
Abstract
Recent advances in high-throughput sequencing of nucleic acids have led to the fascinating insight that the majority of the human genome is transcribed. This includes tens of thousands of RNAs sized larger than 200 nucleotides that are not translated into proteins and are referred to as long noncoding RNAs (lncRNAs). Until now, only a few of these lncRNAs have been functionally characterized. Here we highlight lncRNAs related to cardiovascular physiology and disease (CVD). We start with an overview of lncRNA classification schemes and of molecular functions of lncRNAs, giving examples of lncRNAs with cardiovascular function in each class. The main focus then is to systematically review 57 lncRNAs implicated in atherosclerosis, myocardial infarction, aortic aneurysm, cardiomyopathy, angiogenesis, arrhythmia, and stroke. We discuss the evidence how these lncRNAs partake in the regulation of cell lineage specification, differentiation potential, cell proliferation, and cell survival in the cardiovascular cell lineages. Specific emphasis is put on recently published lncRNA knockout approaches, and on lncRNAs that have been implicated as important regulators in animal in vivo models of cardiovascular diseases and/or identified in human patient cohorts.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


7.1 Introduction
For a long time, proteins have been considered the main functionally active molecules in mammalian cells. Yet, in a few selected cases, functionally important non-protein-coding cellular RNAs had been identified. Most prominently, the long noncoding RNA (lncRNA) XIST has been known for decades to be essential for establishing X-chromosome inactivation in female mammals, and studies on XIST have become blueprints for approaches in studying lncRNA function (see [1] for review). Similarly, several noncoding RNAs have been identified early on in several important imprinted gene clusters, among them RNAs with relevance for cardiovascular disease, like H19, Meg3, or Kcnq1ot1. Their study has inspired more recent investigations on how lncRNAs interact with chromatin regulators and affect transcription. But only with the advent of high-throughput nucleic acid sequencing analyses in the 2000s, and soon after the detection of the large class of regulatory small interfering RNAs and microRNAs, lncRNAs have entered the focus of the investigation on a genome-wide scale. It is becoming clear by work of the ENCODE or FANTOM consortia that protein-coding genes account for only as little as 1.5% of the genome. In recent years, thousands of lncRNAs have been identified. LncRNAs are transcribed from thousands of previously unannotated non-protein-coding genes in our genomes [2,3,4,5]. This raises the question if and to what extent the many thousands of lncRNA transcripts are functional. Whether only few or many of these lncRNA transcripts carry cellular functions is a matter of currently ongoing research.
Overall, on a molecular level, it seems that lncRNAs do not have catalytic ribozyme functions. Only ribosomal RNAs (rRNA) and small nuclear RNAs (snRNA) function as catalytic entities in ribosomes and the spliceosome, respectively. Rather, lncRNAs serve as regulators of other molecules by binding to them. In this function, and as will be reviewed in detail in this book chapter, lncRNAs guide protein complexes to specific DNA sequences, or scaffold multiprotein complexes, or increase or inhibit the activity of enzymes, or affect mRNA stability and translation capacity. In functional terms, a growing number of lncRNAs are being found to impact the development of cardiovascular cell types and organs in the vertebrate embryo. To name a few, Braveheart [6] Fendrr [7], Upperhand [8], and HoxBlinc [9] are important recently identified lncRNAs in this class. Other lncRNAs, in some cases first identified in independent systems like in cancer models or in generic cellular screens, were then also found to be misexpressed in diseases of the heart or vasculature and to causally contribute to cardiovascular disease. ANRIL [10,11,12], MALAT1 [13, 14], Myheart [15], Chaer [16], lincRNA-p21 [17], CARL [18], CARMEN [19], Rffl-lnc1 [20], and ROR [21] are prominent examples of this latter class. Extending the compendium of lncRNAs, recent experiments have documented that besides the thousands of linear lncRNAs, thousands of genes also express circular RNAs (circRNAs), most of which are noncoding [22]. circRNAs emerge from unconventional backsplicing (tail-to-head splicing) of a downstream exon to a more upstream-located exon, resulting in covalent linkage of RNA in cis by a covalent 3′–5′ phosphodiester bonding. Compared to linear lncRNAs, an even smaller number of circRNAs has been functionally studied. But from the little existing insight it has become clear that also some circRNAs, though not encoding proteins, can carry regulatory functions and contribute to cardiovascular disease when misregulated, such as circRNAs emerging from the ANRIL locus on the well-known cardiovascular risk region on chromosome 9p21 [23, 24].
7.2 General Characteristics and Classification of lncRNAs
Here we describe the molecular characteristics of lncRNAs, linear and circular (Sects. 7.2.1–7.2.2.8), and their well-known molecular functions (Sects. 7.3.1–7.3.10), always by focusing on the description of those lncRNAs that have been implicated in cardiovascular physiology and disease. Their specific roles in physiology and cardiovascular disease are described later (Sects. 7.4.1–7.4.2.7) following the introductory parts on classification and molecular function.
7.2.1 Characteristics of lncRNAs
lncRNAs have been defined rather arbitrarily by a length >200 nucleotides (nts), a threshold rooting in cutoffs during biochemical separation from shorter RNAs like microRNAs using a commercial DNA/RNA isolation kit [25, 26]. Earlier functionally annotated noncoding RNAs, such as ribosomal RNA, transfer RNAs, small nuclear RNAs, small nucleolar RNAs, microRNAs, endogenous small interfering RNAs, or Piwi-associated RNAs, even when >200 nts, are not classified as lncRNAs [27] (see Box 7.1 for a list of general characteristics of lncRNAs). The key criterion for being a lncRNA is that, firstly, lncRNAs do not carry prominent open reading frames for protein translation. Secondly, lncRNAs are unlikely to be translated into proteins even when carrying open reading frames. In fact, although a significant number (50%) of lncRNAs do associate with ribosomes, this association is not productive, and no translation ensues [28]. Over 27,000 lncRNA genes are predicted to exist in humans, leading to over 100,000 different lncRNA transcripts (https://lncipedia.org) [29]. This number rivals the number of protein-coding genes in our genomes (19,817; www.ensembl.org), feeding the hypothesis that much of our organismic complexity as higher metazoans may be related to the function of noncoding RNAs [30]. A common question is whether lncRNAs are more likely to act in the nucleus or in the cytoplasm. When assessing the transcript abundance of 1339 robustly expressed lncRNAs and of 13,933 mRNAs on a genome-wide scale, 17% of the tested lncRNAs were found to be exclusively localized to the nucleus, a ratio that is slightly but significantly larger than the ratio of exclusively nuclear protein-coding mRNAs among all mRNAs (15%). In contrast, the frequency of exclusively cytoplasmic lncRNAs is small (4%) compared to mRNAs (26%). Still a majority of lncRNAs and mRNAs are present in both nucleus and cytoplasm [31,32,33,34]. One exception is the class of circular lncRNAs (circRNAs, see below), which are mainly cytoplasmatic [22, 35]. Overall, the cellular localization patterns do not indicate a preferred subcellular compartment of function for the class of lncRNAs. Instead lncRNA function appears to be highly diverse. Also, on average, lncRNAs are similarly stable compared to coding mRNAs, and only specific classes of lncRNAs, few in the overall lncRNA number, qualify as being specifically unstable, as specified below [36].
Box 7.1 Characteristics of lncRNAs
-
LncRNAs are a heterogeneous class of tens of thousands of noncoding transcripts [29, 37]
-
Similarity to protein-coding mRNAs [37]:
-
RNA polymerase II transcripts
-
Expressed from genes and organized in exons that are defined by chromatin states alike protein-coding mRNAs
-
Carrying 5′cap (depending on class)
-
Spliced and displaying 3′ polyA tails (depending on class)
-
-
Unique features:
-
Unlikely to carry open reading frames >300 nucleotides (see text for details) [38, 39].
-
Shorter than mRNAs on average, with fewer but longer exons [31].
-
Expressed at lower levels [40].
-
Faster primary sequence evolution than mRNAs, but still often with orthologs in other species [41].
-
Less efficient splicing compared to mRNAs [42].
-
95% of lncRNAs do not productively associate with translating ribosomes [28].
-
Prominent tissue-specificity [31] and potential to regulate gene expression in cis and in trans [37, 43,44,45,46].
-
Sometimes specialized 3′ ends (polyA-independent; triple helices, tRNA/snoRNA-like ends) [47].
-
A large class of lncRNAs can be circular (5′ and 3′ ends are covalently linked; circRNAs do not have 5′ cap or polyA tail) [22].
-
7.2.2 Classification of lncRNAs
An important approach in trying to grasp the diversity of lncRNA function is to classify lncRNAs according to the location of their gene bodies in the genome. This relates to the observation that in many cases the relative positioning of lncRNA genes to other functional elements nearby is important. For example, a common function of lncRNAs is to affect the transcription of nearby genes, either directly or indirectly, by influencing enhancers or the local chromatin state at promoters. However, there is more than one classification scheme, and lncRNAs may also be grouped according to their molecular biogenesis, or their type of functionality, and the group affiliation changes depending on the grouping principle (see Box 7.2 for different classification schemes). In the following paragraphs we present the major classification scheme for lncRNAs that roots in the genomic organization of genes encoding these noncoding RNAs relative to neighboring protein-coding gene elements.
7.2.2.1 Large Intergenic Noncoding RNAs (lincRNAs)
The genetic loci of lincRNAs do not overlap with protein-coding genes. lincRNAs are on average 1 kb long, contain exons, and are polyadenylated and spliced. Compared to mRNAs, lincRNAs are not as efficiently co-transcriptionally spliced and not so well polyadenylated [40, 48]. As a consequence, without efficient end-processing and when still carrying introns, such types of lincRNAs can be unstable and become subject to early degradation by the nuclear exosome, the major RNA degradation complex [48]. The degradation process can become active even before lincRNA transcription is terminated. However, a number of lincRNAs, even when lacking a polyA tail can become stabilized by unconventional molecular features, such as structured RNA folds at their 3′ ends. For example, backfolding into RNA triple helices, tRNA-like or snoRNA-like folds, can stabilize certain lincRNAs [47, 49,50,51]. Several noncoding RNAs have been identified as lincRNAs and have later been implicated in cardiovascular disease, such as MALAT1 [13, 14, 47], lincRNA-p21 [17], Chaer [16], ROR [21], HOTAIR [52], Rncr3 [53], Gas5 [54], Mirt1 [55], UCA1 [55], linc00305 [56], and lincRNA-DYNLRB2–2 [57]. Among these, MALAT1 is an especially well studied case where unconventional 3′ end processing determines the 3′ end: MALAT1 is not polyadenylated after endonucleolytic cleavage as the vast majority of long RNA polymerase II transcripts, but it is end-processed by RNase P, which cleaves off a tRNA-like RNA-fold from the 3′ end. After that cut, an A/U-rich sequence is exposed that stabilizes MALAT1 RNA by folding into a triple helical RNA structure that stabilizes MALAT1 in the absence of a polyA tail [47].
7.2.2.2 Natural Antisense Transcripts (NATs, asRNAs)
A large majority of transcripts in our genomes represent natural antisense transcripts (NATs, asRNAs) relative to neighboring transcripts. NATs are encoded on the strand opposite to the strand transcribed in the primary locus. Mostly on the edges of the host gene, NATs can either overlap the promoter or the terminator of a neighboring primary locus. NATs are less frequently spliced or polyadenylated than mRNAs or lincRNAs [58,59,60,61,62,63,64,65]. Examples with relevance to cardiovascular physiology and disease are ANRIL [66], SENCR [67], MALAT1 [68] MANTIS [69], Myheart [15], HOXC-AS1 [70], HIF1αAS1 [71], KCNQ1OT1 [72], and FosDT [73].
7.2.2.3 Promoter Upstream Antisense Transcripts (PROMPTS, uaRNAs)
There is a rather diverse class of lncRNA emerging from regions 5′ to promoters of established genes, and specifically in antisense orientation to these. These RNAs are termed promoter upstream transcripts (PROMPTS), and are also known as upstream antisense RNAs (uaRNAs) [74,75,76]. They are expressed in an antisense direction because mammalian RNAP II transcription initiation sites, both at promoters and at enhancers, typically contain oppositely oriented core promoter elements within a single nucleosome-depleted region that defines these transcription start sites. These transcripts are diverse in size and are 5′ capped. Transcription upstream of promoters likely occurs because many gene promoters consist of two separate core promoter elements, which drive divergent transcription events. Often, only the major direction of transcription leads to productive elongation and to stable RNAs. PROMPTS/uaRNAs are rather unstable due to the absence of transcription start site (TSS)-proximal 5′ splice sites in sequences upstream of promoters. They are also unstable because of the premature occurrence of TSS-proximal polyA sites. As a consequence, they are degraded rapidly by the nuclear exosome [74]. Before the class of uaRNAs was identified and coined in vertebrate genomes, similar unstable upstream RNAs had been found in yeast and named differently: Yeast has two major types of these transcripts. First, there are cryptic unstable transcripts (CUTs). CUTs are expressed in the opposite direction from nonoverlapping bidirectional promoters and their ends can lie in the starts of other genes [61, 77]. The expression of CUTs is limited by rapid degradation [78]. Secondly, hundreds of stable unannotated transcripts (SUTs) exist in yeast [79]. As such, CUTs and SUTs also belong to the previously mentioned class of antisense RNAs [44, 80]. PROMPTs and uaRNAs occur in all gene classes, in principle, and thus have not been specifically studied in the context of cardiovascular disease or cardiovascular genes or lncRNAs. An exception is the lncRNA Upperhand, whose expression from a bidirectional promoter, shared with the cardiac transcription factor Hand2, is involved in its role in cardiac development [8]. Also ANRIL shares a bidirectional promoter with p14, but no conclusive insight into the relevance of this positioning has yet been published [66].
7.2.2.4 Enhancer RNAs (eRNA) and Activating Noncoding RNAs with Enhancer Function (ncRNA-a)
As the fourth class of lncRNAs, enhancers of genes have been found to be transcribed. As a consequence, enhancer elements in the genome express enhancer eRNAs in 5′ and 3′ direction [81,82,83,84]. eRNAs are 50–2000 nts long and do not carry 5′ cap or polyA tail [85]. The classification of eRNAs is at the moment not satisfactorily resolved, as many eRNAs do not appear in lncRNA databases like GENCODE [31, 85]. This could be due to the fact that eRNAs are rather unstable and often present at even lower levels than other lncRNAs [85]. In fact, eRNA analysis requires different, more sensitive and specialized biochemical preparation methods (CAGE [86, 87], GRO-seq [82], or PRO-seq [88, 89]). Apart from true eRNAs, a number of lncRNAs have been found, called ncRNA-a, that are not classified as eRNAs, but, similar to enhancers, stimulate targets genes in their vicinity [89]. Up to 3000 bona fide lncRNAs from ENCODE databases exist, whose initiation sites overlap predicted enhancers, as determined by the H3K4me1 and H3K27 acetylation during chromatin profiling approaches [90]. With relevance to the cardiovascular system, Carmen [19], Wisper [91], and Upperhand [8] have been proposed to function by enhancing transcription as enhancer RNAs. Also, HOTTIP [92], linc-HOXA1 [93], and SMILR [94] activate genes in their immediate neighborhood, but it is not clear whether as eRNA or ncRNA-a or by another mechanism.
7.2.2.5 Long Intronic ncRNAs
The fifth class of lncRNAs is encoded within introns of some multi-exon genes and these are called long intronic ncRNAs [4, 95]. Overall, their expression is not a passive bystander to the expression of their parental gene, but they display a differential expression. A subclass of intronic ncRNAs is stable intronic sequences (sisRNAs), which is a broad class of noncoding RNAs that comprises both linear as well as circular RNAs [96, 97].
7.2.2.6 Transcribed Pseudogenes
The sixth class of lncRNAs can be defined as transcripts of a subset (2–20%) of pseudogenes [4]. Pseudogenes have originated in evolutionary history through tandem duplication of a parental gene or retrotransposition of an mRNA of a protein-coding gene. Without the selective pressure of encoding a protein, the rate by which mutations were retained in them increased. As a consequence, most pseudogenes have been inactivated and are not expressed anymore. The best-studied example is the Xist RNA, which also has a function in the cardiovascular system, and has emerged through a complex process from a pseudogenized protein-coding gene [98]. On the other hand, when expressed, transcribed pseudogenes can exert regulatory functions as lncRNAs and possibly affect their coding host genes [99].
7.2.2.7 Circular RNAs
Based on advances in RNA/DNA sequencing and in bioinformatics tools to map transcriptomes onto genomes, it has been revealed that thousands of genes express circular circRNAs [22]. As much as 20% of all genes expressed at any point in cells produced circRNAs [100,101,102]. These are produced by spliceosomal action from multiexon host genes: Instead of conventional colinear splicing of exons in their genomic 5′ → 3′ order, where introns are excised and later degraded as intronic lariats, an atypical form of splicing (backsplicing) occurs during the biogenesis of circRNAs: A downstream exon is ligated to a more upstream exon, causing a covalent 3′ → 5′ linkage of exonic sequences (see [103] for review). circRNA molecules are rather stable because they are not accessible anymore by cellular RNA degradation machineries, which are primarily exonuclease based (see [104] for review). circRNAs vary in size, but are >500 nucleotides on average [100], and thus classify as lncRNAs, though not usually reviewed together. In contrast to linear lncRNAs, circRNAs are mostly cytoplasmic [22]. One important function of circRNAs is to control transcriptional initiation and splicing in the nucleus, but also cytoplasmic functions have been documented (see in detail below). So far, only a few circRNAs have been part of studies that explored functions in the cardiovascular system in vivo, and these include circANRIL [23, 24], HRCR [105], and MFACR [106].
7.2.2.8 lncRNAs with Translated Small Open Reading Frames (sORFs)
Though generally not encoding classical protein-coding open reading frames (ORFs) much longer than 300 nts, small ORFs do occur by chance in any sufficiently long RNA transcript, and even in the most classical lncRNA, Xist [39]. Only the development of ribosome release profiling technology has since firmly documented that lncRNAs may associate with ribosomes, but that 95% of lncRNAs are indeed not functionally translated [28]. This means also, however, that hundreds of lncRNAs are potentially protein-coding or at least encoding small peptides. Recent studies revealed that several hundred micropeptide-encoding short sORFs are potentially translated from our genomes. Functionally active regulatory and signaling molecules encoded as true micropeptides have already been found in animals [107,108,109,110,111,112] and plants [113]. Together, a continuum seems to exist between noncoding and coding sequence content in RNA messages [114], but the number of functional translated sORFs in bona fide lncRNAs is considered rather small overall [115]. Some of the best understood micropeptide-generating lncRNAs have indeed a function in the cardiovascular system, namely, in regulating heart muscle contractility [110, 112, 116].
Since different lncRNA classification schemes are used in the literature, key concepts in lncRNA classification are summarized in Box 7.2.
Box 7.2 Classification of lncRNAs
-
Classification based on neighborhood to protein-coding genes:
-
Linear intergenic lncRNAs originating from enhancers (bidirectionally transcribed and unstable)
-
Linear lncRNAs originating from host gene promoters (transcribed opposite to host mRNAs, unstable)
-
Linear lncRNAs as stand-alone genes (more stable, longer)
-
Long intronic ncRNAs
-
Circular lncRNAs (stable, produced from internal exonic and intronic sequences of expressed multiexonic genes)
-
-
Classification based on molecular biogenesis (see text for explanation of acronyms):
-
lincRNAs
-
NATs, asRNAs
-
PROMPTS, uaRNAs, CUTs, SUTs
-
eRNAs, ncRNA-a
-
-
Classification based on function (majority of lncRNAs are functional; see Fig. 7.1):
-
lncRNAs with functions as RNA molecules
-
lncRNAs for which transcriptional act executes functionality
-
Small minority of lncRNAs that are translated to polypeptides
-
Transcription noise
-

Molecular functions of lncRNAs in eukaryotic cells. Roles of linear and of circular long noncoding RNAs in the nucleus (a–e) and in the cytoplasm (f–j). Names of lncRNAs with cardiovascular relevance are indicated for each class, as far as known. Chromatinized DNA (dark grey), lncRNAs (light green), and protein-coding genes (light blue). (a) Tethering/Scaffolding/Regulating chromatin modifiers. As an example, a lncRNA is shown to tether a repressive chromatin regulator (Polycomb complex) via a lncRNA hairpin motif to the transcription start site of a protein-coding target gene to inhibit transcriptional activation. (b) Long-range chromatin folding/looping. As an example, Xist-dependent chromatin compaction is shown on the female inactive X-chromosome, whose inactivation is promoted by localization to a heterochromatic territory close to the nuclear membrane (light grey). (c) Enhancing transcription from gene promoters. Transcription of an enhancer region, which leads to the production of eRNAs, to chromatin fiber looping towards a promoter, and to the activation of the RNAP II preinitiation complex through the Mediator complex (Top). At the promoter, certain classes of intron-containing circRNAs (EIciRNAs, ciRNAs) independently stimulate RNAP II (Bottom). (d) Antisense transcription: transcription of lncRNAs in antisense to a protein-coding gene, shown to inhibit sense target mRNA transcription. (e) Regulation of mRNA splicing by lncRNAs: linear and circular lncRNAs affect the spliceosome (and associated splicing factors) during alternative splicing. The processes of circRNA formation (“backsplicing”) and linear mRNA splicing mutually impair each other. circRNAs can independently affect splicing through R-loop formation and by blocking RNAP II progression. (f) Linear lncRNAs (top) and circRNAs (bottom) can harbor multiple microRNA seed sequences, which leads to sequestration and inactivation of microRNA:Argonaute 2 complexes without lncRNA degradation (termed microRNA sponge or competing endogenous RNA effect). Reducing the availability of free microRNAs de-represses mRNAs that are natural targets of these microRNAs. (g) Binding of lncRNAs to a stability-regulating motif in a target mRNA, thereby regulating mRNA stability. (h) Regulation of mRNA translation: lncRNAs interacting with translation initiation factor (grey) shown as an example. (i) Regulation of cytoplasmic proteins: as an example circANRIL is shown, which inhibits the PeBoW protein complex in its activity of rRNA processing upstream of ribosome maturation. (j) Rare cases where the translation of ORF encoded on linear or circular lncRNAs is observed. A ribosome is depicted to translate a micropeptide from a small ORF after associating via an internal ribosome entry site (IRES). AGO Argonoute 2-containing RNA-induced silencing complex, cheRNA chromatin-enriched RNAs with enhancer function, circRNA 5′3′-linked exonic circular RNA, ciRNA intronic circular RNA, EIciRNA exon-and-intron-containing circular RNA, eRNA enhancer RNA, IRES internal ribosome entry site, me H3 lysine K9/K27 trimethylation of histone tails, ORF open reading frame, pA polyA tail of RNAs, RNAP II RNA polymerase II transcription apparatus
7.3 Molecular Functions of lncRNAs
Here we review lncRNA functions that have been studied in the specific context of cardiovascular disease (Fig. 7.1). We start with functions in the nucleus (Fig. 7.1a–e) and then describe functions in the cytoplasm (Fig. 7.1f–j). A majority of lncRNAs can also directly affect transcriptional initiation and elongation by RNA polymerase II at promoters of protein-coding genes by scaffolding, tethering, and regulating the activity of enzyme complexes that modify histone tails or reposition nucleosomes (Fig. 7.1a). Related to this function, lncRNAs that interact with chromatin regulators can regulate large-scale chromatin fiber folding and influence repositioning of fibers relative to heterochromatic subnuclear domains (Fig. 7.1b). Another large fraction of lncRNAs emerge from enhancer regions of protein-coding genes and determine enhancer-dependent activation of the promoter of protein-coding genes (Fig. 7.1c). Many other lncRNAs are positioned in the genome in antisense to protein-coding genes and thereby affect their transcription (Fig. 7.1d). Yet other lncRNAs, but much fewer in number, are known to participate in splicing regulation (Fig. 7.1e). In the cytoplasm, some lncRNAs sequester and inactivate mRNA-regulating microRNAs (Fig. 7.1f). Likely for technical reasons in the experimental assessment, much fewer lncRNAs are known to bind mRNAs and thereby affect mRNA stability (Fig. 7.1g) or translation potential of mRNAs (Fig. 7.1h). lncRNAs can also bind protein complexes in the cytoplasm and affect their function, as shown in the example of circANRIL in inhibiting rRNA and ribosome maturation (Fig. 7.1i). Finally, a small minority of linear and circular lncRNAs actually encode small ORFs and are translated to micropeptides or larger parts of proteins, depending on the size of the ORF (Fig. 7.1j). Since no dedicated studies have been performed on the specific cardiovascular role of lncRNAs functioning as regulators of centromeres or of telomeres, or in DNA repair, even if important for general cell functionality, we will not specifically review these latter lncRNAs. In Sects. 7.3.1–7.3.10, we review each functional lncRNA class in detail. We list cardiovascular lncRNAs in each functional class (Fig. 7.1), and refer to these classes also in the following paragraphs on the roles of lncRNAs in cardiovascular physiology and diseases (Sects. 7.4.1–7.4.2.7; Tables 7.1, 7.2, and 7.3).
7.3.1 Tethering/Scaffolding/Regulating Chromatin Modifiers
One of the most intensively studied function of lncRNAs is their interaction with chromatin regulatory proteins during the control of gene transcription (Fig. 7.1a). This includes interactions with proteins involved in chromatin fiber regulation by DNA methylation systems or histone tail-modifications, in nucleosome-remodeling, and in chromatin fiber folding and long-range looping in the 3D architecture of the nucleus.
The interaction with chromatin regulators is one of the best-understood function of nuclear lncRNAs. Some of the best-known lncRNAs have been shown to tether chromatin-regulating complexes to specific sites in the genome, and thereby to promote or repress transcription of protein-coding genes at selected target genes (Fig. 7.1a). Besides tethering, another central task in this interaction is the ability of lncRNAs to scaffold (link together) chromatin-regulating factors, for example, to link several different chromatin readers and writers to specific genomic loci [221,222,223,224,225,226]. Recent experiments show, however, that the picture is more complex than that: lncRNAs not only specifically activate chromatin regulators, but can also inhibit them. The hypothesis has formed that at least some of the long noncoding transcripts in our genome are used to inhibit the activity of chromatin regulators. The aim is to inhibit aberrant, low-affinity misassociation of chromatin regulators on the genome. With such a strategy, many nascent RNA transcripts from many types of genes are employed as noncoding RNAs in making gene expression more accurate [227].
