
Abstract
Renal cell carcinoma (RCC) and its treatment have consistently remained in the foreground as one of the most rapidly evolving areas in the ever-expanding field of solid tumor oncology. Significant developments have occurred over the past two decades in the clinical landscape that have vastly enhanced comprehension of etiopathogenesis of RCC and its modalities of management including advancements in minimally invasive surgical techniques, employment of focal therapy, increased renal biopsy-based approach, advancements in immunotherapy, adoption of active surveillance strategies, and the use of targeted treatment strategies for patients with advanced disease. Efforts aimed at morphologically grouping specific cancers into distinct pathologic subtypes have not only allowed a common descriptive language, but are helping to crystallize the understanding of RCC’s molecular origins and its clinical behavior. It is these improved insights into the similarities and differences among RCC variants that offer clinical and therapeutic opportunities to improve patient care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Renal cell carcinoma
- WHO classification of renal tumors
- Collecting duct carcinoma
- Acquired cystic disease-associated RCC
- Renal medullary carcinoma
- Clear cell renal cell carcinoma
- Papillary renal cell carcinoma
- Chromophobe renal cell carcinoma
- Mucinous spindle and tubular cell carcinoma
Renal cell carcinoma (RCC) and its treatment have consistently remained in the foreground as one of the most rapidly evolving areas in the ever-expanding field of solid tumor oncology. Significant developments have occurred over the past two decades in the clinical landscape that have vastly enhanced comprehension of etiopathogenesis of RCC and its modalities of management including advancements in minimally invasive surgical techniques, employment of focal therapy, increased renal biopsy-based approach, advancements in immunotherapy, adoption of active surveillance strategies, and the use of targeted treatment strategies for patients with advanced disease. Efforts aimed at morphologically grouping specific cancers into distinct pathologic subtypes have not only allowed a common descriptive language, but are helping to crystallize the understanding of RCC’s molecular origins and its clinical behavior. It is these improved insights into the similarities and differences among RCC variants that offer clinical and therapeutic opportunities to improve patient care.
Renal neoplasms represent a group of heterogeneous tumors with various genetic and epigenetic abnormalities that are reflected in their histopathologic features and molecular profiles [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16]. Improved comprehension of the morphology, immunohistochemistry, genomics, and epidemiology of renal cell tumors has resulted in the identification of novel morphologic as well as molecular features. Therefore, the classification of renal cell tumors has recently been revised and published in the 2016 World Health Organization (WHO) classification [1]. This review briefly summarizes the pathologic, molecular, and epidemiologic features of the major subtypes of renal cell tumors.
WHO 2016 Classification of Renal Tumors
The revised WHO classification is based on advances in the understanding of newly identified characteristics of the molecular pathological epidemiology of renal cell tumors. The majority of the International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia [17] was adopted for the revised 2016 WHO classification of renal cell tumors [1].
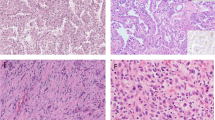
The various subtypes of renal cell tumors are based on characteristic morphologic features (Table 5.1) [1]. The major subtypes are clear cell RCC (CCRCC) (Fig. 5.1a), papillary RCC (PRCC) (Fig. 5.1b), and chromophobe RCC (ChRCC) (Fig. 5.1c) comprising 65–70%, 15–20%, and 5–7% of all RCCs, respectively. The designations of these various subtypes are based on their predominant cytoplasmic staining and cellular features (e.g., CCRCC, ChRCC, and renal oncocytoma), architectural features (e.g., PRCC), and combinations of these features (e.g., clear cell papillary RCC [CCPRCC]). Other subtypes of renal cell tumors are based on the anatomical location of the tumor (e.g., collecting duct and renal medullary carcinomas), association with renal disease (e.g., acquired cystic disease-associated RCC [ACD-associated RCC]), defining molecular alterations (e.g., microphthalmia transcription factor [MiT] family translocation RCC and succinate dehydrogenase-deficient RCC [SDH-deficient RCC]), and familial predisposition (e.g., hereditary leiomyomatosis and RCC-associated RCC [HLRCC-associated RCC]) [1].

Well-circumscribed clear cell renal cell carcinoma confined to kidney parenchyma
Clear Cell Renal Cell Carcinoma
CCRCC arises in epithelial cells lining the proximal tubule [19]. Although it can affect people of all ages including children, most of these tumors develop in patients older than 40 years of age with a male predominance and a male-to-female ratio of approximately 1.5: 1 [20].
The vast majority of cases of CCRCC have characteristic cytogenetic abnormalities that involve loss of genetic material from the short arm of chromosome 3 (3p) and mutations in the VHL gene [21,22,23,24,25,26,27]. The VHL gene, which is located at 3p25-26 and serves as a tumor suppressor gene, has been identified through studies of patients with VHL disease [23, 27,28,29]. One copy of VHL is either mutated or silenced in 90% of sporadic CCRCCs, whereas another copy is typically lost through 3p deletions, according to the comprehensive molecular profiling of CCRCCs by The Cancer Genome Atlas (TCGA) [2]. The biallelic loss of VHL allows for the inappropriate stabilization of hypoxia-inducible factors (HIFs), which results in a proangiogenic gene expression signature that is an important step in the carcinogenesis of CCRCC [30, 31].
According to the TCGA, CCRCCs are characterized by recurrent mutations in the PI3K/AKT/MTOR pathway (a potential therapeutic target), mutations in SETD2 (associated with widespread DNA hypomethylation), and mutations involving the SWI/SNF chromatin remodeling complex (PBRM1, ARID1A, and SMARCA4). Aggressive CCRCCs demonstrate a metabolic shift [2]. Other genes may be involved as tumor suppressors and lead to development of CCRCC, particularly 3p14.2 deletions, likely resulting in inactivation of the FHIT gene, as well as tumor suppressor genes at 3p12.176. A continuous deletion from 3p14.2-p25, including the FHIT and VHL genes, can be identified in up to 96% of cases [32]. Following the initiating event involving the 3p gene, additional genetic alterations occur in clonal tumor cell populations resulting in tumor progression and metastatic disease. Consequently, these additional genetic abnormalities, when detectable, are often associated with higher histologic grade, higher pathologic stage, and an adverse prognosis. The genetic abnormalities associated with these effects are loss of 9p, 14q, and loss of heterozygosity on chromosome 10q around the PTEN/MAC locus [33].
CCRCC may arise in a familial setting, especially in cases of VHL disease. In addition, 35–45% of affected patients with VHL disease develop bilateral multifocal CCRCC. Onset of renal carcinoma in patients with VHL disease is often early; clinically evident renal cancer has been reported in adolescence, and the mean age at diagnosis is 39 years. Historically, without treatment, up to 40% of patients with VHL disease died of advanced renal carcinoma. In VHL disease, patients are born with a germline defect in one of the two alleles of the VHL gene, located on chromosome 3p9.25-26, which functions as a tumor suppressor. Loss of the second allele results in clinical disease expression [34]. Additional heritable settings in which clear cell RCC may develop include families segregating constitutional chromosome 3 translocations as well as families with succinate dehydrogenase B (SDHB)-associated heritable paraganglioma [35, 36].
On gross examination, CCRCC ranges in size from subcentimeter lesions to large masses weighing several kilograms and an average diameter of 7 cm. These tumors are usually unilateral and solitary; bilaterality and multicentricity are reported in familial cases. Imaging studies frequently show a bosselated mass protruding from the external surface. The cut surface often demonstrates a characteristic golden-yellow appearance due to abundant cholesterol and other phospholipids within the tumor cells. The cut surface is typically heterogeneous with areas of gray-white fibrosis and recent or remote hemorrhage. These tumors exhibit an expansile pushing growth pattern and are either well demarcated from the adjacent uninvolved parenchyma by a variably thick fibrous pseudocapsule or widely infiltrate the adjacent renal parenchyma. Cystic change and foci of calcification are commonly present, often in association with areas of necrosis. Figures 5.1, 5.2, 5.3, 5.4, 5.5, 5.6, and 5.7 illustrate different growth patterns and gross features that are helpful in staging renal carcinomas in nephrectomy specimens.

Polycystic disease with characteristic gross appearance of bivalved kidney and solitary circumscribed clear cell renal cell carcinoma identified in upper right corner

Clear cell renal cell carcinoma widely infiltrating kidney parenchyma with invasion into perinephric adipose tissue

High-grade clear cell renal cell carcinoma with direct invasion and effacement of overlying adrenal gland

Clear cell renal cell carcinoma penetrating through capsular surface and extending into perinephric adipose tissue (pT3)

Widespread invasion of renal sinus adipose tissue in clear cell renal cell carcinoma (pT3)

Large tumor thrombus occluding lumen of renal vein (arrow) in a case of clear cell renal cell carcinoma
The microscopic appearance of CCRCC includes a variety of architectural patterns; tumor cells are arranged in a variable combination of compact nests, alveolar, acinar, solid sheet-like and cystic patterns, separated by an arborizing network of thin-walled blood vessels (Fig. 5.8). Cystic areas are filled with extravasated erythrocytes or eosinophilic fluid. Occasional, small papillary structures lined by clear cells may be present focally, but they almost always represent a minor component of the tumor.

Clear cell renal cell carcinoma demonstrating characteristic features with nests of tumor cells containing clear cytoplasm and an arborizing network of thin-walled blood vessels
In CCRCC, the tumor cells contain distinct cell membranes and optically clear cytoplasm owing to loss of cytoplasmic lipids and glycogen during histologic processing. Some cases of CCRCC comprise of varying areas demonstrating tumor cells with granular eosinophilic cytoplasm; such foci are more often seen in high-grade cancer or near areas of hemorrhage or necrosis. The nuclei of CCRCC show considerable variation in size, shape, and nucleolar prominence, as discussed in the section on grading, and these features are assessed when assigning a nuclear grade to an individual tumor.
Sarcomatoid and rhabdoid differentiation are seen in approximately 5% of cases and carry prognostic significance (Figs. 5.9, 5.10, and 5.11) [37,38,39]. Other uncommon histologic variations with unknown prognostic significance have been described in RCC, including intra- or extracellular hyaline globules, basophilic cytoplasmic inclusions, abundant multinucleated giant cells, sarcoid-like granulomas, myospherulosis, and dense inflammation [40,41,42,43,44].

