
Abstract
Predicting the functions of the proteins from their structure is an active area of interest. The current trends of the secondary structure representation use direct letter representation of the specific secondary structure element of every amino acid in the linear sequence. Using graph representation to represent the protein sequence provides additional information about the structural relationships within the amino acid sequence. This study outlines the protein secondary structure with a novel approach of representing the proteins using protein secondary structure graph where nodes are amino acids from the protein sequence, and the edges denote the peptide and hydrogen bonds that construct the secondary structure. The developed model for protein function prediction Structure2Function operates on these graphs with a defined variant of the present idea from deep learning on non-Euclidian graph-structure data, the Graph Convolutional Networks (GCNs).
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
- Protein function prediction
- Protein secondary structure
- Protein secondary structure graphs
- Deep learning
- Graph convolutional networks
1 Introduction
Proteins are large complex molecules that play incredibly crucial roles in organisms. These molecules carry the bulk of the work in cells and are needed for the structure, function, and regulation of tissues and organs in the organisms. The role of the protein in a particular cell depends on the DNA sequence of the gene synthesizing the protein, that is, from the resulting primary sequence of amino acids. However, in organisms under given optimal conditions, proteins are not found as a chain of amino acids but have their unique form in the three-dimensional space.
Today there are many databases of proteins, their primary, secondary and tertiary structures, as well as their functions, clusters, and other information [7]. The protein databases that represent the proteins with their sequence of amino acids, such as UniProt Knowledgebase [3], grow with high rates as the protein sequencing is getting chipper and chipper. However, annotations of proteins are still a bottleneck in the proteomics research area, so the computational protein function annotation methods are mainly developed to solve this problem.
Protein function prediction is an important application area of bioinformatics that aims to predict the Gene Ontology functions (terms) associated with the proteins. Gene Ontology (GO) represents an ontology (vocabulary) consisted of terms that describe the functions of the gene products (GO terms) and how these functions are connected between them (relationships) [4]. Currently developed methods in this field make predictions by homology searching, protein sequence analysis, protein interactions, protein structure, etc. [18]. Many proteins with similar sequences have similar functions, but there are also exceptions, such that proteins with very different sequences/structures have very similar functions [20].
Predicting the functions of the proteins from their structure is an active area of interest, closely connected to predicting the structure of the proteins. One drawback of this method is the limited number of proteins with known structures, unlike the models that utilize only the protein sequence. The solution for this problem is predicting the structure of the protein using its known amino acid sequence, which is already investigated with several existing methods. The efforts in this area are mainly divided into two sub-fields, the prediction of the secondary structure and the tertiary structure of the proteins [1, 15].
As presumed from the complexity of the problem, predicting the secondary structure is a more investigative problem with methods that produce competent results. There is a clear trend of a slow but steady improvement in predictions over the past 24 years [24]. The latest techniques are approaching the theoretical upper limit of 88%–90% sufficient accuracy of predictions [23]. Hence, predicting the protein function from the secondary structure at this moment is more supportive than prediction using the ternary structure directly, mainly because of the promising solutions of using predictive models for the problem of limited structure data.
The secondary structure of the protein in currently developed models is represented as a sequence of letters indicating the affiliation of a specific amino acid from the linear sequence to one secondary structure element [10]. Consequently, the structure is described as a linear sequence of letters, similar to the primary structure - the linear sequence of amino acids. This representation of the secondary structure cannot give information about the connectivity of the amino acids, which means that we only know the particular denoted secondary element for the individual amino acid without additional information about the hydrogen bonds. This fact can be considered as a deficiency of information that is needed to extract the necessary information to predict the function by utilizing the secondary structure.
In this study, we explore the idea of generating an intelligent system capable of annotating proteins with their functions given the protein secondary structure. The secondary structure is interpreted in a novel approach, as a protein secondary structure graph. The nodes in this graph are the amino acids with their type, and these nodes are connected by two types of edges: peptide bonds and hydrogen bonds. The nodes and the peptide bonds are a representation of the primary structure, while the addition of the hydrogen bonds supplements the information with the secondary structure. This graph comprises the knowledge of the primary structure, the peptide sequence, and also the local connection between the residues.
