
Abstract
The interplay between innate and adaptive immunity strongly influences the pathobiology of neurodegenerative, neuroinflammatory, and neuroinfectious diseases. Specific and sustained immune responses can induce disease by affecting neuronal injury and death. Disease progression parallels glial proliferation, proinflammatory cytokine production and adaptive immune responses against the inciting misfolded protein or infectious agent. All affect neuronal demise. Neuroprotective immune transformation remains a therapeutic avenue being developed by several research groups towards the shared goal of sustaining a nourishing brain microenvironment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Human immunodeficiency virus
- Alzheimer’s disease
- Adaptive immunity
- Parkinson’s disease
- Innate immunity
- Neuroprotection
- Neurodegeneration
1 Introduction
Multifaceted disease mechanisms characterize the pathobiology of neurodegenerative and neuroinfectious disorders. One common pathway affecting neuronal vitality in all diseases states is disordered innate and adaptive immunity [1]. Innate microglial and astrocyte responses are considered early signs of disease as is antigen-driven T cell proliferative responses. Such immune responses affect multiple disease components including neuronal loss, peripheral blood cell extravasation across the blood brain barrier (BBB) and lymphocyte surveillance of pathogenic proteins or microbes [2, 3]. During disease, both innate and T cell responses be come operative and are considered to be detrimental for a spectrum of diseases. These include, but are not limited to, Alzheimer’s and Parkinson’s diseases (AD and PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), and infectious diseases including human immunodeficiency virus type one (HIV-1) and its associated neurodegeneration [4, 5]. Furthermore, immune-incited neurodegeneration can affect both disease onset and progression [6, 7]. Indeed, mounting evidence shows that the interplay between the peripheral immune system and resident central nervous system (CNS) immune cells amplifies neuroinflammatory responses and exacerbates neurodegeneration [8]. This chapter examines the role of immunity in neurodegenerative and neuroinfectious disorders. Particular focus rests in the interactions between the innate and adaptive immune responses that affect neurodegenerative and neuroprotective responses.
2 Immune Interplay for Neurodegenerative Diseases
Neurodestructive immune responses can be harnessed or even transformed to control disease onset and progression [9]. Our laboratories and others have investigated the role of immunity in affecting the onset and progression of Alzheimer’s and Parkinson’s disease (AD and PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Huntington’s disease, stroke, traumatic brain and spinal cord injuries, and drug-related nervous system damage [10,11,12,13,14,15,16]. With this in mind, the pathobiology of neurodegeneration is required [17]. Neurodegeneration is a pathological condition in which the nervous system loses structure or function characterized by synaptic loss and neuronal death. Clinically, this leads to progressive cognitive decline and motor dysfunction [18]. While the precise cause(s) have not yet been fully elucidated for each disease, there is no cure, and disease progression is unavoidable. While neurodegenerative diseases affect the nervous system differently [19] common disease mechanisms do exist. First, all are associated with the death of specific neuronal cell subpopulations, resulting in the degeneration of specific brain regions often leading to disease-specific manifestations [18]. Second, neuronal loss is linked to the formation and spread of protein aggregates. These occur during advanced age but can also be present sporadically or due to defined genetic mutations [20]. Each neurodegenerative disease is further classified based on the kind and type of protein deposition seen in brain sub regions. Third, neurodegenerative disorders are linked, in measure, to immune responses that trigger overt neuroinflammatory responses that can affect disease [21]. For most neurodegenerative disorders, the pathways of neuronal demise are similar. Common mechanisms include oxidative stress, mitochondrial damage, excitotoxicity, and misfolded or post-translationally modified protein aggregation [20, 22,23,24]. To counteract these events, therapies have been developed to elicit neuroprotective responses with the intent to preserve already damaged neuronal and synaptic structure [25]. Such treatments serve to attenuate inflammation, oxidative stress, and excitotoxicity [9, 26, 27].
2.1 Innate and Adaptive Immunity and Neurodegeneration
Both innate and adaptive immune responses are important for mounting the body’s defense against a pathogen or foreign microorganism [28]. The innate response is the first line of defense. It is rapid, does not require immune memory, and is characterized by phagocytic activity of macrophages, dendritic cells, or microglia. While serving as a first line of defense against microbial infections and injuries, it also perpetuates tissue and wound healing and repair [28]. Within the brain, microglia are the resident innate immune cell with similar functions to macrophages [29]. Apart from cell ontogeny, both brain macrophages and microglia maintain CNS homeostasis. Morphologically, microglia have long, branched processes that are constantly surveying the environment for homeostatic changes [30]. They are in contact with neurons, astrocytes, endothelial cells, and other surrounding microglia. When a change in the CNS microenvironment occurs, microglia become amoeboid and rounded in appearance [31, 32]. This morphological change reflects a reaction to injury or infection with increased phagocytic capacity and production of proinflammatory cytokines. As a result of aging and/or neurodegeneration, microglia become functionally impaired leading to an overactive neuroinflammatory response that further contributes to neural injuries [17]. In the aged brain, there is evidence for increased number of reactive microglia and increased proinflammatory microglial function [33]. Likewise, evaluation of cerebral spinal fluid (CSF), serum, and brains of individuals suffering from neurodegeneration also indicate increased levels of tumor necrosis factor alpha (TNF-α), IL-1β, and IL-6 [29, 34]. These secretory products are from resident microglia themselves [35] and display a link between disease progression and microglial immunity.
The adaptive immune response is specific [36]. To mount an immune response, the innate arm of the immune system must be activated [37]. Antigen is taken up by antigen presenting mononuclear phagocytes (MP) such as macrophages, dendritic cells or microglia, processed, and then presented to cells of the adaptive immune system generating an effective, robust, and specific immune response. Because of this, antigen presenting cells (APCs) are the bridge between the innate and adaptive immune system [38]. They directly activate T cells during antigen presentation, causing them to proliferate and migrate to areas of injury or infection [39]. Specifically, APCs activate T cells through presentation of antigen in conjunction with major histocompatibility complex (MHC) molecules and interaction with T cell receptors (TCRs) and co-stimulatory molecules such as CD80, CD86, CD70, CD40, and CD200 [8]. Because of the ability to recognize specific antigens, T cells comprise the cell population that is responsible for unique immune specificity. Once activated, T cells undergo clonal expansion to increase their cell number and potential to eliminate pathogens [8, 39]. Such activation causes T cell differentiation, expansion, and proliferation with associated cytokine production within a surrounding environment. Likewise, APCs themselves deliver many cytokine signals including IL-12, IL-4, IL-6, and transforming growth factor beta (TGF-β) to polarize naïve T cells into activated T cells with specific effector functions [40].
There are major T cell subsets that can be generated from both lymphoid tissues such as thymus, spleen, and lymph node, or in the periphery [40]. Upon activation by innate immunity, CD4+ T cells differentiate into different subsets such as T helper 1 (Th1), Th2, Th17, and regulatory T cells (Tregs) [41]. Classically, Th1 and Th17 cells mount active immune responses through the secretion of proinflammatory cytokines and mediators, including interferon gamma (IFN-γ) and IL-17A [42, 43]. On the other hand, Th2 and Tregs are responsible for anti-inflammatory responses [44]. Specifically, Tregs maintain suppression of an immune response [45]. Tregs mediate this function by diminishing antigen presentation and secreting anti-inflammatory cytokines including IL-10, IL-35, and TGF-β. These cause suppression of activated MP and T effector cells (Teffs) [46]. Each of these T cell subsets play crucial yet independent roles in mounting a robust and effective adaptive immune response. Following activation, T cells are recruited to sites of disease and promote inflammation [47]. To enter sites of disease, cells undergo extravasation. This process allows circulating lymphocytes to migrate across cell barriers such as the BBB to gain entry to sites of inflammation [48]. Once inside the brain, cell-mediated immune responses can affect neurodegeneration. The cross-talk between T cells and glia mediate effector functions by either cell-cell contact or cytokine-mediated mechanisms, including direct cytotoxicity by proinflammatory cytokines, activation of microglia or diminished suppressive function of Tregs [49].
This interplay between the innate and adaptive immune arms is essential for the development of neuroinflammation as it affects neurodegeneration or neuroprotection. Findings from multiple neurological disorders have provided insight into common disease outcomes [50]. Although neuroinflammation and T cell interactions play a prominent role in disease progression or protection against disease, it should be noted that the type of immune response are commonly specific [8].
2.2 Immunity in Alzheimer’s Diseae (AD)
Recent research findings in studies of human and animal models of neurodegenerative disorders have shown direct involvement of T cells in disease initiation and progression [51]. An example of such immune-linked disease effects is linked to the pathobiology of AD. AD is notable as it is the most common neurodegenerative disorder affecting anywhere from 10–30% of individuals over 65 years of age [52]. Cognitive loss is associated with impairment in short term memory that eventually leads to profound cognitive and memory deficits. Pathologically, the disease is characterized by loss of neurons in the hippocampus and cortical regions. The key neuropathological features include senile plaques containing beta-amyloid (Aβ) protein and the formation of neurofibrillary tangles (NFT) containing tau protein [53]. Aβ is processed by the sequential cleavage of amyloid precursor protein (APP) into smaller peptides [54]. The majority of the processed peptides consist of either Aβ40 or Aβ42 forms. These peptide forms can cluster into monomers, oligomers, protofibrils, or fibrils resulting in the formation of protein aggregates [55, 56]. Normally, extracellular Aβ peptides are removed from the brain and drained into the CSF, where they are degraded by microglia within the parenchyma [55, 57]. However, in a diseased state degradation is impaired. Tau is a microtubule-associated protein that can be phosphorylated at multiple serine, tyrosine, or threonine residues [58]. The mechanism of tau aggregation is thought to be mediated through abnormal phosphorylation leading to atypical conformations that can aggregate together [59]. Therefore, the loss of functional peptide clearance is proposed as a disease inciting event [60].
Post-mortem evaluation of AD brains reveals a relationship between neuron loss and memory [61]. This finding is associated with brain inflammation characterized by microgliosis, astrocyte activation, edema, and infiltration of MP across the BBB [62]. Activated microglia are shown to integrate deep into senile plaques, along with the detection of increased levels of proinflammatory cytokines [63, 64]. The associated glial activation and neurotoxicity is due to the formation of reactive nitrogen and oxygen species, increased proinflammatory cytokine production, and changes in excitatory amino acids in a diseased microenvironment [65]. The enhanced proinflammatory state decreases phagocytosis of Aβ plaques and inhibits intracellular Aβ degradation [66]. The resulting Aβ aggregates preferentially activate surrounding microglia launching signaling cascades needed to initiate clearance [55]. Resident microglia mediate such Aβ clearance, displaying the ability to phagocytize and ingest Aβ through a range of surface receptors. These pattern recognition receptors include CD14, TLRs, and CD47 [67,68,69]. Immune stimulation with Aβ enhances microglial phagocytosis. Microglia internalize Aβ through interactions with Aβ-scavenging receptors such as SR-A, CD36, and RAGE [70, 71]. However, even with this uptake, studies show that phagocytized Aβ can remain within the activated microglia for up to one month [72]. Aβ protein accumulation results from the failure of microglia to successfully remove the aggregated protein [73].
Post-mortem assessment of AD brains shows microglia surrounding Aβ plaques [74, 75]. These microglia were determined to be functionally impaired, lacking the capacity to properly uptake Aβ. Furthermore, Aβ can induce inflammatory responses involving inflammasome activation, resulting in increased proinflammatory cytokine production, including IL-1β and IL-18 [76]. These cytokines, along with IL-12, TGF-β, TNF-α, and IL-6, have been implicated in the progression of AD [77]. Increased IL-1β in serum is linked to cognitive impairment, and IL-12 is important for regulating the innate and adaptive immune response [78, 79]. Likewise, increased TGF-β levels have been noted in senile plaques, as well as in the CSF of individuals with AD [80, 81]. Presence of TGF-β is also associated with NFT formation [82]. Similarly, there is evidence showing that IL-1β and IL-6 can lead to hyperphosphorylation of tau, further contributing to tangle formation [83]. Apart from microglial cytokine production, there are also increases in reactive nitrogen and oxygen species, leading to direct neuron cytotoxicity [65]. Therefore, to assess the neuroinflammatory condition within the living AD patient, positron emission tomography (PET) scans have been utilized [77, 84, 85]. Scans indicate that, when compared to age-matched controls, there are increased numbers of activated microglia near primary disease areas [77]. Similarly, microglia that were collected post-mortem were biased toward a proinflammatory phenotype following immunological challenge.