Examples for lncRNAs that bind chromatin-regulating complexes at protein-coding gene promoters, and where relevance to cardiovascular pathophysiology has been established, are: Fendrr binding to both the repressive PRC2 and the activating TrxG/MLL complexes [7]; Braveheart binding to the repressive PRC2 [6]; Malat1 binding the activating MLL, LSD1, BAF57, and unmethylated Polycomb 2 proteins in their complexes [206] and indirectly binding to nascent transcripts of active genes [228]; HoxBlinc binding to the activating Setd1a/MLL1 [9]; Xist to at least 10 repressive chromatin-binding and chromatin-regulating protein complexes including hnRNPK/U, noncanonical repressive PRC1, SHARP/Spen, SAF-A, and LBR [229, 230]; H19 binding the repressive MBD1 [200]; ANRIL binding to activating as well as repressive PRC1 and PRCR2 complexes [191, 192]; lincRNA-p21 binding to repressive hnRNPK [17]; Meg3 binding to repressive JARID2-PRC2 complexes [204]; Tug1 binding to repressive PRC2, RIZ1, Sin3A, and JARID1A [206]; Kcnq1ot1 binding to repressive PRC2 [210]; MANTIS binding an activating BRG1-containing SWI/SNF chromatin remodeling complex [69]; HOTTIP binding activating WDR5/MLL complexes [92]; Carmen binding PRC2 complexes with unclear outcome [19]; Myheart binding and inhibiting the BRG1-containing SWI/SNF chromatin remodeling complex [15]; Chaer binding and inhibiting PRC2 [16]; and HOTAIR binding the repressive PRC2 [208]. By interacting with these chromatin regulators, such lncRNAs can scaffold multiprotein complexes, tether them to specific genomic sites, serve as decoys during target site association, and change their activity. In the example of the study of HOTAIR it becomes obvious, however, that a careful follow-up analysis is required to confirm the functional relevance of a previously observed physical interaction with a chromatin regulator: While the HOTAIR lncRNA does bind the repressive PRC2 complex without doubt, and while there is a correlation between the presence of HOTAIR at a genetic locus and the repression of this locus, later decisive experiments challenged the simple picture that HOTAIR silenced target genes through PRC2. In fact, it was revealed that PRC2 was not primarily required for HOTAIR-mediated target gene repression and that PRC2:HOTAIR binding was likely a consequence of later PRC2 recruitment to an already silenced locus [209]. Therefore, care is advised in drawing conclusions on effector mechanisms from the many studies that report a binding of a lncRNA to Polycomb or Trithorax complexes, which are often recruited to loci to maintain but not to induce chromatin states.
What decides if a lncRNAs acts only in cis on few neighboring genes, and/or also in trans (on loci on other chromosomes on many targets in the genome)? This question has begun to be addressed also by studying lncRNAs with cardiovascular relevance. For example, MALAT1, representing one class of lncRNAs, binds with high affinity to hundreds of target genes [228, 231], as opposed to a different class of lncRNAs that bind only to a single locus in the genome [232]. It is thought that the local concentration of a lncRNA on chromatin is decisive for this differential behavior: Low-abundance lncRNAs interact rather with few genes in close proximity (like HOTTIP) (Fig. 7.1a), while XIST with 100 copies/cell can spread further using low-affinity sites, and MALAT1 with thousands of stable RNA copies can diffuse also in trans throughout the nucleus and gain access to hundreds of targets [233]. The presented studies offer blueprints for thinking about how any novel lncRNAs may dynamically interact with chromatin.
7.3.2 lncRNAs Regulate Long-Range Chromosomal Looping in the Nuclear Space
How lncRNAs first bind to selected focal genomic points and then spread over larger domains on the genome to impose long-range gene expression control is currently being investigated (Fig. 7.1b). The Xist RNA has been instructive to understand lncRNA spreading over large chromosomal domains, which coincides with establishing a unique chromatin state over the domain, and with affecting the three-dimensional folding of the chromosome fiber. Through these processes, the lncRNA transduces a repressed or active chromatin state over large domains also by repositioning the chromosomal domain to preexisting repressive or activating nuclear subdomains. Intranuclear domains of relevance are different repressive domains (Polycomb bodies, heterochromatic regions close to the nuclear membrane, etc.) and active domains (interchromatin granules, transcription factories, etc.). How lncRNAs mediate chromosome fiber folding is a matter of current research, and this topic is probably best understood for Xist-dependent X-chromosome inactivation. There, unexpectedly, a small number of only 50 Xist RNA/PRC2 complexes are at work to compact the nucleosomes over the >150 megabases long X-chromosome. It has been concluded that lncRNA-chromatin interaction is highly dynamic, and a dynamic hit-and-run model has been invoked to explain spreading and coincident compaction of long chromosomal domains [233,234,235]. For spreading over such long distances, Xist RNA is initially locally stabilized by co-transcriptional binding to proteins at a nucleation within its own gene body [236]. Only after that it can start spreading on chromatin in cis. Xist uses the 3D architecture of the chromosome to spread to noncontiguous loci [237]. In the course of spreading, Xist changes chromatin loop structures through evicting cohesin rings which entrap fibers [238] (Fig. 7.1b).
Conceptually similarly, at least three other cardiovascular lncRNAs affect gene activity of target genes by determining looping of chromosomal domains encompassing the target genes. MALAT1 was found to reposition target genes in the 3D nuclear space. In particular, MALAT1 associated with the Polycomb complex member Pc2, inhibited its preferred binding of repressive chromatin marks inside heterochromatic Polycomb bodies, and thereby led to looping of target genes into more active interchromatin granules in the nucleus [206]. Kcnq1ot1 is involved in regulating chromatin loops, in particular by binding two distinct sequences that are 200 kb apart from each in the Kcnq1 imprinting locus, an interaction that may contribute to imprinted monoallelic repression of the Kcnq1 promoter [211]. HOTTIP expression and long-range chromosomal looping between the HOTTIP locus and the 5′ HOXA sites correlate with the recruitment of the activating WDR5/MLL complexes and expression of the 5′ HOXA cluster. Thus, lncRNA function can involve aspects that apply to single loci as well as to large chromosomal domains.
Paramount for interacting with chromatin regulatory proteins, for example, during folding chromatin fibers, lncRNAs use short conserved and functional subsegments or secondary RNA structure folds [41, 230, 239,240,241] (Fig. 7.1a). As a second general feature, lncRNAs biophysically have more flexible joints than proteins linkers [242, 243]. Thirdly, lncRNAs can often be highly modular, exhibiting a number of RNA folds in order to scaffold multiple effectors at once. The cardiovascular ANRIL lncRNA is such a case, and it can interact with different repressive Polycomb complexes [192], while using other RNA domains to bind and regulate other effector proteins [24], or to interact with specific genomic regions [191]. In an exemplary case that may be generally instructive for the study of lncRNAs, Xist has been found to be able to interact with an unexpectedly large (80–250) number of proteins [238]. In these interactions, Xist uses a dozen different RNA segments that separately recruit, for example, histone deacetylases [229], methyltransferases [244], ubiquitin ligase complexes [245], and corepressors [117, 230, 246] and tie the RNA to the nuclear lamina [247], all of which contributes to the heterochromatinization of Xist RNA-painted chromosomal regions. In the near future it can be anticipated that some of the recently identified cardiovascular lncRNAs will be biochemically studied in more detail, and one can expect that the number of interacting factors and possible effector mechanisms will significantly increase for each noncoding RNA.
7.3.3 Regulation of the RNAP II Preinitiation Complex at Gene Promoters
The regulation of transcriptional initiation is a multidimensional problem, and involves DNA, RNA, and chromatin-dependent regulation, all of which are mutually influencing each other [248, 249]. A major role of lncRNAs in this context is to regulate the activation of the promoter-bound RNA polymerase II preinitiation complex. In this context, lncRNAs can contribute to RNAP II regulation at several levels (Fig. 7.1c): (1) eRNAs stimulate enhancer function, (2) cheRNAs and ncRNA-a promote enhancer:promoter looping, (3) specialized circular RNAs (EIciRNAs and ciRNAs) activate the RNAP II holocomplex at promoters, and (4) other lncRNAs control transcription factor activity.
7.3.3.1 Activation of Promoters by Noncoding Transcription of Enhancers
Enhancers are known to loop over large distances to allow contact with promoter regions for specifying spatial and temporal or signal-dependent control of gene activation [248]. lncRNAs have taken a central role in how enhancers become active (Fig. 7.1b). The major finding in this respect was that enhancers are transcribed and express specific enhancer RNAs (eRNAs) [46, 81,82,83, 250,251,252,253,254,255]. Functionally, these eRNAs have been found to function, for example, as decoys for the NELF complex, a known repressor of the core RNAP II complex, thus promoting transcription [251]. Secondly, eRNAs bind and activate the Mediator complex, the central protein connector between enhancers and promoters, which also results in activating the RNAP II preinitiation complex at the promoter [256]. One has to be cautious, however, because whether enhancer transcription is a cause or consequence of promoter activation needs to be carefully tested in each case [257]. Finally, it may be the transcriptional act over an enhancer that activates a nearby gene [250, 258]. To understand this phenomenon, one must consider that during transcription, DNA is partly unwrapped from nucleosomes [259] and such chromatin opening can affect neighboring genes [43, 250]. As such, much remains to be learned about how eRNA-like lncRNAs affect looping between enhancers and promoters. Recent work has revealed how DNA-interacting proteins like CTCF and cohesin contribute to organizing chromosomes into 1–5 megabase-sized domains, so-called topologically associated domains (TAD), within which specific enhancer-promoter interactions can occur. But details of how specificity in the interaction between enhancer and promoter are constrained, and where lncRNAs can become active, are not yet fully understood (see [260] for a recent review on TADs).
7.3.3.2 cheRNAs and ncRNA-a Exert Enhancer-Like Functions
Surprisingly, also conventional genes, either coding or noncoding, can behave as enhancers [89]. For example, a group of lncRNA genes have been identified by virtue of the encoded lncRNA to reach out to about 3 kb to their neighborhood by acting as eRNA-like molecule (called ncRNA-a). In a similar way, but identified in a separate study, a class of chromatin-enriched cheRNAs have been identified as eRNA-like RNAs that associate with the chromatin fraction [261, 262] (Fig. 7.1c). In fact, many well-known lncRNAs with cardiovascular relevance like HOTTIP [92], Kcnq1ot1 [263], or linc-HOXA1 [93] activate genes in their immediate neighborhood [92, 264, 265], and it is not always clear whether as classical eRNA or as ncRNA-a or as cheRNA.
7.3.3.3 Circular EIciRNAs and ciRNAs Activate the RNAP II Holoxomplex
lncRNAs can also rather directly stimulate the RNAP II complex at promoters, and two classes of circular RNAs have been implicated: 3′-5′-linked circular intronic RNAs (ciRNAs) and Exon-Intron-containing circular RNAs (EIciRNAs) [103, 104]. Both co-immunoprecipitate with RNAP II and stimulate its activity [266, 267]. The molecular details of how RNAP II is activated in each case are mostly unclear, but in the case of EIciRNAs the activation depends on the small nuclear U1 snRNA and leads to the activation of TFIIH and P-TEFb within the RNAP II preinitiation complex (Fig. 7.1c).
7.3.3.4 Transcriptional Regulation Through lncRNA:DNA R-Loop Formation
Compared to proteins, RNAs exhibit efficient and easily evolvable modes to both interact with other nucleic acids, as well as be a template for nucleic acid synthesis. Single-stranded RNAs, including nascent lncRNAs and circRNAs, can hybridize with base-complementary DNA sequences by threading in and partially opening the DNA helix through complementary base pairing. This can form an R-loop in the form of a rigid A-type-like RNA:DNA double helix [268]. Firstly, R-loops affect transcription: they can decompact nucleosome arrays and stimulate the formation of histone modifications conducive to transcription [268]. Secondly, R-loops cause RNAP II stalling, and associated with this an enhancer looping, or they can affect any type of chromatin-dependent process, such as splicing [269]. In fact, some lncRNAs induce genes in trans by establishing R-loops by threading into actively transcribed genes that already offer partially single-stranded DNA regions [231, 270]. Conversely, also the opposite can happen: degradation of R-loop-forming eRNAs on enhancer regions has been found to activate enhancers [252]. The functions of R-loops are still intensively studied, and the role of R-loop formation as a lncRNA effector mechanism is novel. No relevant studies have explored this effector mechanism for cardiovascular lncRNAs yet.
7.3.3.5 Transcriptional Regulation Through Binding Transcription Factors
Finally, different lncRNAs can also directly modulate transcription by binding to certain transcription factors and thereby modulating their binding ability. For example, lncRNA RMST was found to interact with the SOX2 transcription factor and to be important for its binding to SOX2 target gene promoters [271]. Similar interactions allow other lncRNAs to co-activate steroid hormone receptor targets or SP1 targets. Not all lncRNAs stimulate transcription though. For example, Gas5, a lncRNA with cardiovascular relevance, was found to bind and inhibit SMAD3 during TGFβ signaling [272], and the cardiovascular Braveheart lncRNA bound and inhibited the nucleic-acid-binding ZNF9, a factor previously implicated in dilated cardiomyopathy [273].
7.3.4 Antisense Transcription
Genome sequencing of many eukaryotes has shown that as much as 30% of human genes have antisense noncoding transcription partner genes that can overlap in part with the sense gene (Fig. 7.1d). These NATs or asRNAs are a rather diverse class of lncRNAs that function through different effector mechanisms and affect partner genes depending on the relative positioning of their gene bodies.
Transcriptomic analyses indicated that an antisense transcript is often orders of magnitude less abundant than its partner and remains nuclear [274]. Historically, after focusing on selected candidate NATs, a number of studies have begun to address the function of asRNAs on a global scale. Initial studies indicated that repression of the sense coding partner gene was a common function [80] (Fig. 7.1d). Subsequent genome-wide tests suggested, however, that only a minority (one quarter) of stable asRNAs was functional in competitively silencing their sense gene, and that many asRNAs may not be functional, at least under standard conditions [44]. Mechanistically, silencing via asRNAs can occur through two principal mechanisms: direct transcriptional interference modes and DNA methylation, or chromatin compaction, or combinations thereof (see [80] for review). Generally, the grade of asRNA-dependent silencing was found to be more pronounced when asRNA transcription reached over the start of the sense gene [61, 80, 208, 275,276,277]. Most recent experiments suggest that many lncRNAs only weakly affect the magnitude of expression of their sense partners, but that they rather reduce noisy spurious transcription events [44].
7.3.5 Regulation of mRNA Splicing
Several lncRNAs are known to be nuclear, and some of these have been shown to play important regulatory roles in splicing regulation (Fig. 7.1e). As a best understood example with cardiovascular relevance, MALAT1 was found to localize in nuclear speckles, sites of coordinated transcription and splicing. This lncRNA binds splicing regulators of the SR splicing factor family and is essential for their localization in nuclear speckles and for alternative splicing of specific target genes therein [126, 127, 217, 218]. The exact regulatory mechanism has recently been investigated in detail: MALAT1 interacts with the 3′ ends of actively expressed and alternatively spliced pre-mRNAs, likely the RNA-binding SR splicing factors. Other cardiovascular lncRNAS implicated in regulating splicing are Miat [195] and, possibly, Wisper [91]. By affection splicing indirectly by influencing chromatin structure at gene loci, or as transcriptional regulators of splicing factors, many lncRNAs are expected to be involved in splicing regulation, but only a few mammalian lncRNAs have been identified as clear splicing regulators, and the details of their effector mechanism are still investigated [278,279,280]. Also, circRNAs have been implicated in regulating splicing. In fact, several independent studies suggest that co-transcriptional generation of circRNAs by the spliceosome, termed backsplicing, competes with linear splicing of mRNAs inside protein-coding genes. The picture emerges that on a genome-wide level, the function of most circRNAs might be to fine-tune the expression of their host mRNA. Underlying this mutual inhibition, several scenarios have been suggested: First, co-transcriptional backsplicing is thought to block the progression of the elongating RNAP II, which affects alternative exon skipping in mRNAs. Secondly, co-transcriptional backsplicing causes an intramolecular covalent linkage in the linear mRNA molecule, an internal junction with a single-strand RNA overhang. This may be the entry site for RNA exonucleases that degrade such mRNA molecules. For a detailed description of the different models that try to explain how backsplicing negatively affects coinciding linear splicing of mRNAs, see a recent review [104].
7.3.6 microRNA Sponging
An intensely studied and highly discussed function of lncRNAs is their capacity to regulate the stability of coding mRNAs by sequestering microRNAs that would usually target mRNAs for destruction (Fig. 7.1f). During sequestering, lncRNAs bind microRNAs only over limited stretches of 6–8 nucleotides in length, and therefore lncRNA sponges are spared from degradation [214, 215]. lncRNAs that sponge microRNAs are also called competing endogenous RNAs (ceRNAs). Sponging can occur in all classes of lncRNAs, in linear lncRNAs, in transcribed pseudogenes, and in circRNAs [99, 281,282,283]. Regarding cardiovascular-relevant lncRNAs, this relates to H19, LINC00305, Meg3, Tug1, CHRF, ROR, CARL, Rncr, and TCONS_00075467 as well as to the circRNAs HRCR and CDR1-as. Since microRNA-binding regions are short and abundant, theoretically many microRNA sponging events may occur simultaneously on one lncRNA, and this greatly expands gene regulatory complexity [281]. Yet, given the comparatively low abundance of a given single lncRNA, and the relatively high abundances of miRNA and/or target mRNA(s), there is still some controversy about details if and when sponging actually occurs as a sufficiently potent gene-regulator mechanism in vivo [284,285,286,287,288]. A large fraction of lncRNAs, and therefore also of lncRNAs with cardiovascular function, have been implicated in microRNA sponging. We list sponging as a potential effector mechanism in Tables 7.1 and 7.3, but note that experimental evidence on sponging is not equally conclusive in each report. In particular, regarding circRNAs, CDR1-as may be one of the very few circRNAs that are actually effective as microRNA sponge in vivo [103].
7.3.7 Regulating mRNA Stability or RNA/DNA Editing
Certain noncoding RNAs pair with target RNA molecules. Thereby, lncRNAs can promote or impair the stability of mRNAs in the cytoplasm. This can be due to direct lncRNA:mRNA binding or by influencing the stability of protein:mRNA complexes (Fig. 7.1g). For example, the well-known Staufen 1 protein leads to nonsense-mediated RNA decay when it binds to double-stranded RNA regions in the 3′ UTR of mRNAs [289]. lncRNAs were found to bind such stability-determining sequences in a target mRNA and protect from decay [289, 290]. Currently, the first mapping approaches by sequencing explore the entirety of all RNA:RNA duplex interactions in the transcriptome, including lncRNA:mRNA interactions [228, 291, 292]. More than 8000 intermolecular RNA interactions have been described therein, >3000 duplex structures are shared between human and mouse, and >100 lncRNA:mRNA interaction exist in single-cell types, indicating a potentially huge regulatory space [292]. As RNAs with cardiovascular relevance, H19 has been described to bind the RNA-binding protein KSRP, and thereby promote KSRP-dependent target mRNA decay [201].
Secondly, lncRNAs bind to other mRNAs to promote posttranslational modifications at double-strand RNA regions (Fig. 7.1g). For example, hydrolytic deamination of adenosine leads to the production of the base inosine, referred to as A-to-I editing. Causal for this editing, ADAR, the responsible enzyme, requires double-stranded RNA folds as substrate. A-to-I editing has roles in many cellular processes, and first insights also showed a role in vascular smooth muscle cells [293] and misregulation in atherosclerosis [294]. Conceptually related, special noncoding RNAs have been identified that serve as guides for posttranscriptional modification of DNA bases by other enzymes in cells of the immune system [295].
7.3.8 Translational Regulation of mRNAs
In the cytoplasm, both linear and circular lncRNAs exhibit a number of regulatory roles relating to the activity of protein complexes as well as to the translation capacity of coding mRNAs (Fig. 7.1h).
A number of regulatory mechanisms have been identified for how lncRNAs impact the protein translation machinery. For example, some lncRNA are thought to positively stimulate the association of protein-coding mRNAs with translating ribosomes under conditions of translation stress [296]. Regarding lncRNAs with cardiovascular relevance, lincRNA-p21 has been found to bind specific mRNAs and to impair their translation by interacting with the mRNAs and translation factors inside polysomes [202].
7.3.9 Protein Activity Control
Concerning circRNAs with cardiovascular relevance, two circRNAs have been suggested to interact with proteins in the cytosol, and, thereby, to impact central cellular functions (Fig. 7.1i). First, circANRIL has been found to bind to the protein PES1 and inhibit its function in a dominant-negative way [24]. PES1 is a member of the evolutionarily conserved PeBoW complex, consisting of Pes1 (Pescadillo), Bop1 (block of proliferation) and WDR12 (WD-repeat protein). This complex is essential from yeast to mammals to instruct the endonucleolytic excision of the internal and external transcribed spacer elements from the pre-rRNA and the formation of mature 28S and 5.8S rRNAs, failure of which denies the formation of a functional 60S large ribosomal subunit and, consequently, the proper translation of any kind of protein. circANRIL stems from the ANRIL locus at chromosome 9p21, the most prominent genetic factor of atherosclerotic cardiovascular disease identified by genome-wide association studies [12, 23, 129, 191]. ANRIL is transcribed as a linear and a circular lncRNA (linANRIL and circANRIL, respectively). While linANRIL is thought to be a major effector of CVD risk, circANRIL appears to be protective. The function of linANRIL will be described in detail separately below, but it is not involved in translational regulation, as far as known. In contrast, circANRIL-dependent PeBoW complex regulation impairs cellular protein translation capacity, and this function has been suggested to underlie circANRIL’s protection from atherosclerosis [24] (Fig. 7.1i). In a different type of effector mechanism, a second circRNA, circPABPN1, is thought to function as a decoy for the HuR protein. By sequestering HuR, circPABPN1 has been suggested to inhibit the translation of a subset of mRNAs that depended on this RNA-binding protein [297]. In fact, HuR is known to promote the stability of many mRNAs and noncoding RNAs [298]. As a direct or indirect consequence of sponging HuR, the protein translation from HuR target mRNAs is affected.
7.3.10 Translation Potential of lncRNAs
Linear lncRNAs do not contain open reading frames (ORFs) that are longer than 300 nucleotides (encoding for more than 100 amino acids), which is the central definition of their character [38, 299]. The distinction between noncoding and coding is not so clear after all in RNAs, and this for several reasons: First, many lncRNAs do contain small ORFs just by chance, and more sensitive mass-spectroscopic analyses and genetic screens revealed that some of these are translated to micropeptides that are as small as 24 amino acids in length on average (Fig. 7.1j). A relatively well-studied class of micropeptides is the family of structurally conserved sarcolamban peptides. These are 34–46 amino acids in length, and are encoded on classically defined lncRNAs. They are functionally important because they bind and regulate the activity of the sarcoplasmic Ca2+ pump SERCA and regulate its activity. SERCA is an ATPase important for contraction-relaxation coupling in myocytes by pumping Ca2+ from the cytosol into the lumen of the sarcoplamatic reticulum, which is important also in the heart muscle [112]).
On the other hand, circRNAs do stem from protein-coding genes, and thus contain one or several exons. Consequently, circRNAs can contain longer open reading frames. A number of independent ribosome profiling studies have concurred that the large majority of the many thousands of circRNAs are not translated [35, 100, 300]. From the thousands of circRNAs only a few dozen had the potential to encode a polypeptide [301] (Fig. 7.1j), and so far, translation of only two circRNA molecules, circMbl [301] and circZNF609, have been documented with confidence [302]. Paramount for their translation was that RNA circularization led to the inclusion of the endogenous start codon, as well as of 5′ untranslated regions (UTRs) that folded into specific secondary RNA structures with internal ribosome entry site (IRES)-like properties [301, 302]. Translation from circRNAs can usually not happen, as the circularization that occurs in the internal regions of the gene is likely a consequence of circularization from internal portions of genes and, consequently, the absence of a 5′ Cap, of linear ends, and of the Kozak sequence for ribosome entry and translation initiation in linear 5′ capped mRNAs (see [303] for review). In the rare case of translation from circRNAs, usually the protein produced suffers from premature truncation compared to the native full-length protein of the endogenous linear host mRNA. Whether truncated proteins translated from circRNAs are functional is still unknown.
Summarizing, as heterogeneous as their biogenesis are the functions exerted by lncRNAs. But a common overarching feature is that many lncRNAs participate in the expression control of protein-coding genes. For this, lncRNAs can act in cis and in trans and affect gene expression on multiple levels, such as by regulating transcription, or affecting pre-mRNA splicing, mRNA stability, and mRNA translational control. As such, many lncRNAs function because of engaging in molecular interactions of the lncRNA with proteins or mRNAs directly, but also the transcriptional act over a lncRNA gene body can per se have functional consequences on neighboring genes, while the lncRNA product formed in this case may be a side product without any function [43, 52, 304]. Lastly, lncRNA genes may even be functional because the length of their gene bodies determines the relative distance between the left and right neighboring genes or regulatory DNA sequences, as learned, for example, by multiple knockout experiments in the complex Hox gene cluster [305]. Thus, functional studies of lncRNAs have to be carefully designed to take account of all possible levels of functions of a lncRNA.
7.4 LncRNAs in Cardiovascular Health and Disease
Compared to protein-coding genes, the function of lncRNAs is less well studied. This is especially true for lncRNAs implicated in cardiovascular diseases because many disease-relevant lncRNAs have only been identified within the recent 1–5 years. Consequently, only a few knockout studies on lncRNAs have been reported, and most of these were performed on lncRNAs that had been studied already before in more general cellular functions or in cancer biology.
In the following we first review developmental roles of lncRNAs at the organ level in the context of the embryonic cardiovascular system. These studies used classical knockouts and transgenic overexpression, and assessed the embryonic development of the heart, angiogenesis, or more specifically, lineage specification of the cardiovascular system, for example, from early mesoderm precursor cells. Table 7.1 summarizes the 17 lncRNAs studied by knockout analysis, which were found to have a function in cardiovascular physiology. We then continue by highlighting lncRNAs that have been linked to cardiovascular disease in rodent disease models in vivo (Table 7.2). For a good part of these lncRNAs, evidence exists on differential gene expression of the orthologous human lncRNAs in patients. For only a very few, additional genome-wide association studies (GWAS) in humans have determined single nucleotide polymorphisms (SNPs) in the lncRNA genes, which are associated with disease risk and with differential lncRNA expression (Table 7.2). In Table 7.3, we summarize the evidence on cellular and molecular functions of lncRNAs from Table 7.2.
