
Abstract
Driver mutations in the BRAF oncogene occur in 2–4% of non-small cell lung cancers (NSCLC). Approximately half of these BRAF mutations are characterized by a glutamic acid substitution for valine at position 600 within the BRAF kinase domain (V600E or class I). The remaining non-V600E mutations are a heterogeneous group that can be further subdivided into mutations that activate BRAF kinase activity (class II) and mutations that remain dependent on upstream signaling through Ras-GTPase (class III). In normal cells, the BRAF kinase functions as an intermediary within the MAPK/ERK signaling pathway. Activating mutations in the BRAF oncogene lead to downstream signaling through the MAPK/ERK pathway, resulting in an increased risk of malignant transformation in preclinical models. Based on these findings, as well as the need for more effective treatment options for patients with BRAF-mutated NSCLC, there has been a significant interest in developing targeted therapies to inhibit the MAPK/ERK pathway. In a series of phase 2 clinical trials enrolling patients with metastatic BRAF V600E-mutated NSCLC, Planchard et al. established a role for combined BRAF and MEK tyrosine kinase inhibition (TKI) with dabrafenib and trametinib, respectively. While this represents an important new treatment strategy, a number of questions still remain including how best to sequence targeted therapy with other available treatment options and how to effectively overcome acquired resistance to TKI therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
Following the earliest description of BRAF mutations in human cancer nearly two decades ago, an improved understanding of the pathogenesis of these mutations has enabled the development of targeted therapies [1]. Although BRAF mutations occur infrequently in non-small cell lung cancer (NSCLC), the recent approval of targeted therapies for patients with metastatic disease represents an important milestone [2, 3]. With new therapeutic options to choose from, it is becoming increasingly important to understand the biology and clinical behavior of BRAF-mutated NSCLC. In this chapter, we aim to provide a comprehensive review of this topic, focusing on (1) the molecular mechanisms involved in tumorigenesis, (2) clinical features and outcomes associated with BRAF mutations, and (3) the role of targeted therapies. We conclude by describing what questions still remain in the field and how answers to these questions might improve the future management of patients with BRAF-mutated NSCLC.
Molecular Foundations
MAPK Signaling Pathways
BRAF-mutated cancers rely on signaling through the mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathway, which regulates the differentiation, growth, and survival of normal cells [4, 5]. It is one of four related signaling pathways, each of which is driven by a specific group of serine-threonine kinases from the MAPK family, including ERK, JNK (c-Jun N-terminal kinase), p38, and ERK5/BMK1 (extracellular signal-related kinase 5/Big MAP kinase 1). Although MAPK signaling pathways have gained attention for their role in cancer, they have also been linked in preclinical models to a number of normal biological processes ranging from cytokine production to embryonic development [6,7,8]. The function and specific molecular players vary from one MAPK pathway to another but the basic structure of the signaling cascade is highly conserved [9]. At the center of each pathway is a MAPK enzyme that phosphorylates and activates a series of downstream targets through its serine/threonine kinase activity. Two additional kinase families lie upstream of MAPK: the dual-specificity MAPK serine/threonine and tyrosine kinase (MAPKK) and the MAPKK serine/threonine kinase (MAPKKK). Each of these kinases is activated sequentially, beginning with MAPKKK which activates MAPKK and ending with MAPK which is activated by MAPKK [10].
The structure of the MAPK/ERK signaling pathway has been particularly well described (Fig. 1). In its simplest form, the pathway begins upstream with extracellular growth signals that bind to and activate transmembrane receptor tyrosine kinases [11]. Once activated, these receptor tyrosine kinases recruit multiple intracellular proteins to the membrane surface including Ras-GTPases (KRAS, NRAS, HRAS). When bound to GTP, Ras-GTPases are capable of activating proteins of the rapidly accelerated fibrosarcoma (RAF) family, which belong to the MAPKKK group of kinases. RAF in turn phosphorylates downstream MAPK/ERK kinase (MEK), which is a member of the MAPKK group of kinases. The activated form of MEK then phosphorylates ERK, which has numerous terminal targets including transcription factors (e.g., c-Myc), transcription repressors (e.g., ERF), and components of the cytoskeleton (e.g., myosin light chain kinase) [12,13,14]. Like other signaling pathways, the MAPK/ERK pathway is also tightly regulated at multiple levels by a series of negative feedback loops and inhibitory proteins [11, 15, 16].