Clear cell renal cell carcinoma with sarcomatoid and rhabdoid growth patterns as demonstrated by the presence of fleshy nodular areas in the tumor on gross examination

Sarcomatoid RCC with a malignant spindled-tumor cell morphology

Rhabdoid differentiation in renal cell carcinoma with tumor cells containing eccentric nuclei and abundant eosinophilic cytoplasm
Immunohistochemically, clear cell carcinoma typically shows positive immunostaining for vimentin, cytokeratin (CK) AE1/AE3, RCC antigen, CD10, PAX2, PAX8, and carbonic anhydrase-IX (CA IX). Immunostaining reactions for HMWCK, CK7, CK20, mucin 1, cell surface associated (MUC1), parvalbumin, AMACR, kidney-specific cadherin, and CD117 are negative in most cases [45,46,47,48,49,50,51,52,53,54,55,56,57].
Pathologic staging most accurately predicts the prognosis for patients with CCRCC [58,59,60]. Major factors outlining the prognosis in tumors with same stage include tumor grade, tumor necrosis, and the presence or absence of sarcomatoid or rhabdoid differentiation. Tumors with higher grades are associated with progressively worsening prognosis [58]. Tumor necrosis accounting for more than 10% of the total tumor volume is more likely to have an adverse outcome. Sarcomatoid and rhabdoid differentiation are seen in less than 10% of cases of CCRCC and associated with a worse prognosis [59, 60]. Cases with sarcomatoid differentiation have a 5-year survival rate of 15–22%, and in cases with rhabdoid differentiation, the median survival ranges from 8 to 31 months [59, 60].
Multilocular Cystic Renal Neoplasm of Low Malignant Potential
This tumor was previously referred to as multilocular cystic RCC and encompasses renal neoplasms with a fibrous pseudocapsule that are composed entirely of cysts and septa with no expansile solid nodules; the septa should contain aggregates of low-grade tumor cells with clear cytoplasm [61,62,63,64,65,66]. Combined experience with more than 200 patients with follow-up times longer than 5 years indicates no recurrence or cancer-related mortality in these patients; however, the natural history of this tumor is unknown because all these reported cases were treated with definitive surgery. Consequently, multilocular cystic renal neoplasm of low malignant potential (MCRNLMP) is now the WHO-recommended term for this lesion.
It constitutes approximately up to 5% of all RCCs and has a male predominance with a male-to-female ratio of 2–3: 1. The age ranges from 20 to 76 years; most patients are above 50 years of age, and women tend to present at a younger age than do men [61,62,63,64,65,66].
Most patients in this cohort are asymptomatic and these neoplasms are incidental lesions with few cases presenting with a palpable mass, gross hematuria, abdominal or back pain. Most patients have no biochemical abnormalities. Imaging studies usually outline a complex cystic mass that may have focal calcification.
Grossly, MCRNLMP ranges from subcentimeter lesions to large tumors measuring over 10 cm in greatest dimension. The tumor is usually a unilateral and solitary well-circumscribed mass composed entirely of variably sized cysts separated from each other by thin fibrous septae and from adjacent renal parenchyma by a fibrous wall (Fig. 5.12); however, it can be multifocal as well as bilateral [61,62,63,64,65,66]. The cysts contain clear or hemorrhagic fluid. Necrosis is not seen and there are no grossly identifiable nodules expanding the septa, a feature that differentiates this tumor from extensively cystic conventional CCRCC.

Multilocular cystic renal cell neoplasm of low malignant potential, a well-circumscribed tumor composed entirely of variably sized cysts
Microscopically, the cysts are lined by a single layer of clear tumor cells with occasional multilayered epithelium and rare papillary structures; some cysts lack any lining epithelium (Fig. 5.13a, b). The tumor cells have variable amounts of cytoplasm that may be clear or lightly eosinophilic. Many of these tumors show calcifications within the septa, and metaplastic bone formation is occasionally encountered. Within the septa in all cases are clusters of low-grade tumor cells with clear cytoplasm (Fig. 5.13b). These cells are often difficult to distinguish from histiocytes or from lymphocytes with surrounding retraction artifact. Increased vascularity is sometimes associated with septal tumor cell clusters, and this feature may be helpful in recognition of this entity. There should be no nodular expansion of clear tumor cells in the septa, a feature that serves to mainly distinguish it from extenisvely cystic CCRCC. In challenging cases, the epithelial nature of the tumor cells can be confirmed by their immunoreactivity to antibodies against cytokeratin and EMA; results of immunostains for histiocytic markers are negative. Tumor cells are also strongly immunoreactive for PAX8 and CA-IX aiding in this diagnosis [61,62,63,64,65,66].

Multilocular cystic renal neoplasm of low malignant potential. Note the low-power appearance of variably sized cysts filled with eosinophilic fluid (a) and clusters of low-grade tumor cells with clear cytoplasm within the septae (b)
VHL gene mutations have been reported in about 25% in MCRNLMP, and there is no difference in the status of chromosome 3p deletion between low-grade CCRCC and this neoplasm, supporting the hypothesis that it is a subtype of CCRCC [64,65,66].
Papillary Renal Cell Carcinoma
PRCC, the second most common type of RCC, bears characteristic cytogenetic, gross, and histologic features that distinguish it from other types of RCC [67].
Most of these tumors are sporadic, but some occur in a familial setting with hereditary papillary renal carcinoma (HPRCC), an inherited renal cancer characterized by mutations in the MET oncogene at 7q31 and by a predisposition to develop multiple bilateral papillary renal tumors. MET mutations have been detected in approximately 13% of patients with PRCC who have no family history of renal tumors [68,69,70,71].
The mean age of patients with PRCC ranges from 52 to 66 years and the male-to-female ratio of 2.4: 1 [67, 72]. The common presenting signs and symptoms of RCC are noted. PRCC is more likely to be multifocal and necrotic than other common RCC subtypes and exhibits an association with end-stage renal disease. Each papillary tumor arises independently in cases of multiple PRCCs without a family history of renal tumors [73].
Grossly, PRCC is typically well circumscribed, the vast majority of cases are confined to the renal parenchyma [67, 72,73,74]. Multifocality is identified particularly in cases of HPRC [68,69,70,71]. A thick fibrous pseudocapsule is present in up to two-thirds of mirroring the extent of hemorrhage and necrosis present in the tumor. The cut surface ranges from light gray tan to golden yellow to red brown, depending upon the hemorrhage and hemosiderin accumulation in accompanying macrophages as well as stroma (Fig. 5.14) [ 67, 72,73,74].

Well-circumscribed papillary renal cell carcinoma with hemorrhage and hemosiderin deposition grossly imparting a brown appearance to the cut surface
Microscopically, PRCC is composed of varying proportions of papillary and tubular structures and also contains variable cystic change with papillary excrescences or with tumor infiltrating the cyst wall [67, 72,73,74]. “Papillary” morphology is also encountered in a host of other kidney tumors and is thus not specific or entirely representative of this neoplasm. Papillae are lined by a single layer of tumor cells with variable pseudostratification [74]. The papillary stalks contain fibrovascular cores and a variable degree of macrophages that is not linked to the extent of accompanying hemorrhage and/or necrosis. The tumor papillae may be slender and easily recognizable or compact and tightly packed resulting in a solid appearance; and in some tumors, the papillae are arranged in long parallel arrays, imparting a trabecular appearance [72].
A few cases of papillary carcinoma exhibit solid areas composed of spindle cells with low-grade nuclear features, admixed with variable amounts of tumor with tubular or papillary architecture [75, 76]. In one series of such cases, all were confirmed as PRCC by molecular studies [75]. The spindle cell components of such tumors do not meet the criteria for sarcomatoid differentiation and are therefore not associated with a worse prognosis.
PRCC can be categorized into two morphologic classes, which have been designated types 1 and 2, based on morphologic criteria, immunohistochemical staining, clinicopathologic staging parameters, and survival outcome analysis [74, 77,78,79]. Tumors in both categories share several features including tumor-associated inflammatory infiltrate, extensive necrosis, psammoma bodies, cholesterol clefts, hemorrhagic background, and foci of dystrophic calcification yet exhibit significant differences in molecular profiles and overall outcomes [74, 78].
Type 1 PRCC is composed of papillae covered by a single layer of low-grade tumor cells containing small round to ovoid nuclei with inconspicuous nucleoli and minimal pale to clear cytoplasm (Fig. 5.15). Tubular profiles in these neoplasms have similar lining cells. The short, complex papillae sometimes impart a glomeruloid appearance. The papillae of type 1 tumors are usually thin, delicate, and often short and are frequently edematous [74, 78]. Aggregates of foamy macrophages are commonly seen within the papillary cores or in between aggregates of tumor cells. This morphological pattern is seen in both sporadic and in HPRC cases [71].

Papillary renal carcinoma type 1 with characteristic morphologic features including delicate fibrovascular cores lined by single layer of tumor cells
Type 2 PRCC demonstrates significantly higher nucleolar grade and overall larger tumor size [74, 78]. In these cases, tumor cells exhibit prominent nucleoli and varying degrees of nuclear pseudostratification with abundant eosinophilic cytoplasm (Fig. 5.16). The fibrovascular cores of most cases of type 2 PRCC tend to be dense and fibrous instead of thin and delicate, and edema and glomeruloid bodies are less prevalent than in type 1 tumors. Macrophages are more likely identified near necrotic tumor foci. ISUP nucleolar grading system is validated for reporting of PRCCs. Sarcomatoid and rhabdoid changes are categorized as grade 4 tumors, usually associated with an adverse prognosis.