In the brain, Aβ also interacts with resident astrocytes. Astrocytes uptake and remove Aβ in a CCL2-dependent manner [86]. This primary innate immune response is mediated through a variety of inflammatory factors including proinflammatory cytokines, proinflammatory chemokines, acute phase proteins, and complement factors [87]. Upregulating these systems results in enhanced cytokine production, including increases in IFN-γ, IL-1β, IL-6, TNF-α, CD40L, and macrophage inflammatory protein 1-alpha (MIP-1α). In response to the enhanced neuroinflammatory state, increased APP production occurs in surrounding neurons, causing overall Aβ production to be upregulated [88]. Resulting Aβ deposits can form, which may be the cause of plaque formation [89]. It has also been shown that autoantibodies bound to neurons can induce Aβ internalization and deposition, leading to further neuronal damage [90,91,92].
Under normal physiological conditions, few T lymphocytes cross the BBB and survey the brain [3]. In AD patients, there is an increase in the number of T lymphocytes within the hippocampus and cortex [88, 91]. This infiltration arises due to chemoattractants originating from activated microglia and astrocytes within injured brain sub regions. The ensuing immune cross-talk can influence immune cell populations and their mediators in the periphery. Therefore, peripheral changes in the function of immune populations may have an effect on the CNS microenvironment. Notably, there are a variety of changes in lymphocyte distribution, signature and specific cytokine levels and signatures within whole blood and plasma of AD patients [93,94,95]. However, the exact peripheral immune dysregulation observed varies. For instance, peripheral blood mononuclear cells (PBMCs) from AD patients produce increased levels of IL-1β, when compared to controls [96, 97]. Other studies, however, show decreased amounts of naïve T cells, increased memory T cells, increased CD4+ T cells, reduced CD4+CD25+ Treg populations, and decreased total B cell populations [93, 98]. Other studies indicated a significant reduction in CD3+ T cells, but CD4+ and CD8+ levels remained unchanged [99]. A fourth evaluation confirmed the decrease in CD3+ populations, but also observed a decrease in CD8+ populations and a modest increase in CD4+ T cells [100]. Along with decreased Treg numbers, one investigative group noted a decrease in CD8+ suppressor cells and a decrease in IL-10, suggesting that the immunosuppressive capacity is diminished during AD [101]. This immune dysfunction decreases the ability to control detrimental Teff responses. Such Teff responses are characterized by increased activities of Th17 and Th9 subsets in AD [102]. Saresella and colleagues observed increased levels of proinflammatory cytokines associated with Th17 and Th9 subsets, including IL-21, IL-6, and IL-23, and the Th17-associated transcription factor, RORγ, in lymphocytes isolated from AD patients. Similarly, PBMCs recovered from AD patients, and consequently activated, exhibit significantly increased production of IL-1β, IL-6, TNF-α, and IFN-γ [102]. Even though a consensus has not been reached, these immune profiling studies do indicate significant aberrations in adaptive immune populations associated with disease that may result in decreased ability to regulate immune responses. Taken together, this data may suggest a profound skewing of systemic immune populations affecting the brain microenvironment.
2.3 Immunity in Parkinson’s Disease (PD)
PD is the second most common form of neurodegeneration, yet it is the most common movement disorder [103]. It is characterized by the formation of proteinaceous inclusions termed Lewy bodies. Lewy bodies contain modified and misfolded forms of alpha-synuclein (α-syn) along with ubiquitin [104]. The main clinical features include resting tremor, postural instability, rigidity, and bradykinesia [104]. Most often, the clinical presentation of PD is sporadic, with a small fraction of individuals actually inheriting the disease. The clinical manifestations of the disease are preceded by a loss of dopaminergic neuronal cell bodies within the substantia nigra pars compacta along with their projections into the striatum [105]. Although post-mortem investigations indicate that other ascending dopaminergic pathways within PD brains are affected, they are not affected as profoundly as the nigrostriatal pathway [106]. Apart from neuronal loss and the formation of proteinaceous inclusions, there is also an immune imbalance and proinflammatory response associated with disease and disease progression [107].
PD progression is linked, in measure, to neuroinflammation [108]. Loss of dopaminergic neurons is associated with both microgliosis and astrogliosis. Morphologically, microglia within affected brain regions are reactive, exhibiting ameboid cell bodies and thick, elongated processes and altered immune control [109, 110]. Likewise, the number of reactive microglia is much greater in PD than in age-matched controls [111]. Diffuse microglial activation is located near dead or dying neurons within the substantia nigra, as well as within the striatum [109]. This indicates the possibility that microglial activation could be initiated by a change in the neuronal state. This change triggers the release of soluble factors or mediators into the surrounding microenvironment. For instance, release of cyclooxygenase-2 or neuromelanin from neurons can activate microglia [109, 112, 113]. It is also hypothesized and suggested that misfolded, aggregated, and post-translationally modified proteins, such as nitrated alpha-synuclein, are released from dying neurons [17]. Biochemically, PD brains show increased levels of post-translationally modified proteins, lipid peroxidation, DNA damage, and reduced glutathione levels, all indicative of an aberrant response and neurotoxic milieu [114,115,116,117]. There are also elevated levels of nitrated proteins in both the brain and CSF of PD patients [118]. The most prevalent form is comprised of a 3-nitrotyrosine modification [119, 120]. Similarly, the expression of markers of reactive microglia correlates with the deposition of α-syn within the substantia nigra of PD patients [121]. The resulting reactive microglia become potent generators of reactive oxygen and nitrogen species, proinflammatory cytokines, and prostaglandins, all contributing to the inflammatory state and continued neuronal death. Nitric oxide, NAPDH-oxidase, TNF-α, and IL-1β are some of the major oxidative and inflammatory mediators released by reactive glia [122,123,124,125]. All are increased in the substantia nigra and CSF of PD patients [123]. Resulting interactions with cytokine receptors trigger intracellular death-related pathways, involving translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Interestingly, PD patients display 70 times more NF-κB than controls within dopaminergic neurons, suggesting the presence of neuronal death activation [126]. Collectively, these observations indicate an aberrant innate immune response that is associated with disease progression and PD pathology.
Besides chronic innate immune activation, there is compelling evidence that cell-mediated adaptive immune responses also play a role in PD progression. While T cells generally remain outside the CNS, the neuroinflammatory response results in the recruitment and extravasation of lymphocytes from the periphery to sites of active neurodegeneration [3]. The response is associated with disruption of the BBB due to the secretion of toxic mediators into the environment [127]. This dysfunction allows peripheral immune populations to readily enter the normally “immune-privileged” brain. Within the substantia nigra of PD patients, there are increased numbers of CD8+ and CD4+ T cells at levels exceeding 10-fold when compared to controls [128]. These peripheral T cell populations are found in close proximity to reactive microglia and degenerating neurons. Of note, the increased levels of T cells are not detected in any non-lesioned brain regions, suggesting that infiltration is site-specific and related to the neuronal injury itself. Upon microarray analysis of infiltrating T cell populations, it was determined that cells displayed gene changes associated with Th17-mediated immune reactions, indicating that PD may be a Teff-mediated immune disorder. Additionally, increased levels of Th17 cells have been noted in newly diagnosed PD patients, further suggesting their involvement in disease initiation [129]. A second group confirmed higher frequencies of Th17 cells in the blood of PD patients, as well as an increase in the number of CD3+ T lymphocytes within the midbrain of PD brains [130]. The study confirmed that infiltrating lymphocytes induce neuronal death through IL-17 receptor ligand interactions. The observed increased infiltration could potentially be due to increased BBB permeability. In vivo evidence for this phenomenon is observed using PET scans in PD patients [131, 132]. Scans indicate increased BBB permeability through the detection of albumin within the CSF. However, whether T cell infiltration occurs prior to neuronal cell death or after degeneration has occurred is not yet defined.
Apart from direct immune cell infiltration into the brain, peripheral immune populations and mediators are affected in PD patients as well. Compared to controls, levels of total lymphocytes, both B and T cells, are decreased in PD patients [133]. Specifically, CD19+, CD3+, and CD4+ levels are significantly reduced, whereas CD8+ levels remain relatively unchanged. Likewise, a correlation study indicates a decrease in CD3+ and CD4+ T lymphocytes within peripheral blood isolated from PD patients [134]. Work from our own group also indicates a shift in T cell phenotypes [135, 136]. Our cohort of PD patients had increased effector memory T cell subsets and decreased CD4+CD25+FoxP3+ Treg numbers. Similarly, the Tregs that were present were functionally inadequate in suppressing the proliferation of other Teff immune populations [135]. This deficit correlated with an increase in disease severity, which indicates that Treg dysfunction leads to an unbalanced and overactive immune response that ultimately speeds disease progression. These findings were verified in numerous animal studies using neurotoxin models of PD [15, 137,138,139]. A second recent study noted that PD patients have a Th1-biased immune response [140]. This study indicates increased levels of IFN-γ-producing cells within the periphery, with an overall decrease in CD4+ T cells in total. Along the same vein, increased proinflammatory cytokine levels including, IL-1β, TGF-β, IFN-γ, and IL-6, are detected in the substantia nigra and the CSF following post-mortem analyses [123,124,125, 141, 142]. Increased levels of IL-6 and TNF-α within the serum of PD patients is also correlated with increased disease severity based on Hohn and Yarr staging [143]. Increases in complement proteins are also observed, indicating an overall immune dysfunction both inside and outside of the brain. Importantly, it is shown that dopaminergic neurons exhibit enhanced sensitivity to cytokines such as TNF-α and IFN-γ, so increases within the periphery may be indirectly affecting neuronal survival within the brain [65]. Together, the majority of human observations suggest a clear pathogenic role of inflammation on disease severity, indicating that neuroinflammation could be targeted to modify disease progression.
2.4 Neuroimmunity in HIV-1 Infection
HIV-1-associated neural dysfunction is characterized by chronic CNS infection [144]. Infection results in notable cognitive impairments, leading to HIV-associated neurocognitive disorders (HAND) [145]. HAND can affect the frontal cortex, subcortical regions, hippocampus, and putamen of the brain [146]. Development of cognitive impairment is accompanied by motor and behavioral impairments including slowed movement, decreased motor coordination, decreased learning, and impaired memory [147]. Overt and unregulated viral infection leads to brain inflammation termed HIV encephalitis (HIVE). Neuropathology of viral encephalitis is characterized by the presence of HIV-1-infected macrophages within the brain, resulting in enhanced microgliosis and reactive microglia formation [148, 149]. Likewise, there is an increased occurrence of multi-nucleated giant cells and astrogliosis. Both macrophages and microglia are the primary viral targets; however, astrocytes have been shown to be infected, but at much lower levels [150]. Clinical manifestations correlate to the number of activated microglia and macrophages within the CNS, implicating them in disease pathogenesis [151]. Virus is thought to enter the brain through the “trojan-horse” method. Infected monocytes, macrophages, and/or lymphocytes crossing the BBB carry the virus into the CNS with them since virus does not readily cross the barrier itself [152]. This viral entry occurs relatively early after primary infection and maintains itself at low levels within the CNS due to the general immune privileged nature of the brain. However, there is a significant correlation between the amount of viral burden in the brain and the neuro-cognitive deficit [146]. Once inside the brain, the small number of infiltrating cells still secrete viral factors and neurotoxins, leading to neuronal damage by direct and indirect methods. Multiple studies indicate that virus-infected macrophages and microglia secrete neurotoxic metabolites such as arachidonic acid, TNF-α, IL-1β, nitric oxide, glutamate, and viral particles such as tat and gp120 [153,154,155,156].
Initial control of viral infection is mediated by cytotoxic CD8+ T lymphocytes (CTLs) [157, 158]. CTLs mediate their immune function by selectively targeting virus-infected cells through interaction with viral particles presented on infected cells [159]. There is a strong association with the lack of effective T cell responses and HIVE development [158]. Analysis of brain tissue from HIVE individuals reveals increased numbers of CD8+ CTLs near virus-infected mononuclear phagocytes when compared to brain tissue of diseased patients that did not succumb to HIVE [160, 161]. Here, CTLs release perforins and granzymes into the microenvironment that may contribute to the neurological insult resulting from HIV infection itself. Infiltrating CD8+ CTLs are also shown to be a source of CD40L and IFN-γ, further activating mononuclear phagocytes within the brain [160]. Individuals suffering from this disease have a profound loss in peripheral lymphocyte populations as well, making it hard to fight against the virus. Not only is viremia inhibited by CD8+ T cells, but HIV-1-specific CD4+ T cells appear to play a role too [162]. However, limited attention has been paid to CD4+ T cell control of viral replication due to the fact that they are major viral targets [163]. During primary HIV-1 infection, there is a massive infection of both resting and activated CD4+ T cells, reaching levels as high as 60% [164]. Initially, there is a rise in CD4+ T cell numbers; however, after a few months of infection, these numbers begin to decrease. This may be due to a natural contraction following viral infection or due to preferential infection and death of this cell type.