To predict the function using the developed protein graph, an algorithm that operates with data in the form of graphs is needed. The proposed model in this study employs the recently developed deep learning approaches for non-Euclidian graph-structured data, Graph Convolutional Networks (GCNs) [6]. The architecture of the developed method combines the GNCs with a conventional neural network, to construct a model for protein function prediction called Structure2Function.
The rest of the paper is organized as follows. Section 2 includes the details of the problem, the approach for modeling the protein structure and the proposed model for protein function annotation. Section 3 presents the experimental study and evaluation of the results and Sect. 4 concludes the study.
2 Problem Definition and Framework
The annotation of the protein is reflected as a protein classification task. The proteins are expressed with secondary structure graphs, so the proposed method for protein classification performs graph classification.
The Protein Data Bank (PDB) archive [5] is a worldwide resource for 3D structure data of biological macromolecules, which includes proteins and amino acids. Protein structure inferred through experiments is preserved in a Protein Data Bank (PDB) file. This file represents every atom of the protein molecule with relative coordinates in three-dimensional space. Hence, this file describes the protein ternary structure. The DSSP program [17] is applied to derive the information about the secondary structural elements of the folded protein. DSSP is the de facto standard for the assignment of secondary structure elements in PDB entries. The hypothesis tested here determines an intelligent system proficient for function prediction of the proteins, utilizing their secondary structure represented as a graph.
2.1 Construction of Protein Secondary Structure Graphs
The protein secondary structure can be interpreted as a graph with nodes denoting amino acids and edges outlining the chemical bonds between these amino acids. The amino acid sequence of the protein is a chain of amino acids connected by peptide covalent bonds. Therefore, one type of chemical bond between amino acids is the peptide bond. Alternatively, amino acids can form hydrogen noncovalent bond with the CO and NH groups, chemical connections that participate in the formation of secondary structure. These two types of chemical bonds are the edges in the protein graph.
The hydrogen bond that forms the secondary structure is a partially electrostatic attraction between the NH group from one residue as a donor and the other residue considered as an acceptor with its CO group. Hence, the hydrogen bond can be modelled as a directed linkage between the donor residue to the acceptor residue. One residue can be a donor in one hydrogen bond, and acceptor in another hydrogen bond. Accordingly, one node in the defined graph model can have indegree and outdegree of the hydrogen edges equal to one. Notwithstanding, a single hydrogen atom can participate in two hydrogen bonds, rather than one, bonding called “bifurcated” hydrogen bond. Consequently, some of the nodes could have more than one incoming or outgoing edges.
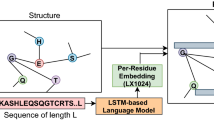
The initial backbone of the protein graph is formed utilizing the peptide sequence of the protein, through the insertion of the amino acid nodes and the peptide bond edges in the graph. DSSP method is used to construct the protein secondary structure graph. The hydrogen bonds forming the secondary structure elements identified with this program are incorporated in the primary protein graph, and the outcome is the protein secondary structure graph. Figure 1 illustrates the generation procedure of the initial protein graph and the extraction of the protein secondary structure graph. This procedure is used for proteins with known tertiary structure.

Illustration of the procedure for generating the protein secondary structure graphs. The example is the PDB entry 1EOT. Circles in the protein graph represent the amino acid residues as nodes, grey arrows are the peptide bond edges, and the orange arrows are the hydrogen bond edges. (Color figure online)
2.2 Identification of Hydrogen Bonds Forming the Secondary Protein Structure
DSSP recognizes the elements of the secondary structure through hydrogen bonding patterns [17]:
-
n-turn—represents a hydrogen bond between the group CO of the residue at the position i and the group NH of the residue \(i+n\), where \(n=3,4,5\);
-
bridge—signifies the hydrogen bonding between residues which are not near in the peptide sequence.