7.4.1 lncRNAs Regulating Cardiovascular Development in the Embryo
For the heart to form, and be induced by signaling cues, pluripotent embryonic stem cells progressively differentiate into mesodermal and cardiac precursor cells that subsequently terminally differentiate [306]. Transcriptional networks are directed to control lineage commitment and cardiac cell specification [307]. By participating in gene expression regulation, lncRNAs are an intricate part of heart development. A large number of lncRNAs (>200) were found to be differentially expressed in different steps of cardiac commitment during in vitro differentiation from embryonic stem cells [308]. Moreover, hundreds of lncRNAs are known to be differentially abundant at different points in fetal heart development in vivo based on whole-tissue profiling [309]. Also, about 300 lncRNAs are cardiac-specific in the adult heart. Based on coexpression analysis and considering which known protein-coding genes were the closest neighbors of the relevant lncRNAs, some lncRNAs were prioritized as candidates in regulating key developmental determinants of heart development [310].
As a first example of a lncRNA studied by knockout analysis, the mouse lncRNA Fendrr has been found to be rather specifically enriched in the lateral plate mesoderm during mid-gestation, and its deletion by insertion of polyA signals, known to cause a transcriptional stop, disrupted the development of ventral structures, including the heart and body wall [7]. As a consequence of myocardial dysfunction, the mutant embryos died. A mere depletion down to 40% of lncRNA levels by RNA interference caused no mutant phenotype in this case, which may be an important consideration for similar developmental studies of other lncRNAs [7]. Mechanistically, Fendrr was found to scaffold a repressive Polycomb group complex and, separately, also an activating Trithorax-MLL complex, and to tether it to gene promoters by triple helix formation. This coincided with a long-term effect on target gene expression in the descendants of cells of the cardiac mesoderm [7]. Likely in this function Fendrr affected the transcriptional regulators of heart development, such as Gata6, and lateral plate mesoderm control genes like Foxf1, Irx3, and Pitx2. Some of the many target genes were regulated in cis, some in trans [7]. When studied in an independent knockout where internal lncRNA sequence was replaced by a lacZ cassette, a slightly delayed perinatal mutant phenotype was observed, and these Fendrr mutants showed lung defects [311]. Together, the discrepancy in mutant phenotypes between different knockouts is not without precedence and may even be expected given that both the RNA transcript and transcription through the lncRNA locus may be relevant [304, 312].
Similar to Fendrr, the mouse lncRNA Braveheart (Bvht) interacts with a chromatin regulator. Bvht binds and inhibits the repressive Polycomb complex [6]. Bvht also binds and inhibits the transcription factor ZNF9 [273]. Thereby, Bvht regulates an entire cardiac transcription factor network to promote early cardiac cell fate [6]. This network is upstream of master cardiac transcription factors, like MesP1, at least in an in vitro model of cardiomyocyte differentiation from embryonic stem cells (ESCs) [6] [273]. MesP1 activity is known to be essential for the specification of all different cardiac cell types, cardiomyocytes, vascular smooth muscle cells (VSMCs), and endothelial cells (ECs). Consistent with the proposed function in cultured cells, in vivo, Bvht is essential for early heart development and for maintenance of neonatal cardiac cell fate [6]. No clear Bvht ortholog was found in humans, but the possibility exists that different lncRNAs have taken over a conserved role in establishing a cardiogenic transcription factor network also in other vertebrate species.
HoxBlinc is a lincRNA residing in the Hoxb gene locus and has been associated with lineage commitment during cardiovascular development by knockout studies [9]. HoxBlinc tethers the activating trithorax Setd1a/MLL1 histone methylating complex to HoxB. Loss of function experiments showed that HoxBlinc is required to activate HoxB during embryogenesis when precursor cells initiate mesoderm formation in the primitive streak. In the mutant, cardiogenic and hemangiogenic mesoderm cell fates are not specified.
Based on high-throughput RNA sequencing of cells differentiating from human ESCs to cardiovascular progenitors and finally to terminally differentiated fetal-like vascular endothelial cells in culture, two independent studies established that several hundred lncRNAs were specific for each investigated stage [118, 119]. From each stage-specific set, one lncRNA was randomly chosen to be functionally analyzed in more detail. Corresponding to their expression during EC differentiation, in vivo expression analysis in both mouse and zebrafish embryos confirmed that Terminator was specifically expressed early after fertilization, DEANR1/Alien only later in the lateral plate mesoderm and Punisher only when the vasculature had formed [118]. Injection of antisense morpholinos binding and degrading these lncRNAs in zebrafish showed stage-specific requirements consistent with the roles inferred from cell culture profiling: Terminator was important for gastrulation, with survivors showing defects in the vasculature. Alien was important for mesoderm specification, including subsequent vascular patterning, dorsal and intersegmental blood vessel and cardiac chamber formation. And Punisher was important for vascular vessel formation, extension, and branching as well as for cardiac development [118]. The molecular effector mechanisms of Terminator and Punisher remain unknown. DEANR1/Alien was also the focus of another study that investigated stage-specific lncRNA expression during definitive endoderm and pancreatic cell specification from hESCs [119]. DEANR1/Alien was shown to be encoded close to FOXA2, an endoderm marker gene, and to stimulate FOXA2 expression. It was at the same time also important for endoderm specification. The latter function was at least in part due to FOXA2 regulation by DEANR1/Alien, as confirmed by genetic rescue experiments in cells [119]. While the mechanism of regulation remains to be confirmed, the first experiments suggested that in the forming endoderm, DEANR1/Alien may be important to recruit or stabilize SMAD2/3 at the FOXA2 locus [119].
Tie-1AS is a NAT that binds and downregulates the sense mRNA of its host gene, Tie-1. Based on overexpression of Tie-1AS and on inhibition of Tie-1 in zebrafish, the lncRNAs was suggested to have a mild impact on the proper organization of cell-cell junction markers in vivo, including those between vascular endothelial cells [121]. Corroborating these data, the human orthologous lncRNA inhibited human Tie-1 and was important for tube formation of HUVECs in collagen gels in vitro [121]. These observations are relevant because an earlier mouse knockout for Tie-1 had documented that this gene was required for vessel integrity [120]. Also, mutations in the related TIE2/TEK are known in humans to lead to different defects in venous morphogenesis based on a role of TIE2 in EC:VSMC (vascular smooth muscle cell) interaction [122].
Upperhand (Uph) is a lncRNA gene that has been found to be important for heart morphogenesis in a mouse knockout study. The mouse Uph has a human orthologue, called HAND2-AS1, and both mouse and human Uph/UPH share a bidirectional promoter with Hand2/HAND2, a well-known and important transcription regulator of heart development [8]. Experiments on the mouse Uph locus revealed that Uph functioned as an enhancer for Hand2 in embryonic heart tissue and was essential for Hand2 activation. TALEN-mediated insertion of premature polyA signals that stopped Uph transcription led to the embryonic death of knockout mice. Uph KO death was ascribed to a failure in forming a right ventricular chamber, a phenotype identical to the Hand2 KO, and corroborating that Uph functioned through activating Hand2 in vivo [8]. For its enhancer activity towards Hand2, the lncRNA transcribed from the Uph locus was not essential. Instead it was the transcriptional act over the heart enhancers in the Uph locus that seemed to make these enhancer elements active.
Xist is the central regulator of dosage compensation (X-inactivation), which is inherently essential for female survival, and will not be discussed here (see [1] for a review). A recent conditional Xist knockout using Sox2-Cre drivers allowed some Xist mutant females to survive to adulthood. Surprisingly, the survivors showed rather specific organ defects, and these included defects in heart and spleen maturation [123]. Specifically, perinatal heart growth was delayed and mutants had smaller hearts, as cardiomyocytes did not sufficiently mature by cytoplasmic enlargement [123]. Separately, another study had deleted Xist in the fetal hematopoietic lineage and had also found a rather specific phenotype. Xist mutant hematopoietic stem cells (HSCs) in females were impaired in differentiating to all lineages [124]. The latter manifested in multilineage blood cell defects, and in adulthood, mutants eventually died from aggressive blood cancers, especially in the myeloid lineage [124]. Together, although the available evidence is skewed by which drivers have been used for Xist deletion in the two knockout studies [123, 124], it is surprising that such a general pathway like Xist-dependent X-inactivation is somewhat selective for regulating blood stem cells, cardiac, and spleen development. There is a tangible explanation at least for why HSCs are affected: Hematopoietic precursor cells are special among other cell types because they selectively regain the capacity to initiate de novo X-inactivation, while other cell types at this advanced stage of embryogenesis lack the silencing factors [125].
Bioinformatics screens had initially identified muscle-specific human LINC00948 and mouse AK009351. These were later found to encode small protein-coding ORFs that were translated to myoregulin (MLN), a micropeptide that bound and activated the sarcoplasmic Ca2+ pump SERCA [112]). SERCA is an ATPase important for contraction-relaxation coupling in myocytes by pumping Ca2+ from the cytosol into the lumen of the sarcoplamatic reticulum. These micropeptides belong to the larger family of structurally conserved sarcolamban peptides. LncRNAs of this class have been studied by knockout approaches in vivo. It has been found that micropeptides in this family, like MLN, phospholamban (PLN), or sarcolipin (SLN), can repress the activity of SERCA to terminate muscle contraction [110, 112]. In contrast, the related micropeptide DWORF, which is encoded on human LOC100507537 and mouse NONMMUG026737 lncRNAs, displaces these SERCA repressors and thereby enhances contraction [116]. SERCA regulation is of relevance also for heart muscle function, due to its regulation of cardiomyocyte contractility.
The Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) is one of the molecularly best-studied lncRNAs. Based on assays in standard cell lines, MALAT1 has been implicated in regulating alternative splicing [217] and in promoting selective transcriptional activation of target genes. The latter involves binding of MALAT1 to Pc2, a central member of the Polycomb group silencing complex. Upon binding to MALAT1, Pc2 loses its preference for reading heterochromatic histone modifications and gains preference for binding to active chromatin, causing reactivation of Pc2-marked cell-cycle stimulating genes [206]. Beyond that, MALAT1 interacts with dozens of other proteins [313] and is even the origin of functional small RNAs excised from its 3′ end during maturation (mascRNAs) [51], indicating a possibly wide range of still elusive functions. In the face of all this knowledge, it was surprising that three independent knockouts of the lncRNA MALAT1 in mouse showed that MALAT1 was not essential for survival or for organ development and function under normal growth conditions in embryos or adults [126,127,128]. Neither was global transcription or splicing affected in these mice, which could be due to redundancy with yet unknown different lncRNAs. Only a selective role in regulating genes encoded close to the MALAT1 locus was found [126, 127]. In contrast to the lack of clear in vivo phenotypes, in vitro MALAT1 had some functions under stress conditions: MALAT1 was upregulated under hypoxia, and exerted proangiogenic effects, as will be described in detail in Sect. 7.4.2.3.
Human MIAT, also known as Gomafu/MIAT/Rncr2, has been linked to cardiovascular disease as will be summarized in the following section on lncRNAs in disease [314]. Separate studies showed that it binds to several splicing regulators and regulates a relatively small number of genes. MIAT was implicated before in stem cell, neuronal, and retinal cell differentiation [195]. A recent mouse knockout was established for Miat but did not reveal any obvious anatomical defects. Only selective behavioral defects were observed, and only a small number of splicing alterations were documented in primary neuron cultures from these mutant mice [219]. How the lncRNA regulates selective neuronal functions in specific brain parts, and by which molecular mechanisms, is still unknown.
Among 150 lncRNAs deregulated by pressure overload after transaortic constriction in a mouse model, Chaer, cardiac hypertrophy associated epigenetic regulator, is a mouse lncRNA conserved also in humans, which shows enriched expression in the heart [16]. It will be described in detail in the context of lncRNAs with roles in cardiovascular diseases in Sect. 7.4.2.1. In the course of studying Chaer, a genomic deletion was inserted into the Chaer lncRNA locus. The mutant mice did not show obvious morphological organ deficits or functional heart problems in normal conditions. Only in experimental pressure-overload models in the mouse was a function in cardiomyocyte growth control revealed [16].
Rncr3, retinal noncoding RNA 3, also known as LINC00599, is a lncRNA with cardiovascular relevance as will be described in detail in the description of lncRNAs involved in atherosclerosis in Sect. 7.4.2.1. A knockout of the Rncr3 locus exists, but the mutants did not show any morphological or functional abnormalities of the heart or of the vasculature, as far as reported [205]. Instead, the initially viable Rncr3-deficient mice become debilitated and later die, likely because of defects observed in multiple types of neurons in the central nervous system.
CDR1-as, cerebellar degeneration-related protein 1 antisense RNA, is a circular lncRNA. It was the first circRNA that was found to have a dedicated function in eukaryotes [214, 215]. It will be described in detail in Sect. 7.4.2.1 because reports exist that suggest this circRNA to be misregulated and misfunctioning in cardiovascular disease. Inconsistent with these reports, loss-of-function studies have been performed in mouse and in zebrafish in vivo, but CDR1-as showed no obvious function in hearts of the vasculature. Instead, and consistent with the expression pattern in vivo, a requirement for neuronal functions was revealed. Recently, the CDR1-as circRNA was knocked out in mouse, which was technically possible, because of the peculiarity of this locus to only express a circRNA but no linear RNA. The CDR1-as circRNA knockout did not show anatomical alterations, and also no heart defects, but defects in central nervous system function [213].
The H19 lncRNA is located in the H19/Igf2 imprinted locus, where H19 and Igf2 are reciprocally imprinted, such that the two genes are expressed from maternal and paternal chromosomes, respectively, but not from both alleles in a cell. Relevant for the cardiovascular system, H19 and Igf2 are known to be expressed in muscle and other mesodermal organs in the embryo. Igf2 has been studied in the context of embryonic heart development, where it was found to be an epicardial mitogen that was important for ventricular wall proliferation. Heart-specific functions of H19 remain unknown, but it is peculiar that H19 is downregulated in adult tissues except in skeletal muscle and heart and that H19 is reactivated in the cardiovascular system during stress signaling, as will be described in Sect. 7.4.2.1. H19’s major function has been found to be limiting for the growth of the placenta and to inhibit cell proliferation, for example, during tumorigenesis. One major effector mechanism is the production of microRNAs from the H19 sequence. In this function, H19 was the parent molecule for a microRNA that repressed an IGF receptor for maintaining quiescence in long-term quiescent hematopoietic stem cells (HSCs) [198], and for another microRNA that promoted muscle differentiation from myoblasts and muscle regeneration.
7.4.2 Cardiovascular Disease-Associated lncRNAs
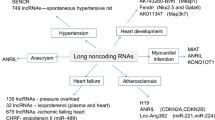
There are three principal approaches to how lncRNAs have been implicated in cardiovascular disease. First, lncRNAs may reside in genomic regions, which have been associated with cardiovascular disease (CVD) in genome-wide association studies (GWAS). This approach has so far been reported in only a few cases because genetic variants identified through GWAS are often found in regulatory sequences in some distance to genes, and it is far from trivial to establish causal links in the functionality (expression, splicing, and sequence) of a lncRNA. Secondly, and following a different rationale, a number of lncRNAs have been linked to CVD because they were found to be differentially regulated in disease conditions through genome-wide RNA expression profiling. In a third strategy, lncRNAs were investigated in candidate gene approaches because they regulated disease-relevant processes or resided in or close to protein-coding genes with already established disease relevance. Accordingly, here we will highlight lncRNAs implicated in vivo as causal effectors of the following seven disease entities: atherosclerosis, myocardial infarction, aortic aneurysm, cardiomyopathies and congenital heart disease, vascularization and angiogenesis, arrhythmia, and stroke and cerebrovascular aneurysms. Especially for the more established and better-studied lncRNAs, in vivo functions in more than one disease entity have emerged, all of which will be discussed in separate sections. Table 7.2 summarizes all lncRNAs, their change in expression in disease (up/down), and whether their normal function is to protect from disease (protective) or to exacerbate disease (detrimental), as far as determined from genetic knockdown or overexpression in animal disease models. Table 7.3 lists the cellular and molecular functions of all lncRNAs from Table 7.2, as determined from accompanying experiments in relevant cell culture systems.
7.4.2.1 Atherosclerosis and Myocardial Infarction
Briefly, atherosclerosis is characterized by the formation of fibro-fatty lesions (plaques) in arteries, whereby vascular wall cells (SMCs, fibroblasts, ECs) and diverse immune cells (neutrophils, macrophages, T cells, B cells) aberrantly proliferate, undergo cell death, change cell fate, and trigger nonresolving immune reactions. The lesions accumulate cells and lipoprotein material and are covered by a fibrous cap. Upon necrosis in the lesions, and extracellular matrix (ECM) remodeling, the cap thins, plaques can rupture, and plaque material ends up in the bloodstream and trigger luminal thrombosis also at distant sites, leading to myocardial infarction (MI, heart attack) or stroke in brain tissue. In this book chapter we are reviewing lncRNAs that have been linked by functional analysis to the onset and severity of atherosclerosis, and to myocardial infarction, which occurs as acconsequence of atherosclerotic processes. Here we will not review lncRNAs associated with CAD risk factors such as dyslipidemia, diabetes, obesity, metabolic syndrome, or systemic blood pressure regulation. So far, 16 lncRNAs have been implicated in the pathogenesis of atherosclerosis in animal disease models in vivo, of which 3 were also found to play a role when tested in myocardial infarction models in vivo. Among CAD-related lncRNAs, as of date, only five lncRNAs emerging from GWAS have been functionally studied in the context of CAD. There are, however, dozens of other known CAD risk loci from GWAS datasets, which contain noncoding RNAs and have not yet been explored in the same way. How lncRNAs determine risk at GWAS loci will be interesting to explore. In our review, nine different lncRNAs have been associated only with myocardial infarction in relevant models, without being implicated in atherosclerosis through in vivo evidence. We start out with describing the CAD-related lncRNAs: First, a number of tightly clustering SNPs have been found on the Chr9p21 locus in several independent genome-wide association studies testing the susceptibility for coronary artery disease (CAD) [130, 131], peripheral artery disease [136], and myocardial infarction (MI) [135]. A separate haplotype block associated with type II diabetes risk is encoded nearby [315,316,317]. Regarding CAD/MI, the dosage of the risk SNPs was found to be associated with atherosclerosis severity. Also, homozygosity for the risk SNPs correlated with a twofold increased risk for CAD/MI, which was relevant for approximately 20% of the population [131, 318]. As explained before, the major effector of this locus is not a protein-coding gene but a long noncoding RNA, human ANRIL. ANRIL expression levels are increased in more severe atherosclerosis phenotypes [10, 12, 129, 131,132,133,134]. Its molecular effector mechanism is under intensive investigation, not least because this lncRNA is tightly linked also to different diseases, including cancer. Relevant for one proposed effector mechanism, ANRIL is expressed adjacent to the human INK4-ARF tumor suppressor gene locus on chromosome 9p21, which encompasses p16 INK4a, P14 ARF, and P15INK4b, whereby transcription of ANRIL overlaps in antisense with transcription of P15INK4b, hence its name Antisense noncoding RNA in the INK4 locus.
ANRIL has become a paradigm for physiological control of cell proliferation and survival by a lncRNA. For example, by binding to the PRC1 and PRC2 complex, ANRIL has been suggested to impose selective repressive histone modifications at the INK4-ARF locus, a central tumor suppressor locus whose repression is known to result in a cell cycle G0-G1 entry in specific contexts [192, 319, 320]. Such a cell-cycle stimulating role may hypothetically be relevant for ANRIL’s described roles in activating atherosclerosis [321]. Indeed overexpression of ANRIL in several cell types, including PBMCs as well as cells of the vessel wall, was found to be proproliferative [191]. On the other hand, ANRIL can also interact with transcriptional coactivators to alter the transcription of target genes in trans and, thereby, affect cell adhesion and apoptosis in ways that stimulate atherosclerotic plaque formation [191, 322]. Although the ANRIL sequence is primate specific, the structure of the 9p21 locus is conserved in mice, where this locus is also a gene desert with a lncRNA. AK148321, the mouse lncRNA in this locus, has a conserved exon structure. Corroborating the importance of the locus for disease, deletion of the entire region led to cardiac-specific misregulation of the INK4-ARF orthologs Cdkn2a/b, to the misregulated proliferation of mutant aortic VSMCs [323], and to increased vascular aneurysm formation [324]. However, the knockout was not associated with altered atherosclerosis plaque formation, thus raising the question whether the mouse locus is a lncRNA-dependent atherosclerosis risk factor in vivo. In any case, the noncoding region of human Chr9p21, and likely its transcription, is important for CAD susceptibility in humans.
Making the regulation even more complex, the ANRIL locus also expresses circular ANRIL (circANRIL) RNA isoforms [23, 24]. circANRIL abundance was decreased, while linear ANRIL levels were increased, in carriers of CAD/MI risk alleles in an association study of 1000–2000 patients with suspected coronary artery disease in PBMCs and whole blood. These findings were recapitulated also in more than 200 human carotid endarterectomy tissue specimens [24]. Moreover, circANRIL was decreased in CAD patients also in blood T-lymphocytes [23]. The notion emerged that circANRIL protects from CAD [24]. Specifically, circANRIL impairs rRNA processing in cells by inhibiting the rRNA-processing PeBoW protein complex [24]. This results in nucleolar stress, p53 activation, and reduced translation capacity, functions that are antiproliferative and, conceptually, would antagonize the proproliferative functions of linear ANRIL. Indeed, in cultured cells circANRIL functions independently of linear ANRIL and impairs proliferation and sensitizes for apoptosis [24], functions that have been associated as to be anti-atherogenic [24]. Together, the ANRIL locus highlights a situation, where both the linear and the circular RNA products are functional, and where linear and circular RNA may have antagonistic functional outputs (pro- and antiproliferative, respectively).
GAS5, growth arrest-specific 5, has been known as a noncoding transcript before genome-wide detection of lncRNAs became routine. GAS5 is conserved in mouse and human and was found to be upregulated during conditions of cell growth arrest, and contribute to cell cycle arrest and apoptosis when induced in lymphocytes and many other cell types. GAS5 also harbors 10 snRNAs in its introns, some of which give further rise to small piRNAs that induce activating chromatin remodeling at the locus encoding the proapototic TRAIL gene. Beyond GAS’s role as tumor suppressor in cancers, GAS5 has also been studied in atherosclerosis. GAS5 was found to be more abundant in atherosclerotic plaques in patients, and in a rat CAD model [144]. Consistent with its role in other cell types, GAS5 contributed to apoptosis in cultured macrophages and SMCs [325]. In which cells and at which stage of plaque development GAS5 contributes to atherosclerosis will be important to test in the future.
Another lncRNA has been associated with cardiovascular disease in a GWAS case-control study of over 3000 patients: six SNPs in a previously unknown lncRNA on human Chr22q12.1were found to be associated with myocardial infarction, and the lncRNA was, therefore, named Gomafu, MIAT, myocardial infarction associated transcript, later also designated as Rncr2. Of these SNPs, one was linked to increased MIAT transcription [314]. MIAT is also known to be upregulated in atherosclerotic plaques [145]. The mouse MIAT orthologous lncRNA was found to be aberrantly upregulated in the myocardium shortly after MI in a mouse model for induced infarction [146]. MIAT knockdown reduced cardiac fibrosis and improved cardiac function, which indicated that overactivation of MIAT was detrimental and contributed to heart fibrosis [146]. The underlying molecular mechanisms remained unknown in this study. Molecular effector functions of mouse Miat have, however, been delineated in a number of unrelated mouse models, which give an idea of how Miat might function in organs in general: For example, mouse Miat was found to be one of the few highly conserved noncoding targets of Oct4, and it was required for mouse embryonic stem cell maintenance. Conversely, increased levels promoted mesodermal and ectodermal lineage specification [196]. This may be of relevance also for the heart and progenitor cells therein. Also in other cell lineages, this proposed role in switching from progenitor to differentiated cell state was corroborated: It could be shown that Miat was involved in prosurvival signaling of differentiated neuronal cell progeny in mouse [195]. Apart from binding and potentially sponging microRNAs, in all these studies its only other described molecular function was in splicing regulation, possibly in guiding splicing of cell fate determinants [195]. How this function in progenitor cell differentiation relates to the function of progenitor cells in damaged tissues and organs remains to be tested.
H19, an imprinted maternally expressed transcript, is a lncRNA known as a model for studying the regulation of an imprinted locus, and for how imprinted growth-regulating loci are implicated in tumorigenesis. Two SNPs in H19 on human Chr11p15.5 have been associated with CAD in a GWAS of 700 individuals [149]. Increased H19 levels have been found in the plasma of atherosclerosis patients [150, 151]. It is also known already for long that H19 is low expressed but that it can be reinduced in the aorta or VSMCs upon stress signaling or in atherosclerotic plaques. Also, when tested in cultured macrophages upon treatment with oxidized LDL, H19 contributed to the inflammatory transcription response [151]. How H19 contributes to cardiovascular diseases on a molecular level is still to be determined.
HAS2-AS1, HAS2 antisense RNA 1, is located at the HAS2 gene locus [155]. This locus is interesting, because it is neighboring another cardiovascular lncRNA, SMILR (smooth muscle–induced lncRNA enhances replication, also known as RP11-94a24.1). SMILR and HAS2 were transcribed from the same strand and in the same direction, while HAS2-AS1 is transcribed as antisense RNA. HAS2-AS1 expression has been found to be stimulated by inflammatory signaling and to activate HAS2 expression. HAS2 shows increased levels in human plaque tissue as compared to healthy controls. Thereby, HAS2, a hyaluronan synthetase, is thought to promote atherosclerotic neointima formation. On the other hand, also SMILR has been found to be increased in human plaque tissue. Additionally, HAS2, but not HAS2-AS1 levels dropped after SMILR siRNA-mediated knockdown. Together, by a still elusive molecular mechanism, SMILR is thought to be required for the full transcriptional stimulation of HAS2 by opening the chromatin compaction state of the HAS2 locus [155]. Thus two lncRNAs may independently relay the detrimental overactivation of HAS2.
Some members of the evolutionarily conserved HOX gene family have been specifically associated with regulatory roles in the cardiovascular system. For example, several selected HOX genes are involved in cardiac progenitor proliferation and differentiation; others must be repressed for angiogenesis to proceed. Two human HOX gene-regulating lncRNAs have been studied: HOTTIP and HOXC-AS1. HOTTIP, HOXA distal transcript antisense RNA, is a well-studied lncRNA that had originally been found as a cis-regulator of the HOXA locus from where it is transcribed, particularly as an activator that recruited stimulating histone-modifying WDR5/MLL complexes for long-range chromatin activation. HOTTIP has recently also been found to be increased in arterial tissues samples of CAD patients [156]. Overexpression and knockdown experiments showed that HOTTIP levels regulated EC proliferative activity. HOTTIP also affected migration capacity, as determined by experiments in cell culture. Whether and how HOTTIP contributed to atherosclerotic plaque formation, however, remained unclear. A more general role of HOTTIP is likely, since overactivation of HOTTIP has also been found in cancers, and was implicated in prostate cancer hormone-independent growth, likely in large parts via HOX gene activation.
