoinductive molecule, bone morphogenetic protein (BMP)-2 [23]. In a rat calvarial defect model, hydrogels that were not degradable but loaded with BMP-2 led to similar low amounts of bone formation as degradable hydrogels without BMP-2 (approximately 20% bone coverage); however, the hydrogels that were degradable and loaded with BMP-2 resulted in similar amounts of bone formation as the standardly used collagen sponge with BMP-2 (approximately 90% bone coverage) [23].

Fibroblast migration into PEG hydrogels prepared through a Michael-type addition reaction with the MMP-sensitive GPQGIWGQ peptide cross-linker. (a) Phase contrast images after 1, 3, 5, and 7 days with scale bar = 250 μm. (b) Confocal image showing cell membranes and nuclei with scale bar = 150 μm. (Reprinted with permission from [22]. ©2003 National Academy of Sciences, USA)
Library Screening Approaches
It is also possible to design peptides completely from scratch, and several methodologies have been developed to screen libraries of peptides for specific biological activity. In fact, half of the 2018 Nobel Prize in Chemistry was jointly awarded to George P. Smith and Sir Gregory P. Winter for developing the technique of phage display of peptides [24] and antibodies [25]. Phage display utilizes gene editing of the coat proteins of bacteriophage to incorporate peptide sequences that are displayed as part of the coat proteins. These phage can then be screened against target molecules for an interaction of interest and further amplified. The DNA within the selected phage can finally be sequenced to determine the relevant peptide sequence. Phage display can be useful in determining substrate specificity of enzymes. For example, to determine mutant substrates that are not cleaved by an enzyme, the coat proteins are modified with the library of peptide sequences, each with a tag on the terminus to allow for separation using antibody-coated beads (Fig. 3). After incubation with the enzyme, the phage that are still attached to the beads can be separated, washed, amplified, treated with enzyme, and sorted again. After several rounds of sorting and amplification, the individual phage can be isolated and sequenced. This approach has been used to identify substrates that are less susceptible to cleavage by the metalloproteinase ADAMTS13 [26], and by changing the method to select the phage of interest, one can identify enzyme substrates that are preferentially cleaved, peptides that bind to specific proteins or other molecules, etc. [24].

Working principle of phage display . In this case, the substrates that are not cleaved by the enzyme ADAMTS13 are selected and amplified. (Reprinted under the terms of the Creative Commons Attribution License from Desch et al. ©2015 [26])
An alternative to phage display for peptide design is mixture-based oriented peptide libraries [27]. The mixture-based oriented peptide library approach screens the peptides in solution and relies on peptide sequencing methods (Edman degradation) to determine the sequence. It has been applied for determining enzyme substrates and protein interaction domains [28,29,30,31]. For example, to determine an enzyme substrate, in a first round, a totally degenerate peptide library with the amino (N-) termini acetylated is treated with the protease. The fragments that are generated have a free amine group on the N-terminus and are subjected to N-terminal sequencing. The amino acids that are most abundant in each position are then fixed in a second-generation library, which have the N-termini as free amines and the carboxy (C-) termini tagged with biotin. After cleavage with the protease, the C-terminal fragments and uncleaved peptides can be separated using immobilized avidin, and the N-terminal fragments can be sequenced. This approach has been used to generate substrates for a variety of MMPs [27]. Another approach to screen for enzyme substrates is a technique called cellular libraries of peptides substrates (CLiPS) [32]. A library of peptides containing potential substrates tagged with a ligand for a fluorescent probe are displayed on the surface of E. coli. After treating the cells with the protease of interest, the intact peptides are able to be detected using fluorescence-activated cell sorting (FACS) , and the clones with hydrolyzed substrates can be enriched by repeating this cycle several times. Compared to phage display and soluble peptide libraries, up to 104 copies of the substrate are displayed on a single cell, which allows quantitation of substrate conversion for an individual clone [32].
These techniques are commonly used to identify peptides that bind to target receptors or to natural ligands, for epitope mapping or mimicking, in drug discovery to find new enzyme inhibitors and receptor agonists and antagonists, for epitope discovery in vaccine development and diagnostics, and selection of DNA-binding motifs, among others [24]. Of relevance to biomaterials development, they have also been used to identify new substrates for various MMPs , such as MMP-2 [33], MMP-3 or -7 [34], MMP-11 [35], MMP-13 [36], and MT1-MMP [37]. These peptide substrates were evaluated with the PEG hydrogel system described above, and sequences resulting in hydrogels with increased susceptibility to MMP-1, MMP-2, or plasmin were identified [38, 39]. The faster degrading hydrogels supported increased proliferation of encapsulated fibroblasts as well as sprouting of endothelial cells in a chick aortic ring outgrowth assay [38, 39]. Similar to the cell adhesion peptides already described, the functionalization of scaffolds with enzyme substrate peptides also provides a generic strategy to support the matrix remodeling, proliferation, and migration of cells within the matrix. More specific peptides that are osteoinductive or anti-biofilm, which have been found by library screening approaches, are detailed in sections “Osteoinductive Peptides” and “Anti-biofilm Peptides” of this chapter.
Chemical Methods
Peptide Synthesis
During protein translation from messenger RNA , the nascent protein chain is synthesized from N- to C- terminus in the ribosome via the action of transfer RNA, which recognizes the codon in the RNA strand. A new amide bond is formed to attach the amino acid to the growing polypeptide chain. Specific codons initiate and terminate the synthesis of the protein. On the other hand, peptides are typically synthesized chemically using a technique called solid-phase peptide synthesis, which allows excess reagents and by-products of the reactions to be easily removed by washing and filtration after each step. They can also be synthesized in solution, although this requires additional work to separate the peptide intermediate after each step. In the case of solid-phase peptide synthesis, the peptide is synthesized on a solid support from the C- to N-terminus. Cleavable protecting groups typically block reactive functionalities on the side chains and amino group of the amino acid. The latter protecting group is often an Fmoc (base-labile) or Boc (acid-labile) group, which can be removed before the addition of the next amino acid. Merrifield was the first to describe the solid-phase peptide synthesis technique in 1963 [40], and the Fmoc/t-Bu orthogonal protecting group strategy has become the most common approach applied today. Solid-phase peptide synthesis typically proceeds by coupling of the first amino acid to the supporting resin followed by a deprotection step to remove the Fmoc group. The next amino acid is added, followed by deprotection, and so on until the last amino acid or other N-terminal group is added. Then, the peptide is cleaved from the resin, and the protecting groups are also removed from the side chains. The peptide is typically purified by reversed-phase HPLC-MS, with the MS used to confirm the molecular mass of the synthesized peptide.
Important considerations when planning to synthesize a peptide include the general synthesis strategy, the choice of protecting groups for the side chains, the resin used as solid support as well as how it is linked to the C-terminus of the peptide, and the choice of coupling reagents [41]. The speed and quality of solid-phase peptide synthesis can be improved by the application of microwave irradiation, which raises the temperature and helps to break up chain aggregation of the peptide intermediates [42]. The microwave energy reduces the reaction time for both the coupling of amino acids and also the removal of the Fmoc protecting group [42, 43]. Nevertheless, synthesis of longer peptides tends to result in lower yields, and some amino acids are more difficult to couple than others. As a result, chemical ligation strategies can be used to join two shorter peptides together in solution phase. These methods result in the formation of amide or other chemical bonds between the two peptide segments, and typically chemoselective functional groups are chosen to allow ligation in the presence of the unprotected side chains on the peptides. Commonly used reactions include thioester ligation, thioether ligation, and imine ligation [44]. Enzyme-catalyzed methods , which have been primarily developed for ligation of proteins, can also be applied to peptides. For example, sortase A derived from Staphylococcus aureus recognizes and cleaves the LPxTG motif, which can be tagged on one peptide. The resulting thioester is then ligated to the N-terminus of a second peptide that starts with a G residue [45, 46]. The enzyme subtiligase, an engineered mutant of subtilisin, can also catalyze the reaction between a peptide thioester or peptide ester and the N-terminus of a second peptide [47, 48].
An advantage of solid-phase peptide synthesis is that it is not limited to the 20 natural amino acids, and in fact any compounds with appropriate reactive groups can be added to the peptide chain. Some examples are provided in Table 3. The functional group on the C-terminus of the peptide is usually determined by the choice of starting resin, and the N-terminus is often acetylated to block the reactivity of the amine group. Fluorescent labels or dyes can be incorporated into the peptides, including them as modifications on side chains or directly within the peptide chain if proper reactive groups are available. Examples of molecules used in this approach are summarized in Table 4. Fluorescent dyes such as fluorescein isothiocyanate (FITC), 5-carboxyfluorescein (5-FAM), Dansyl, 5- (and 6-) carboxytetramethylrhodamine [5(6)-TAMRA], 7-methoxycoumarin-4-acetic acid (MCA), and 5- (and 6-) carboxyrhodamine 6G [5(6)-CR6G] have different excitation and emission wavelengths, and they can be selected to avoid overlap with tissue autofluorescence or with other fluorescent molecules used in a biomaterial formulation. Biotin can be incorporated for later tagging with fluorophores conjugated to streptavidin, and other molecules such as dinitrophenol (Dnp) or DABCYL can be used as part of Forster energy resonance transfer (FRET) pairs. Some of these molecules can be used as an N-terminal modification by reaction of the molecule with the amine group at the end of the peptide as the last step during peptide synthesis. This reaction can be achieved through a carboxylic acid group already present in the molecule or by first modifying the molecule with a succinimidyl ester group, which can then react with the terminal amine group. Alternatively, the fluorescent molecule can be added to the side chain of a lysine residue, and then this modified lysine can be incorporated anywhere in the peptide using standard coupling and deprotection reactions.
Labeling of peptides with fluorescent dyes can be a useful strategy for examining the loading and release of the peptide from a biomaterial carrier. For example, FITC-labeled peptides were used to track release from electrospun nanofibrous membranes and aerogels of poly(lactide-co-glycolide) (PLGA)/collagen/gelatin [49, 50]. A fluorescence plate reader can directly measure the concentration of the released peptide over time without the need for more complicated assays, such as ELISA, for the detection of the peptide. FRET pairs and dye quenching can also be used in peptide design to track spatial information about the peptides or peptide fragments. Fluorescent dyes and molecules that quench their fluorescence when in close proximity, such as the combination of fluorescein with DABCYL, can be incorporated on the opposite ends of enzyme substrate peptides similar to the ones described above. When the peptide is intact, the dye and quencher are close enough that there is no fluorescence; however, once the substrate is cleaved, the two fragments can diffuse away from each other, and the fluorescence is restored. In addition to being useful for determining kinetic parameters of the enzyme substrate interaction in solution [21], this approach has been used to track the local degradation of a PEG hydrogel incorporating such peptides (Fig. 4) [51].

Design and characterization of quenched fluorescent peptides as enzyme substrates. (a) Sequence and design of cleavable peptide and non-degradable control. (b) Degradation kinetics tracked by monitoring fluorescence over time. (Reprinted from Biomaterials, 34, Leight JL et al., Direct measurement of matrix metalloproteinase activity in 3D cellular microenvironments using a fluorogenic peptide substrate, 7344–52, ©2013 with permission from Elsevier [51])
Peptide Functionalization
In addition to synthesis of the desired peptide, it must be able to be chemically bound to or mixed in with the biomaterial while maintaining its biological activity. This can also influence the design of the peptide. For covalent binding of the peptide, an appropriate reactive group or additional amino acids must be added to allow the reaction to occur. One example of this is the use of a peptide substrate, which can be covalently cross-linked through the action of an enzyme. One such substrate is NQEQVSP(L), which is derived from α2-plasmin inhibitor and is a substrate for the transglutaminase enzyme factor XIIIa. This peptide allows covalent cross-linking with some natural biomaterials, such as fibrin [52, 53], or with synthetic biomaterials that have a complementary substrate (FKGG) recognized by the enzyme [54]. For covalently bound peptides that act as a ligand, spacer amino acids may need to be added within the peptide to prevent steric hindrance interfering with receptor recognition. Cyclization of the peptide to constrain its presentation may also help to improve ligand–receptor interactions, as has been demonstrated with the RGD peptide [55]. For peptides that are active in soluble form, an MMP substrate can be added in the peptide sequence between the covalent attachment point and the peptide of interest to allow enzymatic cleavage and release of the peptide. For example, this has been done by adding the MMP substrate sequence PVGLIG to the N-terminus of osteogenic growth peptide (OGP) and then using carbodiimide chemistry to link the peptide to a partially oxidized form of alginate. Release of the peptide from the alginate matrix through the action of MMP-2 was confirmed [56].