These two types of hydrogen bonding identify the possible hydrogen bonds between the amino acid residues. Six structure states are defined through the patterns of the specified hydrogen bonds: 310-helix (G), \(\alpha \)-helix (H), \(\pi \)-helix (I), turn (T), \(\beta \)-sheet (E), and \(\beta \)-bridge (B).
A helical structure is defined by at least two consecutive n-turn bonds of the \(i-1\) and i residues. The ends of the helical structure are left out; that is, they are not given the appropriate state of the helical structure that they start or end. Consequently, the smallest \(\alpha \)-helix structure is defined between the amino acid residues i and \(i+3\) if there are 4-turn hydrogen bonds of the \(i-1\) and i residues, meaning it has a length of at least 4, not counting the start and end residues. The smallest 310-helix structure between the residues i and \(i+2\) and the shortest \(\pi \)-helix structure between the residues i and \(i+4\) are assigned in the same way, with lengths 3 and 5 respectively. Following these rules, the subsequent n-turn H-bonding patterns define the states G, H or I. If one n-tun bond to the residue i does not have a co-occurring n-turn bond of the previous amino acid, then the residues in the range from i to \(i+n\) are denoted by the state T, where n depends on the hydrogen bond itself.
The state \(\beta \)-sheet (E) is defined on residues with at least two consecutive bridge bonds or residues surrounded by two \(\beta \)-bridge hydrogen bonds. The remaining residues with inconsistent bridge hydrogen bonds obtain the \(\beta \)-bridge state (B). For the antiparallel \(\beta \)-bridge for the residues i and j it is necessary to have two connections between \(i \rightarrow j\) and \(j \rightarrow i\) or between \(j+1 \rightarrow i-1\) and \(i+1 \rightarrow j-1\). The parallel \(\beta \)-bridge for the residues i and j is characterized by the hydrogen bonds \(j \rightarrow i-1\) and \(i+1 \rightarrow j\) or \(i \rightarrow j-1\) and \(j+1 \rightarrow i\). The smallest \(\beta \)-sheet comprises two residues in each of the segments of the two strands.
The described six states are defined according to the hydrogen bonds, while the state of the bend (S) is defined geometrically according to the angles of torsion. If an amino acid residue is not found in any of the seven states, then its state is indicated by blank space and denotes a residue that is not in any of the structural elements.
2.3 Protein Functional Annotation
Gene Ontology (GO) is an ontology (vocabulary) whose elements, GO terms, describe the functions of gene products, and how these functions are related to each other [4]. This ontology is organized as a directed acyclic graph (DAG) with relations between one term on one level with one or more terms from the previous level. The ontology is mainly divided into three domains [9]: Biological Process (BP), Molecular Function (MF), and Cellular Component (CC).
One way to get functional protein annotations (current or previous versions) is through GOA annotation files. The GOA annotations annotate the protein structure, identified with their PDB identifiers and chain number, with the GO terms of the three domains BP, MF and CC. The protein annotations were filtered by removing the annotations that are not assigned by experimental methods with experimental annotation evidence code (EXP, IDA, IPI, IMP, IGI, IEP, TAS, and IC). Additionally, the annotations that have a qualifier “NOT”, which indicates that the given protein is not associated with the given GO term, are also removed.
Since the structure of the ontology is designed as a directed acyclic graph (DAG), the task of protein function annotation is analyzed as a hierarchical multi-label classification. This concept indicates that for given predictions, the parent term should have a likelihood of occurrence, at least as the maximum of the probabilities of his descendant terms in the ontology.
2.4 Proposed Method: Structure2Function
The protein secondary structure graph is represented as \(G=\{V,E,X\}\), where V is the set of nodes, E represents the set of edges, X is a matrix for the node content, and \(N=|V|\) indicates the number of nodes. Every node has a label for the amino acid identity. There are 20 amino acids in the standard genetic code, two additional amino acids incorporated with specific processes and three other states for ambiguity (unknown amino acid or undistinguished amino acids). Therefore, every node has a one-hot encoding vector for the amino acid label with length 25. These vectors form the X matrix, where \(X \in \mathbb {R}^{(N \times 25)}\).