The second relevant lncRNA is HOXC-AS1, HOXC cluster antisense RNA. Microarray RNA profiling of carotid atherosclerosis revealed an antisense lncRNA in the human HOXC cluster, HOXC-AS1, HOXC cluster antisense RNA, as downregulated compared to normal arterial intimal tissue [70]. Investigating how this downregulation might occur, the authors turned to cultured monocytes and found that administration of oxidized LDL to cultures decreased HOXC-AS1 expression. In these conditions, overexpression of HOXC-AS1 abolished the accumulation of cholesterol in monocytes as well as the inhibitory effect of ox-LDL on the expression of the nearby HOXC6 gene. The authors suggested that the HOXC-AS1 antisense lncRNA might somehow regulate cholesterol metabolism, and that the possibility existed that this occurred via transcriptionally affecting HOX genes in the HOXC cluster [70]. This finding is of particular importance because independent work on HOXC9 has revealed that this HOX gene can promote EC quiescence and vascular vessel morphogenesis [70]. Therefore, HOX-regulating lncRNAs have been studied with a particular focus on angiogenesis, as described in Sect. 7.4.2.4.
By searching the GWASdb database for SNPs already previously linked to atherosclerosis by GWAS but which are less significant (P < 1.0 × 10–3) and were manually curated from the literature [326], one group focused on one such SNP that resided in the intron of a lncRNA on Chr18q22.1: This lncRNA, LINC00305, had so far not been characterized and its expression was found to be enriched in atherosclerotic plaque samples as well as in blood monocytes of atherosclerosis patients, showing that the SNP associated with lncRNA expression [56]. To study the function of LINC00305, gene expression profiling was performed upon lncRNAs overexpression in THP-1 monocyte cell lines, as well as in human aortic smooth muscle cells cocultured with these. LINC00305 promoted the expression of inflammatory genes by activating NF-κB signaling in monocytes, which translated to shifting SMCs from a contractile to a pathological synthetic state [56]. Using immunoprecipitation experiments and mass spectrometry to screen for lncRNA-interacting proteins, the authors found that LINC00305 bound to membrane-resident lipocalin-interacting membrane receptor and increased its interaction with the aryl hydrocarbon receptor repressor (AHRR). This interaction altered downstream AHRR signaling, such that more nuclear AHRR and increased NF-kB signaling were observed after LINC00305 overexpression. Whether and how AHRR regulated NF-kB signaling components is not yet clear [56].
A recent RNA expression analysis of foam cell formation upon oxLDL administration to cultured human macrophages revealed induction of lincRNA-DYNLRB2-2 [157]. This lncRNA was further studied because it was found that it induced expression of the evolutionarily conserved GPR119. This regulation potentially is of relevance for CAD because GPR119 is a G protein-coupled receptor that is known from earlier work to be involved in metabolic homeostasis by suppressing food intake and reducing body weight gain in rat models [327]. In the study on lincRNA-DYNLRB2-2, it was found that both lincRNA and GPR119 also promoted cholesterol efflux from human foam cells. The efflux driven by lincRNA-DYNLRB2-2 depended on GPR119 [157]. Conversely, lincRNA-DYNLRB2-2 repressed TLR2, a well-known upstream Nf-κB signaling stimulator, and this repression was necessary for cholesterol efflux [57]. These findings were substantiated by the finding that GPR119 overexpression in apoE −/− mice conferred antiatherosclerotic effects, while TLR2 overexpression conferred proatherosclerotic effects [157]. lincRNA-DYNLRB2-2 was not tested in this in vivo context.
Another lncRNA is lincRNA-p21, located on mouse Chr17, 15 kb upstream of the tumor suppressor p21 (cyclin-dependent kinase inhibitor 1A), but transcribed from the opposite strand and in the opposite direction. lincRNA-p21 was not primarily identified by GWAS studies in humans, but by a genomic study of mouse lincRNAs that were direct transcriptional targets of p53 and that were induced by p53 [17]. Only later studies focused on lincRNA-p21’s role in atherosclerosis. Four SNPs were found in human lincRNA-21 on Chr9p21.2, locating in a single haplotype block and correlating with reduced risk for CAD and MI in over 600 patients in a case-control study [158]. The effect of risk alleles on lincRNA-p21 expression was not reported in this study, and the molecular function of lincRNA-p21 not explored. In another study, lincRNA-p21 was also found to be downregulated in atherosclerotic plaques in arteries in an apoE −/− mouse model for CAD, as well as in arterial tissue and in PBMCs of human CAD patients [159]. Knocking-down lincRNA-p21 locally in injured mouse carotid arteries with siRNA technology caused hyperplasia of the neointima in the lesion, coinciding with increased proliferation and reduced apoptosis levels in cells of the vessel [159]. How does this relate to p53? Through decades of earlier research on p53, it has become clear that p53 affects many cellular facets, most notably the switch between cell survival and apoptosis, cell proliferation, and DNA repair. The majority of studies have focused on p53’s role in tumorigenesis, but some studies on its role in atherosclerosis exist: In atherosclerotic tissue, p53 is not mutated, but in mouse models of atherosclerosis, p53 knockouts showed increased disease severity, and VSMCs and macrophages were thought to be affected [328]. This shows that lincRNA-p21 phenocopies p53’s roles, consistent with the notion that this lincRNA was a p53 effector also in the process of atherogenesis [17, 159]. In fact, earlier functional tests had shown that mouse lincRNA-p21 served a function as a corepressor for p53 in its second known function as a transcriptional repressor of a set of target genes, including p21 [17]. It could, thus, be that normally lincRNA-p21 repressed antiproliferation genes like p21 to induce apoptosis and reduce proliferation in an atheroprotective function.
Meg3, maternally expressed gene 3, has been well studied. It is known to inhibit proliferation and migration, in part via inhibiting TGFβ signaling, and promote apoptosis in the context of cancer studies. In a cardiovascular context, a study reported that human MEG3 was downregulated in tissues of CAD patients and that overexpression of MEG3 in endothelial cells suppressed EC proliferation in vitro [160]. It is important to mention in this context that MEG3 is expressed from the conserved DLK1-MEG3 imprinted domain. Studies of this imprinted locus in mouse showed that Meg3 function as a repressor by tethering the Polycomb PRC2 complex at target genes in trans [204]. This is relevant as mouse Dlk1 has been independently linked to atherosclerosis: The suppression of Dlk1 protected from atherosclerosis through promoting regenerative EC division for EC turnover in injured arteries, while disturbed blood flow inhibited this pathway [329]. Thus, MEG3/Meg3 may have several cell-type-specific roles in the cardiovascular system, and EC-related functions may be particularly interesting to study in the future.
RNCR3, retinal noncoding RNA 3, also known as LINC00599, is a lincRNA that is a source of the microRNA miR-124a. RNCR3 was found to be induced in mouse and human aorta atherosclerotic lesions, and specifically also by ox-LDL treatment of ECs and VSMCs [53]. In a mouse model of atherosclerosis, systemic injection of shRNA for RNCR3 increased lesion size. This proceeded with EC apoptosis, reduced VSCM proliferation and migration, and with increased cholesterol levels and higher levels of TNF-α and IL-6 in the circulation [53]. Though the exact underlying mechanism is still unknown, these data suggest that RNCR3 is an atheroprotective factor. Possibly RNCR3 exerts a role in protecting from apoptosis because an earlier independent knockout of RNCR3 had shown a phenotype in the central nervous system, with defects in brain development, axonal morphogenesis of dentate gyrus granule cells, and retinal cone cell death.
SENCR, smooth muscle and endothelial cell-enriched migration/differentiation-associated long noncoding RNA, is a lncRNA that is encoded as NAT of the FLI1 gene. FLI1 is a transcription factor known to regulate EC and blood cell formation. SENCR has described pleiotropic functions in ECs and VSMCs, making it difficult to propose how it specifically functions in CAD [67]. Directed tests revealed that SENCR was downregulated in ECs from vessels of patients with premature CAD, as well as in patients suffering from occlusive peripheral artery disease [163]. Whether any of the cellular roles of SENCR contributed to disease was not further functionally investigated. Although being a NAT, SENCR functions independent of FLI1 regulation, at least while regulating VSMC contractility. How SENCR functions is unclear, but it has been shown that SENCR is cytoplasmic [67].
SMILR, smooth muscle–induced lncRNA enhances replication, also known as RP11-94a24.1, has already been mentioned above in the context of HAS2 locus regulation. SMILR has, however, also been studied on its own: While profiling RNA expression following stimulation of human VSMCs with the inflammatory interleukin-1α and the mitogen and chemoattractant PDGF, 200–300 noncoding transcripts were found to be differentially regulated [94]. SMILR was among the top differentially abundant lncRNAs. SMILR also became the focus of interest for this study because IL-1α and PDGF treatment did not change its abundance in other vascular cell types such as ECs [94]. SMILR was shown to be more abundant in unstable human atherosclerotic plaques, as compared to healthy adjacent vessel tissue and prospectively defined by positron emission tomography imaging. SMILR knockdown in cultured VSMCs reduced VSMC proliferation, while overexpression enhanced it [94]. When investigating how SMILR functioned on a molecular level, the authors found that genes in the genomic neighborhood of SMILR were coordinately upregulated together with SMILR in their cell culture model.
Tug1, taurine upregulated gene 1, is another well-studied lncRNA. It had originally been found in studies of the developing retina, but subsequent studies revealed a multitude of functions in diverse cell types: Overarching some functions, it has been proposed that Tug1 repressed growth control genes though scaffolding and promoting their intranuclear association with repressive Polycomb group complexes in heterochromatic Polycomb bodies in the nucleus [206]. Since Tug1 is also expressed in ECs, and the role of PcG members is of general importance for many cell types, Tug1’s function also in the cardiovascular system was investigated. Here, it was found in cell culture studies that Tug1 activated the enzymatic activity of the PcG member Ezh2 in methylating α-actin as a nonhistone target in the cytoplasm of VSMCs. This type of regulation has been found in earlier independent studies and correlates with increased actin polymerization. Also, Tug1 was shown to regulate EC tight junctions. Lastly, Tug1 was found to be upregulated in an ApoE −/− mouse model of atherosclerosis, and to be less upregulated when these mice were fed the anti-atherosclerotic and antianginal compound Tanshinol, a carbocyclic catechol compound in herbal extracts of sage in traditional Chinese medicine [165]. Whether the human TUG1 orthologue was also differentially expressed in human atherosclerotic lesions was not tested. Nevertheless, Tanshinol was found to repress the induction of mouse Tug1 and of human TUG1 by ox-LDL in mouse and human ECs, in cell culture, respectively. Conversely, TUG1 overexpression inhibited the antiapoptotic effect of Tanshinol in ox-LDL-treated human ECs [165]. This suggested that human TUG1 may be a therapeutically interesting lncRNA in human CAD.
In the following section we highlight lncRNAs that have been implicated in myocardial infarction through the analysis of relevant animal models of MI, but which have not been specifically studied in atherosclerosis models. Data mainly derive from mouse MI models, in many cases experimentally triggered by transverse aortic constriction (TAC). In some cases, differential lncRNA expression data have been obtained from orthologous lncRNAs in humans myocardial infarction.
Among 150 lncRNAs deregulated by pressure overload after transaortic constriction in a mouse model, Chaer, cardiac hypertrophy associated epigenetic regulator, showed enriched expression in hearts and was studied carefully [16]. A human CHAER ortholog exists that is also expressed in the human heart. There is at least a trend for Chaer to be induced in hearts from patients with dilated cardiomyopathy. The Chaer locus was inactivated by deleting exon 2, but the knockout mice did not show obvious morphological organ deficits or heart problems. Yet, under pressure overload, cardiac hypertrophy and pathological fibrosis were reduced in the absence of Chaer [16]. The knockouts also showed overall increased heart function in this model. Molecularly, Chaer was found to interact with Ezh2, the central histone methyltransferase of the PRC2 polycomb complex. Further analyses suggested that Chaer competed with other lncRNAs such as Hotair or Fendrr in binding to PRC2, while leaving the enzymatic activity of Ezh2 intact. This interaction of Chaer and the PRC2 complex seemed to negatively impact the association of PRC2 with selected target genes and thus their repression [16]. The human CHAER ortholog shows similar interactions with the human PRC2 complex and stimulated the expression of hypertrophic genes in human cardiomyocytes. Why Chaer/CHAER gained access to PRC2 over Hotair during a hypertrophic growth phase in cardiomyocytes is not yet fully clear, but a modification of the PRC2 complex by the growth-promoting mTOR complex was indicated in the first analysis. Targeting this switch is of specific interest also from a therapeutic aspect.
While screening for lncRNAs differentially expressed in conditions of cardiomyocyte hypertrophy upon transverse aortic constriction in mice, Chast, cardiac hypertrophy-associated transcript, was identified [167]. This lncRNAs was also upregulated in human hypertrophic hearts when analyzing heart tissue of aortic stenosis patients. Systemic overexpression of Chast ameliorated heart pathology in TAC-operated mice, indicating that it might be exploited as a protective factor in the future.
CHRF, cardiac hypertrophy related factor, was identified in a study on the microRNA-dependent regulation of cardiac hypertrophy. Among other microRNAs, the conserved miR-489 was recently shown to be downregulated in pressure-overload models following transverse aortic constriction (TAC) in mice, as well as in human heart failure samples, and in cultured cardiomyocytes upon treatment with angiotensin II [168]. Ang-II is a vasorestrictive peptide binding to its membrane receptors, known for a long time to promote cardiomyocyte hypertrophy also in vitro. Apart from many other important physiological functions in the kidney and other organs, Ang-II is known to be released upon hemodynamic overload and can stimulate cardiac hypertrophy. Ang-II treatment of cardiomyocytes is an in vitro model to study aspects of the downstream effects. Interestingly, miR-489 depletion by RNAi caused hypertrophy, and further promoted hypertrophy upon Ang-II treatment in vitro [168]. Conversely, the cardiac-specific overexpression of miR-489 in conditional transgenic mice did not show anatomical aberrations, but displayed a reduced hypertrophic response in the heart after in vivo Ang-II treatment. This effect was ascribed, at least in part, to micro-RNA-dependent inhibition of translation of Myd88, an adaptor protein for Nf-κB signaling that was already previously involved in regulation of hypertrophy. CHRF, on the other hand, acted upstream, was induced by Ang-II and in TAC mouse models, sequestered miR-489, and when overexpressed impaired miR-489. Together, CHRF was suggested to promote cardiac hypertrophy and caused apoptosis [168].
Hotair, Hox transcript antisense intergenic RNA, is another famous lncRNA studied in the context of embryonic development as well as in cancer. In a recent study, Hotair was found to be decreased after TAC surgery in mouse hearts [169, 170]. There is also a human HOTAIR lncRNA, and it is known to be downregulated in left heart ventricle biopsies as well as in PBMCS in dilated ischemic cardiomyopathy patients [170]. Overexpression of mouse Hotair in cultured cardiomyocytes reduced angiotensin II-triggered hypertrophic growth in vitro [169]. The authors suggested that for this function Hotair sequestered miR-19 to derepress PTEN [169], a pair of factors implicated directly and indirectly before in other cells in growth-dependent DNA replication control [330]. Whether this is the effector mechanism in vivo is open, also because Hotair has been previously implicated in Hox gene regulation. A number of studies put forward evidence that Hotair repressed transcription of posterior Hox (Hoxd) genes in trans, through tethering several independent repressive chromatin-modifying complexes. Most recently, dedicated knockout analyses of the entire Hotair locus in vivo challenged the trans-regulation model that based on near-complete or partial exonic knockouts of Hotair. The knockouts showed that Hotair only had a very subtle role, and this was in cis-regulation of nearby Hoxc genes [52].
KCNQ1OT1, KCNQ1 opposite strand/antisense transcript 1, is known as a classical example of a lncRNA expressed at an imprinted growth regulating locus. In a recent study, human KCNQ1OT1 levels were also found to be increased in neutrophils and monocytes in whole blood preparations in patients with myocardial infarction (MI), where it was associated with hypertension, and not so much with inflammation markers [171]. This study showed that KCNQ1OT1 together with ANRIL might be useful in prediction of left ventricular dysfunction after MI. Extending this study, a recent report tested the role of KCNQ1OT1 in infarcted tissue. Silencing of KCNQ1OT1 antagonized cell death in myocardial cells upon oxygen/glucose deprivation in vitro [172]. Thus, KCNQ1OT1 has heart-specific functions, and may not only be a marker of MI, but also actively promote injury in ischemic hearts when overacted [172]. The effector mechanisms of KCNQ1OT1 during MI are still unknown. KCNQ1OT1’s molecular roles are only known from unrelated studies in other organ systems. Knockout analyses showed that dysregulation of mouse Kcnq1ot1 correlated with embryonic overgrowth phenotypes and mental disabilities but also with some cancers. In these studies it became clear that Kcnq1ot1is paternally expressed and recruits the repressive PRC2 and the G9a histone methyltransferase for silencing Kcnq1 and other protein-coding genes in the locus in cis. In the heart, Kcnq1ot1 represses Kcnq1 during cardiac development, but whether this is the causal determinant for becoming detrimental during MI remains open.
Mirt1, myocardial infarction-associated transcript 1 or NR_028427,) and Mirt2 (ENSMUST00000100512) were identified in a group of 30 lncRNAs that were differentially expressed in a mouse model of myocardial infarction based on coronary ligation [55]. Within the 24 h after permanent occlusion of the anterior interventricular artery, Mirt1 and Mirt2 were upregulated 10–20-fold. Mirt1 levels then decreased to baseline levels by 48 h. Mirt1 located in fibroblasts more than in cardiomyocytes. When Mirt1 was depleted by injecting shRNA-expressing constructs into the myocardium before MI, cardiac functions were ameliorated after MI and the infarction size slightly reduced, suggesting that Mirt1 upregulation contributed to pathological changes in the infarcted heart [173]. Similarly, in vitro, hypoxia-induced Mirt1 expressed in cultured mouse cardiac fibroblasts, and Mirt1 contributed to their apoptosis, to Nf-κB activation, and to the expression of inflammatory cytokines [173].
Myheart (Mhrt), is a NAT conserved in mouse and humans, and is expressed antisense to the Myosin Heavy Chain 7 RNA transcript, a molecular motor protein allowing heart muscles to contract. Mhrt is a myocardial-specific lncRNA and expressed with several isoforms in nuclei of cardiomyocytes [15]. Mouse Mhrt lncRNAs were found to be downregulated in a mouse TAC-induced heart pressure overload model, which was used as a trigger for cardiac hypertrophy and fibrosis and impairing hearts functionally. Likewise, human MHRT levels were downregulated in heart tissue of patients suffering from left ventricular hypertrophy, or from ischemic cardiomyopathy or from idiopathic dilated cardiomyopathy. Conditional forced transgenic expression of mouse Mhrt in cardiomyocytes of adult mouse hearts in vivo reduced stress-induced cardiac hypertrophy and fibrosis, as well as left ventricular dilation and fractional shortening [15]. This offered evidence for the notion that Mhrt is a lncRNA that protects from hypertrophy-induced heart injury. Mechanistically, mouse Mhrt was shown to bind SWI/SNF chromatin remodeling complex, which is well known to regulate a number of target genes by modulating nucleosome positioning and occupancy at genetic loci. The binding of Mhrt to Brg1 was found to repress Brg1 activity, since Mhrt was shown to compete with binding of Brg1 to chromatinized DNA. Since Brg1 was additionally found to repress Mhrt1 lncRNA expression, a feedback loop was suggested that may fine-tune Mhrt RNA levels and Brg1 activity levels to protect hearts from hypertrophy of cardiomyocytes [15]. Given its pronounced therapeutic effects in the mouse model, Myheart may become one of the first lncRNAs that might be used for therapeutic purposes in humans in the future.
ROR, regulator of reprogramming, or previously known as lincRNA-ST8SIA3, has been identified in unrelated work as lincRNA that is upregulated during derivation of human-induced pluripotent stem cells from fibroblasts and contributed to gaining an undifferentiated proliferative cellular phenotype in pluripotent cells [21]. At least part of this role was ascribed to functioning as a sponge for miR-145, miR-181, and miR-99 [212]. This allowed the core pluripotency transcription factors OCT4, SOX2, and NANOG, which were otherwise targeted by these three microRNAs, to become more stably expressed, especially to withstand low-level aberrant differentiation signals. Since these transcription factors also stimulated ROR expression, a feed-forward regulatory loop had been proposed that stabilized stem cell self-renewal, but also made differentiation progress more robust since ROR levels decreased during programmed progenitor cell differentiation [212]. These findings were noticeable because they placed, for the first time, a lncRNA in the core pluripotency transcription factor network. ROR has more recently also been found to be upregulated in a mouse heart injury model following transverse aortic constriction, as well as in models of cardiomyocytes hypertrophy in cell culture [174]. siRNA-mediated depletion of ROR impaired hypertrophic growth in vitro. Since ROR could be depleted by miR-133, a known antihypertrophic microRNA, the authors suggested a pathological role of ROR in promoting hypertrophic growth downstream of this microRNA [174]. How the stimulation of hypertrophy in cardiomyocytes of the diseased heart relate to the hypothetical reactivation of pluripotency-related genes, as suggested from the previous unrelated studies, remains to be addressed.
The authors of a recent study focused on a set of lncRNAs that were heart-enriched, conserved also in human and differentially induced after myocardial infarction in mouse and human [331]. Five percent of these lncRNAs could be mapped to heart-selective super-enhancers [91]. Super-enhancers, also known as stretch-enhancers, represent a subgroup of enhancers that have been identified by a body of recently published work, particularly influential enhancer elements in the genome: Super-enhancers have a high multiplicity and density of enhancer elements, and locate close to genes that encode for important cell-fate-determining factors, master microRNAs, and for central signaling pathway components with relevance for development and disease (see a recent publication for characteristics of super-enhancers [332]). Among the top induced lncRNAs in the class of heart-enriched lncRNAs, a recent study found the mouse Wisper lncRNA, named after the location close to the protein-coding gene Wisp2, a nonstructural signaling protein in the ECM [91]. Wisper was shown to be present in cardiac fibroblasts and to peak in expression two weeks after MI in mice in an LAD mouse MI Model, as well as in the proliferative fibrotic phase in hearts in a model of cardiac fibrosis induced by hypertension due to left renal artery clipping. Depletion of Wisper upregulated Wisp2, decreased proliferation and migration, impaired the transdifferentiation of cardiac fibroblasts to myofibroblasts, at least based on marker gene expression profiling in vitro, and induced proapoptotic genes. Since Wisp2 depletion alone did not trigger all these effects, [91] must have additional effectors. Among many possible mechanisms, the authors found four RNA-processing factors and splicing regulators, TIAR, PTB3, DIS3L2, and CELF2, to specifically bind the lncRNA Wisper [91]. The relevance of these interactions is not yet clear, but using GapmeRs to deplete Wisper in vivo reduced the extent of cardiac fibrosis and the infarction size. A human ortholog of Wisper exists, and shows fibrosis-associated induction in AOS (Adams Oliver syndrome) patients, who characteristically show myocardial fibrosis and LV dysfunction. These data suggest that Wisper/WISPER has a conserved detrimental function in MI [91]. It is interesting to note that Wisper is not the only example of a lncRNA residing at a super-enhancer locus. Also Carmen and Upperhand (see above) may be super-enhancer RNAs, and have been investigated in relation to the cardiovascular system.
7.4.2.2 Aortic Aneurysms
Aortic aneurysms are focal asymmetrical dilations of the vessel. They occur most commonly in the infrarenal abdominal aorta, but can also be found elsewhere, for example in the chest. The biomechanical integrity of the vessel is in large parts determined by fibers, such as collagen and elastin fibers, in the ECM, and the ECM of the vessel depends in large parts on the VSMCs. An SNP in ANRIL has been linked not only to abdominal aortic aneurysms, but also to sporadic and familial intracranial aneurysm [136, 333, 334]. A nearby SNP in ANRIL that associated with diabetes did not associate with aneurysms in this context. How ANRIL molecular contributes to changes in the extracellular matrix or in the contractility of VSMCS has not been tested in the context of aneurysm formation. Yet, in studies of cancerous cells, ANRIL was implicated in promoting metastasis, which may point to an underlying role in promoting ECM remodeling, a hypothesis that remains to be tested in more detail [335,336,337,338].
Also HIF1aAS1, HIF1A antisense RNA 1, became the focus of interest because it was found to be differentially abundant in the serum of patients with thoracoabdominal aortic aneurysms (TAA) [71]. HIF1aAS was more abundant in TAA. The authors tested its role in affecting the survival of vascular cell types in culture. Knockdown of HIF1aAS1 partially protected from experimentally induced apoptosis in VSCMs and ECs in vitro. Whether and how this function in VSMCS and ECs related to aneurysm formation was not further tested [71].
7.4.2.3 Cardiomyopathies and Congenital Heart Disease
So far, knockout analyses have shown that 10 lncRNAs have roles in embryonic heart development, and that a further 6 lncRNAs do not show any obvious morphological or functional abnormalities in the embryo but develop dysfunction under stress conditions in the adult (Table 7.1). These latter lncRNAs are part of the group that will be described in the following: In this section we review 12 lncRNAs linked to cardiomyopathies and heart diseases, some of which are genetically inherited (congenital). LncRNAs in this group have often been experimentally studied in rodent ischemia-reperfusion (I/R) heart injury models, and specifically in the context of hypertrophic overgrow of cardiomyocytes, which is triggered thereby. When cardiomyocyte hypertrophy is sustained it may eventually increase the risk of heart failure. So, the functional separation of this class from the class of MI-related lncRNAs (see above) is not always clear-cut. Beyond the lncRNAs that have been functionally studied and are reported here, many more lncRNAs have been found to be differentially expressed in human heart diseases but have so far not been functionally explored. For these we refer to the relevant studies that profiled lncRNA expression in dilated cardiomyopathy [170, 339], ischemic heart failure, inherited hypertrophic cardiomyopathy [340], and congenital heart defects that range from ventricular septal defects over atrial septal defects to tetralogy of Fallot [341, 342]. The following lncRNAs have, however, been studied in functional terms and we present them in alphabetical order:
Apf, the lncRNA autophagy promoting factor, was found to be induced in infarcted hearts in ischemia-reperfusion (I/R)-mediated mouse models of heart injury [175]. Apf was found to overactivate autophagy by sequestering miR-188-3p and, thus, inhibiting miR-188-3p-dependent repression of ATG7 [175]. A balanced repression of ATG7 seems, however, important, as ATG7 is one of the central known components in forming the autophagosome vesicle. Consistent with this idea, Apf RNAi resulted in a reduced size of infarcted heart tissue in the I/R model and ATG7 overexpression rescued this effect. Therefore, the model was proposed that Apf induction led to a detrimental overactivation of autophagy that mediated cell death in the infarcted heart, possibly in cardiomyocytes [175]. Autophagy is normally operating in physiology for the homeostasis of cells’ molecular building. During metabolic stresses like starvation or hypoxia, autophagy is especially important, also to remove dysfunctional molecules and organelles. Both overactivation and blockage of autophagy have been linked by multiple earlier studies to many diseases and contribute to cancer, aging, degenerative diseases, and cardiovascular diseases (see [343] for review). Although Apf-dependent autophagy was detrimental in the current study in the I/R model [175], the proposed mechanism should not be generalized to other cardiovascular diseases, where autophagy also plays a role. For example, autophagosome number was found to be increased in macrophages of atherosclerotic plaques, but there autophagy is thought to curb plaque necrosis and to stabilize plaques by reducing macrophage apoptosis [344].