In the early stages of infection, there is a Th1-predominant profile, characterized by a high production of IL-2 and IFN-γ [165]. Late stage HIV infection is generally regarded as a Th2- predominant profile, indicated by increased production of IL-4 and IL-10 [166,167,168]. The exact role of CD4+ T cell subsets and their ability to control infection and viral replication is still under debate. For instance, Th17 cells have been implicated as being proinflammatory and immune activating in this disease [169]. However, similar to their role observed in AD and PD, this immune activation may not be beneficial in the context of HIV-1 infection. On the other hand, several studies have linked a protective role to HIV-specific CD4+ T cells with regard to viremia and disease progression [170,171,172]. These studies indicate that gag-specific CD4+ T cells and granzyme-producing CD4+ T cells are important for viral inhibition [170, 171]. Similarly, lack of these types of cell responses can be associated with disease progression [172]. In a contradictory human study, levels of CD4+ T cell activation correlated directly to viral load [173]. Characterization of these activated cells indicated an effector memory phenotype that was inversely associated with Treg phenotypes, and this dysregulation was found to drive the pathological immune activation in HIV-1 infection. Nonetheless, the growing body of evidence does support a specific role for CD4+ cells in HIV infection. Conversely, it still remains unclear how viral replication and peripheral immune activation shape CD4+ T cell responses and whether or not these responses may actually contribute to early immune activation with infection.
Similar to shifting immune cell phenotypes, cytokine alterations can be observed over the course of HIV disease progression [165]. Dysregulation is thought to contribute to HIV-associated immune deficiency. Increases in soluble factors and cytokines such as TNF-RII, neopterin, and β2-microglobulin are observed with HIV infection and indicate cellular activation [174]. They are also associated with disease progression and viral load measurements. When compared to uninfected controls, HIV-infected individuals have significantly higher levels of IL-2, IL-6, and IFN-γ [175]. Increases in IL-1β and TNF-α levels within HIV-infected brains and CSF are also been reported [176]. Their presence and mechanism of action can be detrimental on surrounding neurons, implicating these cytokines in the development of HAD. For instance, TNF-α and IL-1β increase the permeability of the BBB and induce an over-stimulation of NMDA-receptors on neurons resulting in fatal increases of Ca2+ [176]. TNF-α is also reported to induce translocation of NF-κB to the nucleus, causing upregulation of many other potent inflammatory cytokines, further contributing to disease progression [177]. Likewise, exposure of microglia to gp120 viral particles results in the upregulation of IL-1β and reactive oxygen species [178, 179]. Together, these findings indicate the presence of an overactive peripheral and central immune response occurring with disease, justifying the need for neuroprotective targets in this disease.
3 Neuroprotective Immune Responses
As discussed, activated microglia and Teffs are thought to be the main mediators of neuroinflammatory processes in these disease states. Left uncontrolled, these mediators support an inflammatory cascade that affects the tempo of disease. However, there are neuroprotective immune responses available that counterbalance the inflammatory milieu observed with disease progression. Current neuroprotective strategies are focused on modulation of microglial responses, alteration of Teff responses, induction of immunosuppressive cell populations, formation of antibodies, and enhancement of misfolded protein or viral clearance [9, 180,181,182,183]. Targeting the immune response to elicit a protective mechanism would diminish the extent of neuroinflammation and therefore increase the number of surviving neurons in the CNS of patients with neurodegenerative disorders. Here, we discuss the role of neuroprotective immunity and the current clinical and preclinical strategies being utilized to modulate the inflammatory immune response into one that is neurotrophic and protective.
Healing in response to injury is orchestrated by numerous factors and processes working together or sequentially. Therefore, it involves specific interactions between resident immune populations and peripheral immune cells [184]. Outside of the brain, tissue damage triggers infiltration of circulating immune cells to the site of injury. Initially, this immune population is mainly comprised of circulating monocytes that become activated and converted into macrophages. The primary job of these activated cells is wound healing and debris clean up [185]. This is done through the secretion of cytokines and growth factors. Without the function of this cell type, wound healing occurs much slower [186]. However, in the CNS, invading monocytes are not as prevalent. Macrophage infiltration is also delayed so resident microglia become the major phagocytic populations at the injury site. As discussed previously, once activated, microglia can become over-reactive [187]. This results in a neurodestructive cascade furthering damage [188]. In vitro work indicates that production of proinflammatory cytokines and growth factors can decrease the ability of astrocytes to support neuronal survival and increase the formation of tissue scarring [189]. Other studies suggest that these factors have a cytotoxic effect on oligodendrocytes as well [190,191,192,193,194]. Therefore, shifting microglial phenotype from proinflammatory to anti-inflammatory would potentially decrease these cytotoxic effects.
Microglia are a unique cell type that maintain two main functions within the CNS. Microglia are both supportive glial populations and immunocompetent defense cells [195]. During an infection with foreign antigen, microglia act as potent generators of proinflammatory mediators and reactive oxygen species that help drive the immune response needed to clear the brain of foreign invaders [196]. On the other hand, many studies indicate that microglia can support a neuroprotective and potentially proregenerative role in the injured CNS environment depending on their activation state [195, 197, 198]. Microglia have been found on or near the cell surface of neurons that do not undergo cell death but eventually regenerate axons [199]. This data suggests that microglia may be enhancing and supporting the recovery and regeneration of damaged neurons. Upon activation, microglia are also shown to upregulate their release of neurotrophic molecules and protective cytokines and/or chemokines [196]. Increased production of protective mediators into the microenvironment results in recruitment of neural progenitor cells to help regenerate previously lost neurons [200, 201]. Mediators can also act on surviving neurons, resident astrocytes, and other reactive microglia, shifting the brain microenvironment to one that is anti-inflammatory and restorative rather than proinflammatory and destructive [17]. For instance, early downregulation of TNF and increased levels of IL-10 have been linked to decreased scarring, decreased tissue and cell loss, and increased functional capacity following CNS injury [202, 203].The exact mechanism in which this occurs is still under debate. However, some investigators propose the idea of “protective autoimmunity,” in which having a controlled and localized proinflammatory immune response may be required for neuronal repair [204].
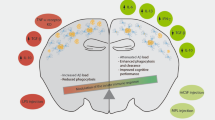
Classically, microglia can exhibit an activated inflammatory and neurotoxic phenotype called M1, but they can also acquire a neuroprotective phenotype termed M2 (Fig. 1) [17]. The M1 phenotype is generated in response to harmful stimuli and inflammatory cytokines such as TNF-α, IL-6, IL-1β, and IFN-γ [205]. Generally, Th1 cells produce the cytokines necessary for this polarization, but microglia have been shown to secrete them as well, allowing them to regulate in an autocrine fashion [206]. In most cases, this response is downregulated once the damage has been cleared but in many neurodegenerative diseases, this does not occur. This leads to an uncontrolled and prolonged immune activation further exacerbating disease. The neurosupportive and protective phenotype is characterized by the production of anti-inflammatory mediators and neurotrophic factors such as insulin-like growth factor 1 (IGF-1), brain-derived neurotrophic factor (BDNF), and glial cell-derived neurotrophic factor (GDNF) [207, 208]. Therefore, in order to shift microglial populations into an M2-like anti-inflammatory and proregenerative phenotype, researchers are focusing on agents known to directly modulate these responses [209,210,211]. To do so, studies have been focused on utilizing M2-inducing molecules such as IL-10, resolvin D, peroxisome proliferator-activated receptor (PPAR-y) agonists, and minocycline to directly modulate microglial responses [9]. Their exact protective effects and mechanisms are discussed later in “Neurotrophic mediators, endogenous neuropeptides, and cytokines as immunomodulators.” However, these two separate states may be an oversimplification. Microglia within the brain are plastic, resulting in a range of microglial phenotypes [206]. For instance, two-photon microscopy indicates that microglia within the CNS are constantly sampling the environment in order to maintain homeostasis, suggesting that they are never truly resting [206, 212]. A second target and source of neuroprotective immunity lies in modulating the adaptive immune response associated with disease initiation and progression. Currently, research is focused on the induction of immunosuppressive cell types within the periphery, such as regulatory T cells and/or tolerogenic dendritic cells. Researchers are also focused on vaccination strategies and antibody formation against proteins of interest in order to help clear protein plaques or virus associated with neuronal loss. Lastly, there have been numerous studies concentrated on the use of anti-inflammatory drugs, known immune modulators, neuropeptides, and cytokines as neuroprotective agents for the treatment of neurodegenerative and neuroinflammatory diseases. These neuroprotective targets are outlined below.

Immune modulation in neurodegenerative disease. In neurodegenerative diseases, neuronal death occurs through either environmental or genetic insult. Damaged neurons undergo apoptosis, leading to microglial activation and phenotypic shift into proinflammatory M1 microglia (red arrows). This activation occurs through events such as ingestion of misfolded protein aggregates containing beta-amyloid or alpha-synuclein or through direct viral infection or ingestion of viral particles. Either way, activation leads to production of proinflammatory and neurotoxic mediators, resulting in additional neuronal death and damage of healthy neurons in the surrounding area. Therapeutic intervention through the use of immune-modulating agents can shift the M1 phenotype into a neurosupportive and neuroprotective M2 phenotype (green arrows). M2 microglia can act on damaged neurons and support neuronal growth and regeneration through the production of neurotrophic and anti-inflammatory mediators. The presence of M2 microglia also provide a neuroprotective microenvironment allowing healthy neurons to remain viable. Modulating microglial phenotypes ultimately shifts the microenvironment from neurotoxic to neurotrophic
Both CD4+ and CD8+ T cells can play a dual role in neurodegeneration and neuroprotection during CNS disorders depending on their phenotype and environmental signals [8]. Therefore, targeting this portion of the adaptive immune system would provide a potential strategy to halt neurodegenerative disease progression. Treg are potent modulators of the immune system, have distinct immunosuppressive capabilities, and are characterized by the positive expression of CD4, CD25, and FoxP3 and negative expression of CD127 [213]. They maintain the ability to suppress inflammation through multiple mechanisms including inhibition of Teff differentiation and proliferation, secretion of anti-inflammatory cytokines such as IL-10, IL-35, and TGF-β, direct killing of Teff subsets through granzyme and perforin release, blockade of T cell co-stimulation, and metabolic disruption of Teffs and APCs via uptake of IL-2 and use of CTLA-4 [44, 137] (Fig. 2). Anti-inflammatory cytokines produced by Tregs, such as IL-4, IL-10, and TGFβ, are crucial anti-inflammatory mediators that diminish neuroinflammation and increase neuroprotection [214]. Induction of Tregs contributes to development of M2 anti-inflammatory microglial phenotypes, leading to the release of neurotrophic factors, including IGF-1 and BDNF, ultimately promoting neuronal protection [206]. Similarly, from our own animal studies, we demonstrated that Tregs elicit neuroprotection of dopaminergic neurons along the nigrostriatal pathway in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced lesions and in hippocampal neuron populations within an AD mouse model [12, 14, 15, 138]. Other analyses indicate that Tregs have the capacity to act directly on activated microglia, resulting in an attenuation of reactivity, decreased phagocytosis and migration, and decreased production of neurotoxic factors [138, 215]. In vitro studies suggest that Tregs can mediate inhibition of proinflammatory microglial functions through the suppression of NF-κB pathways via direct cell-cell contact [215]. Specifically, Tregs elicit a potent down-regulation of proinflammatory mediators such as iNOS, TNF-α, IL-1β, IFN-γ [216]. Coincidently, this was associated with decreased levels of ROS production and NF-κB activation [217]. Utilization of this regulatory population would shift microglial responses from a neurotoxic M1 response to a neurotrophic M2 response [138]. In further support, adoptive transfer of CD3-activated Treg resulted in the attenuation of both astrogliosis and microgliosis in HIV-1-associated neurodegeneration [218]. This attenuation was associated with neuroprotection mediated by upregulation of BDNF and GDNF and downregulation of proinflammatory mediators.