Papillary renal cell carcinoma type 2 with broad papillae containing tumor cells exhibiting prominent nucleoli and nuclear pseudostratification with abundant eosinophilic cytoplasm
Type 1 PRCC reveals loss of Y chromosome and gains in chromosomes 7, 17, 16, and 20 [79]. Activation of the MET pathway is a recognized finding in up to 80% of type 1 PRCC tumors [80, 81]. Type 2 PRCC tumors demonstrate a heterogeneous pattern of chromosomal gains and losses, involving chromosomes 1, 3, 4, 5, 6, 8q, 9, 14, and 15 [80,81,82,83,84]. Type 2 PRCCs are more frequently associated with aggressive clinicopathologic parameters than type 1 PRCCs, including higher TNM stage, larger tumor size, and an overall worse prognosis [77, 78, 85]. Although some studies have documented a worse prognosis in type 2 PRCCs based on univariate analysis, other studies have not reported the same findings on multivariate analysis upon factoring of tumor grade and stage [77, 78, 82, 85,86,87].
PRCCs exhibit positivity for cytokeratin AE1/AE3, CK7, CAM5.2, EMA, high-molecular-weight cytokeratin, racemase (AMACR), CA-IX, RCC antigen, CD10, and vimentin. Type 2 PRCC demonstrates more variable immunoreactivity for the above markers, including reduced expression of CK7.
Subtyping PRCCs is difficult in a notable subset of cases, further complicating the classification schemata as some examples exhibit nuclear features typical of type 1 PRCC, but cytoplasmic features characteristic of type 2 PRCC, whereas other cases comprised tumor cells with high nuclear grade with variable cytoplasmic features [77,78,79,80,81,82,83,84,85,86,87]. Across the board, the proportion of both type 1 and type 2 PRCC tumors varies between 30% and 70% [77,78,79,80,81,82,83,84,85,86,87]. A significant proportion of PRCC cases do not meet the documented histologic parameters for typing in either category, and such tumors have been designated as mixed, unclassified, overlapping, or not otherwise specified tumors [77,78,79,80,81,82,83,84,85,86,87]. Additionally, an oncocytic low-grade variant of PRCC has been identified, composed predominantly of tumor cells with oncocytic cytoplasm and round, nonoverlapping low-grade nuclei with inconspicuous nucleoli and a linear arrangement toward cell apices (Fig. 5.17). These tumors exhibit molecular features akin to type 1 PRCC, with gains in chromosomes 7 and 17. These tumors carry a good prognosis owing to their indolent behavior [88,89,90,91,92].

Oncocytic low-grade variant of papillary RCC composed of tumor cells with oncocytic cytoplasm, papillary architecture, and round, nonoverlapping low-grade nuclei
The Cancer Genome Atlas (TCGA) Research Network showed that types 1 and 2 PRCCs are actually different types of renal cancer based on comprehensive molecular analysis of a cohort of 161 tumors. PRCC1 is associated with MET alterations. Multiple molecular subgroups were identified within the type 2 PRCC cohort that was otherwise characterized by CDKN2A silencing, SETD2 mutations, transcription factor E3 (TFE3) fusions, and increased expression of the NrF2-antioxidant response element pathway. A CpG island methylator phenotype was observed in a distinct subgroup of type 2 PRCC that was characterized by poor survival and mutation of the gene-encoding fumarate hydratase (FH) [81]. It is now established that type 2 PRCCs represent a heterogeneous group of tumors that requires better characterization based on molecular characterization and morphologic correlates.
The cancer-specific survival rates at 5 years after surgery for a large series of patients with CCRCC, PRCC, and chromophobe RCC were 72%, 91%, and 88%, respectively [93]. PRCC has a more favorable outcome than other aggressive subtypes like collecting duct carcinoma and HLRCC-RCC [94, 95]. Type 1 PRCC cases are associated with a significantly better survival rate than type 2 PRCCs, though this difference is linked to the grade and stage at presentation rather than tumor subtyping [86].
Chromophobe Renal Cell Carcinoma
ChRCC was first reported in 1985 and derived its name from the morphologically similar tumor cells identified in the experimentally produced rat kidney tumor [96]. The eosinophilic variant of ChRCC was described in 1988 [97]. ChRCC has its postulated origin in the intercalated cells of the collecting duct system [98]. Most of these tumors are sporadic, but also occur in a familial setting in patients with Birt–Hogg–Dubé syndrome [99]. A germline mutation of PTEN in Cowden syndrome also predisposes to development of ChRCC [100].
Patients range in age from 23 to 86 years, and a slight male preponderance is seen with a greater incidence in Middle Eastern nations [101,102,103,104,105,106]. The presenting signs and symptoms are not different from other renal tumors and a palpable mass is rarely noted. Radiologically, no features reliably distinguish chromophobe RCC from other kidney tumors, including oncocytoma.
Tumors are well circumscribed and vary widely in size, from 1.5 to 25 cm in diameter, mean diameter ranges from 6.9 to 8.5 cm [101,102,103,104,105,106]. The cut surface is usually solid, homogeneous, and tan brown (Fig. 5.18). Hemorrhage and necrosis, when present, are limited and seen in a minority of cases. Central scar formation is infrequently present.

Chromophobe renal cell carcinoma. Note the well-circumscribed tumor with a characteristic tan brown appearance
Microscopically, the tumor cells are typically arranged in solid sheets with focal tubulocystic pattern in some cases. There are fibrous septa of variable thickness with relatively larger caliber blood vessels in contrast to the delicate vasculature seen in clear cell carcinoma. Variable populations of two types of tumor cells are seen – the typical ChRCC tumor cell is a large polygonal cell with abundant, almost transparent, and slightly flocculent cytoplasm and prominent plant-like cell membranes that is commonly seen abutting vascular channels (Fig. 5.19a). Usually these are admixed with a second population of smaller cells with less abundant cytoplasm that is granular and eosinophilic (Fig. 5.19b) [101,102,103,104,105,106].

Chromophobe renal cell carcinoma with (a) tumor cells containing abundant flocculent cytoplasm and well-defined plant-like cell membranes and (b) tumor cells with more granular eosinophilic calcification, perinuclear haloes, and raisinoid nuclei. Microcalcifications are frequently seen in these tumors
A variant of ChRCC that is virtually entirely composed of intensely eosinophilic cells with prominent cell membranes has been designated the eosinophilic variant of this neoplasm (Fig. 5.20a) [101,102,103,104,105,106]. The nuclei of both cell types are typically hyperchromatic with irregular wrinkled nuclear contours; binucleation is commonly present. Perinuclear halos are more commonly identified in the eosinophilic cells, a diagnostically helpful finding, but often it is not possible to distinguish eosinophilic ChRCC from oncocytoma particularly in biopsy specimens. Immunostaining for CK7 showing strong and diffuse positivity in tumor cells is noted in eosinophilic variant of ChRCC (Fig. 5.20b) [101,102,103,104,105,106,107]. Sarcomatoid transformation, a feature common to other types of RCC, may be identified in less than 10% of cases [37, 38, 108, 109]. Rare cases with osteosarcoma-like differentiation, rhabdoid morphology, or extensive calcification and ossification have been reported [110, 111].

(a) Eosinophilic variant of chromophobe renal cell carcinoma with sheets of tumor cells resembling an oncocytoma. (b) Strong and diffuse cytokeratin 7 expression on immunostaining serves to distinguish this tumor from oncocytoma
Some kidney tumors demonstrate mixed morphologic patterns, with features overlapping between oncocytoma and ChRCC in the same tumor, and are designated hybrid oncocytic chromophobe tumors [112,113,114,115]. Such tumors may arise in different settings: as tumors present in cases of renal oncocytosis, as tumors arising in patients with Birt–Hogg–Dubé syndrome, and as sporadic tumors [112,113,114,115]. These tumors either show a gradual transition from one pattern to another or appear as distinctly separate areas adjacent to one another, or the two patterns are intimately admixed with one another. In spite of overlapping morphologies, tumors from all three groups have different molecular genetic makeup, and their molecular features are different from those of oncocytoma and ChRCC. The tumors in all three groups exhibit indolent behavior in the reported literature and are currently subcategorized under ChRCC [112,113,114,115].
The genetic abnormality most frequently identified in ChRCC has been loss of one copy of the entire chromosome for most or all of the chromosomes 1, 2, 6, 10, 13, 17, and 21 (in >80% of cases), as well as losses of various other chromosomes [98, 116, 117]. In regard to the eosinophilic variant of ChRCC, approximately 50% of cases have different chromosomal abnormalities than the classic type [98]. Sarcomatoid ChRCC analysis often shows multiple gains (polysomy) of chromosomes 1, 2, 6, 10, and 17, and distant metastases in ChRCC cases show the same genetic alterations as reported in the primary tumors [118].
ChRCC demonstrates positive immunohistochemical staining for PAX8, CD117 (c-kit), pancytokeratin, and EMA and negative immunostaining for vimentin, CK20, and racemase. The most helpful immunostain in distinguishing between oncocytoma and ChRCC is CK7 that is very frequently diffusely positive in ChRCC, in contrast with oncocytomas, which usually show focal scattered positivity in tumor cells [101,102,103,104,105,106].
Renal oncocytomas do not demonstrate combined losses of heterozygosity at chromosomes 1, 2, 6, 10, 13, 17, and 21 that are characteristically associated with ChRCC. Fluorescence in situ hybridization (FISH) studies detect the aforementioned cytogenetic findings and therefore are helpful distinguishing between the two entities [119].
A recent study demonstrated that approximately 70% of ChRCCs carry either a hemizygous deletion of RB1 or ERBB4, and oncocytomas do not show deletions of these genes. FISH testing may therefore be useful to detect deletion of these genes and furnish an assay with high sensitivity and specify to distinguish between ChRCC and oncocytoma [120].
The prognosis for ChRCC has been shown in several sizable case series to be significantly better than for CCRCC. There is no significant difference in outcome between the classical and the eosinophilic variants of this neoplasm [101,102,103,104,105,106]. Stage at presentation with ChRCC is significantly lower than with CCRCC. Metastatic ChRCC is seen in less than 5% of cases at presentation compared with one-fourth of cases of CCRCC. Sarcomatoid differentiation and histologic tumor necrosis confer a worse prognosis. The cancer-specific 5-year survival rate for ChRCC postnephrectomy is about 90% [28]. Median cancer-specific survival for patients with metastatic ChRCC is 0.6 years, which is not unlike cases of metastatic RCC of other subtypes. Metastatic ChRCCs demonstrate the presence of TP53 and PTEN mutations apart from imbalanced chromosome duplications (duplication of three or more chromosomes) [121].
Collecting Duct Carcinoma
Collecting duct carcinoma (CDC) is an uncommon albeit well-recognized aggressive subtype of RCC that has its purported origin from the principal cells in the collecting ducts of Bellini. It comprises less than 1% of renal malignancies with over 250 reported cases in the literature, the first case being reported in 1986 [122].
This RCC affects all ages, with a mean age of 55 years, and occurs more commonly in male patients, with a male-to-female ratio of 2: 1 [123,124,125,126,127,128,129,130,131,132]. Although up to a quarter of cases are discovered incidentally, most patients present with abdominal or constitutional symptoms or even with metastatic disease [123,124,125,126,127,128,129,130,131,132]. Radiological examination reveals a predominantly solid kidney mass. Urine cytology can also rarely be positive for malignancy in these cases [128].
CDC arises at any location in the renal parenchyma, and identification of a particular site of origin is difficult, especially as the tumor increases in size. Tumor size is variable with cases ranging up to 16 cm in greatest dimension. The cut surface of CDC is usually gray white and firm, and foci of overt necrosis are often present. Tumors are frequently infiltrative and multifocal involvement of the kidney may be exhibited as satellite tumor nodules (Fig. 5.21). Involvement of the adrenal gland, perinephric fat, renal sinus fat, renal pelvis, Gerota’s fascia, renal vein, and regional lymph nodes is grossly identified in the majority of cases on radiological and/or pathologic examination [123,124,125,126,127,128,129,130,131,132].