In the PEG hydrogels prepared by a Michael-type addition reaction described above, a covalent bond was formed between the polymer and the peptides through the functionalization of the PEG with vinyl sulfone groups and the incorporation of cysteine residues, which have a reactive thiol group, in the peptides. This allowed both the functionalization of the PEG with the cell adhesive RGD peptide and the cross-linking of the PEG into an enzymatically degradable network using the substrate peptides [22, 23, 38, 39]. Similar conjugation reactions can also be performed between maleimide groups and thiol groups [57]. This strategy has been used to functionalize surfaces with peptides, for example, with layer-by-layer films of peptide-grafted poly(allylamine hydrochloride) (PAH) [58] and maleimide self-assembling monolayers on quartz substrates [59].
Depending on the reactive groups present on the material, peptides can also be covalently attached via an amide bond using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS) as coupling reagents to materials such as maleic anhydride-modified poly(lactic acid) (PLA) [60] or the carboxyl groups of partially oxidized alginate [56]. Typically, this approach relies on the reaction of an amine group coming from the N-terminus or a lysine side chain of the peptide with a carboxyl group on the material. Further, copper-catalyzed azide alkyne cycloaddition reactions can be used with azide-functionalized peptides and, for example, propargyl functionalized l-phenylalanine-based poly(ester urea)s [61]. Alternatively, the reactive groups can be switched between peptide and polymer, as in the case of azidopropyl hyperbranched poly(arylene oxindole) functionalization with 5-Hexynoic-RGDS [62].
For noncovalent binding of peptides, the peptides may be allowed to adsorb to the material or be mixed in during processing, so that they are entrapped in the matrix. Alternatively, the peptide can be modified with an affinity domain that promotes noncovalent interactions with the material. For example, the addition of a highly negatively charged E7 domain, consisting of seven glutamic acid residues in a row, enables peptides to bind to calcium-based materials [63,64,65,66] or to pre-mineralized materials, such as PLGA/collagen/gelatin nanofibers [49]. For an anorganic bovine bone graft material, coupling of a BMP-2-derived peptide to E7 led to increased loading and greater retention, even after 8 weeks of implantation in vivo [63]. Alternatively, the hydroxyapatite binding domain of statherin (N15 domain) was used to bind either PGRGDS or PDGEA (a cell adhesive peptide derived from type I collagen) to hydroxyapatite surfaces. The peptide-coated materials bound MC3T3-E1 osteoblasts via the αVβ3 integrin (for PGRGDS) and α2β1 integrin (for PDGEA) [67].
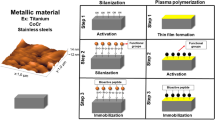
Other linkers, such as polydopamine, can be used to mediate peptide attachment to surfaces [68,69,70,71,72]. In one approach, the surface can be coated with polydopamine, and then the peptide can be bound to the polydopamine coating [68]. Polydopamine coatings can be formed by dip-coating of a material in an aqueous solution of dopamine, which self-polymerizes, and they allow for further functionalization via secondary reactions [73]. This methodology is inspired by the adhesive proteins in mussels, which contain 3,4-dihydroxy-l-phenylalanine and lysine amino acids, and the covalent and noncovalent interactions that can occur with catechol compounds, and it is particularly interesting because it is compatible with a number of different inorganic and organic materials, such as metals, oxides, polymers, and ceramics [73]. Alternatively, peptides can be functionalized with a short polydopamine tag that can then interact with surfaces via the catechol groups (Fig. 5) [70]. This approach has been used to functionalize otherwise inert titanium (Ti) surfaces with RGDS and OGP, which enhanced the attachment, proliferation, and osteogenic differentiation of bone marrow-derived mesenchymal stem cells (BM-MSCs) in vitro as well as improved osteogenesis and mechanical stability of peptide-coated screws implanted in the femoral condyles of New Zealand White rabbits in vivo [70].

Cell adhesive (DOPA)4-G4-GRGDS and osteoinductive (DOPA)4-G4-YGFGG peptides that mimic mussel-derived proteins with catechol groups to enable coordination between the catechol groups and titanium oxide for surface functionalization of Ti cortical bone screws. (Reprinted with permission from [70]. ©2016 American Chemical Society)
Osteoinductive Peptides
Peptide-functionalized materials provide an interesting approach to stimulate bone regeneration. In the fields of orthopedics and dentistry , peptides are relevant in two major ways. The first is to provide surface functionalization of the frequently Ti-based implants that are used as permanent implants. The use and characterization of Ti implants [74] as well as their combination with biopolymers [75] have been previously reviewed. The second is to use peptides in tissue engineering approaches. Typically, tissue engineering aims to combine cells, biomaterials, and biological factors to create living implants that can repair or regenerate tissue function [76], and peptides are one means of providing these biological signals. In this case, tissue engineering scaffolds of a variety of materials have been modified and examined for their effects both in vitro and in vivo.
This section focuses on the application of peptides with osteoinductive capabilities, that is, peptides that can promote the osteogenic differentiation of progenitor cells in vitro and can lead to de novo bone formation in vivo. While the cell adhesive and enzymatically degradable peptides discussed above as well as other peptides, such as pro-angiogenic peptides, can support tissue remodeling and repair more generally and thus are interesting to consider as part of treatments to regenerate bone, as considered in recent reviews [77, 78], in this section, peptides that influence bone progenitor cells are specifically considered. These peptides are derived from a number of sources including identification of active domains in BMP-2 , BMP-7, and other osteogenic proteins. A number of osteogenic peptides , their sequences and variants, and their effects are briefly summarized in Table 5.
OGP Peptides
Osteogenic growth peptide (OGP; ALKRQGRTLYGFGG) is a 14 amino acid peptide derived from the C-terminus of histone H4. It is found naturally in soluble form in serum and has been shown to influence osteoblast proliferation and differentiation [79]. OGP increased alkaline phosphatase (ALP) activity of MC3T3-E1 pre-osteoblasts in vitro and improved bone remodeling in vivo in a rat mandibular condyle model when delivered systemically [79]. In addition, systemic administration of OGP has been shown to improve the structural and mechanical properties of the fracture callus in a rat model [93].
OGP has also been used with several biomaterials, both as a 2D surface modification and for incorporation in a 3D scaffold [94, 95]. When OGP and its shorter variant, OGP(10–14) (YGFGG) [80], were tethered to 2D surfaces via click chemistry between an azide-functionalized peptide and an alkyne-terminated self-assembled monolayer (SAM), they led to increases in MC3T3-E1 attachment and proliferation in vitro [96]. A similar approach has been used with azide-functionalized OGP and poly(ester urea) scaffolds via propargyl groups on the surface of the scaffolds. This functionalization increased the differentiation of human mesenchymal stem cells (hMSCs), as demonstrated by increases in ALP activity, calcium deposition, and expression of osteogenic genes [61]. Carbodiimide chemistry has been utilized to link OGP(10–14) to maleic anhydride-modified PLA scaffolds, improving the proliferation, differentiation, and mineralization of neonatal rat calvarial osteoblasts [60]. Moreover, OGP has been used to functionalize the surface of Ti implants through a polydomamine linker. In combination with the cell adhesive peptide RGD, the OGP-functionalized surfaces promoted cell attachment and mineralization in vitro, and functionalized Ti screws led to increased osseointegration, as shown by increased bone volume, bone-implant contact, and pull-out force (Fig. 6) [70].

Effect of OGP surface treatment of Ti bone screws on osteogenesis and mechanical stability in a rabbit femoral condyle. (a) Reconstructed microCT images. (b) Quantitation of bone volume (BV)/total volume (TV) from microCT images. (c) Representative histological images stained with toluidine blue with 1 = untreated control, 2 = OGP, 3 = RGD, and 4 = OGP/RGD (3,1). (d) Quantification of bone-implant contact (BIC) from histological images. (e) Biomechanical pull-out testing. (Reprinted with permission from [70]. ©2016 American Chemical Society)
Additionally, hydrogels have been formed using OGP(10–14) tethered to alginate matrices by a protease-sensitive linker. Exposure to enzymatic treatment led to the release of the peptide and osteogenic differentiation of treated hMSCs. The MSC-laden, OGP-functionalized alginate hydrogels also resulted in ectopic bone formation in vivo [56]. Moreover, functionalization with OGP increased MC3T3-E1 proliferation and alkaline phosphatase activity for cells cultured on hydrogels formed from the self-assembling peptide RADA16 [97].
Peptides Derived from BMP-2
As BMP-2 is one of the most potent osteoinductive growth factors [98], peptides derived from BMP-2 have also been studied for their ability to stimulate osteogenic differentiation in vitro and bone formation in vivo. One of the first peptides derived from BMP-2 was a 20-amino-acid-long sequence (NSVNSK-IPKACCVPTELSAI), which led to ectopic bone formation in the calf muscle of rats when linked to an alginate hydrogel [81]. A peptide containing the motif DWIVA has also been demonstrated to promote proliferation and osteogenic differentiation of MC3T3-E1 cells in vitro and to increase bone growth when conjugated to the surface of Ti implants used in mandibular bone defects in beagle dogs [83].
The peptide KIPKASSVPTELSAISTLYL, derived from residues 73–92 of the knuckle epitope of BMP-2, has promoted ALP activity and osteocalcin gene expression by C3H10T1/2 cells in culture and, when combined with an alginate hydrogel, has led to ectopic bone formation in rat muscle tissue [82, 99] and accelerated bone formation in a rat tibial defect [100]. Entrapping the peptide in chitosan microspheres that were then embedded in nano-hydroxyapatite/collagen/poly(lactic acid) (PLA) scaffolds provided controlled release of the peptide, which still retained its activity [101]. Further, grafting the peptide to self-assembled monolayers in combination with the cell adhesive RGD peptide led to the upregulation of bone sialoprotein expression as well as promotion of mineralization, even in the absence of other osteogenic supplements [102]. As has been shown with OGP, polydopamine has also been used to link the BMP-2-derived peptide to PLGA scaffolds [69, 71]. For example, the modified scaffolds promoted osteogenic differentiation of human adipose-derived stem cells (hADSCs) in culture and resulted in bone formation in calvarial defects in vivo [69].
The BMP-2-derived KIPKASSVPTELSAISTLYL has also been incorporated into surface coatings using electrostatic interactions. One way this has been achieved is by forming layer-by-layer films of PAH and poly(sodium 4-styrenesulfonate), with the peptide grafted on the PAH. The films were coated on electrospun membranes of nano-hydroxyapatite and PLGA, and they promoted alkaline phosphatase activity by MSCs in vitro and bone formation in a rat calvarial defect in vivo [58]. A second method to immobilize the peptide is through the E7 calcium binding domain described earlier. E7-modified BMP-2-derived peptides have been bound to anorganic bovine bone [63], α-tricalcium phosphate (α-TCP) scaffolds [64], and even nanofibrous membranes of PLGA/collagen/gelatin that were pre-mineralized [49]. Nanofibrous aerogels of electropsun PLGA/collagen/gelatin and Sr-Cu-doped bioactive glass fibers were also functionalized with E7-BMP-2 peptides. The addition of the peptide increased bone formation in vivo in rat calvarial bone defects (Fig. 7) [50].