Carl, cardiac apoptosis-related lncRNA, is a lncRNA that has come to attention in the context of studying the regulation of mitochondrial energy production in energetically active cardiomyocytes. Carl is a mouse lncRNA that is expressed in the heart, and is repressed by hypoxia in cultured mouse cardiomyocytes. The authors showed that Carl sequestered miR-539, while miR-539 was found to represses PHB2 [18]. PHB2 is a mitochondrial protein belonging to the class of prohibitins and its function is to limit mitochondrial fission in anoxic conditions. Limiting fission is a proproliferative adaptation necessary to survive anoxia. Since Carl expression is, however, downregulated in hypoxia, and miR-539-dependent PHB2 inhibition begins to prevail, the end result is myocardial death. In vivo, overexpressing mouse Carl in the aortic root in a mouse ischemia/reperfusion model reduced myocardial death and limited heart infarction size [18]. Carl, thus, seems to ensure sufficiently high levels of PHB2, which has protective consequences in the post-infarcted heart and is due to ensuring a correctly balanced level of mitochondrial energy production [345]. How the situation is in the human heart is not known. Conceptually, the regulation of the mitochondrial fission-fusion cycle for balancing energy production is involved in a number of diseases, and in particular also in cardiovascular diseases, since the heart is the most metabolically active organ, carries the highest content of mitochondria, and since mitochondrial capacity is impaired, for example, in heart failure (see [346] for review).
Carmen, cardiac mesoderm enhancer-associated noncoding RNA, is a lncRNA that is required for the specification of the cardiac lineage [19]. It was found upon profiling lncRNAs that were induced upon differentiation of fetal human cardiac precursor cells in culture. Carmen’s gene body overlapped with a previously characterized fetal and adult human heart enhancer that had been determined by chromatin-IP experiments. RNAi experiments showed a requirement for Carmen in the specification to cardiac SMCs and differentiation to cardiomyocytes in a differentiation model of mouse embryonic stem cells in culture [19]. The authors suggested that Carmen functioned as an enhancer RNA to potentiate enhanced expression of genes close to the lncRNA locus in cis. But genetic rescue experiments showed that additional effectors had to exist elsewhere, and that Carmen, maybe, also had a trans-acting role on still unknown target genes. Since Carmen bound the PRC2 complex, its trans-repressive function may be tethering this repressive chromatin regulator to target loci, but whether this was indeed the case and how this related to its eRNA function remains to be shown [19]. How Carmen impacts human heart diseases is less clear. Specific isoforms of Carmen were found to be induced in hearts after myocardial infarction, in human hearts of dilated cardiomyopathy (DCM), and aortic stenosis patients [19]. Since RNAi-mediated depletion of Carmen led to a reduction of structural heart proteins and of heart transcription factors Gata4 and Nkx2–5 in neonatal murine cardiomyocytes, a role in maintaining the differentiated fate of cardiomyocytes has been proposed [19].
The evidence that misregulated levels of these two circRNAs contribute to heart disease is rather solid, at least in the experimental mouse models. The effector mechanism is, however, disputed. microRNA sponging by circRNA is not considered to be a physiological function for the vast majority of circRNAs. Only a few circRNAs are expressed at such high levels and with such a large amount of microRNA binding seed regions that microRNA binding could possibly occur at relevant rates. CDR1-as, cerebellar degeneration-related protein 1 antisense RNA, is a classic example of a microRNA-sponging circRNA, but likely also the exception. It contains 74 microRNA seed regions for the miR-7 microRNA. Since CDR1-as lacks full sequence complementarity with this microRNA, CDR1-as degradation is thought to be avoided [214, 215]. Indeed, CDR1-as does form stabilized microRNA:AGO2 endonuclease complexes, as would be expected for a microRNA sponge [215]. That sponging was indeed a biological function for CDR1-as was concluded from genetic experiments [213, 214]. Due to its expression domain, and as evidenced by the loss of function studies in mouse and zebrafish in vivo, CDR1-as had a neuronal function in the CNS. A knockout exists in mouse, but it does not show anatomical alterations, and also no heart defects [213]. Despite this evidence, CDR1-as has also been studied in the cardiovascular context. CDR1-as circRNA was found to be upregulated in infarcted heart tissue in a mouse model for myocardial infarction. Following the previously proposed model that CDR1-as served as a sponge for miR-7a, they suggested that in this function CDR1-as inhibited the antiapoptotic effects of miR-7a in cardiomyocytes in vitro [176]. Further experiments have to substantiate the proposed model in the disease model in vivo.
The well-known H19 lncRNA was found to be induced after chemical induction of dilated cardiomyopathy in rats. Depletion of H19 by intracoronary injection of shRNA-expressing constructs improved heart function and reduced cardiomyocyte apoptosis [197]. Induction of H19 seems to be also involved in calcific aortic valve disease (CAVD) in humans [152]. This disease is characterized by the abnormal mineralization of the aortic valve, which leads to its thickening, impaired leaflet motion and stenosis. In this case, H19 was pathologically upregulated and this induction promoted cultured human valve interstitial cells to start expressing genes, like osteocalcin, BGLAP, BMP2, or RUNX2, which are normally only expressed during osteogenesis [152]. H19-depended repression of NOTCH1 expression was implicated in the upstream regulation of these genes because H19 was found to inhibit NOTCH1, and NOTCH1 was known from earlier studies to repress the osteogenic genes. The authors offered further evidence for a SNP in the H19 promoter of CAVD patients, which did not cause loss of imprinting as in other H19-dependent diseases, but which correlated with reduced DNA methylation and, thus, increased H19 expression [152].
The Heartbrake lncRNA 1 (HBL1) is a special case of a human-specific cardiac regulator that is highly conserved in primates but not in other vertebrates. Human HBL1 became the focus of interest because it is a cardiovascular lineage-specific lncRNA, as determined by expression profiling in differentiating human pluripotent stem cells in culture [177]. HBL1 was shown to become deactivated after cells exited from pluripotency during the onset of cardiomyocyte differentiation. HBL1 was found to sequester miR-1, a microRNA known from before to be important for cardiomyocyte development. The authors of the current study showed that HBL1 restrained miR-1’s ability in promoting ventricular cardiomyocyte expansion. This is likely of relevance also for humans because one target of mouse miR-1 is the cardiogenic transcription factor Hand2 [347], and a heterozygous missense mutation in human HAND2 has been linked to tetralogy of Fallot syndrome, a disease showing ventricular heart septal defects and ventricular heart hypertrophy [348]. This connection indirectly implicates HBL1 as a mediator of congenital heart defects in humans, possibly in a function in undifferentiated cardiomyocyte progenitors and at the onset of cardiomyocyte cell fate differentiation [177].
Also, some circRNAs have been implicated in hypertrophic growth control in the heart. mm9-circ-012559 is a 5′-3′-linked circRNA in the mouse. Based on its role it has later been renamed HRCR, heart-related circular RNA [105]. HRCR was identified through a circRNA profiling upon TAC-induced cardiac hypertrophy and heart failure, and in parallel also in a heart injury model based on the infusion of isoproterenol. In these models HRCR abundance was reduced compared to controls. Since seed regions for the microRNA miR-223 were found on HRCR, experiments were performed to test HCRC’s role in sequestering this microRNA. miR-223 is relevant for heart physiology because it serves a role as an inhibitor of the known apoptosis repressor with Card domain (ARC), which is known to protect from hypertrophic effects in injured hearts. Since overexpression of HRCR reduced the degree of cardiac hypertrophy depending on ARC, the authors concluded that reducing the levels of HRCR contributed to heart failure because miR-223 became active to deplete ARC, and ARC could, thus, no longer protect against heart hypertrophy [105].
The function of Malat1, metastasis-associated lung adenocarcinoma transcript 1, in cardiomyopathy was recently tested in a rat model. Malat1 was found to be induced in cardiac tissue in diseased hearts of diabetic rats. Depleting Malat1 by intracoronary injected shRNA-expressing constructs improved heart function as measured in left ventricular function [349].
Specific isoforms of the mouse Meg3 lncRNA were among the most abundant lncRNAs specific to cardiac fibroblasts. Meg3 was further enriched after TAC-induced heart injury. Silencing Meg3 systemically by injecting inhibiting oligonucleotides in mice ameliorated cardiac fibrosis and diastolic dysfunction, and reduced cardiomyocyte hypertrophy in the TAC-injury model [161]. Thus, Meg3 is cardioprotective. There is a human MEG3 ortholog, and we have described its functions in CAD. The role and regulation of human MEG3 in cardiomyopathies, and independent of CAD, have so far not been studied.
MFACR, mitochondrial fission and apoptosis-related circRNA, is another 5′-3′-linked circRNA in the mouse. Its levels were found to be upregulated in a heart ischemia/reperfusion injury model in mouse [106]. In this study it was suggested that miR-652-3p was sequestered by MFACR. miR-652-3p had the deleterious capacity to repress MTP18, a factor important for cellular viability by facilitating mitochondrial fission. In the injury model in vivo, and following systemic manipulation of MFACR levels, the authors showed that MFACR activation derepressed MTP18, mitochondrial fission, and that the unbalanced overshooting of mitochondrial apoptosis led to heart dysfunction [106].
MIAT has been described in the sections on CAD-related lncRNAs and will be described also for problems in angiogenesis. MIAT has also been linked to cardiac hypertrophy, and the underlying roles are not necessarily the same. First, MIAT one was found to be induced in heart ventricle samples of patients suffering from noninflammatory dilated cardiomyopathy (DCM), and even more so in Chagas disease patients [147]. This disease typically is triggered by infection with Trypanosomes and develops a form of aggressive inflammatory (DCM) with myocarditis, hypertrophy, and fibrosis. MIAT upregulation was corroborated in a mouse model of Chagas disease [147], as well as in Angiotensin II-triggered cardiac hypertrophy models in mice [180] and in a rat diabetic cardiomyopathy model [181]. In the latter model, depleting MIAT by shRNAs in vivo ameliorated heart function in diabetic mouse cardiomyopathy and antagonized high glucose-mediated apoptosis in cultured cardiomyocytes. How MIAT upregulation affected cardiac hypertrophy in each case is unclear, and whether the different diseases share a common function of MIAT has so far not been addressed.
UCA1, urothelial cancer associated 1, is a lncRNA that is the top induced noncoding RNA in the heart in a rat model of partial cardiac ischemia/reperfusion. Overexpression increased proliferation and depletion decreased viability in cultured cardiomyocytes [179]. Thus, UCA1 has antiapoptotic, proliferative functions. The relevance for humans has not been tested.
7.4.2.4 Vascularization and Angiogenesis
Also in this section on lncRNAs linked to vascularization and angiogenesis we will only review lncRNAs that have been implicated through evidence from animal models in vivo. Depending on lncRNA, additional evidence may exist that a lncRNAs is differentially expressed in diseased human tissue under hypoxic conditions or in animal cell culture models of vascularization. On a cellular level, these lncRNAs are involved in aspects of endothelial cell biology, to vessel formation and branching in normal conditions, or in ischemic conditions such as during atherosclerosis or heart infarction. LncRNAs implicated in cerebrovascular stroke, where vasculariziation plays a role, will be presented in Sect. 7.4.2.6. Relevant lncRNAs in section encompass HOTTIP, Malat1, MANTIS, and SENCR. Secondly, and although we do not consider lncRNAs linked to CAD risk factors like diabetes or high blood pressure per se, in this section we also consider reports on lncRNAs related to studies of ECs during vascularization in diabetic retinopathy (Miat, Malat1) or in end-stage renal disease (Myoslid). The lncRNAs are discussed in alphabetical order.
Given the role of some HOX genes in EC biology, HOTTIP has been investigated in the context of angiogenesis. Overexpression of HOTTIP was found to promote EC proliferation and migration in vitro, while siRNA-mediated knockdown had the opposite effects [156]. This proliferative and migrative function resembled the role of HOTTIP in other previously tested cells. With relevance to the cardiovascular system, so far it is also known that Hoxa13 is important for endothelial cell specification in the vasculature connecting embryo and placenta. Thus, whether HOTTIP would be also functioning in ECs in the adult vascular wall, or be misregulated in disease, remains to be seen.
Malat1 has a number of molecular functions as far as studied in cultured cells, but no obvious anatomical defect when the gene was knocked out in mice in vivo (see the previous section). Human MALAT1 was, however, found to be specifically upregulated in stress conditions, such as in cultured human umbilical vein endothelial cells (HUVECs) exposed to hypoxia, which is a pro-angiogenic condition. Malat1, MEG3, TUG1, and SNHG5 were among the top induced lncRNAs. In this condition Malat1 knockdown by siRNAs or GapmeRs decreased hypoxic EC proliferation, and impaired EC migration and sprouting in spheroid and scratch assay in vitro [13]. Based on these results, the mouse Malat1 knockouts were reinvestigated and a specific deficit in vascularization was found also in vivo, and specifically in the neonatal retina [13]. Stress-specific roles may also underlie Malat1’s roles in cancer cells, where Malat1 is upregulated in tumors, while knockout impaired cancer cell migration and metastasis, as well as alternative splicing in mammary tumor models [216]. The relevance of MALAT1/Malat1 has since been studied also in ischemic disease models in vivo. When GapmeRs directed against Malat1 were intraperitoneally injected into mice during experimentally triggered hindlimb ischemia, blood flow recovery was found to be reduced [13]. Thus, Malat1 was suggested to be required for vascular growth also in this context, and its role in stimulating EC proliferation and sprouting, as determined in cultured cells, may underlie vascularization. In contrast to its beneficial proangiogenic role in CAD models, during diabetic retinopathy, retinal vessels growth instructed by Malat1 can also be detrimental when uncontrolled: Malat1 was found to be induced in hypoxic and in diabetic conditions, as shown, for example, in rat and mouse models. In the context of diabetic retinal pathology, Malat1 contributed to pathological vascular retinal pathology [14]. MALAT1 is also induced in ischemic conditions in cerebrovascular stroke models and will be treated in Sect. 7.4.2.6 in more detail.
A lncRNA encoded in antisense within an intron of Annexin A4 was recently investigated in some more detail and named MANTIS, n342419 [69]. So far, MANTIS expression has been observed in endothelial cells, and was tested in several cardiovascular diseases. MANTIS was found to be downregulated in lung tissue of idiopathic pulmonary arterial hypertension in humans, a small-vessel disease characterized by endothelial apoptosis and proliferation. Secondly, MANTIS was found to be upregulated in vessel tissue in atherosclerosis regression in a monkey CAD disease model, which could be due to a hypothetical role of MANTIS in promoting vascular regeneration [69]. Though not specifically expressed only in endothelial cells (ECs), a recent study focused on ECs and studied functions of human MANTIS based on a CRISPR/Cas9-mediated knockout in human umbilical vein endothelial cells (HUVECs) or depletion by siRNA in other endothelial cell lines [69]. Molecularly, MANTIS localized to the nucleus, where it bound BRG1, known as the catalytic subunit of the SWI/SNF ATP-dependent chromatin remodeling complex [69]. MANTIS was suggested to contribute to BRG1 helicase activity and, thus, to the chromatin-remodeling activity of BRG1-containing complexes. For this function it was suggested that MANTIS stabilized the interaction of BRG1 with selected members of the SWI/SNF chromatin remodeling complex. In this function, MANTIS might activate BRG1-dependent target genes, but this model remains to be experimentally corroborated. In ECs, human MANTIS was shown to be pleiotropically important for proper vascular tube formation in matrigel assays, growth factor-dependent spheroid outgrowth, and cell migration of ECs in vitro [69]. Together, MANTIS is a trans-acting lncRNA that activates angiogenic genes like SOX18 or SMAD6 by nucleosome remodeling.
MIAT, myocardial infarction-associated transcript, is a lncRNA that is involved in pathological angiogenesis. MIAT is upregulated in retinas and in the diseased fibrovascular membranes of type 2 diabetes patients. This induction was confirmed in cell culture in a number of different endothelial cell types, including in HUVECs under glucose stress. In a hyperglycemic rat model in vivo, Miat knockdown ameliorated retinal function in the disease state, for example, by reducing apoptosis of retinal cells [148]. On a cellular level, Miat was implicated in promoting cell death triggered by hyperglycemic stress and in stimulating proliferation and migration of ECs, and on a tissue level in advancing vessel leakage. It is likely that multiple molecular roles underlie these diverse functions. As tested in macaque retinal endothelial cells, one attractive model is that Miat de-represses VEGF by microRNA sponging, which may account for Miat’s role in ECs, and, therein, in angiogenesis and neovascularization [148].
SENCR which has already been described during in vivo studies in the context of CAD (see above) was also found to be relevant for angiogenesis. In limb muscle samples of human critical limb ischemia patients, and in vessel wall ECs of patients with premature CAD, SENCR levels were shown to be reduced [164]. In this study it was also found that human SENCR promoted endothelial commitment and differentiation and angiogenesis-related cell functions, like proliferation migration and tube formation, in a differentiation model of human embryonic stem cells in vitro. This suggested that SENCR’s functions are more complex and potentially cell type specific [164].
The differentiation of vascular smooth muscle cells is known to require the transcription factor serum response factor (SRF) and its coactivator Myocardin. In a sequencing approach, dozens of lncRNAs were found to be induced after overexpression of Myocardin in MYOCD in human coronary artery SMCs [184]. Among them, Myoslid, myocardin-induced smooth muscle lncRNA, was identified, a cytoplasmically enriched lncRNA. Myoslid is VSMC-specific, expressed in blood vessels and bladder, is encoded in overlapping patterns with three other uncharacterized lncRNAs, and is a NAT of at least one of them. Knockdown experiments suggested a role in promoting the contractile, antiproliferative, and antimigrative phenotype of VSMCs, which is consistent with a role in promoting Myocardin-dependent differentiation [184]. Myoslid was subsequently found to be downregulated in arteriovenous tissue samples from patients with end-stage renal disease. Therein, a hyperplastic and stenotic response has been associated with disease. This suggests that reduction of Myoslid in vascular disease may contribute to the disease-promoting de-differentiation of VSMCs.
7.4.2.5 Arrhythmia
In the fifth functional class we summarize lncRNAs linked to heart electrophysiology, heartbeat, and rhythm. Compared to other cardiovascular disease entities, few lncRNAs have been associated with this disease type and the depth of functional insight is limited. Cardiac arrhythmias can be, but do not need to be, associated with structural heart diseases, with defects involving aspects of the conductions system. Heart structures relevant for rhythm include the sinus node, the specialized atrial tracts, the atrioventricular node, and the bundle of His. Characteristically shaped electrical currents reach the myocytes of the heart via ion channels, specialized proteins and gap junctions. Most SNPs associated with arrhythmias through GWAS affect one of the multiple ion channel subunits. Also structural heart defects can lead to heart arrhythmias. In this case it is most often cardiac fibrosis or hypertrophy that impairs proper conduction in the heart. In the following paragraphs, we only focus on those lncRNAs that have been functionally investigated in relation to heart rhythm by in vivo models.
One study investigated lncRNAs differentially expressed in patients with atrial fibrillation when comparing two left atrial regions, one being the region of the pulmonary vein and the surrounding left atrial area (LA-PV), and the other being the area of the left atrial appendage (LAA) [185]. The reason for comparing these two regions lies in the earlier observations that the LA-PV junction is implicated as most important for arrhythmogenicity based on catheter ablation experiments, while the LAA is involved in other functions, it is, for example, a site of thrombus formation. Among more than 90 such lncRNAs, AK055347 was found to be enriched in the LA-PV, and was the most significantly altered lncRNA there. In cultured rat cardiomyocyte cell lines, AK055347 knockdown decreased viability [185]. Whether and how the proproliferative function of AK055347 related to arrhythmia remained, however, open.
A recent study performed quantitative trait locus (QTL) mapping in two rat strains that differed in how their blood pressure was affected by salt levels with the aim to identify genes regulating blood pressure. The researchers identified a QTL responsible for high blood pressure, which was, coincidently, also responsible for shortening the cardiac QT-interval [20]. It was mapped to the Rffl locus, and specifically the noncoding RNA therein, Rffl-lnc1. Interestingly, these two phenotypes have also been co-associated within the synthetic genomic region in humans, as shown by data from human GWAS studies [350]. A pathologically shortened Q-wave/T-wave interval, as recorded in the electrocardiogram, often associates with mutations in cardiac ion channels, and even without involving structural changes in the heart tissue is known to correlate with an increased risk for atrial and ventricular arrhythmia in humans. In the QTL study in the rat, the researchers found that 19 bp indel in a previously undescribed lncRNA, Rffl-lnc1, was responsible for the mutant phenotype in the rat. Rffl-lnc1 locates in the 5′ UTR of the single protein-coding gene in this region, and Rffl is a ring finger and FYVE-like domain containing E3 ubiquitin protein ligase [20]. Deletion of the Rffl-lnc1 and, independently, CRISPR/Cas9-mediated reintroduction of the missing 19 nucleotides into the mutant rat strain showed that Rffl-lnc1 indeed was the causative gene [20]. Together, how Rffl-lnc1 polymorphisms partake in the control of cardiac features will be interesting to investigate. At the moment it is thought that Rffl-lnc1 may either directly regulate the cardiac conduction system or the neural autonomic pathways in the heart, or it may indirectly affect heart rhythm through the regulation of cardiomyocyte hypertrophy in the context of increased blood pressure [20].
Another study performed lncRNA expression profiling at sites of enervation on the surface of the heart, where collections of autonomous nerves form ganglia plexus, mainly in island-like fat pad structures or in the adipose tissue below the epicardial membrane [186]. Autonomic nervous system remodeling (ANR) at such sites is known to contribute to arrhythmia. Canine models have been traditionally used to study this aspect. After receiving heart tachypacing via electrodes, dogs developed ANR, and TCONS_00032546 and TCONS_00026102 were found to be downregulated [186]. Injecting knockdown constructs for the two lincRNAs into the relevant fat pads in dogs was sufficient to slightly shorten, or prolong, respectively, the atrial effective refractory period and increase, or inhibit, respectively, the onset of tachycardia and atrial fibrillation [186]. How these two lncRNAs differentially impact neuronal remodeling in the onset of arrhythmia remained unknown, as well as whether human orthologs existed that carried conserved functions.
In conceptually similar experiments in a rabbit model for studying conduction remodeling in atrial fibrillation, lncRNA expression profiling was performed in right atrial tissue samples [187]. The authors then used coexpression network analysis to focus on pathologically potentially interesting lncRNAs, such as the downregulated TCONS_00075467. A human sequence with 70% conservation exists, opening the possibility that the lncRNA was conserved. Depleting TCONS_00075467 in the right atrium in the rabbit model shortened the atrial effective refractory period and increased the frequency of fibrillation onset in the animal model [187]. The authors suggested that this lncRNA functioned as a protector by inducing the expression of the calcium voltage-gated CACNA1C ion channel as a microRNA sponge. Thereby, this work related to earlier published insight on the role of CACNA1C downregulation by microRNAs in human patients with fibrillation [351] and to the earlier identification of a loss-of-function mutation in CACNA1C in Brugada syndrome patients, a disease with characteristic ST-segment elevation [352].
7.4.2.6 Stroke and Cerebrovascular Aneurysms
In the sixth functional class, lncRNAs related to brain stroke are reviewed. This class is not treated separately to indicate a separation in terms of atherosclerosis pathways leading to thrombus and ischemia formation, but because brain neurons are the cell type hit most by ischemia, and because specialized pathogenic mechanisms occur in the brain involving distinct sets of lncRNAs. The occlusion of the middle cerebral artery is the most common route to cerebral stroke, and lack of oxygen and glucose rapidly inflicts neurons during hypoxia-ischemia. Depolarization and impaired neurotransmitter uptake subsequently cause toxic amounts of extracellular glutamate to accumulate, which further contributes to mitochondrial dysfunction and oxidative stress. Finally, ischemia also triggers inflammatory signaling, which can further increase brain tissue damage. To date, nine lncRNAs have been associated with stroke. For two of them there is some evidence that the relevant lncRNAs were also important in human stroke patients. Of the nine lncRNAs, two had protective functions in stroke-induced brain damage, while seven aggravated neuronal problems after brain stroke.
Chr9p21 is well known as a major CAD risk locus. Different risk genotypes in the human Chr9p21 locus have been found to associate with different cardiovascular disease entities. Initial analyses offered conflicting evidence whether Chr9p21 genotypes were also associated with stroke, with some studies reporting an association [137,138,139], and some studies showing no significant association [136, 353, 354]. A meta-analysis of relevant GWAS explored the association and did document a robust but relatively weak association [140]. The investigation also suggested a subtype-specific association of Chr9p21 with ischemic stroke, namely, with large artery stroke [140]. Also, a more recent study corroborated this notion by showing that specific SNPs in ANRIL lncRNA associated with susceptibility and recurrence rate of atherothrombotic and hemorrhagic cerebrovascular stroke even in those patients that did not have a history of heart disease [141]. Most recently, another study performed an eQTL analysis of five previously identified risk SNPs in ANRIL [12], and identified genes whose expressions associated with at least one of the five risk SNPs. Among 87 of such potential eQTL hits, CARD8 was selected as a potential ANRIL effector, which was further substantiated as CARD8 expression also dropped when ANRIL was knocked down in cultured cells [142]. Since an inactivating mutation in CARD8 was associated with ischemic stroke, and since CARD8 is known from before as negative regulator of NF-κB signaling, the possibility exists that ANRIL impacts ischemic stroke also via CARD8-dependent NF-kB regulation [142]. While this hypothetical pathway remains to be rigorously tested, an implication of ANRIL in stroke is rather well documented by multiple studies. Studies investigating a direct role of ANRIL in stroke are so far limited: In a single study, the role of ANRIL was studied in a model for type 2 diabetes in rats under a high-fat diet and after middle cerebral artery occlusion to trigger stroke [143]. ANRIL was found to be increased in brain tissue in this context. Overexpression and knockdown of ANRIL systemically in the rat model showed that ANRIL augmented endothelial microvessel density, which correlated with increased VEGF and NF-κB expression levels. Since neuronal functionality post stroke was not tested after ANRIL overexpression or knockdown, one can yet only speculate whether the observed ANRIL-promoted angiogenesis aggravated complications of a stroke in this disease model.