Collecting duct carcinoma demonstrating an infiltrative growth pattern comprising multiple tumor nodules with irregular borders invading the renal medulla
Microscopically, CDC shows ill-defined borders with prominent infiltration of adjacent parenchyma and an interstitial growth pattern with relatively preserved glomeruli is commonly noted. Marked stromal desmoplasia is almost always present [94]. It is frequently accompanied by an acute and chronic inflammatory cell infiltrate at the interface between tumor and normal parenchyma. A number of different growth patterns may be seen within the same tumor including solid sheets/cords/nests, tubulopapillary structures, or infiltrating small- to medium-sized malignant glands/tubules (Fig. 5.22a, b) along with surrounding desmoplastic stroma (Fig. 5.23). An extensive component of tubulocystic growth pattern that typifies tubulocystic carcinoma excludes tumors from being considered as CDC [94]. Some cases demonstrate a microcystic pattern with intracystic high-grade malignant papillary proliferations. Another pattern is characterized by intratubular extension with microscopic subcapsular deposits distant from the main tumor. The epithelium lining uninvolved ducts adjacent to or far from the main tumor may exhibit severe cytologic atypia amounting to carcinoma in situ-like growth. Tumor cells are uniformly high cytologic grade, with nuclear pleomorphism and prominent nucleoli and varying amounts of eosinophilic cytoplasm. Cells lining malignant tubular profiles may often exhibit hobnailing, a finding that is not usually encountered in other RCC subtypes. Sarcomatoid differentiation is identified in up to one-third of the cases. Lymphovascular invasion is frequently seen, and renal vein invasion is present in a significant number of cases (20–44%) [94, 123,124,125,126,127,128,129,130,131,132].

Collecting duct carcinoma composed of high-grade carcinoma demonstrating areas of distinctly tubular (a) and tubulopapillary (b) growth patterns with a prominent admixed inflammatory infiltrate

Malignant glandular differentiation in collecting duct carcinoma-exhibiting presence of tumor cells with high-grade nuclei, prominent nucleoli, and an accompanying desmoplastic stromal reaction
Intra- or extracellular mucin production in these tumors can often be demonstrated using mucicarmine, Alcian blue, or PAS stains [132, 133]. Exclusion of metastatic adenocarcinoma and high-grade urothelial carcinoma by obtaining clinical history of extrarenal primary tumor and extensive sampling of the pelvicalyceal system is crucial prior to making a diagnosis of CDC, as these are the major entities in the differential diagnosis. To that effect, an immunohistochemical panel including PAX8, GATA3, and p63 is helpful as urothelial carcinomas are more likely to be positive for GATA3 and p63, whereas CDC does not stain with these markers [134, 135]. Of note, PAX8 may be positive in both urothelial carcinomas involving the renal pelvis and CDCs, a finding that should be considered prior to establishing any further diagnosis [136].
The reported molecular features in CDC are notably variable and limited, and no distinctive molecular mechanism or pathway has been proposed for collecting duct carcinoma [130]. A recent study highlights that a significant number of cases (up to 25%) previously reported as CDC are in fact examples of fumarate hydratase (FH)-deficient RCC [94]. These tumors are described in further detail in the Hereditary Leiomyomatosis and Renal Cell Carcinoma-Associated RCC section. Therefore, immunohistochemical stains for FH and 2SC should be performed in high-grade carcinomas with a diagnostic consideration of CDC, along with germline mutational testing for FH mutations, if deemed clinically necessary. Another entity in the differential diagnosis is renal medullary carcinoma that is discussed in the section below. CDC is thus diagnosed upon excluding the aforementioned entities in the differential diagnosis.
CDC cases often present at an advanced disease stage and the overall prognosis is poor. Almost 50% of patients have regional nodal and/or distant metastases at the time of initial diagnosis [94, 123,124,125,126,127,128,129,130,131,132]. Approximately two-thirds of patients with CDC die of disease-related causes within 2 years and mortality rates are extremely high. Chemotherapy and immunotherapy are of very little benefit in managing collecting duct carcinoma [94, 123,124,125,126,127,128,129,130,131,132].
Renal Medullary Carcinoma
Renal medullary carcinoma (RMC) is a relatively rare and highly aggressive renal malignancy, first reported in 1995. The terminal collecting duct epithelium is the proposed site of origin [137,138,139,140]. There is a very strong association of this tumor occurring in conjunction with sickle cell hemoglobinopathies, and according to various hypotheses it is the terminal collecting duct epithelium that undergoes chronic ischemic damage due to accumulating drepanocytes (sickled erythrocytes) resulting in tumorigenesis. Most affected patients have been African Americans with sickle cell trait (HbAS) or hemoglobin SC disease (HbSC), but this tumor has also been reported in a patient with sickle cell disease (HbSS) as well as in white patients without evidence of sickle cell hemoglobinopathies [96, 137,138,139,140,141].
Most patients are diagnosed in the second or third decades of life, ranging from 5 to 58 years, with a mean age at diagnosis of 20 years. The male-to-female ratio is at least 2: 1, and approximately 75% of tumors occur in the right kidney and 25% occur on the left. The vast majority of cases occur in those with African ancestry, but Central and South American and Mediterranean individuals are also at risk for RMC. Almost 90% of reported cases occurred in patients with sickle cell trait. Common presenting symptoms include abdominal or flank pain, gross hematuria, weight loss with constitutional symptoms of relatively short duration, and a minority of patients (10%) have a palpable abdominal mass [137,138,139,140,141,142,143].
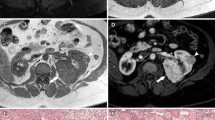
Grossly, these tumors usually involve the renal medulla and are poorly circumscribed, lobulated, firm, gray-tan, with variable hemorrhage and necrosis. They range widely in size (1.8–13 cm in greatest dimension, with a mean of 7 cm) and frequently present at an advanced stage with satellite nodules, perinephric extension, and sinus fat invasion (Fig. 5.24) [137,138,139,140,141,142,143,144]. A host of morphologic patterns is noted on microscopic examination including the characteristic finding of a reticular or microcystic growth pattern resembling yolk sac tumor of the testis (Fig. 5.25). Areas similar to adenoid cystic carcinoma of salivary glands with a cribriform or sieve-like growth are often noted (Fig. 5.26). Drepanocytes (sickled erythrocytes) are also seen within and surrounding the tumors. Other common patterns include tubule formation and growth in diffuse sheets or solid nodules. Tumor cells are commonly pleomorphic with enlarged nuclei, prominent nucleoli, and variable amounts of eosinophilic cytoplasm. Squamoid or rhabdoid appearance of tumor cells in solid sheet-like areas is often noted. Numerous aggregates of neutrophils may be seen within the tumor (Fig. 5.27), and there is often a dense inflammatory response at the interface between tumor and the adjacent renal parenchyma. A prominent desmoplastic stromal reaction is another constant feature seen in most cases (Fig. 5.25). Mucin production is variably seen in most of these tumors (Fig. 5.26).

Renal medullary carcinoma with sinus fat invasion and perinephric extension in an African American patient with sickle cell trait

Renal medullary carcinoma with a microcystic growth pattern and marked stromal desmoplasia

Microcystic cribriform or sieve-like growth pattern with wispy blue mucin secretion in renal medullary carcinoma. Scattered sickled erythrocytes are present in the background