Calvarial bone regeneration induced by E7-BMP-2 peptide loaded aerogels. Radiographs of (a) untreated controls, (b) aerogel only control, (c) E7-BMP-2 peptide loaded aerogel group. Quantification of (d) regenerated bone volume and (e) bone formation area. (Reprinted with permission from [50]. ©2018 John Wiley and Sons)
Peptides Derived from BMP-7
Bone forming peptide-1 (BFP-1; GQGFSYPYKAVFSTQ) is derived from residues 100–114 of BMP-7. Treatment with soluble BFP-1 led to increased ALP activity and calcium deposition by bone marrow stromal cells, and further these BFP-1-treated cells increased bone formation when injected subcutaneously in mice [85]. Likewise, hMSCs also showed increased ALP activity, calcium deposition, and osteogenic gene expression when cultured on nanofibrous scaffolds consisting of polycaprolactone (PCL) functionalized with the peptide via a polydopamine linker. This enhancement in osteogenic differentiation was also seen when the cells were cultured in medium lacking other osteoinductive factors [68]. Further, human-induced pluripotent stem cells (hiPSCs) also increased ALP activity, calcium deposition, and osteogenic gene expression when cultured on 2D surfaces that had been functionalized with BFP-1 and carboxymethyl chitosan [72]. A second osteoinductive peptide, BFP-3, was also determined from BMP-7 residues 250–265 (ETLDGQSI-NPKLAGL ). It increased in vitro osteogenic differentiation of bone marrow stromal cells by increasing ERK1/2 and Smad1/5/8 phosphorylation [86].
Peptides Derived from BMP-4 and BMP-9
Osteoinductive peptides have also been identified within BMP-9, including from the knuckle epitope, residues 68–87 (KVGKACCVPTKLSPISVLYK) [103]. In vitro, this peptide-induced gene expression of Runx2, osterix, type I collagen, and osteocalcin by MC3T3-E1 pre-osteoblast cells, and it activated the Smad pathway [89]. The phosphorylation of Smad1/5/8 was further enhanced when this peptide was presented in combination with a fibronectin-derived peptide containing both PHSRN and RGD ligands and functionalized on films of PCL [104]. When combined with a chitosan-based delivery system, the peptide induced ectopic bone formation in the quadriceps of mice. Interestingly, a collagen-based delivery system abrogated this response [88].
A heparin binding domain from residues 15–24 of BMP-4, RKKNPNCRRH, has been shown to have osteogenic activity. Administered in soluble form, the peptide promoted the osteogenic differentiation of hMSCs, as shown by increased matrix mineralization as well as upregulation of the osteogenic genes, ALP, osteopontin, and osteonectin. Phosphorylation of Smad 1/5/8 and MAPK was demonstrated by Western blotting and confirmed the activation of the ERK1/2 pathway. Further, when combined with an alginate hydrogel matrix, the peptide was able to stimulate in vivo bone formation in a rabbit calvarial defect model [87].
Peptides Derived from Parathyroid Hormone
A peptide derived from parathyroid hormone residues 1–34 (PTH1–34; SVSEIQL-MHNLGKHLNSMERVEWLRKKLQDVHNF) has been used clinically as treatment for osteoporosis [105]. For stimulating bone regeneration, it has been covalently bound to an RGD-functionalized PEG hydrogel and been shown to lead to increases in in vivo bone formation, at levels similar to treatment with autologous bone, when implanted into alveolar bone defects surrounding standard Ti implants in a dog model [90]. Similarly, PTH1–34 was covalently bound to a fibrin matrix via an enzymatically degradable peptide linker, and this led to increased bone formation in defects of the femur and humerus of sheep [106]. The soluble form of PTH1–34 was also administered via injection to determine dosing amount and frequency to improve bone healing in a mouse femoral allograft model [107]. Moreover, it was shown to protect against radiotherapy-induced trabecular bone loss in a rat model [108].
Other Peptides
A number of other peptides have shown osteoinductive activity. Bone marrow homing peptide 1 (BMHP1; PFSSTKT) , which was discovered from screening a phage display library, led to osteogenic differentiation of MSCs cultured on quartz substrates functionalized with the peptide [59]. The peptide P-15 derived from residues 766–780 of the type I collagen α chain (GTPGPQGIAGQRG-VV) accelerated bone formation in a rabbit calvarial defect model when it was coated on deproteinized bovine bone [91]. A collagen-binding motif (CBM) from osteopontin (GLRSKSKKFRRPDIQYPDATDEDITSHM) in combination with a type I collagen matrix increased mineralization by cultured bone marrow stromal cells, as shown by calcein, Alizarin Red S, and ALP staining, as well as phosphorylation of Smad1/5/8 (Fig. 8). The peptide functionalized matrices also led to increased bone formation in rabbit calvarial defects [92].

Effect of CBM on bone marrow stromal cells. (a) Micrographs of calcein staining (left panel), Alizarin Red S staining (middle panel), and ALP expression (right panel). (b) Quantification of ALP activity. (c) Western blot analysis for phosphorylated-Smad expression. (Reprinted from Biomaterials, 28, Lee J-Y et al., Assembly of collagen-binding peptide with collagen as a bioactive scaffold for osteogenesis in vitro and in vivo, 4257–67, ©2007 with permission from Elsevier [92])
Use of Osteoinductive Peptides Clinically
The majority of peptides used to stimulate bone healing and regeneration have only been evaluated in vitro or in preclinical animal studies [109, 110]. Of the osteoinductive peptides discussed in this chapter, two have seen more extensive clinical use, PTH1–34 and P-15. In addition, chrysalin, also known as thrombin peptide 508 (TP508) , has been evaluated in Phase I/II clinical trials to treat distal radial fractures, showing reduced time to healing [111]. PTH1–34 has been approved for the prevention and treatment of osteoporosis clinically [105]. In a clinical study for the treatment of distal radial fractures, PTH1–34 shortened the time to healing with the lower of two doses tested [112] and appeared to improve early callus formation [113]. The use of PTH1–34 in bone healing has also been documented in several case reports, including for sternal nonunion [114] and hip fracture [115], among others [110]. P-15 was combined with anorganic bone mineral and a hydrogel carrier material and tested in a clinical study for anterior cervical discectomy and fusion in comparison with autograft. The radiographic fusion rates, clinical and patient-reported outcomes, and safety profile of the P-15 formulation were all similar to autograft bone [116]. In a first example of its use for orthopedic applications, again combined with anorganic bone mineral, P-15 has been tested in a pilot clinical trial to treat patients with mal-union or delayed union fractures, leading to full consolidation in 90% of the cases [117]. In the oral cavity, P-15 in combination with anorganic bone mineral has been shown to lead to improved defect fill results when compared to anorganic bone mineral alone in a multicenter trial with 33 patients [118].
Anti-biofilm Peptides
Biofilms are formed by the colonization of bacteria on surfaces and represent a leading cause of chronic and implant-associated infections clinically [119, 120]. They are characterized by the formation of aggregates of bacteria that encapsulate themselves in a dense extracellular matrix composed of polysaccharides, extracellular DNA, proteins, and lipids [121]. As a result, bacteria in biofilms are 10- to 1000-fold more resistant to antibiotics than planktonic (motile) bacteria [122, 123]. Of relevance to biomaterials, biofilms can form on the surface of implanted medical devices, such as catheters, valves, stents, and orthopedic prostheses, and even of contact lenses [124]. In dentistry , the plaque that develops on the surface of teeth is also a biofilm [125]. Peptide-based treatments for preventing or eliminating biofilms are becoming interesting due to the development of antimicrobial peptides, which demonstrate broad-spectrum activity against planktonic Gram-positive and Gram-negative bacteria. A subset of these antimicrobial peptides have also demonstrated activity against bacteria in biofilms, both on their own and in combination with other antibiotics [126,127,128,129,130,131,132], and a number of examples are summarized in Table 6.
Antimicrobial peptides, a subset host defense peptides, are naturally produced by organisms including animals, fungi, plants, and bacteria, resulting in the identification of more than 2000 such peptides [152]. Antimicrobial peptides typically act rapidly and have a broad-spectrum antimicrobial activity against planktonic cells, while host defense peptides also include those peptides that act as innate immune modulators through anti-infective, anti-inflammatory, wound healing, and/or anti-biofilm activities [153, 154]. Thus, these natural peptides are interesting to explore as potential therapeutics against biofilms. Alternatively, peptide screening methods can be used to identify peptides with antibacterial activity [155]. As the literature on antimicrobial peptides is vast, this chapter will focus on a few key examples of antimicrobial peptides that are naturally derived or identified from peptide screening studies and have shown anti-biofilm activity. Additional emphasis will be placed on peptides that have shown anti-biofilm activity in vivo in animal models or that have been used to modify biomaterial surfaces. Anti-biofilm activity is typically demonstrated in vitro by the ability to kill multiple species of bacteria that can be present in biofilms, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species [131]. In vivo activity has primarily been tested in mouse skin wound or cutaneous abscess models as well as rat ureteral catheterization models. In addition to killing planktonic bacteria that can form or detach from biofilms as well as killing embedded bacteria, antimicrobial peptides can also act to interfere with bacterial adhesion and gene expression as well as influence the host response to the biofilm [127]. These latter activities can occur at concentrations much lower than the minimum inhibitory concentration (MIC) , which is usually used to characterize the effects of antimicrobial agents on planktonic bacteria [127] and provides a measurement of anti-biofilm activity.
LL-37, P10, AS10, and hep20 Peptides (Naturally Derived)
Of the many antimicrobial peptides , cathelicidin LL-37 (LLGDFFRKSKEKIG-KEFKRIVQRIKDFLRNLVPRTES) was one of the first human-derived peptides to demonstrate anti-biofilm activity. It can be found at mucosal surfaces and in the granules of phagocytes, and its concentration at sites of chronic inflammation is higher than baseline levels in most bodily fluids [146]. It is able to prevent biofilm formation in vitro at concentrations well below its MIC, and it can also reduce existing P. aeruginosa biofilms [146]. Shorter peptides derived from LL-37, such as P10 (LAREYKKIVEKLKRWLRQVLRTL-R), have also shown antimicrobial activity. P10 was more effective at killing the methicillin-resistant S. aureus (MRSA) strain LUH14616 than LL-37, and it could eradicate MRSA strains LUH14616 and LUH15051 from thermally wounded human skin equivalents in vitro without showing toxicity towards the fibroblasts and keratinocytes within the tissue models [147].
Likewise, the mouse cathelicidin-related antimicrobial peptide (CRAMP) has been shown to inhibit fungal biofilm formation [156]. Shorter peptides based on CRAMP, such as AS10 (KLKKIAQKIKNFFQKLVP), were also able to inhibit Candida albicans biofilm growth on Ti disks as well as formation of biofilms from other bacteria [134]. The study also tested the effect of AS10 on human osteoblasts, mesenchymal stromal cells, and endothelial cells in vitro and demonstrated no negative effects on cell viability. Further, AS10 did not interfere with calcium deposition by the osteoblasts or mesenchymal stromal cells or with tube formation by the endothelial cells, which suggests that AS10 could be used as an anti-biofilm coating on implants without affecting cells in the surrounding tissues [134].