There are two types of edges in the graph, hydrogen and peptide bond, so every edge has a label indicating the edge type. These labels are one-hot encoded; therefore, the edges have a discrete feature vector for their class. In general, the graph is directed, with no isolated nodes and no self-loops.
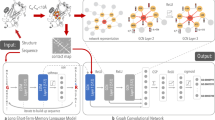
The architecture of the proposed method for protein classification is illustrated in Fig. 2. The model mainly consists of two major parts: graph convolutional network and fully connected neural network. The graph convolutional network maps the graph into latent representation, whereas the fully connected neural network takes the graph representation and generates the class predictions. Graph Convolutional Networks (GCNs) are already implemented in several studies for solving the task of graph classification [11, 12, 25].
The convolutional neural network is defined following the Message Passing Neural Networks (MPNN) framework [13]. The framework defines two phases, a message passing phase, and a readout phase. The message passing phase updates the node representation for one node by aggregating the messages from the neighborhood of the node.
The message \(m_{uv}\) of node v from neighboring node u in our model is defined as a function from the hidden representation of node u from the previous layer \(h_u\) and the edge vector \(e_{uv}\). This function is a neural network that takes the concatenation of \(h_u\) and \(e_{uv}\) as input and generates the message:
where \(W_1\) is a weight matrix, \(b_1\) is a bias vector, \(\sigma \) denotes an activation function, and \([\cdot ]\) represents concatenation. Since the graph is directed, there are two separate message channels for incoming edges \(m_v^{in}\) and outgoing edges \(m_v^{out}\), computed as the sum of individual neighboring messages:
where \(N(v)^{in}\) and \(N(v)^{out}\) are the sets of neighbors from incoming and outgoing edges respectively.
The final step in the graph convolution is the node update function that updates the node hidden representation based on the \(m_v^{in}\) and \(m_v^{out}\) messages. This update function is interpreted as a neural network with \(h_v^{'}\), \(m_v^{in}\) and \(m_v^{out}\) as inputs, where \(h_v^{'}\) is the previous node hidden representation:
For the first layer, the node hidden representations are the feature vectors from the matrix X. Three of the defined layers are stacked together to form a more in-depth model. Also, we use residual connections [14] between the layers to enable the transfer of the information from the previous layer:
where l refers to the layer number, \(H^i\) is a matrix with node hidden representations, \(GCN(\cdot )\) represents the graph convolutional layer, and A is the adjacency matrix.
The final step in the first part of the model is the accumulation of the node representations into the graph feature vector, the readout phase. In this process, all vector representations for all nodes are aggregated to make a graph-level feature representation. The final graph vector representation is defined as
where \(h_V^L\) is the hidden representation of node v from the last layer. The node and graph representations are defined as 500-dimensional vectors.

Visualization of the architecture of the proposed method.
The graph latent representation vector is input in the second part of the network, which consists of three fully connected (FC) layers of size 1024, 512 and N neurons, where N represents the number of terms (classes) in the corresponding ontology. The last layer gives the probabilities for each class as output, so a sigmoid function activates it. These settings create a standard multi-label classification, where each output is a value of 0 to 1.
The last layer is the Hierarchical Class Correction layer designed to capture the hierarchical multi-label classification property of the problem. This layer is composed of a 1-0 matrix with rows indicating the encoding of the relationship between the parent term and its descendant terms. That is, each term in the matrix has a vector where the elements at its position and the position of its descendants have value 1, and the remaining elements have value 0. Once the vector with predictions is element-wise multiplied with this matrix, there is an operation of max-pooling that extracts the maximum probability values for each term.