Renal medullary carcinoma with tubular differentiation, tumor cells with high-grade nuclei containing prominent nucleoli, and a dense neutrophilic inflammatory infiltrate
Renal origin of this tumor is confirmed by positive immunostaining for PAX8 in all cases. Loss of expression of SMARCB1 (INI1), a nuclear transcription regulator encoded on chromosome 22, is now a mandatory criterion to make a diagnosis of RMC [94]. It occurs as a result of a loss of heterozygosity or hemizygous deletions at the SMARCB1 locus, rarely due to loss of chromosome 22 or balanced translocation involving chromosome 22 [145, 146]. Another unique finding is that up to two-thirds of RMCs show positive immunostaining for OCT3/4 (POU5F1) [94, 147]. Variable degrees of positive immunostaining are reported for cytokeratins AE1/AE3 and CAM5.2, CK7, CK20, polyclonal carcinoembryonic antigen (CEA), and EMA [94, 130, 145,146,147,148].
This tumor carries a very dismal prognosis with almost all patients presenting with metastatic disease at the time of diagnosis. Common sites of metastasis are lymph nodes, lung, liver, and adrenal glands. Long-term disease-free survival rates are very low; average survival is between 2 and 68 weeks, with a mean survival duration of 19 weeks [130, 143, 144]. Neoadjuvant therapy prolongs survival by a limited duration, but tumor recurrence and death inevitably occur even after a period of remission [149, 150].
There are notable overlapping clinical and pathologic features between collecting duct carcinoma and RMC, raising a consideration that RMC is a subtype of collecting duct carcinoma [130, 151]. Loss of SMARCB1 (INI1) immunostaining and presence of hemoglobinopathy by history and laboratory test confirmation, with accompanying drepanocytes in tumor stroma and/or blood vessels, are required to establish a diagnosis of RMC [94]. On the other hand, rare tumors demonstrating RMC-like histology, INI1-deficient immunophenotype, but arising in patients in whom sickle cell trait or disease has been definitively excluded are designated as RCC, unclassified, with medullary phenotype in the present scenario [94].
Mucinous Tubular and Spindle Cell Carcinoma
This entity was initially described in 1997 by MacLennan et al. and designated as “low-grade collecting duct carcinoma.” Other subsequently reported them as “low-grade myxoid renal epithelial neoplasms” and “low-grade tubular mucinous renal neoplasms” in 2001–2002 [152,153,154]. Most of these tumors are identified incidentally. The tumor is far more commonly seen in females with a ratio of 3:1. The age range is wide (13–82 years, mean age of 58 years).
Tumors range in diameter from 2.2 to 12 cm (average 6.5 cm). They are well circumscribed, gray white, tan, or yellow, with focal hemorrhage or necrosis (Fig. 5.28). Histologic examination of classic tumors shows tightly packed, small elongated tubules separated by abundant basophilic extracellular mucin, sometimes with a “bubbly” myxoid consistency (Fig. 5.29a) [152,153,154]. Areas of spindled cells are also seen more prominently in some of these tumors (Fig. 5.29b) [155]. The mucin stains strongly with Alcian blue at pH 2.5. Tubules are lined by uniform low cuboidal cells with scant cytoplasm and round nuclei of low nuclear grade with absent or inconspicuous nucleoli. Mitotic activity is not significantly elevated.

Mucinous tubular and spindle cell carcinoma of kidney. Note the tan yellow tumor with well-circumscribed borders and a glistening cut surface with bulging contours

Mucinous tubular and spindle cell carcinoma composed of closely packed elongated tubules in a background of “bubbly mucin” (a) with some cases demonstrating prominent spindling of tumor cells (b)
There are several morphologic variations associated with this tumor including relative lack of mucinous matrix, small well-formed papillae, presence of foamy macrophages, focal clear cell change in tubular component, focal necrosis, oncocytic change, small vacuolations, psammomatous calcification, or heterotopic bone formation [156]. High nuclear grade, areas of coagulative tumor necrosis, sarcomatoid differentiation, and aggressive behavior have been documented in a small cohort of these tumors [157,158,159,160]. Tumor cells have a variable immunophenotype, but are usually immunoreactive for CK7, AMACR, and PAX8 (Fig. 5.30) [161,162,163].