A second human-derived antimicrobial peptide is hepcidin 20 (hep20; ICIFCC-GCCHRSHCGMCCKT), a 20-amino-acid peptide that is found in the liver. The related hepcidin 25 (hep25) is involved in the regulation of plasma iron levels by binding ferroportin on macrophages, enterocytes, and hepatocytes [157]. Both hep25 and hep20 have demonstrated antimicrobial activity against several bacterial strains [158] with hep20 also showing antifungal activity [159]. Hep20 inhibited biofilm formation of polysaccharide intercellular adhesion (PIA)-positive and PIA-negative clinical isolates of Staphylococcus epidermis, most likely by inhibiting the accumulation of extracellular matrix within the biofilm [143].
IDR-1018 and 3002 Peptides (from Peptide Library Screenings)
Innate defense regulator peptide 1018 (IDR-1018) and 3002 are two examples of peptides that have been optimized by peptide library screening methods. Starting from the bovine bactenecin derivative Bac2a (RLARIVVIRVAR-NH2), a library of over 100 peptides, each 12 amino acids long, was generated by performing point mutations, scrambling, and deletion of amino acids [160]. The immunomodulatory potential of the peptides from the library was evaluated by measuring the ex vivo induction of monocyte chemotactic protein 1 (MCP-1) and MCP-3 by human peripheral blood mononuclear cells, and IDR-1018 (VRLI-VAVRIWRR-NH2) resulted in a >50-fold increase compared to Bac2a [160]. IDR-1018 also exhibited broad-spectrum anti-biofilm activity towards P. aeruginosa, E. coli, A. baumannii, K. pneumoniae, MRSA, S. typhimurium, and B. cenocepacia when tested with sub-MIC concentrations [161]. It could also inhibit or eliminate biofilms, as demonstrated by growing P. aeruginosa biofilms in continuous-culture flow cells in a minimal medium. The medium flowing through the cells was supplemented with IDR-1018 either during or after biofilm establishment, and in both cases, the treatment led to thinner biofilms that lacked the structural features of mature biofilms or eliminated them entirely (Fig. 9) [161].

Inhibition and eradication/reduction of biofilms of different bacterial strains by sub-inhibitory concentrations of peptide IDR-1018, as demonstrated in a flow cell assay. The scale bars are 30 μm. (Reprinted under the terms of the Creative Commons Attribution License from de la Fuente-Núñez et al. ©2014 [161])
The sequence specificity of IDR-1018 was further explored by generating a second peptide library that contained 96 peptides with single amino acid substitutions [133]. This library was SPOT-synthesized on cellulose arrays, and the ability to prevent MRSA biofilms was evaluated. The resulting data were fed into a quantitative structure–activity relationship (QSAR) model, which predicted new peptides with anti-biofilm activity from an in silico library of 100,000 peptide sequences. The identified peptide 3002 (ILVRWIRWRIQW-NH2) was more effective than IDR-1018 in inhibiting biofilm growth and in eradicating pre-formed biofilms. Further, peptide 3002 was able to reduce abscess size in a mouse cutaneous model of high-density bacterial infection, an MRSA chronic infection model [133].
DJK-5, DJK-6, and D-RR4 Peptides (d-Enantiomeric Peptides)
While peptides represent a potentially powerful therapeutic option, they have some drawbacks for use in vivo including degradation by proteases and inactivation in bodily fluids. The incorporation of non-natural amino acids into synthetic peptides can address some of these limitations by providing more stable variants, which may lead to increased activity [140,141,142, 162]. The synthetic antimicrobial peptides DJK-5, DJK-6, and D-RR4 have been developed including the d-enantiomeric form of amino acids and have shown strong anti-biofilm activity both in vitro and in vivo [140,141,142]. DJK-5 (vqwrairvrvir; d-amino acids) and DJK-6 (vqwrrirvw-vir; d-amino acids) were identified from screening a library of 12-amino-acid-long peptides with the following design constraints: d-enantiomeric forms of only 9 amino acids (A, F, I, K, L, Q, R, V, W), 4 charged residues, 7–8 hydrophobic residues , and 0–1 Q residues. DJK-5 and DJK-6 were identified as being able to inhibit biofilm formation by several common bacterial strains, at concentrations below their MIC, and to eradicate established biofilms [140]. Further, DJK-5 was shown to decrease abscess size and reduce bacterial burden in a mouse cutaneous abscess model induced by P. aeruginosa [141].
The peptide D-RR4 (wlrrikawlrrika-NH2; d-amino acids) was developed to be resistant to proteolytic digestion, as confirmed by stability in the presence of both bacterial (proteinase K) and mammalian (trypsin) proteases, and it remained active in the presence of physiologic concentrations of salt, serum proteins, and acidic pH [142]. It also showed reduced toxicity towards macrophages and keratinocytes when compared to the l-enantiomeric form of the peptide. The in vivo antibacterial activity of D-RR4 was demonstrated by increased survival and decreased bacterial burden of Caenorhabditis elegans worms infected with P. aeruginosa or A. baumannii, after treatment with the peptide [142].
BMAP28, Tachyplesin III, WRL3, and DRGN-1 Peptides (Evaluation In Vivo)
In vitro activity to inhibit or kill relevant bacterial strains as well as to inhibit or eradicate biofilms helps to demonstrate the potential of anti-biofilm peptides. However, ultimately, they must be effective in vivo, and thus the development of relevant animal models is important. As mentioned previously, the survival of C. elegans worms and the reduction of cutaneous abscesses in mice are two examples of such in vivo models. However, they lack some of the complex features of implant-associated biofilm formation and chronic infection. A more challenging model is an infected mouse burn wound model. In this case, a region of skin is first scalded and then infected with MRSA. In addition to the size of the wound and the bacterial burden, the effects on host response [interleukin (IL)-6, IL-10, tumor necrosis factor (TNF)-α, and MCP-1 production] as well as angiogenesis can be evaluated. Using this model, the antimicrobial effects of an engineered amphiphathic peptide, WRL3 (WLRAFRRLVRRLARGLRR-NH2), was demonstrated (Fig. 10). This confirmed its potential as an anti-biofilm agent as also shown by antimicrobial activity against MRSA and inhibition of biofilm activity in vitro [151]. Likewise, DRGN-1 (PSKKTKPVKPKKVA) enhanced healing in a mouse skin wound model that was infected with a biofilm containing both P. aeruginosa and S. aureus [163].

In vivo antimicrobial activity of WRL3 as demonstrated in a mouse infected burn wound model. (a) Viable bacterial counts in colony-forming units (CFU). (b) Wound area measurements. (c) Images of wounded regions. (Reprinted with permission from [151]. ©2016 American Chemical Society)
As catheters frequently develop biofilms , in vivo models have also been developed to study the effects of antimicrobial peptides on ureteral stent infection. These rat models have involved the implantation of ureteral stents either subcutaneously or in the bladder. The subcutaneous model was used to assess the effectiveness of stents coated with Tachyplesin III (KWCFRVCYRGICYRKCR-NH2), which is derived from horseshoe crabs and exhibits antimicrobial activity against P. aeruginosa. After implantation of the Tachyplesin III-coated stent, P. aeruginosa was injected onto the implant surface. The Tachyplesin-III-treated group showed a reduction in bacterial count compared to uncoated stents, and this effect was further enhanced by co-treatment with intraperitoneal injection of piperacillin-tazobactam (TZP) [149]. On the other hand, the bladder implantation model was used with BMAP-28 (GGLRSLGR KILRAWKKYGPIIVPIIRI-NH2), a cathelicidin peptide. After implantation of the BMAP-28-coated stent, S. aureus or E. faecalis was inoculated into the bladder. The BMAP-28 coating reduced the bacterial counts for both strains compared to uncoated controls, and this level was further reduced by coadministration of vancomycin intraperitoneally [137].
Immobilization of Antimicrobial Peptides
A final consideration in the development of anti-biofilm peptides is their delivery mechanism. As biofilms tend to form on the surface of implanted devices, peptide immobilization techniques become relevant [164]. Antimicrobial peptides can simply be adsorbed on the surface of materials by soaking them in solutions of the peptides [137, 149, 165]. Layer-by-layer films provide a next step in complexity of preparation and controlled release of the peptide. These films are formed from alternating deposition of cationic and anionic molecules (polymers or peptides) [166, 167]. For example, the thickness and stability of the coating, amount of peptide loaded, and the release rates of the antimicrobial peptide ponericin G1 (GWKDWAKKAGGWLKKKGPGMAKAALKAAMQ-NH2) were affected by the choice of polyanion used, and the film composition also influenced resistance to attachment of S. aureus [167].
Polymer brushes provide a means for covalent conjugation of antimicrobial peptides. Ti and quartz surfaces have been functionalized by surface-initiated polymerization of N,N-dimethylacrylamide and aminopropyl methacrylamide hydrochloride, resulting in a primary amine functionality. These amine groups were modified to maleimide groups, which then allowed the reaction with cysteine-containing peptides [168, 169]. The presentation of antimicrobial peptide Tet-20 (KRWRIRVRVIRKC) inhibited P. aeruginosa and S. aureus growth in vitro and S. aureus adherence in vivo on subcutaneous implants [169]. Similarly, an allyl glycidyl ether polymer brush with PEG-maleimide spacer was used to bind cysteine-containing peptides to polydimethylsiloxane (PDMS) surfaces [170].
As reduction in biofilm formation and improved osseointegration are important considerations for Ti orthopedic and dental implants, loading of antimicrobial peptides in porous calcium phosphate (CaP) coatings on Ti surfaces has been explored. Coatings containing the peptide Tet213 (KRWWKWWRRC) demonstrated antimicrobial activity against S. aureus and P. aeruginosa bacterial strains while remaining cytocompatible towards MG-63 osteoblast-like cells [171]. A similar coating loaded with HHC36 (KRW-WKWWRR) did not interfere with bone growth around press-fit grafts in a rabbit femoral defect model [172]. A more complex coating combining the CaP with vertically oriented TiO2 nanotubes and a phospholipid film were also able to provide sustained release and antimicrobial activity of HHC36 [173].
Use of Anti-biofilm and Antimicrobial Peptides Clinically
Similar to the osteoinductive peptides, anti-biofilm peptides as well as antimicrobial peptides more generally have not yet resulted in many products for clinical use [174, 175]. Of the peptides discussed herein, LL-37 has been tested in Phase I/II clinical trials to treat hard-to-heal venous leg ulcers. In this case, the LL-37 improved the healing rate constants [176], but the application was a topical treatment for wound healing and not treatment of biofilms. Polymyxin B and Polymyxin E (colistin) have been used clinically to treat drug-resistant infections resulting from Gram-negative bacteria [177,178,179]. However, the majority of antimicrobial peptides tested clinically have been used topically in clinical trials to treat infected diabetic foot ulcers, catheter infections, and skin and fungal infections [175] and not for treatment and/or prevention of biofilms. While several anti-biofilm peptides show promise, clinical translation of antimicrobial peptides has met a number of regulatory hurdles [174].
Conclusion/Summary and Future Perspectives
This chapter started out by introducing basic concepts in peptide design, from learning from nature and deriving peptides from active domains within proteins to utilizing peptide library screening methods to identify peptides with a desired response. Standard methods for peptide synthesis, most commonly solid-phase peptide synthesis, and various techniques for functionalizing biomaterials with peptides were summarized. These chemical methods for peptide synthesis bring about additional opportunities in peptide design, as non-natural amino acids and fluorescent tags, among other molecules, can be incorporated during the synthesis. This allows for the development of peptides that may be more stable or have higher activity than their naturally derived counterparts, and the possibility of adding dyes further provides simplified methods for detecting the peptides. These so-called peptidomimetics have started to be explored for their antimicrobial activities [126, 132, 180], and it would be interesting to explore further osteoinductive peptidomimetics. Covalent and non-covalent binding of peptides to biomaterials also provides a means to localize the peptide and in some cases provide controlled release. It would also be interesting to explore osteoinductive and antimicrobial peptides in the context of other controlled release systems. For example, research is rapidly advancing in the development of stimuli-responsive systems that release their cargo in response to changes in temperature, pH, light, etc., and conjugation to polymers or encapsulation in micro-/nano-particles also provide a means for extended delivery of compounds [181].