Benchmark Dataset. To create the train and test sets two timeframes of protein annotations are used: historical annotations \(t_0\) from 05 July 2017 and current annotations \(t_1\) from 13 February 2019. The approach in CAFA challenge [13] is applied to determine the separation of proteins into the training set and testing set. This method uses the proteins in both time annotations to create two kinds of sets for each GO ontology:
-
No-knowledge data (NK): protein structures annotated with a term in current annotations \(t_1\) and are not annotated in historical annotations \(t_0\) with terms from an ontology;
-
Limited-knowledge data (LK): proteins which are annotated with a term from an ontology in \(t_1\) and are not annotated in the corresponding ontology in \(t_0\).
The protein structures found in the NK and LK sets are part of the test set, that is, the benchmark set, while the remaining proteins are assigned to the train set. Table 1 shows the size of the train and test sets for every type of GO ontology.
The terms from each type of ontology that annotate less than ten protein chains are excluded from consideration in the training phase of the model. The model is not aware of these functional terms; accordingly, it is not able to learn to predict them.
Model Implementation and Optimization. The model was implemented using Tensorflow and Keras deep learning frameworks. The learning of the model was performed using the AMSGrad variant [19] of the Adam optimizer from [22] with a learning rate set to 0.001. The models are trained with 150 episodes with a batch size of 1, and 1000 steps per epoch. The most appropriate model is selected from the epoch with the lowest value of the loss function of the validation set, and accordingly, this version is used to evaluate the predictions of the particular model.
2.5 Evaluation
The evaluation of the protein-annotated method is based on the methods proposed in the Critical Assessment of the Protein Function Annotation Algorithms (CAFA) experiment [16, 21]. Protein-based measures evaluate the results of classification methods that for a given protein predict the terms with which the corresponding protein has been annotated. Measures are needed that will assess the predictions of a model intended for multilabel classification. For this type of evaluation, the \(F_{max}\) measure, \(S_{min}\) measure, as well as the precision-recall curve, will be used.
The precision pr, the recall rc and \(F_{max}\) are calculated with the following formulas:
where t is a decision threshold, m(t) refers to the number of proteins for which at least one term is provided with a confidence greater than or equal to the defined threshold t, n is the number of proteins in the test set, \(I(\cdot )\) is an indicator function, \(P_i (t)\) is a set of predicted terms with a confidence greater than or equal to t for the protein i, while \(T_i\) is the set of real terms for the protein i. Mainly, the precision is the percentage of the predicted terms that are relevant, and the recall is the percentage of the relevant terms that have been predicted.
The other type of measures includes the remaining uncertainty ru, the misinformation mi, and the minimum semantic distance \(S_{min}\). These measures are defined as:
where IC(f) is the informative content of the term f of the ontology.
3 Experiments: Discussion of Results
The main topic of this section is the discussion of the success of the proposed model and its comparison with related models designed to solve the same problem. The Naive method and the BLAST method are applied as baseline models to compare the proposed method in this project. The Naive approach predicts terms with their relative frequency in the training set; that is, each protein has the same predictions [8]. Consequently, if specific terms are more frequent in the train set, then these terms are predicted with higher confidence than other terms that are usually more explicit terms.
The BLAST method uses the Basic Local Alignment Search Tool – BLAST [2], which compares the protein sequences of the test set with the protein sequences of the training set. BLAST’s basic idea is a heuristic search for alignment of sequences that have a high estimation, between the searched protein and the protein sequences from the database. The result after the search consists of the detected protein sequences along with the percentage of identical hits in the alignment. This value is used as a confidence value to annotate the protein with terms of the discovered proteins, that is, the terms are predicted with the corresponding value for identical hits. If one term is predicted multiple times (from various proteins), the highest confidence value is retained.
Table 2 contains the results of the experiments of protein function prediction with the test set. The results from this table are obtained by the same train and test sets defined previously, for training and testing respectively. For all ontologies, both the BLAST and Structure2Function methods are better than the random standard of the Naive approach.
The BLAST method achieved the highest \(F_{max}\) value for the BP ontology, although this value is close to the performance of Structure2Function model. However, according to the evaluation using the \(S_{min}\) metric, the Structure2Function method is a more refined method for this ontology. The \(S_{min}\) metric gives insight to the model that predicts more informative terms which are more desirable since it weights GO terms by conditional information content. Accordingly, the model with the lowest \(S_{min}\) value predicts more precise terms, than general terms.