Immune-mediated neuroprotection. Within the periphery, immature dendritic cells will differentiate into fully mature dendritic cells and elicit an immune response. Naïve T cells interacting with mature dendritic cells undergo clonal expansion and proliferation in response to antigen. Once activated, the effector T cell population will cross the blood-brain barrier and enter the central nervous system. Effector T cells enter the brain and secrete pro-inflammatory cytokines causing resident microglia and astrocytes to become activated. Upon activation, glia cells secrete neurotoxic and proinflammatory mediators, resulting in neuronal cell death. Effector T cells can also mediate cytolysis of neurons directly. Induction of tolerogenic dendritic cells and regulatory T cell populations can counteract this inflammatory milieu. Immature dendritic cells are also differentiated and shifted into tolerogenic dendritic cells in order to regulate immune responses. Tolerogenic dendritic cells can interact with T cells in various ways, resulting in three different end-points. First, tolerogenic dendritic cells can induce apoptosis in activated, effector T cell populations. Second, when interacting with a naïve T cell, tolerogenic dendritic cells can induce T cell anergy. Third, tolerogenic dendritic cells are potent inducers of regulatory T cell populations. Induction of regulatory T cells leads to overall immune suppression in both the periphery and the central nervous system. Regulatory T cells carry out their immunomodulatory cascade through a number of mechanisms, indicated by the green lines and arrows. These include inhibition of antigen presentation, metabolic disruption, inhibition of reactive microglial and astrocytic activation, stimulation of neurotrophin release from neurosupportive astrocytes, cell-mediated cytolysis of effector T cell subsets, and production of anti-inflammatory cytokines and suppressive molecules. Each of these mechanisms provides support for overall neuronal survival and an anti-inflammatory and neuroprotective microenvironment
On the other hand, in many neurodegenerative diseases, there is a dysregulation in the number and/or function of this suppressive cell type. For instance, in preclinical and clinical studies, we found that individuals suffering from PD have decreased levels of Tregs with a decreased ability to suppress Teff proliferation [135]. Likewise, this dysregulation was associated with increased movement disorder, indicating that the induction or enhancement of this cell population is worth investigation. This can be done through the use of immunomodulatory agents such as granulocyte-macrophage colony-stimulating factor (GM-CSF), vasoactive intestinal peptide (VIP), copolymer -1 (Cop-1), or vaccine strategies targeting Treg populations. Many of these agents are being tested in the preclinical and clinical setting. Adoptive transfer of VIP- or GM-CSF-induced Tregs following MPTP intoxication leads to significant dopaminergic neuronal sparing with a parallel decrease in microglial activation [14,15,16]. These findings prompted a phase I clinical trial utilizing sargramostim, a form of human recombinant GM-CSF, in patients suffering from PD [136]. This study supported the notion that Treg populations are decreased and dysfunctional in PD and that modulation and induction of this population is beneficial. Patients receiving treatment displayed increased Treg numbers, increased suppressive T cell function, and decreased motor deficits, when compared to both baseline and placebo-treated controls. Similarly, Cop-1 immunization in a model of HIVE resulted in anti-inflammatory and neuroprotective effects [219]. This immunization strategy yielded the development of T cells secreting IL-10 and IL-4, as well as an increase in the number of Treg. It was later determined that in HIVE, Tregs readily crossed the BBB and migrated to sites of infection and neuroinflammation while still maintaining phenotype and immunosuppressive function [220]. However, other studies have suggested that breaking immune tolerance through Treg targeting can actually mitigate disease-related pathologies, suggesting that the time and extent of induction may play a role in whether the result is either protective or more detrimental [221].
4 Induction of Tolerogenic Dendritic Cells
Dendritic cells (DCs) are a heterogeneous population of APCs that contribute to innate immunity and initiate the adaptive immune response associated with inflammation and autoimmunity [222]. However, apart from this, DCs also play an important role in maintaining immune homeostasis and immune tolerance [223] (Fig. 2). Unlike classical DC function, tolerogenic DCs should not stimulate T cell proliferation or inflammatory cytokine production. Instead, they act by suppressing the immune response and the effector populations required for the response. Their anti-inflammatory response involves roles in tolerance induction and silencing the immune response. This function is mainly carried out through the induction of regulatory T cells, T effector cell apoptosis, and T cell anergy [224]. The ability of DCs to promote tolerogenic and/or inflammatory responses is related to their maturation state [225]. Generally, immature DCs expressing low levels of MHC class II and co-stimulatory molecules are responsible for generating immunosuppressive responses, whereas inflammatory adaptive immune responses are achieved by mature DCs [226]. Immature DCs have the capacity to induce and expand regulatory T cells; however, some studies have also linked mature DCs to the induction of this cell type [225]. Immature DCs can be defined by their surface marker expression. Phenotypic analysis indicates that this suppressive and regulatory population is CD11clowCD11bhighMHCIIlowCD86low and has the capacity to produce high levels of IL-10, ultimately inhibiting Teff proliferation and promoting Treg function [227]. This cell type is now considered to be tolerogenic. Along with the secretion of IL-10, tolerogenic DCs play a significant role in maintaining peripheral and central tolerance through the secretion of TGF-β, indoleamine 2,3-dioxygenase (IDO), and retinoic acid (RA) [228,229,230]. Tregs that come in contact with this subset exhibit parallel tolerogenic functions and anti-inflammatory functions [231]. On the other hand, tolerogenic DC interaction with activated Teff populations results in an inhibitory effect by decreasing CD4+ T cell proliferation and increasing IL-10 production.
In order to maintain the tolerogenic environment, studies show that there are reciprocal interactions between induced Tregs and tolerogenic DCs [231]. Cross-talk between both populations is needed to induce and maintain immune tolerance. Tregs are shown to modulate both the phenotype and function of DCs [232]. For instance, tolerogenic DCs promote the expansion of Tregs through the expression of PDL-1 while Tregs maintain the tolerogenic population through the production of TGF-β and IL-10 [233]. IL-10 producing Tregs can inhibit DC maturation, maintaining an immature and immunosuppressive state [234]. Furthermore, when FoxP3+ Tregs are depleted, DCs have trouble interacting with CD4+ Teffs, indicating that FoxP3+ Tregs are essential for maintaining the immune tolerant and suppressive state of tolerogenic DCs [235]. Therefore, generation of tolerogenic DCs, either naturally or pharmaceutically, would be beneficial in chronic and progressive neuroinflammatory diseases, such as PD, AD, and HIV-1-associated neurodegeneration.
In support of this, treatments with immunomodulatory agents such as VIP, rapamycin, and GM-CSF have been shown to induce tolerogenic DCs and promote immune suppression [10, 232, 236]. VIP treatment regulates DC differentiation by inducing an upregulation of CD86 in immature DCs and a downregulation of CD80 and CD86 in LPS-stimulated DCs [237]. The induced CD4+ T cells generated via VIP-treated immature DCs exhibit an anti-inflammatory Th2 phenotype as well. Similarly, another study reported that VIP induces tolerogenic DCs that cause surrounding CD4+ T cells to release anti-inflammatory cytokines such as IL-10 and TGF-β, indicating the formation of a regulatory subset rather than an effector population [238]. Likewise, in human studies, VIP treatment generated tolerogenic DCs that induced both CD4+ Tregs and CD8+ Tregs, further supporting the idea that signaling via VIP receptors (VIPRs) is involved in the generation of multiple immunosuppressive subsets [239]. Similarly, in our own studies, treatment with GM-CSF resulted in the generation of tolerogenic DCs, as indicated by an alteration of co-stimulatory molecules and the ability to convert naïve CD4+ T cells into a Treg population [10]. Adoptive transfer of the induced tolerogenic DCs attenuated the neuroinflammatory response and spared dopaminergic neurons in a PD model.
These insights may yield potential clinical targets for the treatment of neuroinflammatory conditions. The role of DCs as an immunotherapy has been confirmed in AD and PD studies utilizing mouse models [240,241,242,243]. Administration of DCs tolerized to Aβ peptide slowed the rate of cognitive decline, increased levels of anti-Aβ antibodies, reduced Aβ plaques within the CNS, and increased spatial learning and memory [240, 241]. Intravenous injections of DCs sensitized against α-syn results in the generation of antibodies against the protein coincident with improved motor function and decreased inflammatory response associated with disease progression [242, 243]. However, translating these findings for clinical use may be challenging due to the varying phenotypes of human DCs and the ability to maintain a stable tolerogenic DC population [244]. Secondly, the tolerogenic response must be maintained for a prolonged amount of time. Due to these factors, clinical trials targeting DCs are not as common.
5 Vaccination Strategies
Modulation of the humoral immune response is a vaccination strategy directed at targeting immunogenic and pathogenic epitopes [245]. Ultimately, this therapeutic strategy focuses on ameliorating neuroinflammation by utilizing the immune system to target misfolded or aggregated proteins and/or viral particles. For instance, immunization of transgenic mice containing human α-syn with misfolded α-syn results in the production of high affinity anti-α-syn antibodies [246]. This antibody formation was associated with decreases in α-syn inclusions in neuronal cell bodies and at neuronal synapses [247]. It also results in decreased neuronal loss and overall neurodegeneration. Also, anti-α-syn antibodies supported the active degradation of α-syn aggregates. Another recent study utilizing an AAV-α-syn rat model of PD indicates that formation of anti-human α-syn N-terminal peptide antibodies can elicit neuroprotection and decrease microglial activation [246]. Vaccination led to increased production of circulating IgGs, increased MHCII expression, and augmented CD4+ T cell infiltration into the CNS [246]. Administration of monoclonal antibodies against the C-terminal region of α-syn reduces levels of protein aggregation, improving PD pathology. Monoclonal antibody treatment attenuated dopaminergic neuronal cell death and decreased motor deficits associated with disease [248,249,250]. Based on these findings, Roche and Prothena commercialized this approach and utilized PRX002 to specifically target α-syn (NCT02095171). Analysis from the phase I clinical trial indicated that the vaccine was safe and tolerable and ultimately prompted a second trial assessing dose, immunogenicity, and pharmacokinetics (NCT02157714). Similarly, several other studies entered clinical trials, showing promise in the use of vaccines for the treatment of PD by demonstrating Treg recruitment, increased levels of neurotrophins, and increased antibody formation [251,252,253]. Collectively, these studies show that α-syn-targeted vaccine strategies have been successful and display the potential to delay dopaminergic neurodegeneration and decrease neuroinflammation.
Similarly, vaccination strategies have been pioneered for the treatment of AD. Anti-Aβ antibodies prevent formation of new Aβ plaques and help dissociate existing plaques [254,255,256,257,258]. The presence of these antibodies also improved learning and protected transgenic mice from developing memory loss. Moreover, the presence of naturally occurring antibodies against Aβ is reported in the CSF of AD patients, but levels are significantly lower than healthy controls, suggesting a dysfunction in the ability of AD patients to induce the desired protective humoral immune response [94]. Therefore, active and passive immunization strategies have been researched and explored for the treatment of AD [259,260,261,262,263,264,265]. For example, active immunization with Aβ1-42 peptide (AN1792) was tested in the clinical setting; however, the trial was halted due to unexpected meningoencephalitis and death associated with vaccine [259,260,261,262]. Post-mortem analysis showed a significant drop in the number of plaques, but vaccination did not continue due to the active neuroinflammatory response that ensued with vaccination [259, 261, 263]. Still, those that did not succumb to adverse events were monitored and appeared to benefit from the vaccine [264, 265]. Individuals with the highest antibody titers remained cognitively stable for up to 2 years post-vaccination. Because of the potential adverse events associated with this vaccination strategy, there have been a number of alternative vaccine approaches to enhance the formation of antibodies against Aβ. For instance, a synthetic and truncated form of Aβ, UB-311, is utilized as a vaccine strategy in order to break self-tolerance and limit the possibility of developing a similar T cell reaction as seen with AN1792 vaccination [266]. Other approaches include production of B cell epitopes against Aβ, DNA-based vaccines, and use of monoclonal antibodies as therapeutic options [267, 268]. Likewise, passive immunization using monoclonal antibodies against Aβ is also effective in reducing amyloid deposits in the CNS [269, 270].