Finally, this chapter focused on the development and application of two classes of peptides, those with osteoinductive and anti-biofilm activity. The osteoinductive peptides have been derived for a large part from the active domains of the different BMPs. Their activities have been demonstrated in vitro by stimulating osteogenic differentiation of different types of progenitor cells and in vivo by promoting bone growth, ectopically or orthopically. These peptides show promise to support osseointegration when functionalized on the surface of Ti or other inert implant materials. Further work to combine these peptides with newly developed biomaterial scaffolds [182, 183] would help to advance the field of bone tissue engineering. The anti-biofilm peptides presented have been developed from naturally occurring antimicrobial peptides as well as from screening of peptide libraries. Candidate peptides have shown activity against several strains of bacteria that are involved in biofilm formation, and they have also been shown to reduce bacterial burden or wound size in in vivo models. Development of more complex animal models that mimic the clinical conditions of biofilm infection would help to further identify and characterize peptides with anti-biofilm activity. Last but not least, the combination of osteoinductive and anti-biofilm peptides would allow for the development of materials that stimulate bone formation while simultaneously protecting against chronic infection. Anti-biofilm peptides have clearly been intended for orthopedic [184] and dental applications, as they have been studied while functionalized on Ti surfaces. Full-length BMPs have been combined with antibacterial agents, such as vancomycin and silver (Ag+) [185], so the combination of osteoinductive and anti-biofilm peptides, functionalized on the surface of permanent implants or implemented in tissue engineering scaffolds, is a logical next step.
References
Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH (1993) Protein design by binary patterning of polar and nonpolar amino acids. Science 262:1680. https://doi.org/10.1126/science.8259512
West MW, Wang W, Patterson J, Mancias JD, Beasley JR, Hecht MH (1999) De novo amyloid proteins from designed combinatorial libraries. Proc Natl Acad Sci U S A 96:11211–11216. https://doi.org/10.1073/pnas.96.20.11211
Zhang S, Holmes TC, DiPersio CM, Hynes RO, Su X, Rich A (1995) Self-complementary oligopeptide matrices support mammalian cell attachment. Biomaterials 16:1385–1393. https://doi.org/10.1016/0142-9612(95)96874-Y
Collier JH, Messersmith PB (2004) Self-assembling polymer–peptide conjugates: nanostructural tailoring. Adv Mater 16:907–910. https://doi.org/10.1002/adma.200306379
Hauser CAE et al (2011) Natural tri- to hexapeptides self-assemble in water to amyloid β-type fiber aggregates by unexpected α-helical intermediate structures. Proc Natl Acad Sci U S A 108:1361. https://doi.org/10.1073/pnas.1014796108
Ruoslahti E (1996) RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol 12:697–715. https://doi.org/10.1146/annurev.cellbio.12.1.697
DeForest CA, Polizzotti BD, Anseth KS (2009) Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat Mater 8:659. https://doi.org/10.1038/nmat2473
Andukuri A, Minor WP, Kushwaha M, Anderson JM, Jun H-W (2010) Effect of endothelium mimicking self-assembled nanomatrices on cell adhesion and spreading of human endothelial cells and smooth muscle cells. Nanomedicine 6:289–297. https://doi.org/10.1016/j.nano.2009.09.004
Dalet-Fumeron V, Boudjennah L, Pagano M (1998) Binding of the cysteine proteinases papain and cathepsin B-like to coated laminin: use of synthetic peptides from laminin and from the laminin binding region of the β1Integrin subunit to characterize the binding site. Arch Biochem Biophys 358:283–290. https://doi.org/10.1006/abbi.1998.0868
Massia SP, Hubbell JA (1991) Human endothelial cell interactions with surface-coupled adhesion peptides on a nonadhesive glass substrate and two polymeric biomaterials. J Biomed Mater Res 25:223–242
Gobin AS, West JL (2003) Val-Ala-Pro-Gly, an elastin-derived non-integrin ligand: smooth muscle cell adhesion and specificity. J Biomed Mater Res A 67A:255–259. https://doi.org/10.1002/jbm.a.10110
Chen S et al (2015b) A laminin mimetic peptide SIKVAV-conjugated chitosan hydrogel promoting wound healing by enhancing angiogenesis, re-epithelialization and collagen deposition. J Mater Chem B 3:6798–6804. https://doi.org/10.1039/C5TB00842E
Davel LE, Puricelli LI, Del Carmen M, Vidal C, De Lorenzo MS, Sacerdote de Lustig E, Bal de Kier Joffe ED (1999) Soluble factors from the target organ enhance tumor cell angiogenesis: role of laminin SIKVAV sequence. Oncol Rep 6:907–918
Maeda T, Oyama R, Titani K, Sekiguchi K (1993) Engineering of artificial cell-adhesive proteins by grafting EILDVPST sequence derived from fibronectin the. J Biochem 113:29–35
Moyano JV et al (1997) Fibronectin type III5 repeat contains a novel cell adhesion sequence, KLDAPT, which binds activated α4β1 and α4β7 integrins. J Biol Chem 272:24832–24836
Woods A, McCarthy JB, Furcht LT, Couchman JR (1993) A synthetic peptide from the COOH-terminal heparin-binding domain of fibronectin promotes focal adhesion formation. Mol Biol Cell 4:605–613
Feng Y, Mrksich M (2004) The synergy peptide PHSRN and the adhesion peptide RGD mediate cell adhesion through a common mechanism. Biochemistry 43:15811–15821. https://doi.org/10.1021/bi049174+
Lee ST et al (2010) Engineering integrin signaling for promoting embryonic stem cell self-renewal in a precisely defined niche. Biomaterials 31:1219–1226. https://doi.org/10.1016/j.biomaterials.2009.10.054
Yokosaki Y et al (1998) Identification of the ligand binding site for the integrin α9β1 in the third fibronectin type III repeat of tenascin-C. J Biol Chem 273:11423–11428
Massia SP, Hubbell JA (1992) Vascular endothelial cell adhesion and spreading promoted by the peptide REDV of the IIICS region of plasma fibronectin is mediated by integrin alpha 4 beta 1. J Biol Chem 267:14019–14026
Nagase H, Fields GB (1996) Human matrix metalloproteinase specificity studies using collagen sequence-based synthetic peptides. Biopolymers 40:399–416. https://doi.org/10.1002/(SICI)1097-0282(1996)40:4<399::AID-BIP5>3.0.CO;2-R
Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA (2003a) Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A 100:5413. https://doi.org/10.1073/pnas.0737381100
Lutolf MP, Weber FE, Schmoekel HG, Schense JC, Kohler T, Müller R, Hubbell JA (2003b) Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat Biotechnol 21:513. https://doi.org/10.1038/nbt818
Smith GP, Petrenko VA (1997) Phage display. Chem Rev 97:391–410. https://doi.org/10.1021/cr960065d
Winter GP, James K, Potter G (1989) Antibody engineering. Philos Trans R Soc Lond B Biol Sci 324:537–547. https://doi.org/10.1098/rstb.1989.0066
Desch KC et al (2015) Probing ADAMTS13 substrate specificity using phage display. PLoS One 10:e0122931. https://doi.org/10.1371/journal.pone.0122931
Turk BE, Huang LL, Piro ET, Cantley LC (2001) Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat Biotechnol 19:661–667. https://doi.org/10.1038/90273
Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, Cantley LC (1994) Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr Biol 4:973–982. https://doi.org/10.1016/S0960-9822(00)00221-9
Songyang Z et al (1997) Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 275:73–77
Songyang Z et al (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72:767–778. https://doi.org/10.1016/0092-8674(93)90404-E
Yaffe MB et al (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91:961–971. https://doi.org/10.1016/S0092-8674(00)80487-0
Boulware KT, Daugherty PS (2006) Protease specificity determination by using cellular libraries of peptide substrates (CLiPS). Proc Natl Acad Sci U S A 103:7583. https://doi.org/10.1073/pnas.0511108103
Chen EI, Kridel SJ, Howard EW, Li W, Godzik A, Smith JW (2002) A unique substrate recognition profile for matrix metalloproteinase-2. J Biol Chem 277:4485–4491. https://doi.org/10.1074/jbc.M109469200
Smith MM, Shi L, Navre M (1995) Rapid identification of highly active and selective substrates for Stromelysin and Matrilysin using bacteriophage peptide display libraries. J Biol Chem 270:6440–6449
Pan W, Arnone M, Kendall M, Grafstrom RH, Seitz SP, Wasserman ZR, Albright CF (2003) Identification of peptide substrates for human MMP-11 (Stromelysin-3) using phage display. J Biol Chem 278:27820–27827
Deng S-J et al (2000) Substrate specificity of human collagenase 3 assessed using a phage-displayed peptide library. J Biol Chem 275:31422–31427. https://doi.org/10.1074/jbc.M004538200
Shuichi O, Kazutaka M, Yoshikazu S, Ken-ichi M, Konstanty W, Yuji Y (2001) Substrate phage as a tool to identify novel substrate sequences of proteases. Comb Chem High Throughput Screen 4:573–583. https://doi.org/10.2174/1386207013330788
Patterson J, Hubbell JA (2010) Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials 31:7836–7845. https://doi.org/10.1016/j.biomaterials.2010.06.061
Patterson J, Hubbell JA (2011) SPARC-derived protease substrates to enhance the plasmin sensitivity of molecularly engineered PEG hydrogels. Biomaterials 32:1301–1310. https://doi.org/10.1016/j.biomaterials.2010.10.016
Merrifield RB (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc 85:2149–2154. https://doi.org/10.1021/ja00897a025
Mäde V, Els-Heindl S, Beck-Sickinger AG (2014) Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J Org Chem 10:1197–1212. https://doi.org/10.3762/bjoc.10.118
Vanier GS (2013) Microwave-assisted solid-phase peptide synthesis based on the Fmoc protecting group strategy (CEM). In: Jensen KJ, Tofteng Shelton P, Pedersen SL (eds) Peptide synthesis and applications. Humana Press, Totowa, pp 235–249. https://doi.org/10.1007/978-1-62703-544-6_17
Ramesh S, de la Torre BG, Albericio F, Kruger HG, Govender T (2017) Microwave-assisted synthesis of antimicrobial peptides. In: Hansen PR (ed) Antimicrobial peptides: methods and protocols. Springer, New York, pp 51–59. https://doi.org/10.1007/978-1-4939-6737-7_4
Chen M, Heimer P, Imhof D (2015a) Synthetic strategies for polypeptides and proteins by chemical ligation. Amino Acids 47:1283–1299. https://doi.org/10.1007/s00726-015-1982-5
Clancy KW, Melvin JA, McCafferty DG (2010) Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Pept Sci 94:385–396. https://doi.org/10.1002/bip.21472