The initial results of the Naive method for the ontology MF showed a high dominant \(F_{max}\) value, which is a consequence of a large number of proteins that are annotated only with the term GO: 0005515 (protein binding) and its parent GO: 0005488 (binding). This significant part of the exclusive annotations introduces great bias in the data, to the point where the Naive method has irrationally good results. Therefore, filtering the test set for the MF ontology has been made to remove proteins annotated only with these two terms. The train set applied to train the Structure2Function model was filtered in the same way, since the affinity of the set to these two terms was seen as a problem in the initial testing of the model. The evaluation for this ontology points the Structure2Function method as the best method according to the \(F_{max}\) metric, and \(S_{min}\) opposes the BLAST method as the best approach.
The CC ontology is the smallest ontology; namely, the proteins of the test set are annotated with a small number of relatively more general terms that are often used in the train set. For this ontology, the Structure2Function method is the superior approach according to \(F_{max}\) and \(S_{min}\) metric.
To visualize the results from Table 2, Fig. 3 depicts the curves for the ratio of precision-response measures. The perfect classifier would be characterized by \(F_{max}=1\) corresponding to the point (1, 1) in the precision-recall plane. These curves confirm the results and conclusions drawn from Table 2.

Precision-recall curves of the experiments using the defined test set.
The train and test sets for protein annotation contain information on the annotations that have been confirmed by experiments, but there is a shortcoming in defining annotations that are not plausible, that is negative samples. Thus, in the predictions obtained with the models, new annotations of a given protein are received, and with the proposed evaluation measures they are assessed as wrong predictions, but there are no conditions to determine whether the new predicted terms represent a new knowledge or model error. Additional empirical support is required for new terms to eliminate this problem so that it is possible to determine with higher confidence whether these terms need to be rejected or confirmed.
The analysis of the experimental results demonstrated by the Structure2Function model proposed in this study validates the direction of further research. The GCN proves its ability to interpret protein secondary structure graphs, with amino acids as nodes and bonds as edges. Further work should include the improvement of this model as well as its extension. One drawback of this method is the limited number of proteins with known structures. The solution for this problem is predicting the secondary structure of the protein using its known amino acid sequence, which is already investigated with several existing methods. Therefore, our future work includes the problem of predicting the protein secondary structure using the primary structure, for the proteins with unknown secondary structure, and assigning functions to these proteins.
4 Conclusions
Proteins are vital components chargeable for carrying out functionalities in living organisms. That is why it is necessary to know their functions. Today, in contrast to a large number of known protein sequences, the number of known functional protein annotations are still in a small amount. Therefore, automatic detection of protein functions is an actively investigated area.
Recent research examines the protein function annotation with the aid of the information derived from protein sequences, protein interactions, and their structure. This study reviews the ability for protein annotation of a model consisted of Graph Convolutional Neural Networks (GANs), called Structure2Function. The model tries to map the patterns from protein secondary structure into protein function terms. The protein secondary structure is modeled in a novel way as a graph where the amino acids are represented as nodes, and the edges are the peptide and hydrogen bonds forming the secondary structure. The experiments employed to compare the proposed method to baseline models verify the tested hypothesis of building an intelligent model proficient for protein function prediction, utilizing their secondary structure represented as a graph.