Similar vaccination strategies have been utilized for the treatment of HIV-infection. Antibodies against HIV-associated proteins, such as Tat, are found in the brain and spinal fluid of infected individuals [271, 272]. Anti-Tat antibodies are also detected in the CSF of individuals suffering from HAND [273]. Recent work indicates that antibodies generated against Tat results in the suppression of Tat-induced viral replication and HIV-associated cytotoxic effects [274]. It is suggested that antibodies against Tat are also protective against NMDA-mediated excitotoxicity [275]. Taken together, it is clear that vaccination strategies may hold promise in clearing disease-causing protein inclusions and viral particles.
6 Immunomodulators and Neurotrophins
Progressive neurodegenerative disorders , such as those discussed above, present a challenge for developing treatments because of unknown time and mechanism of disease onset. As noted previously, therapies aimed at targeting neuroinflammation either directly or indirectly are now front and center. Among these therapies, use of non-steroidal anti-inflammatory drugs (NSAIDS), specifically ibuprofen, is associated with a lower risk of PD development , and is protective in MPTP and 6-hydroxydopamine (6-OHDA) induced lesions [276]. These findings suggest that there is an association with anti-inflammatory use and decreasing the probability of being diagnosed with PD. Therefore, many anti-inflammatory agents have been explored, such as minocycline and natural or endogenous compounds including resveratrol, silymarin, resolvins, and apocynin [9]. These compounds act by downregulating glial activation, decreasing proinflammatory cytokine production, suppressing M1 microglial phenotypes, reducing NF-κB activation, and decreasing amounts of reactive oxygen species present in the brain. Additionally, PPAR agonists, such as pioglitazone and rosiglitazone, also possess neuroprotective and anti-inflammatory activities both in vitro and in vivo [277,278,279]. These agents selectively act on decreasing the amount of reactive microglia and their secreted neurotoxic factors.
A second therapeutic target is found by modulating T cell phenotypes and functions with pharmacologic agents. Ideally, enhancing phenotypes that shut down the inflammatory response within the brain microenvironment through the use of potent immune modulating agents such as VIP or GM-CSF would be of benefit [12, 14, 16, 280, 281]. Such therapeutic interventions have been effective in PD and AD, along with other chronic inflammatory conditions and as such, support their ability to restore immune homeostasis and repair tissue injuries. Similarly, due to the wide variety of biological targets and effects of VIP, previous studies have utilized the native peptide for neuroprotection from HIV neurotoxicity [282,283,284,285]. Various studies have shown that VIP treatment prevents HIV-1 induced neuronal death [283, 284]. This protection is mediated through VIP-associated signaling within astrocytes. When astrocyte and cortical neuron cultures are treated with VIP, there is an increase in MIP-1α, beta-chemokine, and RANTES [282, 283]. This chemokine upregulation blocks the receptor interactions that are needed for viral entry and toxicity, resulting in neuronal survival. Likewise, when VIP binds to the VIPR2 on astrocytes, it induces changes in activity-dependent neuroprotective protein (ADNP), which is associated with cell survival and development, further supporting the neuroprotective effects of VIP-targeting [285].
Apart from anti-inflammatory and immune modulating therapies, researchers are also seeking to utilize neurotrophic factors within damaged brain regions [286]. Neurotrophic factors are a family of molecules that support growth, survival, synaptic plasticity, and differentiation of developing and mature neurons [287]. Thus, their use in diseases in which there is neuronal loss is intriguing. Amongst these factors are GDNF, BDNF, neurturin, and neurotrophin [286]. GDNF is neuroprotective and restorative in the dopaminergic neuron system and has been demonstrated in multiple experimental models including rodents and primates [288, 289]. Some of these studies indicated that the degree of neuroprotection observed correlates with the amount of neurotrophin, specifically GDNF, levels present within the brain region [290]. Similarly, neurturin, a homolog of GDNF, has also shown neuroprotective efficacy with no side effects observed within a large margin of doses [291, 292]. A study using BDNF-treated neural stem cells in an AD model indicated an improved transplant effect resulting in increased memory and learning and increased overall cell survival [293]. A study utilizing neurotrophin-3 (NT3) in an ex vivo PD model showed that NT3 treatment led to an increase in cell survival, an overall neuroprotective response, and an increase in dopamine production [294]. Taken together, use of neurotrophic factors in brain diseases has shown promise as a potential clinical therapy .
7 Summary
Alzheimer’s disease, Parkinson’s disease, and HIV-1-associated neurodegeneration are devastating disorders of the CNS with few therapeutic avenues. Collectively, these diseases are linked to neuroinflammation and aberrant immune responses. Each involves altered innate and adaptive immune responses leading to increased glial reactivity associated with altered frequencies of T effector and T regulatory populations. Since both of these populations play an important role in maintaining a successful and healthy immune response, it is likely that their dysfunction controls the tempo of disease progression. Due to this, many laboratories have focused on harnessing the immune system for therapeutic gain. Current strategies aim to shift the neurodestructive immune phenotypes into those that are neuroprotective. The universal goal of such strategies is to suppress neuroinflammation in order to spare neuronal populations normally lost or affected during the course of disease. Throughout this chapter, we have discussed many neuroprotective strategies, including modulation of the innate glial immune response and transformation of the peripheral adaptive immune response through inhibition of proinflammatory cytokine production, induction of regulatory T cells, induction of tolerogenic dendritic cells, increased production of circulating antibodies, and various vaccination strategies. We have also discussed the protective role of anti-inflammatory agents, neurotrophins, and cytokines in diseases of the brain. Overall, researchers utilizing these strategies are attempting to modify the diseased CNS microenvironment by targeting proinflammatory glial populations directly to decrease proinflammatory and neurotoxic mediator production or by targeting them indirectly through the induction of immunosuppressive populations such as regulatory T cells and tolerogenic dendritic cells. The potential neuroprotective effects of these cell types would certainly restore the harmful inflammatory response to its normal homeostatic state. However, the immune system is also needed to clear debris and repair cellular and tissue damage, which would serve to restore homeostasis and lead to neuronal survival and repair. Therefore, it is likely that a timed control of regulating and shifting the immune response is needed in diseases of the brain in order to maintain the highest level of therapeutic gain.
References
Doty KR, Guillot-Sestier MV, Town T. The role of the immune system in neurodegenerative disorders: adaptive or maladaptive? Brain Res. 2015;1617:155–73.
Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci. 2012;15(8):1096–101.
Negi N, Das BK. CNS: not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. 2018;37(1):57–68.
Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (Review). Mol Med Rep. 2016;13(4):3391–6.
Hong S, Banks WA. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav Immun. 2015;45:1–12.
Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1)
Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29(8):357–65.
Anderson KM, Olson KE, Estes KA, Flanagan K, Gendelman HE, Mosley RL. Dual destructive and protective roles of adaptive immunity in neurodegenerative disorders. Transl Neurodegener. 2014;3(1):25.
Olson KE, Gendelman HE. Immunomodulation as a neuroprotective and therapeutic strategy for Parkinson’s disease. Curr Opin Pharmacol. 2016;26:87–95.
Schutt CR, Gendelman HE, Mosley RL. Tolerogenic bone marrow-derived dendritic cells induce neuroprotective regulatory T cells in a model of Parkinson’s disease. Mol Neurodegener. 2018;13(1):26.
Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Zhang G, et al. URMC-099 facilitates amyloid-beta clearance in a murine model of Alzheimer’s disease. J Neuroinflammation. 2018;15(1):137.
Kiyota T, Machhi J, Lu Y, Dyavarshetty B, Nemati M, Yokoyama I, et al. Granulocyte-macrophage colony-stimulating factor neuroprotective activities in Alzheimer’s disease mice. J Neuroimmunol. 2018;319:80–92.
Kelso ML, Elliott BR, Haverland NA, Mosley RL, Gendelman HE. Granulocyte-macrophage colony stimulating factor exerts protective and immunomodulatory effects in cortical trauma. J Neuroimmunol. 2015;278:162–73.
Kosloski LM, Kosmacek EA, Olson KE, Mosley RL, Gendelman HE. GM-CSF induces neuroprotective and anti-inflammatory responses in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine intoxicated mice. J Neuroimmunol. 2013;265(1-2):1–10.
Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE. Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol. 2010;184(5):2261–71.
Olson KE, Kosloski-Bilek LM, Anderson KM, Diggs BJ, Clark BE, Gledhill JM Jr, et al. Selective VIP receptor agonists facilitate immune transformation for dopaminergic neuroprotection in MPTP-intoxicated mice. J Neurosci. 2015;35(50):16463–78.
Mosley RL, Hutter-Saunders JA, Stone DK, Gendelman HE. Inflammation and adaptive immunity in Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(1):a009381.
Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: what is it and where are we? J Clin Invest. 2003;111(1):3–10.
Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7)
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–7.
Amor S, Woodroofe MN. Innate and adaptive immune responses in neurodegeneration and repair. Immunology. 2014;141(3):287–91.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353(6301):777–83.
Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging. 2002;23(5):795–807.
Tilleux S, Hermans E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res. 2007;85(10):2059–70.
Vajda FJ. Neuroprotection and neurodegenerative disease. J Clin Neurosci. 2002;9(1):4–8.
Cummings J. Disease modification and Neuroprotection in neurodegenerative disorders. Transl Neurodegener. 2017;6:25.
Tarawneh R, Galvin JE. Potential future neuroprotective therapies for neurodegenerative disorders and stroke. Clin Geriatr Med. 2010;26(1):125–47.
Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S24–32.
Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018;69:437–49.
Gomez-Nicola D, Perry VH. Microglial dynamics and role in the healthy and diseased brain: a paradigm of functional plasticity. Neuroscientist. 2015;21(2):169–84.
Boche D, Perry VH, Nicoll JA. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013;39(1):3–18.
Hristovska I, Pascual O. Deciphering resting microglial morphology and process motility from a synaptic prospect. Front Integr Neurosci. 2015;9:73.
Koellhoffer EC, McCullough LD, Ritzel RM. Old maids: aging and its impact on microglia function. Int J Mol Sci. 2017;18(4)
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463–77.
Crotti A, Glass CK. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015;36(6):364–73.
Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S33–40.
Janeway CJT, Travers P, Walport M, et al. Principles of innate and adaptive immunity. New York: Garland Science; 2001.
Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–67.
Janeway CJT, Paul Travers; Walport, M; et al.. Antigen presentation to T lymphocytes. Immunobiology: the immune system in health and disease 5th edn. 5. New York: Garland Science 2001.
Romagnani S. T-cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol. 2000;85(1):9–18; quiz, 21.
Golubovskaya V, Wu L. Different subsets of T cells, memory, effector functions, and CAR-T immunotherapy. Cancers (Basel). 2016;8(3)
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517.
Zhou Y, Sonobe Y, Akahori T, Jin S, Kawanokuchi J, Noda M, et al. IL-9 promotes Th17 cell migration into the central nervous system via CC chemokine ligand-20 produced by astrocytes. J Immunol. 2011;186(7):4415–21.
Corthay A. How do regulatory T cells work? Scand J Immunol. 2009;70(4):326–36.
Beissert S, Schwarz A, Schwarz T. Regulatory T cells. J Invest Dermatol. 2006;126(1):15–24.
Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol. 2009;21(10):1105–11.
Hamann A. Syrbe U. T-cell trafficking into sites of inflammation. Rheumatology (Oxford). 2000;39(7):696–9.
Engelhardt B. Molecular mechanisms involved in T cell migration across the blood-brain barrier. J Neural Transm (Vienna). 2006;113(4):477–85.
Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to Tango. Front Immunol. 2017;8:1905.
Shrestha R, Shakya Shrestha S, Millingtona O, Brewer J, Bushell T. Immune responses in neurodegenerative diseases. Kathmandu Univ Med J. 2014;12(45):67–76.
Mosley RL. Adaptive immunity in neurodegenerative and neuropsychological disorders. J Neuroimmune Pharmacol. 2015;10(4):522–7.
Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease – insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol. 2017;13(11):703.
Hyman BT. The neuropathological diagnosis of Alzheimer’s disease: clinical-pathological studies. Neurobiol Aging. 1997;18(4 Suppl):S27–32.
O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204.
Dansokho C, Heneka MT. Neuroinflammatory responses in Alzheimer’s disease. J Neural Transm (Vienna). 2018;125(5):771–9.