Schmohl L, Schwarzer D (2014) Chemo-enzymatic three-fragment assembly of semisynthetic proteins. J Pept Sci 20:145–151. https://doi.org/10.1002/psc.2600
Chang TK, Jackson DY, Burnier JP, Wells JA (1994) Subtiligase: a tool for semisynthesis of proteins. Proc Natl Acad Sci U S A 91:12544. https://doi.org/10.1073/pnas.91.26.12544
Tan X, Yang R, Liu C-F (2018) Facilitating Subtiligase-catalyzed peptide ligation reactions by using peptide thioester substrates. Org Lett 20:6691–6694. https://doi.org/10.1021/acs.orglett.8b02747
Boda SK et al (2019) Mineralized nanofiber segments coupled with calcium-binding BMP-2 peptides for alveolar bone regeneration. Acta Biomater 85:282–293. https://doi.org/10.1016/j.actbio.2018.12.051
Weng L, Boda SK, Wang H, Teusink MJ, Shuler FD, Xie J (2018) Novel 3D hybrid nanofiber aerogels coupled with BMP-2 peptides for cranial bone regeneration. Adv Healthc Mater 7:e1701415. https://doi.org/10.1002/adhm.201701415
Leight JL, Alge DL, Maier AJ, Anseth KS (2013) Direct measurement of matrix metalloproteinase activity in 3D cellular microenvironments using a fluorogenic peptide substrate. Biomaterials 34:7344–7352. https://doi.org/10.1016/j.biomaterials.2013.06.023
Sakiyama SE, Schense JC, Hubbell JA (1999) Incorporation of heparin-binding peptides into fibrin gels enhances neurite extension: an example of designer matrices in tissue engineering. FASEB J 13:2214–2224
Schense JC, Hubbell JA (1999) Cross-linking exogenous bifunctional peptides into fibrin gels with factor XIIIa. Bioconjug Chem 10:75–81. https://doi.org/10.1021/bc9800769
Ehrbar M, Rizzi SC, Schoenmakers RG, San Miguel B, Hubbell JA, Weber FE, Lutolf MP (2007) Biomolecular hydrogels formed and degraded via site-specific enzymatic reactions. Biomacromolecules 8:3000–3007. https://doi.org/10.1021/bm070228f
Zhu J, Tang C, Kottke-Marchant K, Marchant RE (2009) Design and synthesis of biomimetic hydrogel scaffolds with controlled organization of cyclic RGD peptides. Bioconjug Chem 20:333–339. https://doi.org/10.1021/bc800441v
Maia FR, Barbosa M, Gomes DB, Vale N, Gomes P, Granja PL, Barrias CC (2014) Hydrogel depots for local co-delivery of osteoinductive peptides and mesenchymal stem cells. J Control Release 189:158–168. https://doi.org/10.1016/j.jconrel.2014.06.030
Phelps EA et al (2012) Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv Mater 24:64–70. https://doi.org/10.1002/adma.201103574
Gentile P, Ferreira AM, Callaghan JT, Miller CA, Atkinson J, Freeman C, Hatton PV (2017) Multilayer nanoscale encapsulation of biofunctional peptides to enhance bone tissue regeneration in vivo. Adv Healthc Mater 6:1601182. https://doi.org/10.1002/adhm.201601182
Cao F-Y, Yin W-N, Fan J-X, Zhuo R-X, Zhang X-Z (2015) A novel function of BMHP1 and cBMHP1 peptides to induce the osteogenic differentiation of mesenchymal stem cells. Biomater Sci 3:345–351. https://doi.org/10.1039/C4BM00300D
Hou R et al (2018) Novel osteogenic growth peptide C-terminal pentapeptide grafted poly(d,l-lactic acid) improves the proliferation and differentiation of osteoblasts: the potential bone regenerative biomaterial. Int J Biol Macromol 119:874–881. https://doi.org/10.1016/j.ijbiomac.2018.08.010
Li S, Xu Y, Yu J, Becker ML (2017) Enhanced osteogenic activity of poly(ester urea) scaffolds using facile post-3D printing peptide functionalization strategies. Biomaterials 141:176–187. https://doi.org/10.1016/j.biomaterials.2017.06.038
Soultan AH, Verheyen T, Smet M, De Borggraeve WM, Patterson J (2018) Synthesis and peptide functionalization of hyperbranched poly(arylene oxindole) towards versatile biomaterials. Polym Chem 9:2775–2784. https://doi.org/10.1039/C8PY00139A
Bain JL, Bonvallet PP, Abou-Arraj RV, Schupbach P, Reddy MS, Bellis SL (2015) Enhancement of the regenerative potential of anorganic bovine bone graft utilizing a polyglutamate-modified BMP2 peptide with improved binding to calcium-containing materials. Tissue Eng Part A 21:2426–2436. https://doi.org/10.1089/ten.tea.2015.0160
Cao Q, He Z, Sun WQ, Fan G, Zhao J, Bao N, Ye T (2019) Improvement of calcium phosphate scaffold osteogenesis in vitro via combination of glutamate-modified BMP-2 peptides. Mater Sci Eng C Mater Biol Appl 96:412–418. https://doi.org/10.1016/j.msec.2018.11.048
Culpepper BK, Webb WM, Bonvallet PP, Bellis SL (2014) Tunable delivery of bioactive peptides from hydroxyapatite biomaterials and allograft bone using variable-length polyglutamate domains. J Biomed Mater Res A 120A:1008–1016. https://doi.org/10.1002/jbm.a.34766
Sawyer AA, Weeks DM, Kelpke SS, McCracken MS, Bellis SL (2005) The effect of the addition of a polyglutamate motif to RGD on peptide tethering to hydroxyapatite and the promotion of mesenchymal stem cell adhesion. Biomaterials 26:7046–7056. https://doi.org/10.1016/j.biomaterials.2005.05.006
Gilbert M, Giachelli CM, Stayton PS (2003) Biomimetic peptides that engage specific integrin-dependent signaling pathways and bind to calcium phosphate surfaces. J Biomed Mater Res A 67A:69–77. https://doi.org/10.1002/jbm.a.10053
Gao X et al (2015) Osteoinductive peptide-functionalized nanofibers with highly ordered structure as biomimetic scaffolds for bone tissue engineering. Int J Nanomedicine 10:7109–7128
Ko E, Yang K, Shin J, Cho S-W (2013) Polydopamine-assisted osteoinductive peptide immobilization of polymer scaffolds for enhanced bone regeneration by human adipose-derived stem cells. Biomacromolecules 14:3202–3213. https://doi.org/10.1021/bm4008343
Pan G et al (2016) Biomimetic design of mussel-derived bioactive peptides for dual-functionalization of titanium-based biomaterials. J Am Chem Soc 138:15078–15086. https://doi.org/10.1021/jacs.6b09770
Pan H, Zheng Q, Yang S, Guo X (2014) Effects of functionalization of PLGA-[Asp-PEG]n copolymer surfaces with Arg-Gly-Asp peptides, hydroxyapatite nanoparticles, and BMP-2-derived peptides on cell behavior in vitro. J Biomed Mater Res A 102:4526–4535. https://doi.org/10.1002/jbm.a.35129
Wang M et al (2015) In vitro culture and directed osteogenic differentiation of human pluripotent stem cells on peptides-decorated two-dimensional microenvironment. ACS Appl Mater Interfaces 7:4560–4572. https://doi.org/10.1021/acsami.5b00188
Lee H, Dellatore SM, Miller WM, Messersmith PB (2007a) Mussel-inspired surface chemistry for multifunctional coatings. Science 318:426. https://doi.org/10.1126/science.1147241
Geetha M, Singh AK, Asokamani R, Gogia AK (2009) Ti based biomaterials, the ultimate choice for orthopaedic implants—a review. Prog Mater Sci 54:397–425. https://doi.org/10.1016/j.pmatsci.2008.06.004
Tejero R, Anitua E, Orive G (2014) Toward the biomimetic implant surface: biopolymers on titanium-based implants for bone regeneration. Prog Polym Sci 39:1406–1447. https://doi.org/10.1016/j.progpolymsci.2014.01.001
Langer R, Vacanti JP (1993) Tissue engineering. Science 260:920. https://doi.org/10.1126/science.8493529
Moeinzadeh S, Jabbari E (2015) Morphogenic peptides in regeneration of load bearing tissues. In: Bertassoni LE, Coelho PG (eds) Engineering mineralized and load bearing tissues. Springer, Cham, pp 95–110. https://doi.org/10.1007/978-3-319-22345-2_6
Visser R, Rico-Llanos GA, Pulkkinen H, Becerra J (2016) Peptides for bone tissue engineering. J Control Release 244:122–135. https://doi.org/10.1016/j.jconrel.2016.10.024
Bab I et al (1992) Histone H4-related osteogenic growth peptide (OGP): a novel circulating stimulator of osteoblastic activity. EMBO J 11:1867–1873
Gabarin N et al (2001) Mitogenic Gi protein-MAP kinase signaling cascade in MC3T3-E1 osteogenic cells: activation by C-terminal pentapeptide of osteogenic growth peptide [OGP(10–14)] and attenuation of activation by cAMP. J Cell Biochem 81:594–603. https://doi.org/10.1002/jcb.1083
Suzuki Y, Tanihara M, Suzuki K, Saitou A, Sufan W, Nishimura Y (2000) Alginate hydrogel linked with synthetic oligopeptide derived from BMP-2 allows ectopic osteoinduction in vivo. J Biomed Mater Res 50:405–409. https://doi.org/10.1002/(SICI)1097-4636(20000605)50:3<405::AID-JBM15>3.0.CO;2-Z
Saito A, Suzuki Y, Ogata S-i, Ohtsuki C, Tanihara M (2003) Activation of osteo-progenitor cells by a novel synthetic peptide derived from the bone morphogenetic protein-2 knuckle epitope. Biochim Biophys Acta 1651:60–67. https://doi.org/10.1016/S1570-9639(03)00235-8
Seol Y-J et al (2006) Enhanced osteogenic promotion around dental implants with synthetic binding motif mimicking bone morphogenetic protein (BMP)-2. J Biomed Mater Res A 77A:599–607. https://doi.org/10.1002/jbm.a.30639
Zouani OF, Chollet C, Guillotin B, Durrieu M-C (2010) Differentiation of pre-osteoblast cells on poly(ethylene terephthalate) grafted with RGD and/or BMPs mimetic peptides. Biomaterials 31:8245–8253. https://doi.org/10.1016/j.biomaterials.2010.07.042
Kim HK et al (2012) Osteogenesis induced by a bone forming peptide from the prodomain region of BMP-7. Biomaterials 33:7057–7063. https://doi.org/10.1016/j.biomaterials.2012.06.036
Lee JS, Kim ME, Seon JK, Kang JY, Yoon TR, Park Y-D, Kim HK (2018) Bone-forming peptide-3 induces osteogenic differentiation of bone marrow stromal cells via regulation of the ERK1/2 and Smad1/5/8 pathways. Stem Cell Res 26:28–35. https://doi.org/10.1016/j.scr.2017.11.016
Choi YJ et al (2010) The identification of a heparin binding domain peptide from bone morphogenetic protein-4 and its role on osteogenesis. Biomaterials 31:7226–7238. https://doi.org/10.1016/j.biomaterials.2010.05.022
Bergeron E, Leblanc E, Drevelle O, Giguère R, Beauvais S, Grenier G, Faucheux N (2011) The evaluation of ectopic bone formation induced by delivery systems for bone morphogenetic protein-9 or its derived peptide. Tissue Eng Part A 18:342–352. https://doi.org/10.1089/ten.tea.2011.0008
Bergeron E, Senta H, Mailloux A, Park H, Lord E, Faucheux N (2009) Murine preosteoblast differentiation induced by a peptide derived from bone morphogenetic Proteins-9. Tissue Eng Part A 15:3341–3349. https://doi.org/10.1089/ten.tea.2009.0189
Jung RE, Cochran DL, Domken O, Seibl R, Jones AA, Buser D, Hammerle CHF (2007) The effect of matrix bound parathyroid hormone on bone regeneration. Clin Oral Implants Res 18:319–325. https://doi.org/10.1111/j.1600-0501.2007.01342.x
Park J-B et al (2007) Osteopromotion with synthetic oligopeptide–coated bovine bone mineral in vivo. J Periodontol 78:157–163. https://doi.org/10.1902/jop.2007.060200
Lee J-Y et al (2007b) Assembly of collagen-binding peptide with collagen as a bioactive scaffold for osteogenesis in vitro and in vivo. Biomaterials 28:4257–4267. https://doi.org/10.1016/j.biomaterials.2007.05.040