A schematic representation of the MAPK/ERK signaling pathway is shown. Activation of upstream receptor tyrosine kinases (e.g., EGFR, PDGFR) by extracellular growth signals results in the recruitment of multiple intracellular proteins including Ras-GTPase, son of sevenless (SOS)Fig. 1 (continued) homolog protein, Src homology 2 domain containing protein (Shc), and others not shown. When bound to GTP, Ras-GTPases recruit rapidly accelerated fibrosarcoma (RAF) proteins such as BRAF to the membrane surface. RAF proteins phosphorylate and activate MEK, which, in turn, activates ERK. ERK then activates numerous downstream targets, including transcription factors and repressors that regulate cellular growth and differentiation
Molecular Biology of Wild-Type RAF Kinase
The RAF family is comprised of three serine-threonine kinases: ARAF, BRAF, and CRAF, also known as Raf-1 [17, 18]. BRAF is encoded by the V-raf murine sarcoma oncogene homolog B1 (BRAF) gene located on chromosome 7q34 [19]. Expression of the wild-type BRAF gene is fairly ubiquitous across multiple organs and tissues, with highest expression found in the testis, thyroid, bone marrow, and brain [20]. The BRAF protein is 766 amino acids long, and, like other RAF kinases, it is arranged into three highly conserved regions (CR1, CR2, and CR3), each of which serves a distinct function (Fig. 2a) [21]. CR1 resides closest to the N-terminus and is comprised of both a cysteine-rich domain and a Ras-binding domain. The cysteine-rich domain coordinates translocation of RAF kinases from the cytoplasm to the membrane surface where they interact with Ras-GTPase via the Ras-binding domain [22, 23]. CR2 links the CR1 and CR3 domains and also contains a binding site for the 14-3-3 inhibitory protein. CR3, which resides closest to the C-terminus, contains a kinase domain that phosphorylates and activates downstream targets.

(a) The structure of all RAF kinases is highly conserved and consists of three conserved regions (CR1, CR2, and CR3). CR1 contains both a cysteine-rich domain (CRD) and a Ras-binding domain (RBD). CR3 contains the active kinase domain. (b) In the inactive state, RAF kinases exist in an auto-inhibitory conformation whereby CR1 blocks key phosphorylation sites within CR3 that are responsible for activating the kinase. (c) When bound to Ras-GTPases at the intracellular cell surface via the Ras-binding domain, CR1 dissociates from CR3, enabling activation of the kinase and phosphorylation of downstream targets
In addition to containing important binding sites, the CR1 domain also plays a role in regulating RAF kinase activity. Prior to activation by Ras-GTPases, cytoplasmic RAF kinases exist in an auto-inhibited state in which CR1 binds to CR3 and prevents phosphorylation of the kinase domain (Fig. 2b) [24,25,26]. When the MAPK/ERK pathway is activated, binding of RAF to Ras-GTPases at the cellular membrane interrupts the inhibitory interaction between CR1 and CR3. This, in turn, enables phosphorylation of key residues (e.g., T598, S601) that are necessary for kinase activation (Fig. 2c) [27]. Once activated in this manner, BRAF forms homodimers with itself and heterodimers with other RAF kinases, both of which are then capable of phosphorylating and activating a limited set of downstream targets, primarily MEK [28]. Eventually, phosphorylation and dephosphorylation of other key residues within BRAF by ERK-mediated negative feedback facilitates the dissociation of BRAF from Ras-GTPase, loss of RAF dimerization, and downregulation of kinase activity [29].
Oncogenic Potential of BRAF Mutations
Although multiple effectors along the MAPK/ERK signaling pathway have been implicated in tumorigenesis, the oncogenic potential of BRAF mutations was first described in 2002 [30]. In their seminal report, Davis et al. initially sequenced three BRAF mutations from a limited number of melanoma and NSCLC cell lines. Two of the mutations occurred in exon 15, which encodes the kinase domain of the BRAF protein. This included both a T1796A substitution, which led to glutamic acid in the place of valine at position 599 (V599E), and a C1786G substitution, which led to valine in the place of leucine at position 596 (L596 V). A third G1403C substitution was found in exon 11, leading to the replacement of glycine with alanine at position 468 within the glycine-rich pocket of the ATP-binding domain (G468A). To confirm these findings, the authors performed a larger follow-up screen, which identified BRAF mutations in 59% of melanoma cell lines, 18% of colorectal cancer cell lines, and a smaller minority of cell lines derived from glioma, sarcoma, ovarian cancer, and NSCLC, among others. As the majority of BRAF mutations in melanoma involved a T1796A substitution, which is now known to result in the replacement of valine with glutamic acid at position 600 within the BRAF kinase domain, these mutations came to be known as V600E. On the other hand, mutations affecting other sites within the BRAF kinase were subsequently termed non-V600E. Davis et al. showed that mutations from both groups were capable of activating the MAPK/ERK pathway. Specifically, transfecting either BRAF V599E (now known as V600E) or G468A mutations into cell culture resulted in increased activity of the BRAF kinase as well as downstream phosphorylation of ERK.
Following this initial report from Davis et al., it became clear from subsequent studies that BRAF V600E mutations transform the BRAF protein into a constitutively active kinase that functions independently of upstream activation. Brummer et al. demonstrated this by engineering BRAF genes to co-express both V600E (T1796A) and R188L mutations, the latter of which renders BRAF ineffective at binding to upstream Ras-GTPases [31]. While R188L mutations alone prevented BRAF-mediated MAPK/ERK pathway activation, the presence of a concomitant BRAF V600E mutation restored phosphorylation of ERK, suggesting that BRAF V600E mutant kinases act independently of upstream Ras-GTPases. This conclusion was further supported by the finding that EGFR inhibition was not effective against lung cancer cell lines harboring BRAF mutations, despite EGFR being identified as an upstream activator of the MAPK/ERK pathway [32]. Although BRAF V600E mutations have gained more attention due to their frequent occurrence in melanoma, several BRAF non-V600E mutations (e.g., G468A, L596 V) have also been associated with increased kinase activity compared to wild-type BRAF [33]. However, this may not be true of all non-V600E mutations, as we discuss in greater detail below.