Hosoda S, Glick D. Biosynthesis of 5-hydroxytryptophan and 5-hydroxytryptamine from tryptophan by neoplastic mouse mast cells. Biochim Biophys Acta. 1965;111(1):67–78.
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–71.
Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656–64.
Cowan CM, Mudher A. Are tau aggregates toxic or protective in tauopathies? Front Neurol. 2013;4:114.
Wildsmith KR, Holley M, Savage JC, Skerrett R, Landreth GE. Evidence for impaired amyloid beta clearance in Alzheimer’s disease. Alzheimers Res Ther. 2013;5(4):33.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189.
Arvin B, Neville LF, Barone FC, Feuerstein GZ. The role of inflammation and cytokines in brain injury. Neurosci Biobehav Rev. 1996;20(3):445–52.
Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol Aging. 1988;9(4):339–49.
Styren SD, Civin WH, Rogers J. Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer’s disease brain. Exp Neurol. 1990;110(1):93–104.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69.
Kopec KK, Carroll RT. Alzheimer’s beta-amyloid peptide 1-42 induces a phagocytic response in murine microglia. J Neurochem. 1998;71(5):2123–31.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–74.
Ries M, Sastre M. Mechanisms of abeta clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160.
Wilkinson K, El Khoury J. Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int J Alzheimers Dis. 2012;2012:489456.
El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197(12):1657–66.
Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–91.
Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem. 1997;272(46):29390–7.
Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118(4):475–85.
Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther. 2015;7(1):41.
Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. 1992;84(3):225–33.
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–7.
Latta CH, Brothers HM, Wilcock DM. Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience. 2015;302:103–11.
Forlenza OV, Diniz BS, Talib LL, Mendonca VA, Ojopi EB, Gattaz WF, et al. Increased serum IL-1beta level in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord. 2009;28(6):507–12.
Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19(5):641–4.
Chao CC, Ala TA, Hu S, Crossley KB, Sherman RE, Peterson PK, et al. Serum cytokine levels in patients with Alzheimer’s disease. Clin Diagn Lab Immunol. 1994;1(4):433–6.
Zetterberg H, Andreasen N, Blennow K. Increased cerebrospinal fluid levels of transforming growth factor-beta1 in Alzheimer’s disease. Neurosci Lett. 2004;367(2):194–6.
Chalmers KA, Love S. Neurofibrillary tangles may interfere with Smad 2/3 signaling in neurons. J Neuropathol Exp Neurol. 2007;66(2):158–67.
Zilka N, Kazmerova Z, Jadhav S, Neradil P, Madari A, Obetkova D, et al. Who fans the flames of Alzheimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J Neuroinflammation. 2012;9:47.
Royle NJ. Injuries of the ankle. Med J Aust. 1978;1(7):374–8.
Hellwig S, Frings L, Bormann T, Vach W, Buchert R, Meyer PT. Amyloid imaging for differential diagnosis of dementia: incremental value compared to clinical diagnosis and [18F]FDG PET. Eur J Nucl Med Mol Imaging. 2019;46(2):312–323.
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, et al. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med. 2003;9(4):453–7.
McGeer PL, McGeer EG. Inflammation, autotoxicity and Alzheimer disease. Neurobiol Aging. 2001;22(6):799–809.
Marciani DJ. Alzheimer’s disease vaccine development: a new strategy focusing on immune modulation. J Neuroimmunol. 2015;287:54–63.
Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis. 2005;7(3):221–32.. discussion 55–62
Britschgi M, Wyss-Coray T. Systemic and acquired immune responses in Alzheimer’s disease. Int Rev Neurobiol. 2007;82:205–33.
Lopez-Fernandez MF, Gonzalez-Boullosa R, Blanco-Lopez MJ, Perez M, Batlle J. Abnormal proteolytic degradation of von Willebrand factor after desmopressin infusion in a new subtype of von Willebrand disease (ID). Am J Hematol. 1991;36(3):163–70.
Mohamed A, Posse de Chaves E. Abeta internalization by neurons and glia. Int J Alzheimers Dis. 2011;2011:127984.
Pellicano M, Bulati M, Buffa S, Barbagallo M, Di Prima A, Misiano G, et al. Systemic immune responses in Alzheimer’s disease: in vitro mononuclear cell activation and cytokine production. J Alzheimers Dis. 2010;21(1):181–92.
Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. 2003;112(3):415–22.
Pellicano M, Larbi A, Goldeck D, Colonna-Romano G, Buffa S, Bulati M, et al. Immune profiling of Alzheimer patients. J Neuroimmunol. 2012;242(1-2):52–9.
Reale M, Iarlori C, Gambi F, Lucci I, Salvatore M, Gambi D. Acetylcholinesterase inhibitors effects on oncostatin-M, interleukin-1 beta and interleukin-6 release from lymphocytes of Alzheimer’s disease patients. Exp Gerontol. 2005;40(3):165–71.
Di Bona D, Plaia A, Vasto S, Cavallone L, Lescai F, Franceschi C, et al. Association between the interleukin-1beta polymorphisms and Alzheimer’s disease: a systematic review and meta-analysis. Brain Res Rev. 2008;59(1):155–63.
Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103.
Xue SR, Xu DH, Yang XX, Dong WL. Alterations in lymphocyte subset patterns and co-stimulatory molecules in patients with Alzheimer disease. Chin Med J (Engl). 2009;122(12):1469–72.
Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, et al. Altered lymphocyte distribution in Alzheimer’s disease. J Psychiatr Res. 2007;41(1-2):174–8.
Speciale L, Calabrese E, Saresella M, Tinelli C, Mariani C, Sanvito L, et al. Lymphocyte subset patterns and cytokine production in Alzheimer’s disease patients. Neurobiol Aging. 2007;28(8):1163–9.
Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Alberoni M, et al. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain Behav Immun. 2011;25(3):539–47.
Olanow CW, Stern MB, Sethi K. The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology. 2009;72(21 Suppl 4):S1–136.
Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27(5):494–506.
Cheng HC, Ulane CM, Burke RE. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 2010;67(6):715–25.
Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39(6):889–909.
Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S210–2.
Gelders G, Baekelandt V, Van der Perren A. Linking neuroinflammation and neurodegeneration in Parkinson’s Disease. J Immunol Res. 2018;2018:4784268.
McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38(8):1285–91.
Banati RB, Daniel SE, Blunt SB. Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord. 1998;13(2):221–7.
Mirza B, Hadberg H, Thomsen P, Moos T. The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience. 2000;95(2):425–32.
Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci USA. 2003;100(9):5473–8.
Wilms H, Rosenstiel P, Sievers J, Deuschl G, Zecca L, Lucius R. Activation of microglia by human neuromelanin is NF-kappaB dependent and involves p38 mitogen-activated protein kinase: implications for Parkinson’s disease. FASEB J. 2003;17(3):500–2.
Sanyal J, Bandyopadhyay SK, Banerjee TK, Mukherjee SC, Chakraborty DP, Ray BC, et al. Plasma levels of lipid peroxides in patients with Parkinson’s disease. Eur Rev Med Pharmacol Sci. 2009;13(2):129–32.
Chen CM, Liu JL, Wu YR, Chen YC, Cheng HS, Cheng ML, et al. Increased oxidative damage in peripheral blood correlates with severity of Parkinson’s disease. Neurobiol Dis. 2009;33(3):429–35.
Alam ZI, Jenner A, Daniel SE, Lees AJ, Cairns N, Marsden CD, et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69(3):1196–203.
Sian J, Dexter DT, Lees AJ, Daniel S, Agid Y, Javoy-Agid F, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–55.
Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K, Umegae N, et al. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann Neurol. 2000;47(4):524–7.
Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, et al. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol. 2000;157(5):1439–45.
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–9.
Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation. 2005;2:14.
Hald A, Lotharius J. Oxidative stress and inflammation in Parkinson’s disease: is there a causal link? Exp Neurol. 2005;193(2):279–90.
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165(1-2):208–10.
Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci Lett. 1996;211(1):13–6.
Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, et al. Caspase activities and tumor necrosis factor receptor R1 (p55) level are elevated in the substantia nigra from parkinsonian brain. J Neural Transm (Vienna). 2000;107(3):335–41.
Hunot S, Brugg B, Ricard D, Michel PP, Muriel MP, Ruberg M, et al. Nuclear translocation of NF-kappaB is increased in dopaminergic neurons of patients with parkinson disease. Proc Natl Acad Sci USA. 1997;94(14):7531–6.
Desai BS, Monahan AJ, Carvey PM, Hendey B. Blood-brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. Cell Transplant. 2007;16(3):285–99.
Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. 2009;119(1):182–92.
Chen S, Liu Y, Niu Y, Xu Y, Zhou Q, Xu X, et al. Increased abundance of myeloid-derived suppressor cells and Th17 cells in peripheral blood of newly-diagnosed Parkinson’s disease patients. Neurosci Lett. 2017;648:21–5.
Sommer A, Maxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of parkinson’s disease. Cell Stem Cell. 2018; 23(1):123–131 e6.
Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, Willemsen AT, et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol. 2005;57(2):176–9.
Pisani V, Stefani A, Pierantozzi M, Natoli S, Stanzione P, Franciotta D, et al. Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J Neuroinflammation. 2012;9:188.
Bas J, Calopa M, Mestre M, Mollevi DG, Cutillas B, Ambrosio S, et al. Lymphocyte populations in Parkinson’s disease and in rat models of parkinsonism. J Neuroimmunol. 2001;113(1):146–52.
Jiang S, Gao H, Luo Q, Wang P, Yang X. The correlation of lymphocyte subsets, natural killer cell, and Parkinson’s disease: a meta-analysis. Neurol Sci. 2017;38(8):1373–80.
Saunders JA, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ regulatory and effector/memory T cell subsets profile motor dysfunction in Parkinson’s disease. J Neuroimmune Pharmacol. 2012;7(4):927–38.
Gendelman HE, Zhang Y, Santamaria P, Olson KE, Schutt CR, Bhatti D, et al. Evaluation of the safety and immunomodulatory effects of sargramostim in a randomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinsons Dis. 2017;3:10.
Huang X, Reynolds AD, Mosley RL, Gendelman HE. CD 4+ T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol. 2009;211(1-2):3–15.
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J Leukoc Biol. 2007;82(5):1083–94.
Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, et al. Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS One. 2008;3(1):e1376.
Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation. 2018;15(1):205.
Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, et al. Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci. 2007;27(12):3328–37.
Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202(1-2):17–20.
Koziorowski D, Tomasiuk R, Szlufik S, Friedman A. Inflammatory cytokines and NT-proCNP in Parkinson’s disease patients. Cytokine. 2012;60(3):762–6.
Masliah E, Ge N, Mucke L. Pathogenesis of HIV-1 associated neurodegeneration. Crit Rev Neurobiol. 1996;10(1):57–67.
Grant I, Heaton RK, Atkinson JH. Neurocognitive disorders in HIV-1 infection. HNRC group. HIV neurobehavioral research center. Curr Top Microbiol Immunol. 1995;202:11–32.
Moore DJ, Masliah E, Rippeth JD, Gonzalez R, Carey CL, Cherner M, et al. Cortical and subcortical neurodegeneration is associated with HIV neurocognitive impairment. AIDS. 2006;20(6):879–87.
Alfahad TB, Nath A. Update on HIV-associated neurocognitive disorders. Curr Neurol Neurosci Rep. 2013;13(10):387.
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–93.
Budka H, Wiley CA, Kleihues P, Artigas J, Asbury AK, Cho ES, et al. HIV-associated disease of the nervous system: review of nomenclature and proposal for neuropathology-based terminology. Brain Pathol. 1991;1(3):143–52.
Trillo-Pazos G, Diamanturos A, Rislove L, Menza T, Chao W, Belem P, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol. 2003;13(2):144–54.
Cherner M, Masliah E, Ellis RJ, Marcotte TD, Moore DJ, Grant I, et al. Neurocognitive dysfunction predicts postmortem findings of HIV encephalitis. Neurology. 2002;59(10):1563–7.
Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, et al. Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992;42(9):1736–9.
Nottet HS, Jett M, Flanagan CR, Zhai QH, Persidsky Y, Rizzino A, et al. A regulatory role for astrocytes in HIV-1 encephalitis. An overexpression of eicosanoids, platelet-activating factor, and tumor necrosis factor-alpha by activated HIV-1-infected monocytes is attenuated by primary human astrocytes. J Immunol. 1995;154(7):3567–81.