Gabet Y et al (2004) Osteogenic growth peptide modulates fracture callus structural and mechanical properties. Bone 35:65–73. https://doi.org/10.1016/j.bone.2004.03.025
Pigossi SC, Medeiros MC, Saska S, Cirelli JA, Scarel-Caminaga RM (2016) Role of osteogenic growth peptide (OGP) and OGP(10-14) in bone regeneration: a review. Int J Mol Sci 17:1885. https://doi.org/10.3390/ijms17111885
Policastro GM, Becker ML (2016) Osteogenic growth peptide and its use as a bio-conjugate in regenerative medicine applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol 8:449–464. https://doi.org/10.1002/wnan.1376
Moore NM, Lin NJ, Gallant ND, Becker ML (2010) The use of immobilized osteogenic growth peptide on gradient substrates synthesized via click chemistry to enhance MC3T3-E1 osteoblast proliferation. Biomaterials 31:1604–1611. https://doi.org/10.1016/j.biomaterials.2009.11.011
Horii A, Wang X, Gelain F, Zhang S (2007) Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration. PLoS One 2:e190. https://doi.org/10.1371/journal.pone.0000190
Wozney JM (1989) Bone morphogenetic proteins. Prog Growth Factor Res 1:267–280. https://doi.org/10.1016/0955-2235(89)90015-X
Saito A, Suzuki Y, Ogata S-I, Ohtsuki C, Tanihara M (2004) Prolonged ectopic calcification induced by BMP-2–derived synthetic peptide. J Biomed Mater Res A 70A:115–121. https://doi.org/10.1002/jbm.a.30071
Saito A, Suzuki Y, Ogata S-I, Ohtsuki C, Tanihara M (2005) Accelerated bone repair with the use of a synthetic BMP-2-derived peptide and bone-marrow stromal cells. J Biomed Mater Res A 72A:77–82. https://doi.org/10.1002/jbm.a.30208
Niu X, Feng Q, Wang M, Guo X, Zheng Q (2009) Porous nano-HA/collagen/PLLA scaffold containing chitosan microspheres for controlled delivery of synthetic peptide derived from BMP-2. J Control Release 134:111–117. https://doi.org/10.1016/j.jconrel.2008.11.020
Moore NM, Lin NJ, Gallant ND, Becker ML (2011) Synergistic enhancement of human bone marrow stromal cell proliferation and osteogenic differentiation on BMP-2-derived and RGD peptide concentration gradients. Acta Biomater 7:2091–2100. https://doi.org/10.1016/j.actbio.2011.01.019
Bergeron E, Marquis ME, Chrétien I, Faucheux N (2007) Differentiation of preosteoblasts using a delivery system with BMPs and bioactive glass microspheres. J Mater Sci Mater Med 18:255–263. https://doi.org/10.1007/s10856-006-0687-4
Beauvais S, Drevelle O, Lauzon M-A, Daviau A, Faucheux N (2016) Modulation of MAPK signalling by immobilized adhesive peptides: effect on stem cell response to BMP-9-derived peptides. Acta Biomater 31:241–251. https://doi.org/10.1016/j.actbio.2015.12.005
Neer RM et al (2001) Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441. https://doi.org/10.1056/NEJM200105103441904
Arrighi I, Mark S, Alvisi M, von Rechenberg B, Hubbell JA, Schense JC (2009) Bone healing induced by local delivery of an engineered parathyroid hormone prodrug. Biomaterials 30:1763–1771. https://doi.org/10.1016/j.biomaterials.2008.12.023
Takahata M, Schwarz EM, Chen T, O’Keefe RJ, Awad HA (2012) Delayed short-course treatment with teriparatide (PTH1–34) improves femoral allograft healing by enhancing intramembranous bone formation at the graft–host junction. J Bone Miner Res 27:26–37. https://doi.org/10.1002/jbmr.518
Chandra A et al (2014) PTH1–34 alleviates radiotherapy-induced local bone loss by improving osteoblast and osteocyte survival. Bone 67:33–40. https://doi.org/10.1016/j.bone.2014.06.030
Dent-Acosta RE, Storm N, Steiner RS, San Martin J (2012) The tactics of modern-day regulatory trials. JBJS 94:39–44. https://doi.org/10.2106/jbjs.l.00194
Pountos I, Panteli M, Lampropoulos A, Jones E, Calori GM, Giannoudis PV (2016) The role of peptides in bone healing and regeneration: a systematic review. BMC Med 14:103. https://doi.org/10.1186/s12916-016-0646-y
Ryaby JT, Sheller MR, Levine BP, Bramlet DG, Ladd AL, Carney DH (2006) Thrombin peptide TP508 stimulates cellular events leading to angiogenesis, revascularization, and repair of dermal and musculoskeletal tissues. JBJS 88:132–139. https://doi.org/10.2106/jbjs.f.00892
Aspenberg P et al (2010) Teriparatide for acceleration of fracture repair in humans: a prospective, randomized, double-blind study of 102 postmenopausal women with distal radial fractures. J Bone Miner Res 25:404–414. https://doi.org/10.1359/jbmr.090731
Aspenberg P, Johansson T (2010) Teriparatide improves early callus formation in distal radial fractures. Acta Orthop 81:234–236. https://doi.org/10.3109/17453671003761946
Chintamaneni S, Finzel K, Gruber BL (2010) Successful treatment of sternal fracture nonunion with teriparatide. Osteoporos Int 21:1059–1063. https://doi.org/10.1007/s00198-009-1061-4
Yu C-T, Chang C-C, Chen C-L, Wei JC-C, Wu J-K (2008) Early callus formation in human hip fracture treated with internal fixation and teriparatide. J Rheumatol 35:2082–2083
Arnold PM et al (2016) Efficacy of i-factor bone graft versus autograft in anterior cervical discectomy and fusion: results of the prospective, randomized, single-blinded Food and Drug Administration investigational device exemption study. Spine 41:1075–1083. https://doi.org/10.1097/brs.0000000000001466
Gomar F, Orozco R, Villar JL, Arrizabalaga F (2007) P-15 small peptide bone graft substitute in the treatment of non-unions and delayed union. A pilot clinical trial. Int Orthop 31:93–99. https://doi.org/10.1007/s00264-006-0087-x
Yukna RA, Krauser JT, Callan DP, Evans GH, Cruz R, Martin M (2000) Multi-center clinical comparison of combination anorganic bovine-derived hydroxyapatite matrix (ABM)/cell binding peptide (P-15) and ABM in human periodontal osseous defects. 6-month results. J Periodontol 71:1671–1679. https://doi.org/10.1902/jop.2000.71.11.1671
Bjarnsholt T (2013) The role of bacterial biofilms in chronic infections. APMIS 121:1–58. https://doi.org/10.1111/apm.12099
Darouiche RO (2004) Treatment of infections associated with surgical implants. N Engl J Med 350:1422–1429. https://doi.org/10.1056/NEJMra035415
Sutherland IW (2001) The biofilm matrix—an immobilized but dynamic microbial environment. Trends Microbiol 9:222–227. https://doi.org/10.1016/S0966-842X(01)02012-1
Davies D (2003) Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov 2:114. https://doi.org/10.1038/nrd1008
Jefferson KK (2004) What drives bacteria to produce a biofilm? FEMS Microbiol Lett 236:163–173. https://doi.org/10.1111/j.1574-6968.2004.tb09643.x
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318. https://doi.org/10.1126/science.284.5418.1318
Karygianni L, Al-Ahmad A, Argyropoulou A, Hellwig E, Anderson AC, Skaltsounis AL (2016) Natural antimicrobials and oral microorganisms: a systematic review on herbal interventions for the eradication of multispecies oral biofilms. Front Microbiol 6:1529. https://doi.org/10.3389/fmicb.2015.01529
Andrea A, Molchanova N, Jenssen H (2018) Antibiofilm peptides and peptidomimetics with focus on surface immobilization. Biomol Ther 8:27. https://doi.org/10.3390/biom8020027
Batoni G, Maisetta G, Esin S (2016) Antimicrobial peptides and their interaction with biofilms of medically relevant bacteria. Biochim Biophys Acta 1858:1044–1060. https://doi.org/10.1016/j.bbamem.2015.10.013
de la Fuente-Núñez C, Cardoso MH, de Souza Cândido E, Franco OL, Hancock REW (2016) Synthetic antibiofilm peptides. Biochim Biophys Acta 1858:1061–1069. https://doi.org/10.1016/j.bbamem.2015.12.015
Dostert M, Belanger CR, Hancock REW (2018) Design and assessment of anti-biofilm peptides: steps toward clinical application. J Innate Immun 11:193. https://doi.org/10.1159/000491497
Pletzer D, Coleman SR, Hancock REW (2016) Anti-biofilm peptides as a new weapon in antimicrobial warfare. Curr Opin Microbiol 33:35–40. https://doi.org/10.1016/j.mib.2016.05.016
Pletzer D, Hancock REW (2016) Antibiofilm peptides: potential as broad-Spectrum agents. J Bacteriol 198:2572. https://doi.org/10.1128/JB.00017-16
Strempel N, Strehmel J, Overhage J (2015) Potential application of antimicrobial peptides in the treatment of bacterial biofilm infections. Curr Pharm Des 21:67–84. https://doi.org/10.2174/1381612820666140905124312
Haney EF, Brito-Sánchez Y, Trimble MJ, Mansour SC, Cherkasov A, Hancock REW (2018) Computer-aided discovery of peptides that specifically attack bacterial biofilms. Sci Rep 8:1871. https://doi.org/10.1038/s41598-018-19669-4
De Brucker K et al (2014) Derivatives of the mouse cathelicidin-related antimicrobial peptide (CRAMP) inhibit fungal and bacterial biofilm formation. Antimicrob Agents Chemother 58:5395. https://doi.org/10.1128/AAC.03045-14
De Zoysa GH, Cameron AJ, Hegde VV, Raghothama S, Sarojini V (2015) Antimicrobial peptides with potential for biofilm eradication: synthesis and structure activity relationship studies of battacin peptides. J Med Chem 58:625–639. https://doi.org/10.1021/jm501084q
Almaaytah A, Tarazi S, Al-Fandi M, Abuilhaija A, Al-shar’i N, Al-Balas Q, Abu-Awad A (2015) The design and functional characterization of the antimicrobial and antibiofilm activities of BMAP27-melittin, a rationally designed hybrid peptide. Int J Pept Res Ther 21:165–177. https://doi.org/10.1007/s10989-014-9444-6
Orlando F et al (2008) BMAP-28 improves the efficacy of vancomycin in rat models of gram-positive cocci ureteral stent infection. Peptides 29:1118–1123. https://doi.org/10.1016/j.peptides.2008.03.005
Mataraci E, Dosler S (2012) In vitro activities of antibiotics and antimicrobial cationic peptides alone and in combination against methicillin-resistant Staphylococcus aureus biofilms. Antimicrob Agents Chemother 56:6366. https://doi.org/10.1128/AAC.01180-12
Bionda N et al (2016) Identification of novel cyclic lipopeptides from a positional scanning combinatorial library with enhanced antibacterial and antibiofilm activities. Eur J Med Chem 108:354–363. https://doi.org/10.1016/j.ejmech.2015.11.032
de la Fuente-Núñez C et al (2015) d-Enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol 22:1280–1282. https://doi.org/10.1016/j.chembiol.2015.09.004
Pletzer D, Wolfmeier H, Bains M, Hancock REW (2017) Synthetic peptides to target stringent response-controlled virulence in a Pseudomonas aeruginosa murine cutaneous infection model. Front Microbiol 8:1867–1867. https://doi.org/10.3389/fmicb.2017.01867
Mohamed MF, Brezden A, Mohammad H, Chmielewski J, Seleem MN (2017) A short D-enantiomeric antimicrobial peptide with potent immunomodulatory and antibiofilm activity against multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Sci Rep 7:6953. https://doi.org/10.1038/s41598-017-07440-0