AK153573 is a predominantly nuclear lncRNA that partially overlaps in sense the CaMKIIδ gene. Renamed C2dat1, CaMKIIδ-associated transcript I, this lncRNA was found to be induced in neurons surrounding ischemic regions in a stroke model of middle cerebral artery occlusion in mouse and rat in vivo [188]. C2dat1 was also upregulated after experimental oxygen-glucose-deprivation/reoxygenation (OGD/R) in neuronal cells in vitro. CaMKIIδ levels did decrease in the ischemic core, but were induced, similar to C2dat1, in the periphery of the lesion. Knocking down C2dat1 in cultured neurons reduced CaMKIIδ expression and stress-dependent induction of NF-κB signaling and, while neuronal survival was increased. These findings are consistent with the large body of earlier evidence that acute inhibition of CaMKII is neuroprotective [188]. C2dat1 may, thus, likely function via acute CaMKIIδ locus stimulation after stroke, and this pathway may exacerbate apoptosis during stroke.
FosDT, Fos downstream transcript, has been identified as lncRNA that was induced in the rat cerebral cortex after transient middle cerebral artery occlusion [73]. Further, FosDT bound Sin3 and coREST, which are members of the repressive REST chromatin regulating complex, whereby REST is known from independent work to become active and to be necessary for neuronal apoptosis during stroke. In the present work, it was found that FosDT contributed to the repressive function of REST, and that silencing FosDT reduced brain infarction size and ameliorated neurological functions after infarction, as determined after intracerebral injection of siRNAs [73]. One possibility, thus, is that FosDT scaffolds REST to repress selected target genes that are otherwise necessary for their survival.
Beyond the involvement in CAD and in cardiomyopathies, H19 also plays a role in cerebral stroke. The first insight into this association was obtained in rodent models. When focal cerebral ischemia was induced by middle cerebral artery occlusion/reperfusion in mice or rats, H19 levels rose in blood plasma, and also in infarcted brain tissue [153, 154]. Corroborating this finding, H19 was also upregulated in human patients within hours after a stroke [153]. Thereby, H19 expression was higher in plasma, neutrophils, and lymphocytes. Since an intraventricular injection of siRNAs targeting H19 reduced brain infarct volume and ameliorated neurological functions in the mouse stroke model, it can be deduced that H19 induction usually exerts a detrimental function during stroke. How H19 is detrimental is less clear. Current insights into this question base on work with cultured cells: For example, H19 was found to promote neuroinflammatory TNFα and interleukin expression. Secondly, H19 was shown to impair the formation of neuroprotective M2 microglia. Microglial cells are the major cell type in mounting stroke-induced inflammation [153]. And third, albeit only tested in a human neuroblastoma cell line, and not necessarily related, H19 contributed to apoptosis in an OGD/reperfusion model in vitro [154]. H19 has not appeared in GWAS studies on stroke, but selected SNPs in H19 were still found associated to some degree with stroke [154].
Consistent with its antiproliferative functions in many cell types, Meg3 was recently found to be proapoptotic in the context of cerebral stroke in mouse [162]. While mouse Meg3 is known to be downregulated in CAD-affected vessels, it was, however, upregulated in ischemic brain tissue. Meg3 promoted neuronal cell death, and the authors suggested that Meg3 bound to p53’s DNA binding domain. Whether and how such an interaction would activate p53 in a proapoptotic pathway is of interest not only for the cardiovascular field [162]. However, whether Meg3 indeed functioned in neurons in vivo, or also affected angiogenesis in the infarcted brain, as shown in before in CAD model, or both, is still not clear.
N1LR, also known as MRAK051854, was identified by microarray-based transcriptional profiling of ischemic brain tissue after intraluminal middle cerebral artery occlusion to induce a stroke in the rat model system. Based on coexpression network and pathway analysis, N1LR became the focus of interest as a highly connected upregulated lncRNA [189]. Also, this lncRNA was upregulated in a mouse stroke model. Overexpressing N1LR after injection into the mouse cortex reduced the infarct volume and neuronal cell death, while siRNA against N1LR enhanced the lesions, suggestive of protective functions of this lncRNA. The molecular effector mechanism still remains to be interrogated.
In another approach, lncRNAs were bioinformatically screened for harboring seeds for miR-145-5p, a microRNA whose induction had been previously associated with ischemia in the heart. Specifically, SNHG14, also known as small nucleolar RNA host gene 1 or UBE3A-ATS, was such a lncRNA. It is expressed in antisense to the ubiquitin protein ligase E3A gene, and was found to be induced during the first days after stroke in a middle cerebral artery occlusion model in mice, while miR-145-5p levels correspondingly dropped [190]. In cultured microglial cell lines, SNHG14 stimulated expression of inflammatory factors, while it reduced miR-145-5p levels. The inflammatory phenotype was rescued by adding miR-145-5p mimics, consistent with the interpretation that SNHG14 impaired the protective function of miR-145-5p by keeping its levels low [190].
A role in promoting neuronal cell death was ascribed to the lncRNA Tug1 during stroke [166]. Tug1 levels increased in a middle cerebral artery occlusion model in the rat in vivo, as well as in cultured neurons after oxygen/glucose deprivation. Tug1 knockdown reduced apoptosis under OGD conditions in vitro, and this effect correlated with upregulation of miR-9 levels, suggesting a detrimental role in stroke [166]. Tug1 has been linked to apoptosis, but also to other roles in other cellular contexts before, particularly in tumorigenesis. Given the intense research on miR-9 in neurogenesis, it will be interesting to dissect how Tug1 connects to the rather well-understood functions of miR-9 in the CNS, and whether the regulation of apoptosis is indeed the central function of interest. For example, miR-9 is known from earlier work to be expressed in self-renewing neural progenitors and is required for neurogenesis by promoting the timely cell cycle exit of differentiating neurons. At the same time miR-9 has also previously been found to promote angiogenesis to proceed in concordance with neurogenesis by curbing premature neuronal VEGF expression [355]. Thus, the region- and time-specific coordination of neurogenesis and angiogenesis may be how Tug1 determines the manifestation of stroke.
Together, a number of studies have described RNA expression profiles in brain tissue after stroke, and lists of relevant lncRNAs residing close to known stroke risk loci have been compiled as well [356]. Compared to CAD and MI models, the level of mechanistic in vivo insight is, however, still limited. Questions, like in which cell types and via which effector mechanisms lncRNAs function during stroke are still open. Nevertheless, there is a rich literature on transcriptional regulation of neuronal cell types, and fast advances may be possible if relevant lncRNAs can be convincingly linked to already known and well-studied transcriptional effectors.
7.4.2.7 lncRNAs in Basic Cellular Processes that Overarch CVD Entities
Finally, an important set of lncRNAs has been implicated in the regulation of inflammatory signaling, and derailing immune signaling is a central factor in all entities of cardiovascular diseases. We are not reviewing this inflammation-related set of lncRNAs in depth though, as we are not reviewing lncRNAs implicated in generic proliferation control either. Excellent recent reviews have been published on lncRNAs in immune control [357] and in cell cycle and growth control [358]. Also, as of yet, most of the lncRNAs in these two broad classes have not been specifically studied in the context of cardiovascular disease. This has, in part, also to do with the fact that there is limited knowledge about the exact cell lineage transitions contributing to cell fate changes that underlie atherosclerosis.
Box 7.3 Reading Highlights
-
1.
Reports on linear cardiovascular lncRNAs:
-
(a)
Klattenhoff, C. A. et al., (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment, Cell. 152, 570–83.
-
(b)
Han, P. et al., (2014) A long noncoding RNA protects the heart from pathological hypertrophy, Nature. 514, 102–6.
-
(c)
Anderson, K. M., et al., (2016) Transcription of the non-coding RNA Upperhand controls Hand2 expression and heart development, Nature. 539, 433–436.
-
(d)
Wang, Z et al., (2016) The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy, Nat Med. 22, 1131–1139.
-
(e)
Liu, J. et al., (2017) HBL1 Is a Human Long Noncoding RNA that Modulates Cardiomyocyte Development from Pluripotent Stem Cells by Counteracting MIR1, Dev Cell. 42, 333–348 e5.
-
(f)
Hon, C. C. et al., (2017) CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells, Science. 355.
-
(a)
-
2.
Reports on cardiovascular circRNAs:
-
(a)
Holdt, L. M. et al., (2016) Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans, Nat Commun. 7, 12,429.
-
(b)
Wang et al., (2016) A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223, Eur Heart J. 37, 2602–11.
-
(c)
review: Barrett, S. P. & Salzman, J. (2016) Circular RNAs: analysis, expression and potential functions, Development. 143, 1838–47.
-
(a)
-
3.
Reports addressing generally important concepts in lncRNA biology:
-
(a)
Wide-spread role of lncRNAs as enhancers of transcription: Engreitz, J. M. et al., (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing, Nature. 539, 452–455.
-
(b)
The relevance of RNA:PRC2 interaction is under scrutiny: Kaneko, S et al., (2014) Nascent RNA interaction keeps PRC2 activity poised and in check, Genes Dev. 28, 1983–8.
-
(c)
LncRNAs can be functional despite lack of sequence conservation Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H. & Bartel, D. P. (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution, Cell. 147, 1537–50.
-
(d)
Widespread role antisense transcription of lncRNA genes: Huber, F. et al., (2016) Protein Abundance Control by Non-coding Antisense Transcription, Cell Rep. 15, 2625–36.
-
(e)
Translation of micropeptides from lncRNAs: Micropeptides encoded on lncRNAs: Anderson, D. M. et al., (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance, Cell. 160, 595–606.
-
(f)
microRNA sponging may be less common than thought: Denzler, R., et al., (2016) Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression, Mol Cell. 64, 565–579.
-
(a)
7.5 Summary and Outlook
So far 57 lncRNAs have been functionally investigated in the context of cardiovascular physiology and disease. Many more lncRNAs are differentially expressed, but have not yet been functionally assessed. Therefore, and given the number of different cell types and processes implicated in development, homeostasis and regulation of the heart, vasculature, and vascularized tissue, it is expected that many more lncRNAs will be linked to some aspect of coronary artery physiology and disease in the future. As can be seen already from the work presented in this book chapter, a single lncRNA can exhibit different functions in different contexts, and in extreme cases, can engage with dozens of different binding partners through specialized interaction domains. Therefore, although lncRNAs sometimes show low copy numbers, a limited stability, and a low level of evolutionary selection in sequence, thousands of lncRNAs are expressed with high confidence, with a high degree of cell type specificity, and with functional consequences.
The question of how many of the thousands of lncRNAs are indeed functional genetic elements has until recently not been possible to address. Yet, with the emergence of CRISPR/Cas9-based genetic loss of function experiments, genome-wide genetic screens have begun to explore this question experimentally. When testing 16,000 lncRNAs for effects on cell proliferation capacity in cultured cells, nearly 500 individual lncRNA transcripts (or 3% of all lncRNAs) were found to be important for cell division [359]. This number is unexpectedly large when compared to equivalent genetic tests on the overall functionality of protein-coding genes, of which 10–12% are essential for optimal cell proliferation [360, 361]. Since only a few types of cells and only a single functional readout were assayed in this screen, one can project that a large percentage of lncRNAs will be annotated as functional, when carefully tested [29, 362]. Since every single lncRNA may further produce several spliced isoforms, and since ribonucleotides can additionally be modified posttranscriptionally, for example, by methylation [363], there is a huge regulatory space how lncRNAs can affect physiology and disease.
In a study with genome-wide relevance, the evolutionarily conserved coexpression of lncRNAs was measured relative to other transcripts, and colocalization with eQTL-associated SNPs was taken into consideration. Also, this type of approach suggested that more lncRNAs than previously thought (up to 40%) appear to be potentially functional because they associate with at least one known trait [29]. Future experimental approaches will have to take into account lncRNAs and their time- and location-specific transcription as an integral part of gene-regulatory networks.
Based on the already known important roles of lncRNAs in the cardiovascular system as well as in other physiological processes and diseases, linear and circular noncoding RNAs become a medically interesting target molecule in different respects: First, to lncRNAs and circRNAs as biomarkers for pathophysiological states, both in tissue samples, as well in cell-free form in the circulating blood. In the blood, cell-free noncoding RNAs are found inside exosomes or other membrane-contained vesicles, or in association with proteins, and reporting on cellular states in remote internal organs. To date, most technologies to sequence cell-free nucleic acids in preclinical settings focus on DNA and on finding mutations therein. Combined DNA and RNA sequencing from blood may show increased power in monitoring early disease onset, or therapy success and recurrence of disease after therapy. Second, lncRNAs may be directly used to alter cell physiology in situ: Delivering synthetically produced topically applied lncRNAs to diseased organs may be just one option. Especially if technologies are advanced that stabilize lncRNA half-lives, for example, exchanging the phosphodiester linkage by phosphorothioate bonds in the RNA backbone, or that allow to more reliably target diseased cells in a body, RNA therapeutic agents may become a reasonable option for treatment of some conditions [364]. Editing lncRNA transcripts with modified CRISPR-Cas variants directly, instead of editing the genomic template, is another novel therapeutic approach that will gain importance [364, 365]. In an alternative approach, although it is early days in our understanding of noncoding RNAs, the elaborate secondary structures of a lncRNA may allow us to find or design specific inhibitory drugs that enhance or inhibit the function of a specific lncRNA in its interaction with a protein complex.
In any case, already today, the study of lncRNAs in CAD entities has proven highly valuable, especially because it has yielded a major shift in our understanding of genetic networks that regulate cell fate, and because it has helped to functionally annotate disease-associated genetic loci from GWAS, which often reside in the noncoding DNA sequence space. To conclude, the picture emerges that transcription of noncoding RNAs, linear or circular, is in most cases not a failure in our gene expression program, but an integrated regulatory feature that affects all levels of cell function.
References
Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–23.
Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, Gerstein M, Snyder M. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6.
Carninci P, Kasukawa T, Katayama S, Gough J, Frith M, Maeda C, Oyama N, Ravasi R, Lenhard T, Wells B, Kodzius C, Shimokawa R, Bajic K, Brenner VB, Batalov SE, Forrest S, Zavolan AR, Davis M, Wilming MJ, Aidinis LG, Allen V, Ambesi-Impiombato JE, Apweiler A, Aturaliya R, Bailey RN, Bansal TL, Baxter M, Beisel L, Bersano KW, Bono T, Chalk H, Chiu AM, Choudhary KP, Christoffels V, Clutterbuck A, Crowe DR, Dalla ML, Dalrymple E, de Bono BP, Gatta BD, di Bernardo G, Down D, Engstrom T, Fagiolini P, Faulkner M, Fletcher G, Fukushima CF, Furuno T, Futaki M, Gariboldi S, Georgii-Hemming M, Gingeras P, Gojobori TR, Green T, Gustincich RE, Harbers S, Hayashi M, Hensch Y, Hirokawa TK, Hill N, Huminiecki D, Iacono L, Ikeo M, Iwama K, Ishikawa A, Jakt T, Kanapin M, Katoh A, Kawasawa M, Kelso Y, Kitamura J, Kitano H, Kollias H, Krishnan G, Kruger SP, Kummerfeld A, Kurochkin SK, Lareau IV, Lazarevic LF, Lipovich D, Liu L, Liuni J, McWilliam S, Babu SM, Madera M, Marchionni M, Matsuda L, Matsuzawa H, Miki S, Mignone H, Miyake F, Morris S, Mottagui-Tabar K, Mulder S, Nakano N, Nakauchi N, Ng H, Nilsson P, Nishiguchi R, Nishikawa S, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63.
Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–83.
Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, Macura K, Blass G, Kellis M, Werber M, Herrmann BG. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206–14.
Anderson KM, Anderson DM, McAnally JR, Shelton JM, Bassel-Duby R, Olson EN. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature. 2016;539:433–6.
Deng C, Li Y, Zhou L, Cho J, Patel B, Terada N, Li Y, Bungert J, Qiu Y, Huang S. HoxBlinc RNA recruits Set1/MLL complexes to activate Hox gene expression patterns and mesoderm lineage development. Cell Rep. 2016;14:103–14.
Broadbent HM, Peden JF, Lorkowski S, Goel A, Ongen H, Green F, Clarke R, Collins R, Franzosi MG, Tognoni G, Seedorf U, Rust S, Eriksson P, Hamsten A, Farrall M, Watkins H, Consortium P. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17:806–14.
Schaefer AS, Richter GM, Groessner-Schreiber B, Noack B, Nothnagel M, El Mokhtari NE, Loos BG, Jepsen S, Schreiber S. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378.
Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010;6:e1000899.
Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, Boon RA, Dimmeler S. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–97.
Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, Yan B, Jiang Q. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5:e1506.
Han P, Li W, Lin CH, Yang J, Shang C, Nurnberg ST, Jin KK, Xu W, Lin CY, Lin CJ, Xiong Y, Chien HC, Zhou B, Ashley E, Bernstein D, Chen PS, Chen HS, Quertermous T, Chang CP. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102–6.
Wang Z, Zhang XJ, Ji YX, Zhang P, Deng KQ, Gong J, Ren S, Wang X, Chen I, Wang H, Gao C, Yokota T, Ang YS, Li S, Cass A, Vondriska TM, Li G, Deb A, Srivastava D, Yang HT, Xiao X, Li H, Wang Y. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131–9.
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M, Attardi LD, Regev A, Lander ES, Jacks T, Rinn JL. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19.
Wang K, Long B, Zhou LY, Liu F, Zhou QY, Liu CY, Fan YY, Li PF. CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun. 2014;5:3596.
Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, Reverter F, Alexanian M, Gonzales C, Ng SY, Bussotti G, Pezzuto I, Notredame C, Heymans S, Guigo R, Johnson R, Pedrazzini T. CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol. 2015;89:98–112.
Cheng X, Waghulde H, Mell B, Morgan EE, Pruett-Miller SM, Joe B. Positional cloning of quantitative trait nucleotides for blood pressure and cardiac QT-interval by targeted CRISPR/Cas9 editing of a novel long non-coding RNA. PLoS Genet. 2017;13:e1006961.
Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, Manos PD, Datta S, Lander ES, Schlaeger TM, Daley GQ, Rinn JL. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet. 2010;42:1113–7.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7:e30733.
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233.
Holdt LM, Stahringer A, Sass K, Pichler G, Kulak NA, Wilfert W, Kohlmaier A, Herbst A, Northoff BH, Nicolaou A, Gabel G, Beutner F, Scholz M, Thiery J, Musunuru K, Krohn K, Mann M, Teupser D. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, Bell I, Cheung E, Drenkow J, Dumais E, Patel S, Helt G, Ganesh M, Ghosh S, Piccolboni A, Sementchenko V, Tammana H, Gingeras TR. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8.
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol. 2010;28:503–10.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94.
Guttman M, Russell P, Ingolia NT, Weissman JS, Lander ES. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–51.
Hon CC, Ramilowski JA, Harshbarger J, Bertin N, Rackham OJ, Gough J, Denisenko E, Schmeier S, Poulsen TM, Severin J, Lizio M, Kawaji H, Kasukawa T, Itoh M, Burroughs AM, Noma S, Djebali S, Alam T, Medvedeva YA, Testa AC, Lipovich L, Yip CW, Abugessaisa I, Mendez M, Hasegawa A, Tang D, Lassmann T, Heutink P, Babina M, Wells CA, Kojima S, Nakamura Y, Suzuki H, Daub CO, de Hoon MJ, Arner E, Hayashizaki Y, Carninci P, Forrest AR. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature. 2017;543:199–204.
Ulitsky I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet. 2016;17:601–14.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72.
van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, MacInnes AW, Cuppen E, Simonis M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014;15:R6.
Cabili MN, Dunagin MC, McClanahan PD, Biaesch A, Padovan-Merhar O, Regev A, Rinn JL, Raj A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015;16:20.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–57.
Clark MB, Johnston RL, Inostroza-Ponta M, Fox AH, Fortini E, Moscato P, Dinger ME, Mattick JS. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012;22:885–98.
Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7.
Clamp M, Fry B, Kamal M, Xie X, Cuff J, Lin MF, Kellis M, Lindblad-Toh K, Lander ES. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci U S A. 2007;104:19428–33.
Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol. 2008;4:e1000176.
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–27.
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–50.
Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, Curado J, Snyder M, Gingeras TR, Guigo R. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–25.
Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016;539:452–5.
Huber F, Bunina D, Gupta I, Khmelinskii A, Meurer M, Theer P, Steinmetz LM, Knop M. Protein abundance control by non-coding antisense transcription. Cell Rep. 2016;15:2625–36.
Joung J, Engreitz JM, Konermann S, Abudayyeh OO, Verdine VK, Aguet F, Gootenberg JS, Sanjana NE, Wright JB, Fulco CP, Tseng YY, Yoon CH, Boehm JS, Lander ES, Zhang F. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature. 2017;548(7667):343–6.
Lai F, Gardini A, Zhang A, Shiekhattar R. Integrator mediates the biogenesis of enhancer RNAs. Nature. 2015;525:399–403.
Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012;26:2392–407.
Schlackow M, Nojima T, Gomes T, Dhir A, Carmo-Fonseca M, Proudfoot NJ. Distinctive patterns of transcription and RNA processing for human lincRNAs. Mol Cell. 2017;65:25–38.
Wilusz JE. Long noncoding RNAs: re-writing dogmas of RNA processing and stability. Biochim Biophys Acta. 2016;1859:128–38.
Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL. Long noncoding RNAs with snoRNA ends. Mol Cell. 2012;48:219–30.
Zhang B, Mao YS, Diermeier SD, Novikova IV, Nawrocki EP, Jones TA, Lazar Z, Tung CS, Luo W, Eddy SR, Sanbonmatsu KY, Spector DL. Identification and characterization of a class of MALAT1-like genomic loci. Cell Rep. 2017;19:1723–38.
Amandio AR, Necsulea A, Joye E, Mascrez B, Duboule D. Hotair is dispensible for mouse development. PLoS Genet. 2016;12:e1006232.
Shan K, Jiang Q, Wang XQ, Wang YN, Yang H, Yao MD, Liu C, Li XM, Yao J, Liu B, Zhang YY, J Y, Yan B. Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 2016;7:e2248.
Xu C, Zhang Y, Wang Q, Xu Z, Jiang J, Gao Y, Gao M, Kang J, Wu M, Xiong J, Ji K, Yuan W, Wang Y, Liu H. Long non-coding RNA GAS5 controls human embryonic stem cell self-renewal by maintaining NODAL signalling. Nat Commun. 2016;7:13287.
Zangrando J, Zhang L, Vausort M, Maskali F, Marie PY, Wagner DR, Devaux Y. Identification of candidate long non-coding RNAs in response to myocardial infarction. BMC Genomics. 2014;15:460.
Zhang DD, Wang WT, Xiong J, Xie XM, Cui SS, Zhao ZG, Li MJ, Zhang ZQ, Hao DL, Zhao X, Li YJ, Wang J, Chen HZ, Lv X, Liu DP. Long noncoding RNA LINC00305 promotes inflammation by activating the AHRR-NF-kappaB pathway in human monocytes. Sci Rep. 2017;7:46204.
Li Y, Shen S, Ding S, Wang L. LincRNA DYN-LRB2-2 upregulates cholesterol efflux by decreasing TLR2 expression in macrophages. J Cell Biochem. 2018;119:1911–21.
Berretta J, Morillon A. Pervasive transcription constitutes a new level of eukaryotic genome regulation. EMBO Rep. 2009;10:973–82.
Kramer C, Loros JJ, Dunlap JC, Crosthwaite SK. Role for antisense RNA in regulating circadian clock function in Neurospora crassa. Nature. 2003;421:948–52.
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717–20.
Camblong J, Iglesias N, Fickentscher C, Dieppois G, Stutz F. Antisense RNA stabilization induces transcriptional gene silencing via histone deacetylation in S. cerevisiae. Cell. 2007;131:706–17.
Prescott EM, Proudfoot NJ. Transcriptional collision between convergent genes in budding yeast. Proc Natl Acad Sci U S A. 2002;99:8796–801.
Hobson DJ, Wei W, Steinmetz LM, Svejstrup JQ. RNA polymerase II collision interrupts convergent transcription. Mol Cell. 2012;48:365–74.
Lee JT, Lu N. Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell. 1999;99:47–57.
Sado T, Hoki Y, Sasaki H. Tsix silences Xist through modification of chromatin structure. Dev Cell. 2005;9:159–65.
Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007;67:3963–9.
Bell RD, Long X, Lin M, Bergmann JH, Nanda V, Cowan SL, Zhou Q, Han Y, Spector DL, Zheng D, Miano JM. Identification and initial functional characterization of a human vascular cell-enriched long noncoding RNA. Arterioscler Thromb Vasc Biol. 2014;34:1249–59.
Zong X, Nakagawa S, Freier SM, Fei J, Ha T, Prasanth SG, Prasanth KV. Natural antisense RNA promotes 3′ end processing and maturation of MALAT1 lncRNA. Nucleic Acids Res. 2016;44:2898–908.
Leisegang MS, Fork C, Josipovic I, Richter F, Preussner J, Hu J, Miller MJ, Epah JN, Hofmann P, Gunther S, Moll F, Valasarajan C, Heidler J, Ponomareva Y, Freiman TM, Maegdefessel L, Plate KH, Mittelbronn M, Uchida S, Kunne C, Stellos K, Schermuly RT, Weissmann N, Devraj K, Wittig I, Boon RA, Dimmeler S, Pullamsetti SS, Looso M, Miller FJ, Brandes RP. Long noncoding RNA MANTIS facilitates endothelial angiogenic function. Circulation. 2017;136(1):65–79.
Huang C, Hu YW, Zhao JJ, Ma X, Zhang Y, Guo FX, Kang CM, Lu JB, Xiu JC, Sha YH, Gao JJ, Wang YC, Li P, Xu BM, Zheng L, Wang Q. Long noncoding RNA HOXC-AS1 suppresses Ox-LDL-induced cholesterol accumulation through promoting HOXC6 expression in THP-1 macrophages. DNA Cell Biol. 2016;35:722–9.