Gelbard HA, Nottet HS, Swindells S, Jett M, Dzenko KA, Genis P, et al. Platelet-activating factor: a candidate human immunodeficiency virus type 1-induced neurotoxin. J Virol. 1994;68(7):4628–35.
Adamson DC, Wildemann B, Sasaki M, Glass JD, McArthur JC, Christov VI, et al. Immunologic NO synthase: elevation in severe AIDS dementia and induction by HIV-1 gp41. Science. 1996;274(5294):1917–21.
Jiang ZG, Piggee C, Heyes MP, Murphy C, Quearry B, Bauer M, et al. Glutamate is a mediator of neurotoxicity in secretions of activated HIV-1-infected macrophages. J Neuroimmunol. 2001;117(1-2):97–107.
Dalod M, Dupuis M, Deschemin JC, Sicard D, Salmon D, Delfraissy JF, et al. Broad, intense anti-human immunodeficiency virus (HIV) ex vivo CD8(+) responses in HIV type 1-infected patients: comparison with anti-Epstein-Barr virus responses and changes during antiretroviral therapy. J Virol. 1999;73(9):7108–16.
Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283(5403):857–60.
Janeway CJT, Travers P, Walport M, et al. T cell-mediated cytotoxicity. In: Immunobiology; The immune system in health and disease. 5th ed. New York: Garland Science; 2001.
Potula R, Poluektova L, Knipe B, Chrastil J, Heilman D, Dou H, et al. Inhibition of indoleamine 2,3-dioxygenase (IDO) enhances elimination of virus-infected macrophages in an animal model of HIV-1 encephalitis. Blood. 2005;106(7):2382–90.
Poluektova L, Moran T, Zelivyanskaya M, Swindells S, Gendelman HE, Persidsky Y. The regulation of alpha chemokines during HIV-1 infection and leukocyte activation: relevance for HIV-1-associated dementia. J Neuroimmunol. 2001;120(1-2):112–28.
Streeck H, Nixon DF. T cell immunity in acute HIV-1 infection. J Infect Dis. 2010;202(Suppl 2):S302–8.
Okoye AA, Picker LJ. CD4(+) T-cell depletion in HIV infection: mechanisms of immunological failure. Immunol Rev. 2013;254(1):54–64.
Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434(7037):1093–7.
Reuter MA, Pombo C, Betts MR. Cytokine production and dysregulation in HIV pathogenesis: lessons for development of therapeutics and vaccines. Cytokine Growth Factor Rev. 2012;23(4-5):181–91.
Clerici M, Shearer GM. A TH1-->TH2 switch is a critical step in the etiology of HIV infection. Immunol Today. 1993;14(3):107–11.
Stacey AR, Norris PJ, Qin L, Haygreen EA, Taylor E, Heitman J, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83(8):3719–33.
Barcellini W, Rizzardi GP, Borghi MO, Fain C, Lazzarin A, Meroni PL. TH1 and TH2 cytokine production by peripheral blood mononuclear cells from HIV-infected patients. AIDS. 1994;8(6):757–62.
Chevalier MF, Didier C, Girard PM, Manea ME, Campa P, Barre-Sinoussi F, et al. CD4 T-Cell Responses in Primary HIV Infection: Interrelationship with Immune Activation and Virus Burden. Front Immunol. 2016;7:395.
Schieffer M, Jessen HK, Oster AF, Pissani F, Soghoian DZ, Lu R, et al. Induction of Gag-specific CD4 T cell responses during acute HIV infection is associated with improved viral control. J Virol. 2014;88(13):7357–66.
Soghoian DZ, Jessen H, Flanders M, Sierra-Davidson K, Cutler S, Pertel T, et al. HIV-specific cytolytic CD4 T cell responses during acute HIV infection predict disease outcome. Sci Transl Med. 2012;4(123):123ra25.
Frater J, Ewings F, Hurst J, Brown H, Robinson N, Fidler S, et al. HIV-1-specific CD4(+) responses in primary HIV-1 infection predict disease progression. AIDS. 2014;28(5):699–708.
Eller MA, Blom KG, Gonzalez VD, Eller LA, Naluyima P, Laeyendecker O, et al. Innate and adaptive immune responses both contribute to pathological CD4 T cell activation in HIV-1 infected Ugandans. PLoS One. 2011;6(4):e18779.
Shebl FM, Yu K, Landgren O, Goedert JJ, Rabkin CS. Increased levels of circulating cytokines with HIV-related immunosuppression. AIDS Res Hum Retroviruses. 2012;28(8):809–15.
Maharaj NR, Phulukdaree A, Nagiah S, Ramkaran P, Tiloke C, Chuturgoon AA. Pro-inflammatory cytokine levels in HIV infected and uninfected pregnant women with and without preeclampsia. PLoS One. 2017;12(1):e0170063.
Brabers NA, Nottet HS. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006;36(7):447–58.
Han Y, He T, Huang DR, Pardo CA, Ransohoff RM. TNF-alpha mediates SDF-1 alpha-induced NF-kappa B activation and cytotoxic effects in primary astrocytes. J Clin Invest. 2001;108(3):425–35.
Viviani B, Corsini E, Binaglia M, Galli CL, Marinovich M. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience. 2001;107(1):51–8.
Barak O, Goshen I, Ben-Hur T, Weidenfeld J, Taylor AN, Yirmiya R. Involvement of brain cytokines in the neurobehavioral disturbances induced by HIV-1 glycoprotein120. Brain Res. 2002;933(2):98–108.
Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352(6288):1001–4.
Gilgun-Sherki Y, Melamed E, Offen D. Anti-inflammatory drugs in the treatment of neurodegenerative diseases: current state. Curr Pharm Des. 2006;12(27):3509–19.
Cayero-Otero MD, Espinosa-Oliva AM, Herrera AJ, Garcia-Dominguez I, Fernandez-Arevalo M, Martin-Banderas L, et al. Potential use of nanomedicine for the anti-inflammatory treatment of neurodegenerative diseases. Curr Pharm Des. 2018;24(14):1589–616.
Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–93.
Strbo N, Yin N, Stojadinovic O. Innate and adaptive immune responses in wound Epithelialization. Adv Wound Care (New Rochelle). 2014;3(7):492–501.
Snyder RJ, Lantis J, Kirsner RS, Shah V, Molyneaux M, Carter MJ. Macrophages: a review of their role in wound healing and their therapeutic use. Wound Repair Regen. 2016;24(4):613–29.
Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol. 2011;178(1):19–25.
Dheen ST, Kaur C, Ling EA. Microglial activation and its implications in the brain diseases. Curr Med Chem. 2007;14(11):1189–97.
Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1(1):14.
Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16(5):249–63.
Griot C, Burge T, Vandevelde M, Peterhans E. Bystander demyelination through antibody induced macrophage activation in canine distemper virus infection. Schweiz Arch Neurol Psychiatr (1985). 1989;140(1):39–41.
Buntinx M, Gielen E, Van Hummelen P, Raus J, Ameloot M, Steels P, et al. Cytokine-induced cell death in human oligodendroglial cell lines. II: alterations in gene expression induced by interferon-gamma and tumor necrosis factor-alpha. J Neurosci Res. 2004;76(6):846–61.
Andrews T, Zhang P, Bhat NR. TNFalpha potentiates IFNgamma-induced cell death in oligodendrocyte progenitors. J Neurosci Res. 1998;54(5):574–83.
Feldhaus B, Dietzel ID, Heumann R, Berger R. Effects of interferon-gamma and tumor necrosis factor-alpha on survival and differentiation of oligodendrocyte progenitors. J Soc Gynecol Investig. 2004;11(2):89–96.
Pouly S, Becher B, Blain M, Antel JP. Interferon-gamma modulates human oligodendrocyte susceptibility to Fas-mediated apoptosis. J Neuropathol Exp Neurol. 2000;59(4):280–6.
Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–9.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Jin X, Yamashita T. Microglia in central nervous system repair after injury. J Biochem. 2016;159(5):491–6.
Wake H, Moorhouse AJ, Nabekura J. Functions of microglia in the central nervous system—beyond the immune response. Neuron Glia Biol. 2011;7(1):47–53.
Streit WJ. Microglia and the response to brain injury. Ernst Schering Res Found Workshop. 2002;39:11–24.
Belmadani A, Tran PB, Ren D, Assimacopoulos S, Grove EA, Miller RJ. The chemokine stromal cell-derived factor-1 regulates the migration of sensory neuron progenitors. J Neurosci. 2005;25(16):3995–4003.
Turbic A, Leong SY, Turnley AM. Chemokines and inflammatory mediators interact to regulate adult murine neural precursor cell proliferation, survival and differentiation. PLoS One. 2011;6(9):e25406.
Brewer KL, Bethea JR, Yezierski RP. Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Exp Neurol. 1999;159(2):484–93.
Logan A, Green J, Hunter A, Jackson R, Berry M. Inhibition of glial scarring in the injured rat brain by a recombinant human monoclonal antibody to transforming growth factor-beta2. Eur J Neurosci. 1999;11(7):2367–74.
Schwartz M, Raposo C. Protective autoimmunity: a unifying model for the immune network involved in CNS repair. Neuroscientist. 2014;20(4):343–58.
Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–94.
Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98.
Gonzalez H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflammation. 2014;11:201.
Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35.
Subramaniam SR, Federoff HJ. Targeting microglial activation states as a therapeutic avenue in Parkinson’s disease. Front Aging Neurosci. 2017;9:176.
McGeer PL, McGeer EG. Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets. 2015;19(4):497–506.
Jin J, Lam L, Sadic E, Fernandez F, Tan J, Giunta B. HIV-1 Tat-induced microglial activation and neuronal damage is inhibited via CD45 modulation: a potential new treatment target for HAND. Am J Transl Res. 2012;4(3):302–15.
Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–8.
Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19(7):665–376.
Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101(5):455–8.
Reynolds AD, Stone DK, Mosley RL, Gendelman HE. Nitrated {alpha}-synuclein-induced alterations in microglial immunity are regulated by CD4+ T cell subsets. J Immunol. 2009;182(7):4137–49.
Lan Q, Fan H, Quesniaux V, Ryffel B, Liu Z, Zheng SG. Induced Foxp3(+) regulatory T cells: a potential new weapon to treat autoimmune and inflammatory diseases? J Mol Cell Biol. 2012;4(1):22–8.
Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248(1):156–69.
Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE. Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol. 2009;182(6):3855–65.
Gorantla S, Liu J, Sneller H, Dou H, Holguin A, Smith L, et al. Copolymer-1 induces adaptive immune anti-inflammatory glial and neuroprotective responses in a murine model of HIV-1 encephalitis. J Immunol. 2007;179(7):4345–56.
Gong N, Liu J, Reynolds AD, Gorantla S, Mosley RL, Gendelman HE. Brain ingress of regulatory T cells in a murine model of HIV-1 encephalitis. J Neuroimmunol. 2011;230(1-2):33–41.
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun. 2015;6:7967.
Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604.
Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity. 2014;40(5):642–56.
Li H, Shi B. Tolerogenic dendritic cells and their applications in transplantation. Cell Mol Immunol. 2015;12(1):24–30.
Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev. 2011;241(1):206–27.
Barratt-Boyes SM, Thomson AW. Dendritic cells: tools and targets for transplant tolerance. Am J Transplant. 2005;5(12):2807–13.
Wilson HL, Ni K, O’Neill HC. Identification of progenitor cells in long-term spleen stromal cultures that produce immature dendritic cells. Proc Natl Acad Sci USA. 2000;97(9):4784–9.
Yamazaki S, Steinman RM. Dendritic cells as controllers of antigen-specific Foxp3+ regulatory T cells. J Dermatol Sci. 2009;54(2):69–75.
Hwu P, Du MX, Lapointe R, Do M, Taylor MW, Young HA. Indoleamine 2,3-dioxygenase production by human dendritic cells results in the inhibition of T cell proliferation. J Immunol. 2000;164(7):3596–9.
Belkaid Y, Oldenhove G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity. 2008;29(3):362–71.
Luckey U, Schmidt T, Pfender N, Romer M, Lorenz N, Martin SF, et al. Crosstalk of regulatory T cells and tolerogenic dendritic cells prevents contact allergy in subjects with low zone tolerance. J Allergy Clin Immunol. 2012;130(3):781–797 e11.
Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. 2005;17(6):638–42.
Wu C, Zhang Y, Jiang Y, Wang Q, Long Y, Wang C, et al. Apoptotic cell administration enhances pancreatic islet engraftment by induction of regulatory T cells and tolerogenic dendritic cells. Cell Mol Immunol. 2013;10(5):393–402.