Brancatisano FL et al (2014) Inhibitory effect of the human liver-derived antimicrobial peptide hepcidin 20 on biofilms of polysaccharide intercellular adhesin (PIA)-positive and PIA-negative strains of Staphylococcus epidermidis. Biofouling 30:435–446. https://doi.org/10.1080/08927014.2014.888062
Mansour SC, de la Fuente-Núñez C, Hancock REW (2015) Peptide IDR-1018: modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J Pept Sci 21:323–329. https://doi.org/10.1002/psc.2708
Anunthawan T, de la Fuente-Núñez C, Hancock REW, Klaynongsruang S (2015) Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim Biophys Acta 1848:1352–1358. https://doi.org/10.1016/j.bbamem.2015.02.021
Overhage J, Campisano A, Bains M, Torfs ECW, Rehm BHA, Hancock REW (2008) Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun 76:4176–4182. https://doi.org/10.1128/IAI.00318-08
Haisma EM et al (2014) LL-37-derived peptides eradicate multidrug-resistant Staphylococcus aureus from thermally wounded human skin equivalents. Antimicrob Agents Chemother 58:4411. https://doi.org/10.1128/AAC.02554-14
de Breij A et al (2018) The antimicrobial peptide SAAP-148 combats drug-resistant bacteria and biofilms. Sci Transl Med 10:eaan4044. https://doi.org/10.1126/scitranslmed.aan4044
Minardi D et al (2007) The antimicrobial peptide Tachyplesin III coated alone and in combination with intraperitoneal piperacillin-tazobactam prevents ureteral stent Pseudomonas infection in a rat subcutaneous pouch model. Peptides 28:2293–2298. https://doi.org/10.1016/j.peptides.2007.10.001
Almaaytah A, Qaoud MT, Khalil Mohammed G, Abualhaijaa A, Knappe D, Hoffmann R, Al-Balas Q (2018) Antimicrobial and antibiofilm activity of UP-5, an ultrashort antimicrobial peptide designed using only arginine and biphenylalanine. Pharmaceuticals (Basel, Switzerland) 11(1). https://doi.org/10.3390/ph11010003
Ma Z et al (2017) Membrane-active amphipathic peptide WRL3 with in vitro antibiofilm capability and in vivo efficacy in treating methicillin-resistant Staphylococcus aureus burn wound infections. ACS Infect Dis 3:820–832. https://doi.org/10.1021/acsinfecdis.7b00100
Haney EF, Mansour SC, Hancock REW (2017) Antimicrobial peptides: an introduction. In: Hansen PR (ed) Antimicrobial peptides: methods and protocols. Springer, New York, pp 3–22. https://doi.org/10.1007/978-1-4939-6737-7_1
Hancock REW, Sahl H-G (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24:1551–1557. https://doi.org/10.1038/nbt1267
Haney EF, Straus SK, Hancock REW (2019) Reassessing the host defense peptide landscape. Front Chem 7:43. https://doi.org/10.3389/fchem.2019.00043
Hilpert K, Volkmer-Engert R, Walter T, Hancock REW (2005) High-throughput generation of small antibacterial peptides with improved activity. Nat Biotechnol 23:1008. https://doi.org/10.1038/nbt1113
Butts A, Krysan DJ (2012) Antifungal drug discovery: something old and something new. PLoS Pathog 8:e1002870
Ganz T, Nemeth E (2012) Hepcidin and iron homeostasis. Biochim Biophys Acta 1823:1434–1443. https://doi.org/10.1016/j.bbamcr.2012.01.014
Maisetta G, Petruzzelli R, Brancatisano FL, Esin S, Vitali A, Campa M, Batoni G (2010) Antimicrobial activity of human hepcidin 20 and 25 against clinically relevant bacterial strains: effect of copper and acidic pH. Peptides 31:1995–2002. https://doi.org/10.1016/j.peptides.2010.08.007
Tavanti A, Maisetta G, Del Gaudio G, Petruzzelli R, Sanguinetti M, Batoni G, Senesi S (2011) Fungicidal activity of the human peptide hepcidin 20 alone or in combination with other antifungals against Candida glabrata isolates. Peptides 32:2484–2487. https://doi.org/10.1016/j.peptides.2011.10.012
Wieczorek M et al (2010) Structural studies of a peptide with immune modulating and direct antimicrobial activity. Chem Biol 17:970–980. https://doi.org/10.1016/j.chembiol.2010.07.007
de la Fuente-Núñez C, Reffuveille F, Haney EF, Straus SK, Hancock REW (2014) Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog 10:e1004152. https://doi.org/10.1371/journal.ppat.1004152
Hamamoto K, Kida Y, Zhang Y, Shimizu T, Kuwano K (2002) Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol Immunol 46:741–749. https://doi.org/10.1111/j.1348-0421.2002.tb02759.x
Chung EMC, Dean SN, Propst CN, Bishop BM, van Hoek ML (2017) Komodo dragon-inspired synthetic peptide DRGN-1 promotes wound-healing of a mixed-biofilm infected wound NPJ biofilms and microbiomes. NPJ Biofilms Microbiomes 3:9. https://doi.org/10.1038/s41522-017-0017-2
Onaizi SA, Leong SSJ (2011) Tethering antimicrobial peptides: current status and potential challenges. Biotechnol Adv 29:67–74. https://doi.org/10.1016/j.biotechadv.2010.08.012
Forbes S, McBain AJ, Felton-Smith S, Jowitt TA, Birchenough HL, Dobson CB (2013) Comparative surface antimicrobial properties of synthetic biocides and novel human apolipoprotein E derived antimicrobial peptides. Biomaterials 34:5453–5464. https://doi.org/10.1016/j.biomaterials.2013.03.087
Etienne O et al (2004) Multilayer polyelectrolyte films functionalized by insertion of Defensin: a new approach to protection of implants from bacterial colonization. Antimicrob Agents Chemother 48:3662. https://doi.org/10.1128/AAC.48.10.3662-3669.2004
Shukla A, Fleming KE, Chuang HF, Chau TM, Loose CR, Stephanopoulos GN, Hammond PT (2010) Controlling the release of peptide antimicrobial agents from surfaces. Biomaterials 31:2348–2357. https://doi.org/10.1016/j.biomaterials.2009.11.082
Gao G, Cheng John TJ, Kindrachuk J, Hancock Robert EW, Straus Suzana K, Kizhakkedathu Jayachandran N (2012) Biomembrane interactions reveal the mechanism of action of surface-immobilized host defense IDR-1010 peptide. Chem Biol 19:199–209. https://doi.org/10.1016/j.chembiol.2011.12.015
Gao G et al (2011) The biocompatibility and biofilm resistance of implant coatings based on hydrophilic polymer brushes conjugated with antimicrobial peptides. Biomaterials 32:3899–3909. https://doi.org/10.1016/j.biomaterials.2011.02.013
Lim K et al (2013) Immobilization studies of an engineered arginine–tryptophan-rich peptide on a silicone surface with antimicrobial and antibiofilm activity. ACS Appl Mater Interfaces 5:6412–6422. https://doi.org/10.1021/am401629p
Kazemzadeh-Narbat M, Kindrachuk J, Duan K, Jenssen H, Hancock REW, Wang R (2010) Antimicrobial peptides on calcium phosphate-coated titanium for the prevention of implant-associated infections. Biomaterials 31:9519–9526. https://doi.org/10.1016/j.biomaterials.2010.08.035
Kazemzadeh-Narbat M, Noordin S, Masri BA, Garbuz DS, Duncan CP, Hancock REW, Wang R (2012) Drug release and bone growth studies of antimicrobial peptide-loaded calcium phosphate coating on titanium. J Biomed Mater Res B Appl Biomater 100B:1344–1352. https://doi.org/10.1002/jbm.b.32701
Kazemzadeh-Narbat M, Lai BFL, Ding C, Kizhakkedathu JN, Hancock REW, Wang R (2013) Multilayered coating on titanium for controlled release of antimicrobial peptides for the prevention of implant-associated infections. Biomaterials 34:5969–5977. https://doi.org/10.1016/j.biomaterials.2013.04.036
Fox JL (2013) Antimicrobial peptides stage a comeback. Nat Biotechnol 31:379. https://doi.org/10.1038/nbt.2572
Mahlapuu M, Håkansson J, Ringstad L, Björn C (2016) Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol 6:194–194. https://doi.org/10.3389/fcimb.2016.00194
Grönberg A, Mahlapuu M, Ståhle M, Whately-Smith C, Rollman O (2014) Treatment with LL-37 is safe and effective in enhancing healing of hard-to-heal venous leg ulcers: a randomized, placebo-controlled clinical trial. Wound Repair Regen 22:613–621. https://doi.org/10.1111/wrr.12211
Falagas ME, Kasiakou SK, Saravolatz LD (2005) Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis 40:1333–1341. https://doi.org/10.1086/429323
Landman D, Georgescu C, Martin DA, Quale J (2008) Polymyxins revisited. Clin Microbiol Rev 21:449. https://doi.org/10.1128/CMR.00006-08
Zavascki AP, Goldani LZ, Li J, Nation RL (2007) Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother 60:1206–1215. https://doi.org/10.1093/jac/dkm357
Rotem S, Mor A (2009) Antimicrobial peptide mimics for improved therapeutic properties. Biochim Biophys Acta 1788:1582–1592. https://doi.org/10.1016/j.bbamem.2008.10.020
Piluso S, Soultan AH, Patterson J (2017) Molecularly engineered polymer-based Systems in drug delivery and regenerative medicine. Curr Pharm Des 23:281–294. https://doi.org/10.2174/1381612822666161021104239
Moreira Teixeira LS, Patterson J, Luyten FP (2014) Skeletal tissue regeneration: where can hydrogels play a role? Int Orthop 38:1861–1876. https://doi.org/10.1007/s00264-014-2402-2
Van den Broeck L, Piluso S, Soultan AH, De Volder M, Patterson J (2019) Cytocompatible carbon nanotube reinforced polyethylene glycol composite hydrogels for tissue engineering. Mater Sci Eng C Mater Biol Appl 98:1133–1144. https://doi.org/10.1016/j.msec.2019.01.020
Romanò CL, Toscano M, Romanò D, Drago L (2013) Antibiofilm agents and implant-related infections in orthopaedics: where are we? J Chemother 25:67–80. https://doi.org/10.1179/1973947812Y.0000000045
Lu H, Liu Y, Guo J, Wu H, Wang J, Wu G (2016) Biomaterials with antibacterial and osteoinductive properties to repair infected bone defects. Int J Mol Sci 17:334–334. https://doi.org/10.3390/ijms17030334
Acknowledgments
This work in the research group of the author has been partially supported by the Research Foundation Flanders (FWO), grant number G.0B39.14, and the special research fund of the KU Leuven, grant numbers CREA/13/017 and IDO/13/016. The author also gratefully acknowledges the interesting discussions about peptide-functionalized biomaterials and protein engineering over the years with the members of her research group, particularly Dr. Al Halifa Soultan, Dr. Susanna Piluso, Dr. Abhijith Kudva, Burak Toprakhisar, and Christian Garcia Abrego, as well as her former mentors Prof. Jeffrey Hubbell, Prof. Patrick Stayton, and Prof. Michael Hecht.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Patterson, J. (2020). Peptide-functionalized Biomaterials with Osteoinductive or Anti-biofilm Activity. In: Li, B., Moriarty, T., Webster, T., Xing, M. (eds) Racing for the Surface. Springer, Cham. https://doi.org/10.1007/978-3-030-34471-9_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-34471-9_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34470-2
Online ISBN: 978-3-030-34471-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)