Zhao Y, Feng G, Wang Y, Yue Y, Zhao W. Regulation of apoptosis by long non-coding RNA HIF1A-AS1 in VSMCs: implications for TAA pathogenesis. Int J Clin Exp Pathol. 2014;7:7643–52.
Lee MP, DeBaun MR, Mitsuya K, Galonek HL, Brandenburg S, Oshimura M, Feinberg AP. Loss of imprinting of a paternally expressed transcript, with antisense orientation to KVLQT1, occurs frequently in Beckwith-Wiedemann syndrome and is independent of insulin-like growth factor II imprinting. Proc Natl Acad Sci U S A. 1999;96:5203–8.
Mehta SL, Kim T, Vemuganti R. Long noncoding RNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin-modifying proteins. J Neurosci. 2015;35:16443–9.
Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–4.
Ntini E, Jarvelin AI, Bornholdt J, Chen Y, Boyd M, Jorgensen M, Andersson R, Hoof I, Schein A, Andersen PR, Andersen PK, Preker P, Valen E, Zhao X, Pelechano V, Steinmetz LM, Sandelin A, Jensen TH. Polyadenylation site-induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–8.
Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–3.
Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature. 2009;457:1038–42.
Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell. 2013;155:1075–87.
Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Munster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–7.
Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14:880–93.
Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–7.
Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, Glass CK, Rosenfeld MG, Fu XD. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–4.
Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, Oh S, Kim HS, Glass CK, Rosenfeld MG. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–20.
Puc J, Kozbial P, Li W, Tan Y, Liu Z, Suter T, Ohgi KA, Zhang J, Aggarwal AK, Rosenfeld MG. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell. 2015;160:367–80.
Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–23.
Shiraki T, Kondo S, Katayama S, Waki K, Kasukawa T, Kawaji H, Kodzius R, Watahiki A, Nakamura M, Arakawa T, Fukuda S, Sasaki D, Podhajska A, Harbers M, Kawai J, Carninci P, Hayashizaki Y. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc Natl Acad Sci U S A. 2003;100:15776–81.
Murakawa Y, Yoshihara M, Kawaji H, Nishikawa M, Zayed H, Suzuki H, Fantom C, Hayashizaki Y. Enhanced Identification of Transcriptional Enhancers Provides Mechanistic Insights into Diseases. Trends Genet. 2016;32:76–88.
Kwak H, Fuda NJ, Core LJ, Lis JT. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339:950–3.
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010;143:46–58.
Vucicevic D, Corradin O, Ntini E, Scacheri PC, Orom UA. Long ncRNA expression associates with tissue-specific enhancers. Cell Cycle. 2015;14:253–60.
Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, Maric D, Maison D, Nemir M, Young RA, Schroen B, Gonzalez A, Ounzain S, Pedrazzini T. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med. 2017;9
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA, Wysocka J, Lei M, Dekker J, Helms JA, Chang HY. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–4.
Maamar H, Cabili MN, Rinn J, Raj A. linc-HOXA1 is a noncoding RNA that represses Hoxa1 transcription in cis. Genes Dev. 2013;27:1260–71.
Ballantyne MD, Pinel K, Dakin R, Vesey AT, Diver L, Mackenzie R, Garcia R, Welsh P, Sattar N, Hamilton G, Joshi N, Dweck MR, Miano JM, McBride MW, Newby DE, McDonald RA, Baker AH. Smooth muscle enriched long noncoding RNA (SMILR) regulates cell proliferation. Circulation. 2016;133:2050–65.
Nakaya HI, Amaral PP, Louro R, Lopes A, Fachel AA, Moreira YB, El-Jundi TA, da Silva AM, Reis EM, Verjovski-Almeida S. Genome mapping and expression analyses of human intronic noncoding RNAs reveal tissue-specific patterns and enrichment in genes related to regulation of transcription. Genome Biol. 2007;8:R43.
Gardner EJ, Nizami ZF, Talbot CC Jr, Gall JG. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 2012;26:2550–9.
Pek JW, Osman I, Tay ML, Zheng RT. Stable intronic sequence RNAs have possible regulatory roles in Drosophila melanogaster. J Cell Biol. 2015;211:243–51.
Duret L, Chureau C, Samain S, Weissenbach J, Avner P. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006;312:1653–5.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8.
Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9:e1003777.
Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, Herzog M, Schreyer L, Papavasileiou P, Ivanov A, Ohman M, Refojo D, Kadener S, Rajewsky N. Circular RNAs in the mammalian brain are highly abundant. Conserved, and Dynamically Expressed, Mol Cell. 2015;58:870–85.
Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. 2016;143:1838–47.
Holdt LM, Kohlmaier A, Teupser D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell Mol Life Sci. 2018;75(6):1071–98.
Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, Gong Y, Liu J, Dong YH, Li N, Li PF. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37:2602–11.
Wang K, Gan TY, Li N, Liu CY, Zhou LY, Gao JN, Chen C, Yan KW, Ponnusamy M, Zhang YH, Li PF. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24:1111–20.
Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9:660–5.
Hanyu-Nakamura K, Sonobe-Nojima H, Tanigawa A, Lasko P, Nakamura A. Drosophila Pgc protein inhibits P-TEFb recruitment to chromatin in primordial germ cells. Nature. 2008;451:730–3.
Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y, Kobayashi S, Payre F, Kageyama Y. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329:336–9.
Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA, Couso JP. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341:1116–20.
Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, Mitchell A, Ma J, Dubrulle J, Reyon D, Tsai SQ, Joung JK, Saghatelian A, Schier AF. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636.
Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160:595–606.
Rohrig H, Schmidt J, Miklashevichs E, Schell J, John M. Soybean ENOD40 encodes two peptides that bind to sucrose synthase. Proc Natl Acad Sci U S A. 2002;99:1915–20.
Pauli A, Valen E, Schier AF. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. BioEssays. 2015;37:103–12.
Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, Saghatelian A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat Chem Biol. 2013;9:59–64.
Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R, Olson EN. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science. 2016;351:271–5.
Monfort A, Di Minin G, Postlmayr A, Freimann R, Arieti F, Thore S, Wutz A. Identification of Spen as a crucial factor for Xist function through forward genetic screening in haploid embryonic stem cells. Cell Rep. 2015;12:554–61.
Kurian L, Aguirre A, Sancho-Martinez I, Benner C, Hishida T, Nguyen TB, Reddy P, Nivet E, Krause MN, Nelles DA, Esteban CR, Campistol JM, Yeo GW, Belmonte JCI. Identification of novel long noncoding RNAs underlying vertebrate cardiovascular development. Circulation. 2015;131:1278–90.
Jiang W, Liu Y, Liu R, Zhang K, Zhang Y. The lncRNA DEANR1 facilitates human endoderm differentiation by activating FOXA2 expression. Cell Rep. 2015;11:137–48.
Puri MC, Rossant J, Alitalo K, Bernstein A, Partanen J. The receptor tyrosine kinase TIE is required for integrity and survival of vascular endothelial cells. EMBO J. 1995;14:5884–91.
Li K, Blum Y, Verma A, Liu Z, Pramanik K, Leigh NR, Chun CZ, Samant GV, Zhao B, Garnaas MK, Horswill MA, Stanhope SA, North PE, Miao RQ, Wilkinson GA, Affolter M, Ramchandran R. A noncoding antisense RNA in tie-1 locus regulates tie-1 function in vivo. Blood. 2010;115:133–9.
Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, Eklund L, Boon LM, Vikkula M. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118–24.
Yang L, Kirby JE, Sunwoo H, Lee JT. Female mice lacking Xist RNA show partial dosage compensation and survive to term. Genes Dev. 2016;30:1747–60.
Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell. 2013;152:727–42.
Savarese F, Flahndorfer K, Jaenisch R, Busslinger M, Wutz A. Hematopoietic precursor cells transiently reestablish permissiveness for X inactivation. Mol Cell Biol. 2006;26:7167–77.
Zhang B, Arun G, Mao YS, Lazar Z, Hung G, Bhattacharjee G, Xiao X, Booth CJ, Wu J, Zhang C, Spector DL. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111–23.
Nakagawa S, Ip JY, Shioi G, Tripathi V, Zong X, Hirose T, Prasanth KV. Malat1 is not an essential component of nuclear speckles in mice. RNA. 2012;18:1487–99.
Eissmann M, Gutschner T, Hammerle M, Gunther S, Caudron-Herger M, Gross M, Schirmacher P, Rippe K, Braun T, Zornig M, Diederichs S. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9:1076–87.
Holdt LM, Beutner F, Scholz M, Gielen S, Gabel G, Bergert H, Schuler G, Thiery J, Teupser D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler Thromb Vasc Biol. 2010;30:620–7.
Cooper JD, Walker NM, Smyth DJ, Downes K, Healy BC, Todd JA, Type IDGC. Follow-up of 1715 SNPs from the Wellcome Trust Case Control Consortium genome-wide association study in type I diabetes families. Genes Immun. 2009;10(Suppl 1):S85–94.
McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316:1488–91.
Liu Y, Sanoff HK, Cho H, Burd CE, Torrice C, Mohlke KL, Ibrahim JG, Thomas NE, Sharpless NE. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4:e5027.
Jarinova O, Stewart AF, Roberts R, Wells G, Lau P, Naing T, Buerki C, McLean BW, Cook RC, Parker JS, McPherson R. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–7.
Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol. 2012;32:196–206.
Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–3.
Helgadottir A, Thorleifsson G, Magnusson KP, Gretarsdottir S, Steinthorsdottir V, Manolescu A, Jones GT, Rinkel GJ, Blankensteijn JD, Ronkainen A, Jaaskelainen JE, Kyo Y, Lenk GM, Sakalihasan N, Kostulas K, Gottsater A, Flex A, Stefansson H, Hansen T, Andersen G, Weinsheimer S, Borch-Johnsen K, Jorgensen T, Shah SH, Quyyumi AA, Granger CB, Reilly MP, Austin H, Levey AI, Vaccarino V, Palsdottir E, Walters GB, Jonsdottir T, Snorradottir S, Magnusdottir D, Gudmundsson G, Ferrell RE, Sveinbjornsdottir S, Hernesniemi J, Niemela M, Limet R, Andersen K, Sigurdsson G, Benediktsson R, Verhoeven EL, Teijink JA, Grobbee DE, Rader DJ, Collier DA, Pedersen O, Pola R, Hillert J, Lindblad B, Valdimarsson EM, Magnadottir HB, Wijmenga C, Tromp G, Baas AF, Ruigrok YM, van Rij AM, Kuivaniemi H, Powell JT, Matthiasson SE, Gulcher JR, Thorgeirsson G, Kong A, Thorsteinsdottir U, Stefansson K. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24.
Matarin M, BrownWM, Singleton A, Hardy JA, Meschia JF, ISGS Investigators. Whole genome analyses suggest ischemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke. 2008;39:1586–9.
Smith JG, Melander O, Lovkvist H, Hedblad B, Engstrom G, Nilsson P, Carlson J, Berglund G, Norrving B, Lindgren A. Common genetic variants on chromosome 9p21 confers risk of ischemic stroke: a large-scale genetic association study. Circ Cardiovasc Genet. 2009;2:159–64.
Wahlstrand B, Orho-Melander M, Delling L, Kjeldsen S, Narkiewicz K, Almgren P, Hedner T, Melander O. The myocardial infarction associated CDKN2A/CDKN2B locus on chromosome 9p21 is associated with stroke independently of coronary events in patients with hypertension. J Hypertens. 2009;27:769–73.
Anderson CD, Biffi A, Rost NS, Cortellini L, Furie KL, Rosand J. Chromosome 9p21 in ischemic stroke: population structure and meta-analysis. Stroke. 2010;41:1123–31.
Zhang W, Chen Y, Liu P, Chen J, Song L, Tang Y, Wang Y, Liu J, Hu FB, Hui R. Variants on chromosome 9p21.3 correlated with ANRIL expression contribute to stroke risk and recurrence in a large prospective stroke population. Stroke. 2012;43:14–21.
Bai Y, Nie S, Jiang G, Zhou Y, Zhou M, Zhao Y, Li S, Wang F, Lv Q, Huang Y, Yang Q, Li Q, Li Y, Xia Y, Liu Y, Liu J, Qian J, Li B, Wu G, Wu Y, Wang B, Cheng X, Yang Y, Ke T, Li H, Ren X, Ma X, Liao Y, Xu C, Tu X, Wang QK. Regulation of CARD8 expression by ANRIL and association of CARD8 single nucleotide polymorphism rs2043211 (p.C10X) with ischemic stroke. Stroke. 2014;45:383–8.
Zhang B, Wang D, Ji TF, Shi L, Yu JL. Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget. 2017;8:17347–59.
Chen L, Yao H, Hui JY, Ding SH, Fan YL, Pan YH, Chen KH, Wan JQ, Jiang JY. Global transcriptomic study of atherosclerosis development in rats. Gene. 2016;592:43–8.
Arslan S, Berkan O, Lalem T, Ozbilum N, Goksel S, Korkmaz O, Cetin N, Devaux Y, Cardiolinc Network. Long non-coding RNAs in the atherosclerotic plaque. Atherosclerosis. 2017;266:176–81.
Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, Yang T, Zhan L, Yuan Y, Chu W, Pan Z, Wang Z, Yang B, Lu Y. MIAT Is a Pro-fibrotic Long Non-coding RNA Governing Cardiac Fibrosis in Post-infarct Myocardium. Sci Rep. 2017;7:42657.
Frade AF, Laugier L, Ferreira LR, Baron MA, Benvenuti LA, Teixeira PC, Navarro IC, Cabantous S, Ferreira FM, da Silva Candido D, Gaiotto FA, Bacal F, Pomerantzeff P, Santos RH, Kalil J, Cunha-Neto E, Chevillard C. Myocardial infarction-associated transcript, a long noncoding RNA. Is Overexpressed During Dilated Cardiomyopathy Due to Chronic Chagas Disease, J Infect Dis. 2016;214:161–5.
Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–56.
Gao W, Zhu M, Wang H, Zhao S, Zhao D, Yang Y, Wang ZM, Wang F, Yang ZJ, Lu X, Wang LS. Association of polymorphisms in long non-coding RNA H19 with coronary artery disease risk in a Chinese population. Mutat Res. 2015;772:15–22.
Zhang Z, Gao W, Long QQ, Zhang J, Li YF, Liu DC, Yan JJ, Yang ZJ, Wang LS. Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Sci Rep. 2017;7:7491.
Han Y, Ma J, Wang J, Wang L. Silencing of H19 inhibits the adipogenesis and inflammation response in ox-LDL-treated Raw264.7 cells by up-regulating miR-130b. Mol Immunol. 2017;93:107–14.
Hadji F, Boulanger MC, Guay SP, Gaudreault N, Amellah S, Mkannez G, Bouchareb R, Marchand JT, Nsaibia MJ, Guauque-Olarte S, Pibarot P, Bouchard L, Bosse Y, Mathieu P. Altered DNA methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation. 2016;134:1848–62.
Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, Wang R, Feng J, Luo Y. Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke. 2017;48:2211–21.
Wang J, Cao B, Han D, Sun M, Feng J. Long non-coding RNA H19 induces cerebral ischemia reperfusion injury via activation of autophagy. Aging Dis. 2017;8:71–84.
Vigetti D, Deleonibus S, Moretto P, Bowen T, Fischer JW, Grandoch M, Oberhuber A, Love DC, Hanover JA, Cinquetti R, Karousou E, Viola M, D'Angelo ML, Hascall VC, De Luca G, Passi A. Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J Biol Chem. 2014;289:28816–26.
Liao B, Chen R, Lin F, Mai A, Chen J, Li H, Xu Z, Dong S. Long noncoding RNA HOTTIP promotes endothelial cell proliferation and migration via activation of the Wnt/beta-catenin pathway. J Cell Biochem. 2018;119(3):2797–805.
Hu YW, Yang JY, Ma X, Chen ZP, Hu YR, Zhao JY, Li SF, Qiu YR, Lu JB, Wang YC, Gao JJ, Sha YH, Zheng L, Wang Q. A lincRNA-DYNLRB2-2/GPR119/GLP-1R/ABCA1-dependent signal transduction pathway is essential for the regulation of cholesterol homeostasis. J Lipid Res. 2014;55:681–97.
Tang SS, Cheng J, Cai MY, Yang XL, Liu XG, Zheng BY, Xiong XD. Association of lincRNA-p21 haplotype with coronary artery disease in a Chinese han population. Dis Markers. 2016;2016:9109743.
Wu G, Cai J, Han Y, Chen J, Huang ZP, Chen C, Cai Y, Huang H, Yang Y, Liu Y, Xu Z, He D, Zhang X, Hu X, Pinello L, Zhong D, He F, Yuan GC, Wang DZ, Zeng C. LincRNA-p21 regulates neointima formation, vascular smooth muscle cell proliferation, apoptosis, and atherosclerosis by enhancing p53 activity. Circulation. 2014;130:1452–65.
Wu Z, He Y, Li D, Fang X, Shang T, Zhang H, Zheng X. Long noncoding RNA MEG3 suppressed endothelial cell proliferation and migration through regulating miR-21. Am J Transl Res. 2017;9:3326–35.
Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K, Batkai S, Thum T. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res. 2017;121:575–83.
Yan H, Yuan J, Gao L, Rao J, Hu J. Long noncoding RNA MEG3 activation of p53 mediates ischemic neuronal death in stroke. Neuroscience. 2016;337:191–9.
Shahmoradi N, Nasiri M, Kamfiroozi H, Kheiry MA. Association of the rs555172 polymorphism in SENCR long non-coding RNA and atherosclerotic coronary artery disease. J Cardiovasc Thorac Res. 2017;9:170–4.
Boulberdaa M, Scott E, Ballantyne M, Garcia R, Descamps B, Angelini GD, Brittan M, Hunter A, McBride M, McClure J, Miano JM, Emanueli C, Mills NL, Mountford JC, Baker AH. A role for the long noncoding RNA SENCR in commitment and function of endothelial cells. Mol Ther. 2016;24:978–90.
Chen C, Cheng G, Yang X, Li C, Shi R, Zhao N. Tanshinol suppresses endothelial cells apoptosis in mice with atherosclerosis via lncRNA TUG1 up-regulating the expression of miR-26a. Am J Transl Res. 2016;8:2981–91.
Chen S, Wang M, Yang H, Mao L, He Q, Jin H, Ye ZM, Luo XY, Xia YP, Hu B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem Biophys Res Commun. 2017;485:167–73.
Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, Dittrich M, Maetzig T, Zimmer K, Remke J, Just A, Fendrich J, Scherf K, Bolesani E, Schambach A, Weidemann F, Zweigerdt R, de Windt LJ, Engelhardt S, Dandekar T, Batkai S, Thum T. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra22.
Wang K, Liu F, Zhou LY, Long B, Yuan SM, Wang Y, Liu CY, Sun T, Zhang XJ, Li PF. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–88.
Lai Y, He S, Ma L, Lin H, Ren B, Ma J, Zhu X, Zhuang S. HOTAIR functions as a competing endogenous RNA to regulate PTEN expression by inhibiting miR-19 in cardiac hypertrophy. Mol Cell Biochem. 2017;432:179–87.
Greco S, Zaccagnini G, Perfetti A, Fuschi P, Valaperta R, Voellenkle C, Castelvecchio S, Gaetano C, Finato N, Beltrami AP, Menicanti L, Martelli F. Long noncoding RNA dysregulation in ischemic heart failure. J Transl Med. 2016;14:183.
Vausort M, Wagner DR, Devaux Y. Long noncoding RNAs in patients with acute myocardial infarction. Circ Res. 2014;115:668–77.
Li X, Dai Y, Yan S, Shi Y, Han B, Li J, Cha L, Mu J. Down-regulation of lncRNA KCNQ1OT1 protects against myocardial ischemia/reperfusion injury following acute myocardial infarction. Biochem Biophys Res Commun. 2017;491:1026–33.
Li X, Zhou J, Huang K. Inhibition of the lncRNA Mirt1 attenuates acute myocardial infarction by suppressing NF-kappaB activation. Cell Physiol Biochem. 2017;42:1153–64.
Jiang F, Zhou X, Huang J. Long non-coding RNA-ROR mediates the reprogramming in cardiac hypertrophy. PLoS One. 2016;11:e0152767.
Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120:1812–24.
Geng HH, Li R, Su YM, Xiao J, Pan M, Cai XX, Ji XP. The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PLoS One. 2016;11:e0151753.
Liu J, Li Y, Lin B, Sheng Y, Yang L. HBL1 is a human long noncoding RNA that modulates cardiomyocyte development from pluripotent stem cells by counteracting MIR1. Dev Cell. 2017;42:333–348 e5.
Lin X, Zhan JK, Wang YJ, Tan P, Chen YY, Deng HQ, Liu YS. Function, role, and clinical application of microRNAs in vascular aging. Biomed Res Int. 2016;2016:6021394.
Liu Y, Zhou D, Li G, Ming X, Tu Y, Tian J, Lu H, Yu B. Long non coding RNA-UCA1 contributes to cardiomyocyte apoptosis by suppression of p27 expression. Cell Physiol Biochem. 2015;35:1986–98.
Zhu XH, Yuan YX, Rao SL, Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharmacol Sci. 2016;20:3653–60.
Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2929.
Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ. Long noncoding RNA Malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci. 2017;37:1797–806.
Xin JW, Jiang YG. Long noncoding RNA MALAT1 inhibits apoptosis induced by oxygen-glucose deprivation and reoxygenation in human brain microvascular endothelial cells. Exp Ther Med. 2017;13:1225–34.
Zhao J, Zhang W, Lin M, Wu W, Jiang P, Tou E, Xue M, Richards A, Jourd'heuil D, Asif A, Zheng D, Singer HA, Miano JM, Long X. MYOSLID is a novel serum response factor-dependent long noncoding RNA that amplifies the vascular smooth muscle differentiation program. Arterioscler Thromb Vasc Biol. 2016;36:2088–99.
Chen G, Guo H, Song Y, Chang H, Wang S, Zhang M, Liu C. Long noncoding RNA AK055347 is upregulated in patients with atrial fibrillation and regulates mitochondrial energy production in myocardiocytes. Mol Med Rep. 2016;14:5311–7.
Wang W, Wang X, Zhang Y, Li Z, Xie X, Wang J, Gao M, Zhang S, Hou Y. Transcriptome analysis of canine cardiac fat pads: involvement of two novel long non-coding RNAs in atrial fibrillation neural remodeling. J Cell Biochem. 2015;116:809–21.
Li Z, Wang X, Wang W, Du J, Wei J, Zhang Y, Wang J, Hou Y. Altered long non-coding RNA expression profile in rabbit atria with atrial fibrillation: TCONS_00075467 modulates atrial electrical remodeling by sponging miR-328 to regulate CACNA1C. J Mol Cell Cardiol. 2017;108:73–85.
Xu Q, Deng F, Xing Z, Wu Z, Cen B, Xu S, Zhao Z, Nepomuceno R, Bhuiyan MI, Sun D, Wang QJ, Ji A. Long non-coding RNA C2dat1 regulates CaMKIIdelta expression to promote neuronal survival through the NF-kappaB signaling pathway following cerebral ischemia. Cell Death Dis. 2016;7:e2173.
Wu Z, Wu P, Zuo X, Yu N, Qin Y, Xu Q, He S, Cen B, Liao W, Ji A. LncRNA-N1LR enhances neuroprotection against ischemic stroke probably by inhibiting p53 phosphorylation. Mol Neurobiol. 2017;54:7670–85.
Qi X, Shao M, Sun H, Shen Y, Meng D, Huo W. Long non-coding RNA SNHG14 promotes microglia activation by regulating miR-145-5p/PLA2G4A in cerebral infarction. Neuroscience. 2017;348:98–106.
Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gabel G, Bergert H, Bechmann I, Stadler PF, Thiery J, Teupser D. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013;9:e1003588.
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–74.
Smith CM, Steitz JA. Classification of gas5 as a multi-small-nucleolar-RNA (snoRNA) host gene and a member of the 5′-terminal oligopyrimidine gene family reveals common features of snoRNA host genes. Mol Cell Biol. 1998;18:6897–909.
Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8.
Aprea J, Prenninger S, Dori M, Ghosh T, Monasor LS, Wessendorf E, Zocher S, Massalini S, Alexopoulou D, Lesche M, Dahl A, Groszer M, Hiller M, Calegari F. Transcriptome sequencing during mouse brain development identifies long non-coding RNAs functionally involved in neurogenic commitment. EMBO J. 2013;32:3145–60.
Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA. 2010;16:324–37.
Zhang Y, Zhang M, Xu W, Chen J, Zhou X. The long non-coding RNA H19 promotes cardiomyocyte apoptosis in dilated cardiomyopathy. Oncotarget. 2017;8:28588–94.
Venkatraman A, He XC, Thorvaldsen JL, Sugimura R, Perry JM, Tao F, Zhao M, Christenson MK, Sanchez R, Yu JY, Peng L, Haug JS, Paulson A, Li H, Zhong XB, Clemens TL, Bartolomei MS, Li L. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature. 2013;500:345–9.
Kallen AN, Zhou XB, Xu J, Qiao C, Ma J, Yan L, Lu L, Liu C, Yi JS, Zhang H, Min W, Bennett AM, Gregory RI, Ding Y, Huang Y. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell. 2013;52:101–12.
Monnier P, Martinet C, Pontis J, Stancheva I, Ait-Si-Ali S, Dandolo L. H19 lncRNA controls gene expression of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad Sci U S A. 2013;110:20693–8.
Giovarelli M, Bucci G, Ramos A, Bordo D, Wilusz CJ, Chen CY, Puppo M, Briata P, Gherzi R. H19 long noncoding RNA controls the mRNA decay promoting function of KSRP. Proc Natl Acad Sci U S A. 2014;111:E5023–8.
Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol Cell. 2012;47:648–55.
Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, Ansell PJ, Zhao J, Weng C, Klibanski A. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731–42.
Kaneko S, Bonasio R, Saldana-Meyer R, Yoshida T, Son J, Nishino K, Umezawa A, Reinberg D. Interactions between JARID2 and noncoding RNAs regulate PRC2 recruitment to chromatin. Mol Cell. 2014;53:290–300.
Sanuki R, Onishi A, Koike C, Muramatsu R, Watanabe S, Muranishi Y, Irie S, Uneo S, Koyasu T, Matsui R, Cherasse Y, Urade Y, Watanabe D, Kondo M, Yamashita T, Furukawa T. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat Neurosci. 2011;14:1125–34.
Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773–88.