Gabrysova L, Nicolson KS, Streeter HB, Verhagen J, Sabatos-Peyton CA, Morgan DJ, et al. Negative feedback control of the autoimmune response through antigen-induced differentiation of IL-10-secreting Th1 cells. J Exp Med. 2009;206(8):1755–67.
Muth S, Schutze K, Schild H, Probst HC. Release of dendritic cells from cognate CD4+ T-cell recognition results in impaired peripheral tolerance and fatal cytotoxic T-cell mediated autoimmunity. Proc Natl Acad Sci USA. 2012;109(23):9059–64.
Yang X, Yao Q, Hu X, Wang W, Yin H, Ren L, et al. Rapamycin-conditioned dendritic cells induced immune tolerance through the regulation of Treg/Th17 cells in mice. Zhonghua Yi Xue Za Zhi. 2015;95(30):2469–73.
Chorny A, Gonzalez-Rey E, Delgado M. Regulation of dendritic cell differentiation by vasoactive intestinal peptide: therapeutic applications on autoimmunity and transplantation. Ann NY Acad Sci. 2006;1088:187–94.
Chorny A, Gonzalez-Rey E, Fernandez-Martin A, Pozo D, Ganea D, Delgado M. Vasoactive intestinal peptide induces regulatory dendritic cells with therapeutic effects on autoimmune disorders. Proc Natl Acad Sci USA. 2005;102(38):13562–7.
Gonzalez-Rey E, Chorny A, Fernandez-Martin A, Ganea D, Delgado M. Vasoactive intestinal peptide generates human tolerogenic dendritic cells that induce CD4 and CD8 regulatory T cells. Blood. 2006;107(9):3632–8.
Luo Z, Li J, Nabar NR, Lin X, Bai G, Cai J, et al. Efficacy of a therapeutic vaccine using mutated beta-amyloid sensitized dendritic cells in Alzheimer’s mice. J Neuroimmune Pharmacol. 2012;7(3):640–55.
Wang F, Liu H, Shen X, Ao H, Moore N, Gao L, et al. Combined treatment of amyloid-beta(1)(-)(4)(2)-stimulated bone marrow-derived dendritic cells plus splenocytes from young mice prevents the development of Alzheimer’s disease in APPswe/PSENldE9 mice. Neurobiol Aging. 2015;36(1):111–22.
Romero-Ramos M, von Euler Chelpin M, Sanchez-Guajardo V. Vaccination strategies for Parkinson disease: induction of a swift attack or raising tolerance? Hum Vaccin Immunother. 2014;10(4):852–67.
Ludewig P, Gallizioli M, Urra X, Behr S, Brait VH, Gelderblom M, et al. Dendritic cells in brain diseases. Biochim Biophys Acta. 2016;1862(3):352–67.
Collin M, McGovern N, Haniffa M. Human dendritic cell subsets. Immunology. 2013;140(1):22–30.
Sarkander J, Hojyo S, Tokoyoda K. Vaccination to gain humoral immune memory. Clin Transl Immunol. 2016;5(12):e120.
Shahaduzzaman M, Nash K, Hudson C, Sharif M, Grimmig B, Lin X, et al. Anti-human alpha-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-alpha-synuclein rat model of Parkinson’s disease. PLoS One. 2015;10(2):e0116841.
Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46(6):857–68.
Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci. 2014;34(28):9441–54.
Lindstrom V, Fagerqvist T, Nordstrom E, Eriksson F, Lord A, Tucker S, et al. Immunotherapy targeting alpha-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] alpha-synuclein mice. Neurobiol Dis. 2014;69:134–43.
Fagerqvist T, Lindstrom V, Nordstrom E, Lord A, Tucker SM, Su X, et al. Monoclonal antibodies selective for alpha-synuclein oligomers/protofibrils recognize brain pathology in Lewy body disorders and alpha-synuclein transgenic mice with the disease-causing A30P mutation. J Neurochem. 2013;126(1):131–44.
Sanchez-Guajardo V, Annibali A, Jensen PH, Romero-Ramos M. Alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72(7):624–45.
Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014;127(6):861–79.
Schneeberger A, Mandler M, Mattner F, Schmidt W. Vaccination for Parkinson’s disease. Parkinsonism Relat Disord. 2012;18(Suppl 1):S11–3.
Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, et al. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79(3):595–605.
Sigurdsson EM, Wisniewski T, Frangione B. A safer vaccine for Alzheimer’s disease? Neurobiol Aging. 2002;23(6):1001–8.
Nicolau C, Greferath R, Balaban TS, Lazarte JE, Hopkins RJ. A liposome-based therapeutic vaccine against beta -amyloid plaques on the pancreas of transgenic NORBA mice. Proc Natl Acad Sci USA. 2002;99(4):2332–7.
Mohajeri MH, Wollmer MA, Nitsch RM. Abeta 42-induced increase in neprilysin is associated with prevention of amyloid plaque formation in vivo. J Biol Chem. 2002;277(38):35460–5.
Mohajeri MH, Saini K, Schultz JG, Wollmer MA, Hock C, Nitsch RM. Passive immunization against beta-amyloid peptide protects central nervous system (CNS) neurons from increased vulnerability associated with an Alzheimer’s disease-causing mutation. J Biol Chem. 2002;277(36):33012–7.
Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, et al. Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci USA. 2001;98(25):14675–80.
Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma-/- knock-out mice. J Neurosci. 2003;23(24):8532–8.
Tariot PN, Federoff HJ. Current treatment for Alzheimer disease and future prospects. Alzheimer Dis Assoc Disord. 2003;17(Suppl 4):S105–13.
Liu R, Yuan B, Emadi S, Zameer A, Schulz P, McAllister C, et al. Single chain variable fragments against beta-amyloid (Abeta) can inhibit Abeta aggregation and prevent abeta-induced neurotoxicity. Biochemistry. 2004;43(22):6959–67.
Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61(1):46–54.
Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, et al. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8(11):1270–5.
Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38(4):547–54.
Wang CY, Wang PN, Chiu MJ, Finstad CL, Lin F, Lynn S, et al. UB-311, a novel UBITh((R)) amyloid beta peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (NY). 2017;3(2):262–72.
Tabira T. Immunization therapy for Alzheimer disease: a comprehensive review of active immunization strategies. Tohoku J Exp Med. 2010;220(2):95–106.
Lannfelt L, Relkin NR, Siemers ER. Amyloid-ss-directed immunotherapy for Alzheimer’s disease. J Intern Med. 2014;275(3):284–95.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6(8):916–9.
Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, et al. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23(9):3745–51.
Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, et al. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6(2):145–55.
Meeker RB, Poulton W, Markovic-Plese S, Hall C, Robertson K. Protein changes in CSF of HIV-infected patients: evidence for loss of neuroprotection. J Neurovirol. 2011;17(3):258–73.
Bachani M, Sacktor N, McArthur JC, Nath A, Rumbaugh J. Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. J Neurovirol. 2013;19(1):82–8.
Devadas K, Boykins RA, Hewlett IK, Wood OL, Clouse KA, Yamada KM, et al. Antibodies against a multiple-peptide conjugate comprising chemically modified human immunodeficiency virus type-1 functional Tat peptides inhibit infection. Peptides. 2007;28(3):496–504.
Rumbaugh JA, Bachani M, Li W, Butler TR, Smith KJ, Bianchet MA, et al. HIV immune complexes prevent excitotoxicity by interaction with NMDA receptors. Neurobiol Dis. 2013;49:169–76.
Rees K, Stowe R, Patel S, Ives N, Breen K, Clarke CE, et al. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: evidence from observational studies. Cochrane Database Syst Rev. 2011;(11):CD008454.
Pisanu A, Lecca D, Mulas G, Wardas J, Simbula G, Spiga S, et al. Dynamic changes in pro- and anti-inflammatory cytokines in microglia after PPAR-gamma agonist neuroprotective treatment in the MPTPp mouse model of progressive Parkinson’s disease. Neurobiol Dis. 2014;71:280–91.
Carta AR, Pisanu A. Modulating microglia activity with PPAR-gamma agonists: a promising therapy for Parkinson’s disease? Neurotox Res. 2013;23(2):112–23.
Investigators NETiPDF-Z. Pioglitazone in early Parkinson’s disease: a phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015;14(8):795–803.
Kim NK, Choi BH, Huang X, Snyder BJ, Bukhari S, Kong TH, et al. Granulocyte-macrophage colony-stimulating factor promotes survival of dopaminergic neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced murine Parkinson’s disease model. Eur J Neurosci. 2009;29(5):891–900.
Delgado M, Ganea D. Vasoactive intestinal peptide: a neuropeptide with pleiotropic immune functions. Amino Acids. 2013;45(1):25–39.
Brenneman DE, Hauser J, Spong CY, Phillips TM. Chemokines released from astroglia by vasoactive intestinal peptide. Mechanism of neuroprotection from HIV envelope protein toxicity. Ann NY Acad Sci. 2000;921:109–14.
Brenneman DE, Hauser J, Spong CY, Phillips TM, Pert CB, Ruff M. VIP and D-ala-peptide T-amide release chemokines which prevent HIV-1 GP120-induced neuronal death. Brain Res. 1999;838(1-2):27–36.
Brenneman DE, Westbrook GL, Fitzgerald SP, Ennist DL, Elkins KL, Ruff MR, et al. Neuronal cell killing by the envelope protein of HIV and its prevention by vasoactive intestinal peptide. Nature. 1988;335(6191):639–42.
Zusev M, Gozes I. Differential regulation of activity-dependent neuroprotective protein in rat astrocytes by VIP and PACAP. Regul Pept. 2004;123(1-3):33–41.
Weissmiller AM. Wu C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl Neurodegener. 2012;1(1):14.
Xiao N, Le QT. Neurotrophic factors and their potential applications in tissue regeneration. Arch Immunol Ther Exp (Warsz). 2016;64(2):89–99.
Yue X, Hariri DJ, Caballero B, Zhang S, Bartlett MJ, Kaut O, et al. Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience. 2014;258:385–400.
Wakeman DR, Redmond DE Jr, Dodiya HB, Sladek JR Jr, Leranth C, Teng YD, et al. Human neural stem cells survive long term in the midbrain of dopamine-depleted monkeys after GDNF overexpression and project neurites toward an appropriate target. Stem Cells Transl Med. 2014;3(6):692–701.
Emborg ME, Moirano J, Raschke J, Bondarenko V, Zufferey R, Peng S, et al. Response of aged parkinsonian monkeys to in vivo gene transfer of GDNF. Neurobiol Dis. 2009;36(2):303–11.
Cass WA, Peters LE. Neurturin protects against 6-hydroxydopamine-induced reductions in evoked dopamine overflow in rat striatum. Neurochem Int. 2010;57(5):540–6.
Cass WA, Peters LE. Neurturin effects on nigrostriatal dopamine release and content: comparison with GDNF. Neurochem Res. 2010;35(5):727–34.
Li T, Yu Y, Cai H. Effects of brain-derived neurotrophic factor-pretreated neuron stem cell transplantation on Alzheimer’s disease model mice. Int J Clin Exp Med. 2015;8(11):21947–55.
Daviaud N, Garbayo E, Sindji L, Martinez-Serrano A, Schiller PC, Montero-Menei CN. Survival, differentiation, and neuroprotective mechanisms of human stem cells complexed with neurotrophin-3-releasing pharmacologically active microcarriers in an ex vivo model of Parkinson’s disease. Stem Cells Transl Med. 2015;4(6):670–84.
Acknowledgements
This work was supported in part by NIH Grant AG043540, DA028555, NS036126, NS034239, MH064570, NS043985, MH062261, DOD Grant 421-20-09A and the Carol Swarts Emerging Neuroscience Fund to HEG, MH086372, MH083627, DA017618, DA037611, and MH104145. We also thank the INBRE grant from NIH (2P20GM103427) for supporting a site license to EndNote software.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Olson, K.E., Mosley, R.L., Gendelman, H.E. (2020). Neuroprotective Immunity for Neurodegenerative and Neuroinfectious Diseases. In: Jain, P., Ndhlovu, L. (eds) Advanced Concepts in Human Immunology: Prospects for Disease Control. Springer, Cham. https://doi.org/10.1007/978-3-030-33946-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-33946-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33945-6
Online ISBN: 978-3-030-33946-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)