References
Al-Lazikani, B., Jung, J., Xiang, Z., Honig, B.: Protein structure prediction. Curr. Opin. Chem. Biol. 5(1), 51–56 (2001)
Altschul, S.F., et al.: Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res. 25(17), 3389–3402 (1997)
Apweiler, R., et al.: Uniprot: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 (2004)
Ashburner, M., et al.: Gene ontology: tool for the unification of biology. Nat. Genet. 25(1), 25 (2000)
Berman, H.M., et al.: The protein data bank. Nucleic Acids Res. 28(1), 235–242 (2000)
Bronstein, M.M., Bruna, J., LeCun, Y., Szlam, A., Vandergheynst, P.: Geometric deep learning: going beyond euclidean data. IEEE Signal Process. Mag. 34(4), 18–42 (2017)
Chen, C., Huang, H., Wu, C.H.: Protein bioinformatics databases and resources. In: Wu, C.H., Arighi, C.N., Ross, K.E. (eds.) Protein Bioinformatics. MMB, vol. 1558, pp. 3–39. Springer, New York (2017). https://doi.org/10.1007/978-1-4939-6783-4_1
Clark, W.T., Radivojac, P.: Analysis of protein function and its prediction from amino acid sequence. Proteins Struct. Funct. Bioinf. 79(7), 2086–2096 (2011)
Gene Ontology Consortium: The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 32(Suppl. 1), D258–D261 (2004)
Crooks, G.E., Brenner, S.E.: Protein secondary structure: entropy, correlations and prediction. Bioinformatics 20(10), 1603–1611 (2004)
Dai, H., Dai, B., Song, L.: Discriminative embeddings of latent variable models for structured data. In: International Conference on Machine Learning, pp. 2702–2711 (2016)
Duvenaud, D.K., et al.: Convolutional networks on graphs for learning molecular fingerprints. In: Advances in Neural Information Processing Systems, pp. 2224–2232 (2015)
Gilmer, J., Schoenholz, S.S., Riley, P.F., Vinyals, O., Dahl, G.E.: Neural message passing for quantum chemistry. In: Proceedings of the 34th International Conference on Machine Learning, vol. 70, pp. 1263–1272. JMLR. org (2017)
He, K., Zhang, X., Ren, S., Sun, J.: Deep residual learning for image recognition. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp. 770–778 (2016)
Jiang, Q., Jin, X., Lee, S.J., Yao, S.: Protein secondary structure prediction: a survey of the state of the art. J. Mol. Graph. Model. 76, 379–402 (2017)
Jiang, Y., et al.: An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 17(1), 184 (2016)
Kabsch, W., Sander, C.: Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22(12), 2577–2637 (1983)
Kihara, D.: Protein Function Prediction: Methods and Protocols. Humana Press, Totowa (2017)
Kingma, D.P., Ba, J.: Adam: a method for stochastic optimization. CoRR abs/1412.6980 (2014)
Pearson, W.R.: Protein function prediction: problems and pitfalls. Curr. Protoc. Bioinform. 51(1), 4–12 (2015)
Radivojac, P., et al.: A large-scale evaluation of computational protein function prediction. Nat. Methods 10(3), 221 (2013)
Reddi, S.J., Kale, S., Kumar, S.: On the convergence of adam and beyond. In: Proceedings of the International Conference on Learning Representations (2018)
Rost, B.: Protein secondary structure prediction continues to rise. J. Struct. Biol. 134(2–3), 204–218 (2001)
Yang, Y., et al.: Sixty-five years of the long march in protein secondary structure prediction: the final stretch? Briefings Bioinform. 19(3), 482–494 (2016)
Zhang, M., Cui, Z., Neumann, M., Chen, Y.: An end-to-end deep learning architecture for graph classification. In: Thirty-Second AAAI Conference on Artificial Intelligence (2018)
Acknowledgements
This work was partially financed by the Faculty of Computer Science and Engineering at the Ss. Cyril and Methodius University, Skopje, North Macedonia. The computational resources used for this research were kindly provided by MAGIX.AI and the NVIDIA Corporation (a donation of a Titan V GPU to Eftim Zdravevski).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this paper

Cite this paper
Stojanovska, F., Ackovska, N. (2019). Protein Secondary Structure Graphs as Predictors for Protein Function. In: Gievska, S., Madjarov, G. (eds) ICT Innovations 2019. Big Data Processing and Mining. ICT Innovations 2019. Communications in Computer and Information Science, vol 1110. Springer, Cham. https://doi.org/10.1007/978-3-030-33110-8_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-33110-8_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-33109-2
Online ISBN: 978-3-030-33110-8
eBook Packages: Computer ScienceComputer Science (R0)