Chen R, Kong P, Zhang F, Shu YN, Nie X, Dong LH, Lin YL, Xie XL, Zhao LL, Zhang XJ, Han M. EZH2-mediated alpha-actin methylation needs lncRNA TUG1, and promotes the cortex cytoskeleton formation in VSMCs. Gene. 2017;616:52–7.
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, Chang HY. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23.
Portoso M, Ragazzini R, Brencic Z, Moiani A, Michaud A, Vassilev I, Wassef M, Servant N, Sargueil B, Margueron R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017;36:981–94.
Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, Peters AH. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell. 2008;15:668–79.
Zhang H, Zeitz MJ, Wang H, Niu B, Ge S, Li W, Cui J, Wang G, Qian G, Higgins MJ, Fan X, Hoffman AR, Hu JF. Long noncoding RNA-mediated intrachromosomal interactions promote imprinting at the Kcnq1 locus. J Cell Biol. 2014;204:61–75.
Wang Y, Xu Z, Jiang J, Xu C, Kang J, Xiao L, Wu M, Xiong J, Guo X, Liu H. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog, and Sox2 in human embryonic stem cell self-renewal. Dev Cell. 2013;25:69–80.
Piwecka M, Glazar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A, Filipchyk A, Klironomos F, Cerda Jara CA, Fenske P, Trimbuch T, Zywitza V, Plass M, Schreyer L, Ayoub S, Kocks C, Kuhn R, Rosenmund C, Birchmeier C, Rajewsky N. Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science. 2017;357
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, le Noble F, Rajewsky N. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8.
Arun G, Diermeier S, Akerman M, Chang KC, Wilkinson JE, Hearn S, Kim Y, MacLeod AR, Krainer AR, Norton L, Brogi E, Egeblad M, Spector DL. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016;30:34–51.
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38.
Bernard D, Prasanth KV, Tripathi V, Colasse S, Nakamura T, Xuan Z, Zhang MQ, Sedel F, Jourdren L, Coulpier F, Triller A, Spector DL, Bessis A. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082–93.
Ip JY, Sone M, Nashiki C, Pan Q, Kitaichi K, Yanaka K, Abe T, Takao K, Miyakawa T, Blencowe BJ, Nakagawa S. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci Rep. 2016;6:27204.
Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007;13:313–6.
Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–14.
Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9.
Cajigas I, Leib DE, Cochrane J, Luo H, Swyter KR, Chen S, Clark BS, Thompson J, Yates JR 3rd, Kingston RE, Kohtz JD. Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development. 2015;142:2641–52.
Kaneko S, Son J, Bonasio R, Shen SS, Reinberg D. Nascent RNA interaction keeps PRC2 activity poised and in check. Genes Dev. 2014;28:1983–8.
Di Ruscio A, Ebralidze AK, Benoukraf T, Amabile G, Goff LA, Terragni J, Figueroa ME, De Figueiredo Pontes LL, Alberich-Jorda M, Zhang P, Wu M, D'Alo F, Melnick A, Leone G, Ebralidze KK, Pradhan S, Rinn JL, Tenen DG. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503:371–6.
Portoso M, Ragazzinni R, Brenčič Ž, Moiani A, Michaud A, Vassilev I, Wassef M, Servant N, Sargueil B, Margueron R. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J. 2017;36:981–94.
Engreitz JM, Sirokman K, McDonel P, Shishkin AA, Surka C, Russell P, Grossman SR, Chow AY, Guttman M, Lander ES. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–99.
McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, Sweredoski MJ, Shishkin AA, Su J, Lander ES, Hess S, Plath K, Guttman M. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 2015;521:232–6.
Chu C, Zhang QC, da Rocha ST, Flynn RA, Bharadwaj M, Calabrese JM, Magnuson T, Heard E, Chang HY. Systematic discovery of Xist RNA binding proteins. Cell. 2015;161:404–16.
West JA, Davis CP, Sunwoo H, Simon MD, Sadreyev RI, Wang PI, Tolstorukov MY, Kingston RE. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol Cell. 2014;55:791–802.
Ramirez F, Lingg T, Toscano S, Lam KC, Georgiev P, Chung HR, Lajoie BR, de Wit E, Zhan Y, de Laat W, Dekker J, Manke T, Akhtar A. High-affinity sites form an interaction network to facilitate spreading of the MSL complex across the X chromosome in drosophila. Mol Cell. 2015;60:146–62.
Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756–70.
Sunwoo H, Wu JY, Lee JT. The Xist RNA-PRC2 complex at 20-nm resolution reveals a low Xist stoichiometry and suggests a hit-and-run mechanism in mouse cells. Proc Natl Acad Sci U S A. 2015;112:E4216–25.
Simon MD, Pinter SF, Fang R, Sarma K, Rutenberg-Schoenberg M, Bowman SK, Kesner BA, Maier VK, Kingston RE, Lee JT. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504:465–9.
Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146:119–33.
Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973.
Minajigi A, Froberg J, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, Lee JT. Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 2015;349
Ilik IA, Quinn JJ, Georgiev P, Tavares-Cadete F, Maticzka D, Toscano S, Wan Y, Spitale RC, Luscombe N, Backofen R, Chang HY, Akhtar A. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol Cell. 2013;51:156–73.
Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, Pyle AM. HOTAIR forms an intricate and modular secondary structure. Mol Cell. 2015;58:353–61.
Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, Chang HY. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–90.
Martin WJ, Reiter NJ. Structural roles of noncoding RNAs in the heart of enzymatic complexes. Biochemistry. 2017;56:3–13.
Blythe AJ, Fox AH, Bond CS. The ins and outs of lncRNA structure: How, why and what comes next? Biochim Biophys Acta. 2016;1859:46–58.
da Rocha ST, Boeva V, Escamilla-Del-Arenal M, Ancelin K, Granier C, Matias NR, Sanulli S, Chow J, Schulz E, Picard C, Kaneko S, Helin K, Reinberg D, Stewart AF, Wutz A, Margueron R, Heard E. Jarid2 is implicated in the initial xist-induced targeting of PRC2 to the inactive X chromosome. Mol Cell. 2014;53:301–16.
Almeida M, Pintacuda G, Masui O, Koseki Y, Gdula M, Cerase A, Brown D, Mould A, Innocent C, Nakayama M, Schermelleh L, Nesterova TB, Koseki H, Brockdorff N. PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science. 2017;356:1081–4.
Wutz A, Rasmussen TP, Jaenisch R. Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet. 2002;30:167–74.
Chen CK, Blanco M, Jackson C, Aznauryan E, Ollikainen N, Surka C, Chow A, Cerase A, McDonel P, Guttman M. Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science. 2016;354:468–72.
Hnisz D, Day DS, Young RA. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 2016;167:1188–200.
Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:661–78.
Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C, Glass CK. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–25.
Schaukowitch K, Joo JY, Liu X, Watts JK, Martinez C, Kim TK. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56:29–42.
Pefanis E, Wang J, Rothschild G, Lim J, Kazadi D, Sun J, Federation A, Chao J, Elliott O, Liu ZP, Economides AN, Bradner JE, Rabadan R, Basu U. RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell. 2015;161:774–89.
Ounzain S, Pedrazzini T. Super-enhancer lncs to cardiovascular development and disease. Biochim Biophys Acta. 2016;1863:1953–60.
Mousavi K, Zare H, Dell'orso S, Grontved L, Gutierrez-Cruz G, Derfoul A, Hager GL, Sartorelli V. eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell. 2013;51:606–17.
Lam MT, Cho H, Lesch HP, Gosselin D, Heinz S, Tanaka-Oishi Y, Benner C, Kaikkonen MU, Kim AS, Kosaka M, Lee CY, Watt A, Grossman TR, Rosenfeld MG, Evans RM, Glass CK. Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature. 2013;498:511–5.
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature. 2013;494:497–501.
Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1–19.
Gribnau J, Diderich K, Pruzina S, Calzolari R, Fraser P. Intergenic transcription and developmental remodeling of chromatin subdomains in the human beta-globin locus. Mol Cell. 2000;5:377–86.
Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–89.
Dekker J, Mirny L. The 3D genome as moderator of chromosomal communication. Cell. 2016;164:1110–21.
Werner MS, Ruthenburg AJ. Nuclear fractionation reveals thousands of chromatin-tethered noncoding RNAs adjacent to active genes. Cell Rep. 2015;12:1089–98.
Werner MS, Sullivan MA, Shah RN, Nadadur RD, Grzybowski AT, Galat V, Moskowitz IP, Ruthenburg AJ. Chromatin-enriched lncRNAs can act as cell-type specific activators of proximal gene transcription. Nat Struct Mol Biol. 2017;24:596–603.
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232–46.
Mohammad F, Pandey RR, Nagano T, Chakalova L, Mondal T, Fraser P, Kanduri C. Kcnq1ot1/Lit1 noncoding RNA mediates transcriptional silencing by targeting to the perinucleolar region. Mol Cell Biol. 2008;28:3713–28.
Shechner DM, Hacisuleyman E, Younger ST, Rinn JL. Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nat Methods. 2015;12:664–70.
Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806.
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, Zhu P, Chang Z, Wu Q, Zhao Y, Jia Y, Xu P, Liu H, Shan G. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256–64.
Chedin F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016;32:828–38.
Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, Chedin F. Prevalent. Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals, Mol Cell. 2016;63:167–78.
Cloutier SC, Wang S, Ma WK, Al Husini N, Dhoondia Z, Ansari A, Pascuzzi PE, Tran EJ. Regulated formation of lncRNA-DNA hybrids enables faster transcriptional induction and environmental adaptation. Mol Cell. 2016;62:148.
Ng SY, Bogu GK, Soh BS, Stanton LW. The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell. 2013;51:349–59.
Tang R, Zhang G, Wang YC, Mei X, Chen SY. The long non-coding RNA GAS5 regulates transforming growth factor beta (TGF-beta)-induced smooth muscle cell differentiation via RNA Smad-binding elements. J Biol Chem. 2017;292:14270–8.
Xue Z, Hennelly S, Doyle B, Gulati AA, Novikova IV, Sanbonmatsu KY, Boyer LA. A G-rich motif in the lncRNA braveheart interacts with a zinc-finger transcription factor to specify the cardiovascular lineage. Mol Cell. 2016;64:37–50.
Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell. 2010;143:1018–29.
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–78.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40:939–53.
Hongay CF, Grisafi PL, Galitski T, Fink GR. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell. 2006;127:735–45.
Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, Bonilla F, de Herreros AG. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008;22:756–69.
Munroe SH, Lazar MA. Inhibition of c-erbA mRNA splicing by a naturally occurring antisense RNA. J Biol Chem. 1991;266:22083–6.
Krystal GW, Armstrong BC, Battey JF. N-myc mRNA forms an RNA-RNA duplex with endogenous antisense transcripts. Mol Cell Biol. 1990;10:4180–91.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8.
Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, Lieberman J, Rigoutsos I, Pandolfi PP. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–57.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–69.
Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016;17:272–83.
Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 2014;54:766–76.
Bosson AD, Zamudio JR, Sharp PA. Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell. 2014;56:347–59.
Karreth FA, Reschke M, Ruocco A, Ng C, Chapuy B, Leopold V, Sjoberg M, Keane TM, Verma A, Ala U, Tay Y, Wu D, Seitzer N, Velasco-Herrera Mdel C, Bothmer A, Fung J, Langellotto F, Rodig SJ, Elemento O, Shipp MA, Adams DJ, Chiarle R, Pandolfi PP. The BRAF pseudogene functions as a competitive endogenous RNA and induces lymphoma in vivo. Cell. 2015;161:319–32.
Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. Impact of MicroRNA Levels. Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression, Mol Cell. 2016;64:565–79.
Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011;470:284–8.
Damas ND, Marcatti M, Come C, Christensen LL, Nielsen MM, Baumgartner R, Gylling HM, Maglieri G, Rundsten CF, Seemann SE, Rapin N, Thezenas S, Vang S, Orntoft T, Andersen CL, Pedersen JS, Lund AH. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat Commun. 2016;7:13875.
Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, Mesirov JP, Cech TR, Chang HY. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016;165:1267–79.
Aw JG, Shen Y, Wilm A, Sun M, Lim XN, Boon KL, Tapsin S, Chan YS, Tan CP, Sim AY, Zhang T, Susanto TT, Fu Z, Nagarajan N, Wan Y. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol Cell. 2016;62:603–17.
Fei J, Cui XB, Wang JN, Dong K, Chen SY. ADAR1-mediated RNA editing. A Novel Mechanism Controlling Phenotypic Modulation of Vascular Smooth Muscle Cells, Circ Res. 2016;119:463–9.
Stellos K, Gatsiou A, Stamatelopoulos K, Perisic Matic L, John D, Lunella FF, Jae N, Rossbach O, Amrhein C, Sigala F, Boon RA, Furtig B, Manavski Y, You X, Uchida S, Keller T, Boeckel JN, Franco-Cereceda A, Maegdefessel L, Chen W, Schwalbe H, Bindereif A, Eriksson P, Hedin U, Zeiher AM, Dimmeler S. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat Med. 2016;22:1140–50.
Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, Chaudhuri J. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell. 2015;161:762–73.
Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest AR, Carninci P, Biffo S, Stupka E, Gustincich S. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491:454–7.
Abdelmohsen K, Panda AC, Munk R, Grammatikakis I, Dudekula DB, De S, Kim J, Noh JH, Kim KM, Martindale JL, Gorospe M. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017:1–9.
Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–52.
Frith MC, Bailey TL, Kasukawa T, Mignone F, Kummerfeld SK, Madera M, Sunkara S, Furuno M, Bult CJ, Quackenbush J, Kai C, Kawai J, Carninci P, Hayashizaki Y, Pesole G, Mattick JS. Discrimination of non-protein-coding transcripts from protein-coding mRNA. RNA Biol. 2006;3:40–8.
You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, Wang X, Hou J, Liu H, Sun W, Sambandan S, Chen T, Schuman EM, Chen W. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603–10.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, Shenzis S, Samson M, Dittmar G, Landthaler M, Chekulaeva M, Rajewsky N, Kadener S. Translation of CircRNAs. Mol Cell. 2017;66(1):9–21.e7.
Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, Laneve P, Rajewsky N, Bozzoni I. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66(1):22–37.e9.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45.
Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, Higgs DR, Miska EA, Ponting CP. Considerations when investigating lncRNA function in vivo. elife. 2014;3:e03058.
Fabre PJ, Leleu M, Mormann BH, Lopez-Delisle L, Noordermeer D, Beccari L, Duboule D. Large scale genomic reorganization of topological domains at the HoxD locus. Genome Biol. 2017;18:149.
Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–48.
Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5:a008292.
Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, Erwin G, Kattman SJ, Keller GM, Srivastava D, Levine SS, Pollard KS, Holloway AK, Boyer LA, Bruneau BG. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–20.
Zhu JG, Shen YH, Liu HL, Liu M, Shen YQ, Kong XQ, Song GX, Qian LM. Long noncoding RNAs expression profile of the developing mouse heart. J Cell Biochem. 2014;115:910–8.
Matkovich SJ, Edwards JR, Grossenheider TC, de Guzman Strong C, Dorn GW 2nd. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci U S A. 2014;111:12264–9.
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, Liapis SC, Mallard W, Morse M, Swerdel MR, D'Ecclessis MF, Moore JC, Lai V, Gong G, Yancopoulos GD, Frendewey D, Kellis M, Hart RP, Valenzuela DM, Arlotta P, Rinn JL. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. elife. 2013;2:e01749.
Delas MJ, Hannon GJ. lncRNAs in development and disease: from functions to mechanisms. Open Biol. 2017;7
Chen R, Liu Y, Zhuang H, Yang B, Hei K, Xiao M, Hou C, Gao H, Zhang X, Jia C, Li L, Li Y, Zhang N. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017;45:9947–59.
Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Saito S, Nakamura Y, Tanaka T. Identification of a novel non-coding RNA. MIAT, that confers risk of myocardial infarction, J Hum Genet. 2006;51:1087–99.
Diabetes Genetics Initiative of Broad Institute of, H., Mit, L. U., Novartis Institutes of BioMedical, R., Saxena, R., Voight, B. F., Lyssenko, V., Burtt, N. P., de Bakker, P. I., Chen, H., Roix, J. J., Kathiresan, S., Hirschhorn, J. N., Daly, M. J., Hughes, T. E., Groop, L., Altshuler, D., Almgren, P., Florez, J. C., Meyer, J., Ardlie, K., Bengtsson Bostrom, K., Isomaa, B., Lettre, G., Lindblad, U., Lyon, H. N., Melander, O., Newton-Cheh, C., Nilsson, P., Orho-Melander, M., Rastam, L., Speliotes, E. K., Taskinen, M. R., Tuomi, T., Guiducci, C., Berglund, A., Carlson, J., Gianniny, L., Hackett, R., Hall, L., Holmkvist, J., Laurila, E., Sjogren, M., Sterner, M., Surti, A., Svensson, M., Svensson, M., Tewhey, R., Blumenstiel, B., Parkin, M., Defelice, M., Barry, R., Brodeur, W., Camarata, J., Chia, N., Fava, M., Gibbons, J., Handsaker, B., Healy, C., Nguyen, K., Gates, C., Sougnez, C., Gage, D., Nizzari, M., Gabriel, S. B., Chirn, G. W., Ma, Q., Parikh, H., Richardson, D., Ricke, D. & Purcell, S. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6.
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–5.
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, Wellcome Trust Case Control C, McCarthy MI, Hattersley AT. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–41.
Dandona S, Stewart AF, Chen L, Williams K, So D, O'Brien E, Glover C, Lemay M, Assogba O, Vo L, Wang YQ, Labinaz M, Wells GA, McPherson R, Roberts R. Gene dosage of the common variant 9p21 predicts severity of coronary artery disease. J Am Coll Cardiol. 2010;56:479–86.
Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H. Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature. 2008;451:202–6.
Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–62.
Kuo CL, Murphy AJ, Sayers S, Li R, Yvan-Charvet L, Davis JZ, Krishnamurthy J, Liu Y, Puig O, Sharpless NE, Tall AR, Welch CL. Cdkn2a is an atherosclerosis modifier locus that regulates monocyte/macrophage proliferation. Arterioscler Thromb Vasc Biol. 2011;31:2483–92.
Zhou X, Han X, Wittfeldt A, Sun J, Liu C, Wang X, Gan LM, Cao H, Liang Z. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway. RNA Biol. 2016;13:98–108.
Visel A, Zhu Y, May D, Afzal V, Gong E, Attanasio C, Blow MJ, Cohen JC, Rubin EM, Pennacchio LA. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–12.
Meijer-Spierings AH. Is there a relationship between social status and interpersonal behavior? Tijdschr Voor Ziekenverpl. 1966;19:208–12.
Chen L, Yang W, Guo Y, Chen W, Zheng P, Zeng J, Tong W. Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS One. 2017;12:e0185406.
Li MJ, Wang P, Liu X, Lim EL, Wang Z, Yeager M, Wong MP, Sham PC, Chanock SJ, Wang J. GWASdb: a database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2012;40:D1047–54.
Overton HA, Babbs AJ, Doel SM, Fyfe MC, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006;3:167–75.
Guevara NV, Kim HS, Antonova EI, Chan L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat Med. 1999;5:335–9.
Schober A, Nazari-Jahantigh M, Wei Y, Bidzhekov K, Gremse F, Grommes J, Megens RT, Heyll K, Noels H, Hristov M, Wang S, Kiessling F, Olson EN, Weber C. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med. 2014;20:368–76.
Feng J, Liang J, Li J, Li Y, Liang H, Zhao X, McNutt MA, Yin Y. PTEN controls the DNA replication process through MCM2 in response to replicative stress. Cell Rep. 2015;13:1295–303.
Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, Crippa S, Nemir M, Sarre A, Johnson R, Dauvillier J, Burdet F, Ibberson M, Guigo R, Xenarios I, Heymans S, Pedrazzini T. Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J. 2015;36:353–68a.
Suzuki HI, Young RA, Sharp PA. Super-enhancer-mediated RNA processing revealed by integrative MicroRNA network analysis. Cell. 2017;168:1000–1014 e15.
Foroud T, Koller DL, Lai D, Sauerbeck L, Anderson C, Ko N, Deka R, Mosley TH, Fornage M, Woo D, Moomaw CJ, Hornung R, Huston J, Meissner I, Bailey-Wilson JE, Langefeld C, Rouleau G, Connolly ES, Worrall BB, Kleindorfer D, Flaherty ML, Martini S, Mackey J, De Los Rios La Rosa F, Brown RD Jr, Broderick JP, FIA Study Investigators. Genome-wide association study of intracranial aneurysms confirms role of Anril and SOX17 in disease risk. Stroke. 2012;43:2846–52.
Hashikata H, Liu W, Inoue K, Mineharu Y, Yamada S, Nanayakkara S, Matsuura N, Hitomi T, Takagi Y, Hashimoto N, Miyamoto S, Koizumi A. Confirmation of an association of single-nucleotide polymorphism rs1333040 on 9p21 with familial and sporadic intracranial aneurysms in Japanese patients. Stroke. 2010;41:1138–44.
Lin L, Gu ZT, Chen WH, Cao KJ. Increased expression of the long non-coding RNA ANRIL promotes lung cancer cell metastasis and correlates with poor prognosis. Diagn Pathol. 2015;10:14.
Qiu JJ, Lin YY, Ding JX, Feng WW, Jin HY, Hua KQ. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int J Oncol. 2015;46:2497–505.
Sun Z, Ou C, Ren W, Xie X, Li X, Li G. Downregulation of long non-coding RNA ANRIL suppresses lymphangiogenesis and lymphatic metastasis in colorectal cancer. Oncotarget. 2016;7:47536–55.
Zhao JJ, Hao S, Wang LL, Hu CY, Zhang S, Guo LJ, Zhang G, Gao B, Jiang Y, Tian WG, Luo DL. Long non-coding RNA ANRIL promotes the invasion and metastasis of thyroid cancer cells through TGF-beta/Smad signaling pathway. Oncotarget. 2016;7:57903–18.
Haas J, Mester S, Lai A, Frese KS, Sedaghat-Hamedani F, Kayvanpour E, Rausch T, Nietsch R, Boeckel JN, Carstensen A, Volkers M, Dietrich C, Pils D, Amr A, Holzer DB, Martins Bordalo D, Oehler D, Weis T, Mereles D, Buss S, Riechert E, Wirsz E, Wuerstle M, Korbel JO, Keller A, Katus HA, Posch AE, Meder B. Genomic structural variations lead to dysregulation of important coding and non-coding RNA species in dilated cardiomyopathy. EMBO Mol: Med; 2017.
Yang W, Li Y, He F, Wu H. Microarray profiling of long non-coding RNA (lncRNA) associated with hypertrophic cardiomyopathy. BMC Cardiovasc Disord. 2015;15:62.
Song G, Shen Y, Zhu J, Liu H, Liu M, Shen YQ, Zhu S, Kong X, Yu Z, Qian L. Integrated analysis of dysregulated lncRNA expression in fetal cardiac tissues with ventricular septal defect. PLoS One. 2013;8:e77492.
Gu M, Zheng A, Tu W, Zhao J, Li L, Li M, Han S, Hu X, Zhu J, Pan Y, Xu J, Yu Z. Circulating LncRNAs as Novel. Non-Invasive Biomarkers for Prenatal Detection of Fetal Congenital Heart Defects, Cell Physiol Biochem. 2016;38:1459–71.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42.
Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–53.
Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506.
Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ, Gheorghiade M. Expert consensus document: mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238–50.
Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–20.
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ. A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med. 2016;37:445–51.
Zhang M, Gu H, Xu W, Zhou X. Down-regulation of lncRNA MALAT1 reduces cardiomyocyte apoptosis and improves left ventricular function in diabetic rats. Int J Cardiol. 2016;203:214–6.
Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH. Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009;41:399–406.
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378–87.
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP Jr, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haissaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–9.
Ikram MA, Seshadri S, Bis JC, Fornage M, DeStefano AL, Aulchenko YS, Debette S, Lumley T, Folsom AR, van den Herik EG, Bos MJ, Beiser A, Cushman M, Launer LJ, Shahar E, Struchalin M, Du Y, Glazer NL, Rosamond WD, Rivadeneira F, Kelly-Hayes M, Lopez OL, Coresh J, Hofman A, DeCarli C, Heckbert SR, Koudstaal PJ, Yang Q, Smith NL, Kase CS, Rice K, Haritunians T, Roks G, de Kort PL, Taylor KD, de Lau LM, Oostra BA, Uitterlinden AG, Rotter JI, Boerwinkle E, Psaty BM, Mosley TH, van Duijn CM, Breteler MM, Longstreth WT Jr, Wolf PA. Genomewide association studies of stroke. N Engl J Med. 2009;360:1718–28.
Lemmens R, Abboud S, Robberecht W, Vanhees L, Pandolfo M, Thijs V, Goris A. Variant on 9p21 strongly associates with coronary heart disease, but lacks association with common stroke. Eur J Hum Genet. 2009;17:1287–93.
Madelaine R, Sloan SA, Huber N, Notwell JH, Leung LC, Skariah G, Halluin C, Pasca SP, Bejerano G, Krasnow MA, Barres BA, Mourrain P. MicroRNA-9 couples brain neurogenesis and angiogenesis. Cell Rep. 2017;20:1533–42.
Dykstra-Aiello C, Jickling GC, Ander BP, Shroff N, Zhan X, Liu D, Hull H, Orantia M, Stamova BS, Sharp FR. Altered expression of long noncoding RNAs in blood after ischemic stroke and proximity to putative stroke risk loci. Stroke. 2016;47:2896–903.
Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. 2017;18:962–72.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29:452–63.
Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, Attenello FJ, Villalta JE, Cho MY, Chen Y, Mandegar MA, Olvera MP, Gilbert LA, Conklin BR, Chang HY, Weissman JS, Lim DA. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355
Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–101.
Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, Mero P, Dirks P, Sidhu S, Roth FP, Rissland OS, Durocher D, Angers S, Moffat J. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell. 2015;163:1515–26.
Chi KR. The dark side of the human genome. Nature. 2016;538:275–7.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222–9.
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, Zhang F. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–27.
Acknowledgments
This work was funded by the University Hospital Munich and by the German Research Foundation (DFG) as part of the Collaborative Research Center CRC1123 “Atherosclerosis—Mechanisms and Networks of Novel Therapeutic Targets” (project B1). Part of this project was also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, # 394237736).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Not applicable.
Disclosures
None.
Conflicts of Interest
There are no competing interests.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Holdt, L.M., Kohlmaier, A., Teupser, D. (2019). Long Noncoding RNAs in Cardiovascular Disease. In: Erdmann, J., Moretti, A. (eds) Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-030-27371-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-27371-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27370-5
Online ISBN: 978-3-030-27371-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)