Diffuse and strong cytokeratin 7 immunohistochemical staining of tumor cells in mucinous tubular and spindle cell carcinoma, a feature in common with papillary renal cell carcinoma
The characteristic gains of chromosomes 7 and 17 and loss of Y chromosome that are seen in papillary RCC are not seen in mucinous tubular and spindle cell carcinoma [164]. Alterations in the Hippo pathway are present in mucinous tubular and spindle cell carcinoma, but are not seen in PRCC [165]. Cytogenetic analyses and comparative genomic hybridization studies demonstrated multiple genetic alterations that include loss of chromosomes 1, 4, 6, 8, 9, 13, 14, 15, 18, and 22 [166, 167].
Most of these tumors with classic histologic findings are indolent, and surgical excision is curative as they are low stage at the time of resection. However, high nuclear grade and sarcomatoid change in a few cases have been associated with adverse outcomes [168, 169].
Renal Cell Carcinoma, Unclassified
RCC, unclassified, is a diagnostic category for the designation of RCCs that have histologic features that cannot be categorized under any of the well-characterized RCC subtypes. These tumors represent up to 5–6% of RCC cases [1]. Morphologic patterns falling under this category include tumors that show a composite mixture of recognized types, novel or unrecognized cell types, tumors with mucin production, and renal carcinomas with entirely sarcomatoid morphology, lacking recognizable epithelial elements [170]. Low- or high-grade unclassifiable oncocytic neoplasms were also included in this category [1]. However, most genitourinary pathologists including this author team do not classify an RCC with a composite mixture of recognized subypes as an unclassified RCC. We designate such tumors as a composite RCC and list the subtypes identified therein.
Patients of any age can be affected (range: 21–91 years) [21,22,23, 171,172,173,174]. These tumors usually present at an advanced stage, are large, and demonstrate histologic features that correlate with an adverse prognosis including high nuclear grade, tumor necrosis, and lymphovascular invasion [21,22,23, 170, 174]. Data from limited analysis of these tumors show that they exhibit marked genetic instability [21]. In a molecular study of 62 unclassified RCC, approximately 75% of cases were categorized into several subsets of abnormalities of variable prognostic significance that might be useful for either diagnostic or therapeutic implications [23].
Unclassified RCC is a histologically and clinically heterogeneous group of tumors, wherein prognosis depends upon similar clinical and pathologic findings determining outcome in conventional RCCs [21, 170, 173, 174].
Grading and Staging Renal Cell Carcinoma
Cytologic grading is one of the most essential parameters that aid in predicting the biologic behavior of RCC [24,25,26,27,28,29]. The Fuhrman system was widely used up until recently for grading RCCs and presently the four-tiered WHO/International Society of Urologic Pathology (ISUP) grading system has been recommended for use in grading clear cell and papillary RCCs in view of the fact that there is sufficient evidence-based data that can be used for predicting prognosis [175, 176]. Grades 1–3 are based on prominence of nucleoli and grade 4 includes pronounced nuclear pleomorphism, tumor giant cells, and rhabdoid or sarcomatoid changes (Table 5.2) [175]. Grading is performed after assessing the aforementioned criteria within a single high-power field showing the highest nucleolar grade or greatest degree of nuclear pleomorphism. The WHO/ISUP grading system is not currently applicable for predicting prognosis for other types of RCC due to a lack of outcome data for these different subtypes [175, 176].
Incorporation of tumor necrosis, defined as homogeneous aggregates and sheets of nonviable tumor cells, or coalescing groups of cells containing nuclear and cytoplasmic debris is another feature that has been studied as a possible factor that can be included in the grading criteria. Some published studies have indicated that including tumor necrosis in CCRCC cases furnishes additional prognostic information in comparison to sole inclusion of WHO/ISUP nucleolar grade [177].
Tumor stage indicates the extent of involvement by disease and represents the most important factor in predicting the clinical behavior and prognosis of RCC [32]. Currently, the tumor, nodes, and metastasis (TNM) staging system is used all across the world for RCC staging [178]. The eighth edition of the AJCC Cancer Staging Manual was published in 2017 and criteria listed in this latest update to the staging system are being reported [179]. Staging parameters for carcinomas arising in the kidney are provided, and definitions for primary tumor (T), regional lymph nodes (N), and distant metastasis (M) are established in this staging system (Table 5.3).
References
Moch H, Humphrey PA, Ulbright TM, Reuter VE. WHO classification of tumours of the urinary system and male genital organs. 4th ed. Lyon: IARC Press; 2016.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9.
Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, Vocke CD, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45.
Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–30.
Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, Zhang N, Toffessi-Tcheuyap V, Nguyen TT, Pahuja KB, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. 2015;47:13–21.
Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. An integrated metabolic atlas of clear cell renal cell carcinoma. Cancer Cell. 2016;29:104–16.
Gotoh M, Ichikawa H, Arai E, Chiku S, Sakamoto H, Fujimoto H, Hiramoto M, Nammo T, Yasuda K, Yoshida T, et al. Comprehensive exploration of novel chimeric transcripts in clear cell renal cell carcinomas using whole transcriptome analysis. Genes Chromosomes Cancer. 2014;53:1018–32.
Malouf GG, Zhang J, Yuan Y, Comperat E, Roupret M, Cussenot O, Chen Y, Thompson EJ, Tannir NM, Weinstein JN, et al. Characterization of long non-coding RNA transcriptome in clear-cell renal cell carcinoma by next-generation deep sequencing. Mol Oncol. 2015;9:32–43.
Gowrishankar B, Przybycin CG, Ma C, Nandula SV, Rini B, Campbell S, Klein E, Chaganti RS, Magi-Galluzzi C, Houldsworth J. A genomic algorithm for the molecular classification of common renal cortical neoplasms: development and validation. J Urol. 2015;193:1479–85.
Christinat Y, Krek W. Integrated genomic analysis identifies subclasses and prognosis signatures of kidney cancer. Oncotarget. 2015;6:10521–31.
Eckel-Passow JE, Igel DA, Serie DJ, Joseph RW, Ho TH, Cheville JC, Parker AS. Assessing the clinical use of clear cell renal cell carcinoma molecular subtypes identified by RNA expression analysis. Urol Oncol. 2015;33:17–23.
Rathmell KW, Chen F, Creighton CJ. Genomics of chromophobe renal cell carcinoma: implications from a rare tumor for pan-cancer studies. Oncoscience. 2015;2:81–90.
Malouf GG, Ali SM, Wang K, Balasubramanian S, Ross JS, Miller VA, Stephens PJ, Khayat D, Pal SK, Su X, et al. Genomic characterization of renal cell carcinoma with sarcomatoid dedifferentiation pinpoints recurrent genomic alterations. Eur Urol. 2016;70:348–57.
Chen YB, Xu J, Skanderup AJ, Dong Y, Brannon AR, Wang L, Won HH, Wang PI, Nanjangud GJ, Jungbluth AA, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131.
De Velasco G, Culhane AC, Fay AP, Hakimi AA, Voss MH, Tannir NM, Tamboli P, Appleman LJ, Bellmunt J, Kimryn Rathmell W, et al. Molecular subtypes improve prognostic value of international metastatic renal cell carcinoma database consortium prognostic model. Oncologist. 2017;22:286–92.
Seles M, Hutterer GC, Kiesslich T, Pummer K, Berindan-Neagoe I, Perakis S, Schwarzenbacher D, Stotz M, Gerger A, Pichler M. Current insights into long non-coding RNAs in renal cell carcinoma. Int J Mol Sci. 2016;17:573.
Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, Hes O, Moch H, Montironi R, Tickoo SK, et al. The International Society of Urological Pathology (ISUP) Vancouver classification of renal neoplasia. Am J Surg Pathol. 2013;37:1469–89.
Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70(1):93–105.
Yoshida SO, Imam A, Olson CA, et al. Proximal renal tubular surface membrane antigens identified in primary and metastatic renal cell carcinomas. Arch Pathol Lab Med. 1986;110:825–32.
Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol. 2004;28:1117–32.
Hu ZY, Pang LJ, Qi Y, et al. Unclassified renal cell carcinoma: a clinicopathological, comparative genomic hybridization, and whole-genome exon sequencing study. Int J Clin Exp Pathol. 2014;7:3865–75.
Li Y, Reuter VE, Matoso A, et al. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology. 2018;72:588–600.
Chen YB, Xu J, Skanderup AJ, et al. Molecular analysis of aggressive renal cell carcinoma with unclassified histology reveals distinct subsets. Nat Commun. 2016;7:13131.
Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6:655–63.
Goldstein NS. The current state of renal cell carcinoma grading. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 1997;80:977–80.
Usubutun A, Uygur MC, Ayhan A, et al. Comparison of grading systems for estimating the prognosis of renal cell carcinoma. Int Urol Nephrol. 1998;30:391–7.
Ficarra V, Righetti R, Martignoni G, et al. Prognostic value of renal cell carcinoma nuclear grading: multivariate analysis of 333 cases. Urol Int. 2001;67:130–4.
Lohse CM, Cheville JC. A review of prognostic pathologic features and algorithms for patients treated surgically for renal cell carcinoma. Clin Lab Med. 2005;25:433–64.
Lohse CM, Blute ML, Zincke H, et al. Comparison of standardized and nonstandardized nuclear grade of renal cell carcinoma to predict outcome among 2,042 patients. Am J Clin Pathol. 2002;118:877–86.
Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18–25.
Haake SM, Rathmell WK. Renal cancer subtypes: should we be lumping or splitting for therapeutic decision making? Cancer. 2017;123:200–9.
Thrasher JB, Paulson DF. Prognostic factors in renal cancer. Urol Clin North Am. 1993;20:247–62.
Velickovic M, Delahunt B, McIver B, et al. Intragenic PTEN/MMAC1 loss of heterozygosity in conventional (clear-cell) renal cell carcinoma is associated with poor patient prognosis. Mod Pathol. 2002;15:479–85.
Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003;170:2163–72.
Van EF, Van RC, Bodmer D, et al. Chromosome 3 translocations and the risk to develop renal cell cancer: a Dutch intergroup study. Genet Couns. 2003;14:149–54.
Vanharanta S, Buchta M, McWhinney SR, et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. 2004;74:153–9.
Cheville JC, Lohse CM, Zincke H, et al. Sarcomatoid renal cell carcinoma: an examination of underlying histologic subtype and an analysis of associations with patient outcome. Am J Surg Pathol. 2004;28:435–41.
de Peralta-Venturina M, Moch H, Amin M, et al. Sarcomatoid differentiation in renal cell carcinoma: a study of 101 cases. Am J Surg Pathol. 2001;25:275–84.
Gokden N, Nappi O, Swanson PE, et al. Renal cell carcinoma with rhabdoid features. Am J Surg Pathol. 2000;24:1329–38.
Haddad FS, Shah IA, Manne RK, et al. Renal cell carcinoma insulated in the renal capsule with calcification and ossification. Urol Int. 1993;51:97–101.
Bonsib SM. Renal cell carcinoma with lymphomatoid features. J Urol Pathol. 1997;6:109–18.
Jagirdar J, Irie T, French SW, et al. Globular Mallory-like bodies in renal cell carcinoma: report of a case and review of cytoplasmic eosinophilic globules. Hum Pathol. 1985;16:949–52.
Arora K, Divatia MK, Truong L, Shen SS, Ayala AG, Ro JY. Sarcoid-like granulomas in renal cell carcinoma: the Houston Methodist Hospital experience. Ann Diagn Pathol. 2017;31:62–5.
Chau KY, Pretorius JM, Stewart AW. Myospherulosis in renal cell carcinoma. Arch Pathol Lab Med. 2000;124:1476–9.
Mathers ME, Pollock AM, Marsh C, et al. Cytokeratin 7: a useful adjunct in the diagnosis of chromophobe renal cell carcinoma. Histopathology. 2002;40:563–7.
Wu SL, Kothari P, Wheeler TM, et al. Cytokeratins 7 and 20 immunoreactivity in chromophobe renal cell carcinomas and renal oncocytomas. Mod Pathol. 2002;15:712–7.
McGregor DK, Khurana KK, Cao C, et al. Diagnosing primary and metastatic renal cell carcinoma: the use of the monoclonal antibody ‘Renal Cell Carcinoma Marker’. Am J Surg Pathol. 2001;25:1485–92.
Avery AK, Beckstead J, Renshaw AA, et al. Use of antibodies to RCC and CD10 in the differential diagnosis of renal neoplasms. Am J Surg Pathol. 2000;24:203–10.
Ordonez NG. The diagnostic utility of immunohistochemistry in distinguishing between mesothelioma and renal cell carcinoma: a comparative study. Hum Pathol. 2004;35:697–710.
Kim MK, Kim S. Immunohistochemical profile of common epithelial neoplasms arising in the kidney. Appl Immunohistochem Mol Morphol. 2002;10:332–8.
Langner C, Wegscheider BJ, Ratschek M, et al. Keratin immunohistochemistry in renal cell carcinoma subtypes and renal oncocytomas: a systematic analysis of 233 tumors. Virchows Arch. 2004;444:127–34.
Young AN, Amin MB, Moreno CS, et al. Expression profiling of renal epithelial neoplasms: a method for tumor classification and discovery of diagnostic molecular markers. Am J Pathol. 2001;158:1639–51.
Tretiakova MS, Sahoo S, Takahashi M, et al. Expression of alpha-methylacyl-CoA racemase in papillary renal cell carcinoma. Am J Surg Pathol. 2004;28:69–76.
Petit A, Castillo M, Santos M, et al. KIT expression in chromophobe renal cell carcinoma: comparative immunohistochemical analysis of KIT expression in different renal cell neoplasms. Am J Surg Pathol. 2004;28:676–8.
Langner C, Ratschek M, Rehak P, et al. Expression of MUC1 (EMA) and E-cadherin in renal cell carcinoma: a systematic immunohistochemical analysis of 188 cases. Mod Pathol. 2004;17:180–8.
Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the differential diagnosis of renal neoplasms. Clin Lab Med. 2005;25:247–57.
Gobbo S, Eble JN, MacLennan GT, et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol. 2008;32:1780–6.
Shuch BM, Lam JS, Belldegrun AS, et al. Prognostic factors in renal cell carcinoma. Semin Oncol. 2006;33:563–75.
Delahunt B, Cheville JC, Martignoni G, et al. The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol. 2013;37:1490–504.
Leroy X, Zini L, Buob D, et al. Renal cell carcinoma with rhabdoid features: an aggressive neoplasm with overexpression of p53. Arch Pathol Lab Med. 2007;131:102–6.
Nassir A, Jollimore J, Gupta R, et al. Multilocular cystic renal cell carcinoma: a series of 12 cases and review of the literature. Urology. 2002;60:421–7.
Suzigan S, Lopez-Beltran A, Montironi R, et al. Multilocular cystic renal cell carcinoma: a report of 45 cases of a kidney tumor of low malignant potential. Am J Clin Pathol. 2006;125:217–22.
Williamson SR, MacLennan GT, Lopez-Beltran A, et al. Cystic partially regressed clear cell renal cell carcinoma: a potential mimic of multilocular cystic renal cell carcinoma. Histopathology. 2013;63:767–79.
von Teichman A, Comperat E, Behnke S, et al. VHL mutations and dysregulation of pVHL- and PTEN-controlled pathways in multilocular cystic renal cell carcinoma. Mod Pathol. 2011;24:571–8.
Halat S, Eble JN, Grignon DJ, et al. Multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Mod Pathol. 2010;23:931–6.
Williamson SR, Halat S, Eble JN, et al. Multilocular cystic renal cell carcinoma: similarities and differences in immunoprofile compared with clear cell renal cell carcinoma. Am J Surg Pathol. 2012;36:1425–33.
Amin MB, Amin MB, Tamboli P, et al. Prognostic impact of histologic subtyping of adult renal epithelial neoplasms: an experience of 405 cases. Am J Surg Pathol. 2002;26:281–91.
Zbar B, Tory K, Merino M, et al. Hereditary papillary renal cell carcinoma. J Urol. 1994;151:561–6.
Ornstein DK, Lubensky IA, Venzon D, et al. Prevalence of microscopic tumors in normal appearing renal parenchyma of patients with hereditary papillary renal cancer. J Urol. 2000;163:431–3.
Fischer J, Palmedo G, von Knobloch R, et al. Duplication and overexpression of the mutant allele of the MET proto-oncogene in multiple hereditary papillary renal cell tumours. Oncogene. 1998;17:733–9.
Lubensky IA, Schmidt L, Zhuang Z, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–26.
Amin MB, Corless CL, Renshaw AA, et al. Papillary (chromophil) renal cell carcinoma: histomorphologic characteristics and evaluation of conventional pathologic prognostic parameters in 62 cases. Am J Surg Pathol. 1997;21:621–35.
Schraml P, Muller D, Bednar R, et al. Allelic loss at the D9S171 locus on chromosome 9p13 is associated with progression of papillary renal cell carcinoma. J Pathol. 2000;190:457–61.
Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997;10:537–44.
Argani P, Netto GJ, Parwani AV. Papillary renal cell carcinoma with low-grade spindle cell foci: a mimic of mucinous tubular and spindle cell carcinoma. Am J Surg Pathol. 2008;32:1353–9.
Cantley R, Gattuso P, Cimbaluk D. Solid variant of papillary renal cell carcinoma with spindle cell and tubular components. Arch Pathol Lab Med. 2010;134:1210–4.
Delahunt B, Eble JN, McCredie MR, et al. Morphologic typing of papillary renal cell carcinoma: comparison of growth kinetics and patient survival in 66 cases. Hum Pathol. 2001;32:590–5.
Allory Y, Ouazana D, Boucher E, et al. Papillary renal cell carcinoma. Prognostic value of morphological subtypes in a clinicopathologic study of 43 cases. Virchows Arch. 2003;442:336–42.
Brunelli M, Eble JN, Zhang S, et al. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol. 2003;16:1053–9.
Chevarie-Davis M, Riazalhosseini Y, Arseneault M, et al. The morphologic and immunohistochemical spectrum of papillary renal cell carcinoma: study including 132 cases with pure type 1 and type 2 morphology as well as tumors with overlapping features. Am J Surg Pathol. 2014;38:887–94.
Cancer Genome Atlas Research N, Linehan WM, Spellman PT, et al. Comprehensive molecular characterization of papillary renal-cell carcinoma. N Engl J Med. 2016;374:135–45.
Klatte T, Pantuck AJ, Said JW, et al. Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma. Clin Cancer Res. 2009;15:1162–9.
Marsaud A, Dadone B, Ambrosetti D, et al. Dismantling papillary renal cell carcinoma classification: the heterogeneity of genetic profiles suggests several independent diseases. Genes Chromosomes Cancer. 2015;54:369–82.
Yang XJ, Tan MH, Kim HL, et al. A molecular classification of papillary renal cell carcinoma. Cancer Res. 2005;65:5628–37.
Mejean A, Hopirtean V, Bazin JP, et al. Prognostic factors for the survival of patients with papillary renal cell carcinoma: meaning of histological typing and multifocality. J Urol. 2003;170:764–7.
Saleeb RM, Brimo F, Farag M, et al. Toward biological subtyping of papillary renal cell carcinoma with clinical implications through histologic, immunohistochemical, and molecular analysis. Am J Surg Pathol. 2017;41:1618–29.
Sukov WR, Lohse CM, Leibovich BC, et al. Clinical and pathological features associated with prognosis in patients with papillary renal cell carcinoma. J Urol. 2012;187:54–9.
Lefevre M, Couturier J, Sibony M, et al. Adult papillary renal tumor with oncocytic cells: clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol. 2005;29:1576–81.
Furge KA, Chen J, Koeman J, et al. Detection of DNA copy number changes and oncogenic signaling abnormalities from gene expression data reveals MYC activation in high-grade papillary renal cell carcinoma. Cancer Res. 2007;67:3171–6.
Saleeb RM, Plant P, Tawedrous E, et al. Integrated phenotypic/genotypic analysis of papillary renal cell carcinoma subtypes: identification of prognostic markers, cancer-related pathways, and implications for therapy. Eur Urol Focus. 2018;4:740–8.
Kunju LP, Wojno K, Wolf JS Jr, et al. Papillary renal cell carcinoma with oncocytic cells and nonoverlapping low grade nuclei: expanding the morphologic spectrum with emphasis on clinicopathologic, immunohistochemical and molecular features. Hum Pathol. 2008;39:96–101.
Park BH, Ro JY, Park WS, et al. Oncocytic papillary renal cell carcinoma with inverted nuclear pattern: distinct subtype with an indolent clinical course. Pathol Int. 2009;59:137–46.
Leibovich BC, Lohse CM, Crispen PL, et al. Histological subtype is an independent predictor of outcome for patients with renal cell carcinoma. J Urol. 2010;183:1309–15.
Ohe C, Smith SC, Sirohi D, et al. Reappraisal of morphologic differences between renal medullary carcinoma, collecting duct carcinoma, and fumarate hydratase-deficient renal cell carcinoma. Am J Surg Pathol. 2018;42:279–92.
Merino MJ, Torres-Cabala C, Pinto P, et al. The morphologic spectrum of kidney tumors in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) syndrome. Am J Surg Pathol. 2007;31:1578–85.
Thoenes W, Storkel S, Rumpelt HJ. Human chromophobe cell renal carcinoma. Virchows Arch B Cell Pathol Incl Mol Pathol. 1985;48:207–17.
Thoenes W, Storkel S, Rumpelt HJ, et al. Chromophobe cell renal carcinoma and its variants—a report on 32 cases. J Pathol. 1988;155:277–87.
Davis CF, Ricketts CJ, Wang M, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–30.
Zbar B, Alvord WG, Glenn G, et al. Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dube syndrome. Cancer Epidemiol Biomark Prev. 2002;11:393–400.
Shuch B, Ricketts CJ, Vocke CD, et al. Germline PTEN mutation Cowden syndrome: an underappreciated form of hereditary kidney cancer. J Urol. 2013;190:1990–8.
Akhtar M, Kardar H, Linjawi T, et al. Chromophobe cell carcinoma of the kidney. A clinicopathologic study of 21 cases. Am J Surg Pathol. 1995;19:1245–56.
Crotty TB, Farrow GM, Lieber MM. Chromophobe cell renal carcinoma: clinicopathological features of 50 cases. J Urol. 1995;154:964–7.
Moch H, Gasser T, Amin MB, et al. Prognostic utility of the recently recommended histologic classification and revised TNM staging system of renal cell carcinoma: a Swiss experience with 588 tumors. Cancer. 2000;89:604–14.
Onishi T, Oishi Y, Yanada S, et al. Prognostic implications of histological features in patients with chromophobe cell renal carcinoma. BJU Int. 2002;90:529–32.
Peyromaure M, Misrai V, Thiounn N, et al. Chromophobe renal cell carcinoma: analysis of 61 cases. Cancer. 2004;100:1406–10.
Abrahams NA, MacLennan GT, Khoury JD, et al. Chromophobe renal cell carcinoma: a comparative study of histological, immunohistochemical and ultrastructural features using high throughput tissue microarray. Histopathology. 2004;45:593–602.
Abrahams NA, Tamboli P. Oncocytic renal neoplasms: diagnostic considerations. Clin Lab Med. 2005;25:317–39.
Abrahams NA, Ayala AG, Czerniak B. Chromophobe renal cell carcinoma with sarcomatoid transformation. Ann Diagn Pathol. 2003;7:296–9.
Akhtar M, Tulbah A, Kardar AH, et al. Sarcomatoid renal cell carcinoma: the chromophobe connection. Am J Surg Pathol. 1997;21:1188–95.
Itoh T, Chikai K, Ota S, et al. Chromophobe renal cell carcinoma with osteosarcoma-like differentiation. Am J Surg Pathol. 2002;26:1358–62.
Shannon BA, Cohen RJ. Rhabdoid differentiation of chromophobe renal cell carcinoma. Pathology. 2003;35:228–30.
Gobbo S, Eble JN, Delahunt B, et al. Renal cell neoplasms of oncocytosis have distinct morphologic, immunohistochemical, and cytogenetic profiles. Am J Surg Pathol. 2010;34:620–6.
Petersson F, Gatalica Z, Grossmann P, et al. Sporadic hybrid oncocytic/chromophobe tumor of the kidney: a clinicopathologic, histomorphologic, immunohistochemical, ultrastructural, and molecular cytogenetic study of 14 cases. Virchows Arch. 2010;456:355–65.
Klomp JA, Petillo D, Niemi NM, et al. Birt-Hogg-Dube renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression. BMC Med Genet. 2010;3:59.
Latham B, Dickersin GR, Oliva E. Subtypes of chromophobe cell renal carcinoma: an ultrastructural and histochemical study of 13 cases. Am J Surg Pathol. 1999;23:530–5.
Brunelli M, Eble JN, Zhang S, et al. Eosinophilic and classic chromophobe renal cell carcinomas have similar frequent losses of multiple chromosomes from among chromosomes 1, 2, 6, 10, and 17, and this pattern of genetic abnormality is not present in renal oncocytoma. Mod Pathol. 2005;18:161–9.
Speicher MR, Schoell B, du Manoir S, et al. Specific loss of chromosomes 1, 2, 6, 10, 13, 17, and 21 in chromophobe renal cell carcinomas revealed by comparative genomic hybridization. Am J Pathol. 1994;145:356–64.
Brunelli M, Gobbo S, Cossu-Rocca P, et al. Chromosomal gains in the sarcomatoid transformation of chromophobe renal cell carcinoma. Mod Pathol. 2007;20:303–9.
Herbers J, Schullerus D, Chudek J, et al. Lack of genetic changes at specific genomic sites separates renal oncocytomas from renal cell carcinomas. J Pathol. 1998;184:58–62.
Liu Q, Cornejo KM, Cheng L, et al. Next-generation sequencing to detect deletion of RB1 and ERBB4 genes in chromophobe renal cell carcinoma: a potential role in distinguishing chromophobe renal cell carcinoma from renal oncocytoma. Am J Pathol. 2018;188:846–52.
Casuscelli J, Weinhold N, Gundem G, et al. Genomic landscape and evolution of metastatic chromophobe renal cell carcinoma. JCI Insight. 2017;2 https://doi.org/10.1172/jci.insight.92688.
Fleming S, Lewi HJ. Collecting duct carcinoma of the kidney. Histopathology. 1986;10:1131–41.
Chao D, Zisman A, Pantuck AJ, et al. Collecting duct renal cell carcinoma: clinical study of a rare tumor. J Urol. 2002;167:71–4.
Srigley JR, Eble JN. Collecting duct carcinoma of kidney. Semin Diagn Pathol. 1998;15:54–67.
Olivere JW, Cina SJ, Rastogi P, et al. Collecting duct meningeal carcinomatosis. Arch Pathol Lab Med. 1999;123:638–41.
Dimopoulos MA, Logothetis CJ, Markowitz A, et al. Collecting duct carcinoma of the kidney. Br J Urol. 1993;71:388–91.
Peyromaure M, Thiounn N, Scotte F, et al. Collecting duct carcinoma of the kidney: a clinicopathological study of 9 cases. J Urol. 2003;170:1138–40.
Mauri MF, Bonzanini M, Luciani L, et al. Renal collecting duct carcinoma. Report of a case with urinary cytologic findings. Acta Cytol. 1994;38:755–8.
Parker R, Reeves HM, Sudarshan S, et al. Abnormal fluorescence in situ hybridization analysis in collecting duct carcinoma. Urology. 2005;66:1110.
Gupta R, Billis A, Shah RB, et al. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol. 2012;36:1265–78.
Baer SC, Ro JY, Ordonez NG, et al. Sarcomatoid collecting duct carcinoma: a clinicopathologic and immunohistochemical study of five cases. Hum Pathol. 1993;24:1017–22.
Kennedy SM, Merino MJ, Linehan WM, et al. Collecting duct carcinoma of the kidney. Hum Pathol. 1990;21:449–56.
Halenda G, Sees JN Jr, Belis JA, et al. Atypical renal adenocarcinoma with features suggesting collecting duct origin and mimicking a mucinous adenocarcinoma. Urology. 1993;41:165–8.
Albadine R, Schultz L, Illei P, et al. PAX8 (+)/p63 (−) immunostaining pattern in renal collecting duct carcinoma (CDC): a useful immunoprofile in the differential diagnosis of CDC versus urothelial carcinoma of upper urinary tract. Am J Surg Pathol. 2010;34:965–9.
Gonzalez-Roibon N, Albadine R, Sharma R, et al. The role of GATA binding protein 3 in the differential diagnosis of collecting duct and upper tract urothelial carcinomas. Hum Pathol. 2013;44:2651–7.
Tong GX, Yu WM, Beaubier NT, et al. Expression of PAX8 in normal and neoplastic renal tissues: an immunohistochemical study. Mod Pathol. 2009;22:1218–27.
Davis CJ Jr, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma. The seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19:1–11.
Adsay NV, de Roux SJ, Sakr W, et al. Cancer as a marker of genetic medical disease: an unusual case of medullary carcinoma of the kidney. Am J Surg Pathol. 1998;22:260–4.
Dimashkieh H, Choe J, Mutema G. Renal medullary carcinoma: a report of 2 cases and review of the literature. Arch Pathol Lab Med. 2003;127:e135–8.
Yang XJ, Sugimura J, Tretiakova MS, et al. Gene expression profiling of renal medullary carcinoma: potential clinical relevance. Cancer. 2004;100:976–85.
Kalyanpur A, Schwartz DS, Fields JM, et al. Renal medulla carcinoma in a white adolescent. AJR Am J Roentgenol. 1997;169:1037–8.
Simpson L, He X, Pins M, et al. Renal medullary carcinoma and ABL gene amplification. J Urol. 2005;173:1883–8.
Khan A, Thomas N, Costello B, et al. Renal medullary carcinoma: sonographic, computed tomography, magnetic resonance and angiographic findings. Eur J Radiol. 2000;35:1–7.
Selby DM, Simon C, Foley JP, et al. Renal medullary carcinoma: can early diagnosis lead to long-term survival? J Urol. 2000;163:1238.
Amin MB, Smith SC, Agaimy A, et al. Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity. Am J Surg Pathol. 2014;38:871–4.
Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur Urol. 2016;69:1055–61.
Rao P, Tannir NM, Tamboli P. Expression of OCT3/4 in renal medullary carcinoma represents a potential diagnostic pitfall. Am J Surg Pathol. 2012;36:583–8.
Liu Q, Galli S, Srinivasan R, et al. Renal medullary carcinoma: molecular, immunohistochemistry, and morphologic correlation. Am J Surg Pathol. 2013;37:368–74.
Pirich LM, Chou P, Walterhouse DO. Prolonged survival of a patient with sickle cell trait and metastatic renal medullary carcinoma. J Pediatr Hematol Oncol. 1999;21:67–9.
Stahlschmidt J, Cullinane C, Roberts P, et al. Renal medullary carcinoma: prolonged remission with chemotherapy, immunohistochemical characterisation and evidence of bcr/abl rearrangement. Med Pediatr Oncol. 1999;33:551–7.
Lopez-Beltran A, Cheng L, Raspollini MR, et al. SMARCB1/INI1 genetic alterations in renal medullary carcinomas. Eur Urol. 2016;69:1062–4.
MacLennan GT, Farrow GM, Bostwick DG. Low-grade collecting duct carcinoma of the kidney: report of 13 cases of low-grade mucinous tubulocystic renal carcinoma of possible collecting duct origin. Urology. 1997;50:679–84.
Parwani AV, Husain AN, Epstein JI, et al. Low-grade myxoid renal epithelial neoplasms with distal nephron differentiation. Hum Pathol. 2001;32:506–12.
Rakozy C, Schmahl GE, Bogner S, et al. Low-grade tubular-mucinous renal neoplasms: morphologic, immunohistochemical, and genetic features. Mod Pathol. 2002;15:1162–71.
Ferlicot S, Allory Y, Comperat E, et al. Mucinous tubular and spindle cell carcinoma: a report of 15 cases and a review of the literature. Virchows Arch. 2005;447:978–83.
Fine SW, Argani P, DeMarzo AM, et al. Expanding the histologic spectrum of mucinous tubular and spindle cell carcinoma of the kidney. Am J Surg Pathol. 2006;30:1554–60.
Dhillon J, Amin MB, Selbs E, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid change. Am J Surg Pathol. 2009;33:44–9.
Bulimbasic S, Ljubanovic D, Sima R, et al. Aggressive high-grade mucinous tubular and spindle cell carcinoma. Hum Pathol. 2009;40:906–7.
Pillay N, Ramdial PK, Cooper K, et al. Mucinous tubular and spindle cell carcinoma with aggressive histomorphology—a sarcomatoid variant. Hum Pathol. 2008;39:966–9.
Sadimin ET, Chen YB, Wang L, et al. Chromosomal abnormalities of high-grade mucinous tubular and spindle cell carcinoma of the kidney. Histopathology. 2017;71:719–24.
Paner GP, Srigley JR, Radhakrishnan A, et al. Immunohistochemical analysis of mucinous tubular and spindle cell carcinoma and papillary renal cell carcinoma of the kidney: significant immunophenotypic overlap warrants diagnostic caution. Am J Surg Pathol. 2006;30:13–9.
Gupta R, Balzer B, Picken M, et al. Diagnostic implications of transcription factor Pax 2 protein and transmembrane enzyme complex carbonic anhydrase IX immunoreactivity in adult renal epithelial neoplasms. Am J Surg Pathol. 2009;33:241–7.
Molinie V, Balaton A, Rotman S, et al. Alpha-methyl CoA racemase expression in renal cell carcinomas. Hum Pathol. 2006;37:698–703.
Cossu-Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol. 2006;19:488–93.
Mehra R, Vats P, Cieslik M, et al. Biallelic alteration and dysregulation of the hippo pathway in mucinous tubular and spindle cell carcinoma of the kidney. Cancer Discov. 2016;6:1258–66.
Brandal P, Lie AK, Bassarova A, et al. Genomic aberrations in mucinous tubular and spindle cell renal cell carcinomas. Mod Pathol. 2006;19:186–94.
Ren Q, Wang L, Al-Ahmadie HA, et al. Distinct genomic copy number alterations distinguish mucinous tubular and spindle cell carcinoma of the kidney from papillary renal cell carcinoma with overlapping histologic features. Am J Surg Pathol. 2018;42:767–77.
Kuroda N, Hes O, Michal M, et al. Mucinous tubular and spindle cell carcinoma with Fuhrman nuclear grade 3: a histological, immunohistochemical, ultrastructural and FISH study. Histol Histopathol. 2008;23:1517–23.
Simon RA, di Sant’agnese PA, Palapattu GS, et al. Mucinous tubular and spindle cell carcinoma of the kidney with sarcomatoid differentiation. Int J Clin Exp Pathol. 2008;1:180–4.
Perrino CM, Grignon DJ, Williamson SR, et al. Morphological spectrum of renal cell carcinoma, unclassified: an analysis of 136 cases. Histopathology. 2018;72:305–19.
Storkel S, Eble JN, Adlakha K, et al. Classification of renal cell carcinoma: workgroup no. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 1997;80:987–9.
Karakiewicz PI, Hutterer GC, Trinh QD, et al. Unclassified renal cell carcinoma: an analysis of 85 cases. BJU Int. 2007;100:802–8.
Lopez-Beltran A, Kirkali Z, Montironi R, et al. Unclassified renal cell carcinoma: a report of 56 cases. BJU Int. 2012;110:786–93.
Crispen PL, Tabidian MR, Allmer C, et al. Unclassified renal cell carcinoma: impact on survival following nephrectomy. Urology. 2010;76:580–6.
Delahunt B, Sika-Paotonu D, Bethwaite PB, et al. Grading of clear cell renal cell carcinoma should be based on nucleolar prominence. Am J Surg Pathol. 2011;35:1134–9.
Dagher J, Delahunt B, Rioux-Leclercq N, et al. Clear cell renal cell carcinoma: validation of World Health Organization/International Society of Urological Pathology grading. Histopathology. 2017;71:918–25.
Khor LY, Dhakal HP, Jia X, et al. Tumor necrosis adds prognostically significant information to grade in clear cell renal cell carcinoma: a study of 842 consecutive cases from a single institution. Am J Surg Pathol. 2016;40:1224–31.
Leibovich BC, Blute ML, Cheville JC, et al. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. Cancer. 2003;97:1663–71.
Amin MB, Edge S, Greene FL, et al. AJCC Cancer staging manual. 8th ed. New York: Springer International Publishing; 2017.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Divatia, M.K., Guo, C.C., Rehman, A., Ro, J.Y. (2020). Major Subtypes of Renal Cell Carcinoma. In: Divatia, M., Ozcan, A., Guo, C., Ro, J. (eds) Kidney Cancer. Springer, Cham. https://doi.org/10.1007/978-3-030-28333-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-28333-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28332-2
Online ISBN: 978-3-030-28333-9
eBook Packages: MedicineMedicine (R0)