After establishing that many BRAF mutations activate the MAPK/ERK pathway, in vivo studies were then conducted to determine the potential association between BRAF mutations, MAPK/ERK pathway activation, and tumorigenesis. In one of these studies, Ji et al. engineered mice to express BRAF V600E mutations in the lung epithelium [34]. These mice developed lung adenomas at 6 weeks followed by adenocarcinomas at 16 weeks. Subsequent silencing of BRAF gene expression led to dephosphorylation of ERK, decreased expression of cyclin D, and decreased tumor burden. This suggested that BRAF activity and downstream MAPK/ERK signaling were the link between BRAF V600E mutations and the ongoing survival of lung tumors.
In other mouse models, however, BRAF mutations have been found to be necessary but not sufficient for the induction of malignant tumorigenesis. Dankort et al., for example, found that while expression of BRAF V600E in the lung parenchyma of mice led to increased phosphorylation of MEK and ERK as well as the development of lung adenomas, these adenomas entered a state of growth arrest, and very few progressed to adenocarcinoma unless a tumor suppressor gene such as TP53 was also mutated [35]. A two-hit process was also suggested by a second mouse model in which co-expression of both BRAF V600E and PIK3CA H1047R mutations was associated with increased tumor burden, higher risk of developing malignant tumors, and shorter overall survival (OS) compared to mice with BRAF V600E mutations alone [36]. Malignant tumorigenesis in these mice was further enhanced when TP53 mutations were also introduced. Interestingly, in vivo models of melanoma have similarly suggested that BRAF mutations may predispose to the growth of benign nevi but not malignant melanomas [37]. Taken together, these findings suggest that while BRAF mutations predispose to benign, premalignant tumors, other concomitant mutations may be necessary for the full induction of malignancy.
Regardless of the complex molecular interactions by which BRAF mutations initiate tumorigenesis, ongoing survival of BRAF-mutated tumors appears to rely strongly on signaling through the MAPK/ERK pathway. In multiple preclinical studies, inhibition of mutant BRAF V600E itself or its downstream targets such as MEK has been shown to lead to tumor regression in patient-derived xenografts and other mouse models of melanoma and lung cancer [35, 38, 39]. These results suggest that continued activation of the MAPK/ERK pathway is necessary for sustained tumor growth and that use of targeted therapies to block this pathway in BRAF-mutated tumors represents a promising treatment strategy.
Classification of BRAF Mutations
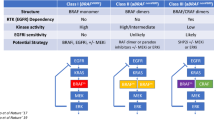
As described above, BRAF mutations have traditionally been classified as either V600E or non-V600E. However, our understanding of the varied mechanisms by which different BRAF mutations activate the MAPK/ERK pathway has evolved. A newer classification system has now been proposed that divides BRAF mutations into three classes (class I, II, or III) based on their effect on BRAF kinase activity and their interaction with upstream Ras-GTPases (Table 1).
In the first of two studies, Yao et al. highlighted the differences between what would become known as class I and class II BRAF mutations. They initially demonstrated that expression of either BRAF V600E mutations or certain BRAF non-V600E mutations (e.g., G469A, L597 V, K601E) in cell culture was associated with increased phosphorylation of MEK, decreased phosphorylation of CRAF, and reduced expression of Ras-GTPase [40]. The authors concluded that these BRAF mutations function in a Ras-independent manner to facilitate both downstream MAPK/ERK activation and upstream feedback inhibition of CRAF and Ras-GTPase. Despite these common endpoints, the authors found that the mechanisms by which different BRAF mutations induced downstream signaling were not the same. Whereas BRAF V600E/K/D/R (V600) mutations produced constitutively active BRAF kinases that functioned as Ras-independent monomers, BRAF mutations affecting sites other than position 600 (non-V600) produced active BRAF kinases that formed Ras-independent dimers. From a treatment perspective, this designation was important, as only BRAF V600 monomers but not BRAF non-V600 mutant dimers were sensitive to vemurafenib in vitro. Based on these findings, the authors defined two groups of BRAF mutations according to their mechanisms of Ras-independent signaling: V600 mutations that signal as constitutively active BRAF monomers (class I) and non-V600 mutations that signal as constitutively active BRAF dimers (class II).
In a second study, Yao et al. broadened their classification system to include a third class of BRAF mutations (class III) [41]. The authors identified a subset of non-V600 mutations, including D594G/N and G466 V/E, that encode for “kinase dead” or “kinase impaired” versions of the BRAF protein. Although these mutations were still capable of activating the MAPK/ERK pathway in vitro through their strong interaction with Ras-GTPase, they were less active than class I or II BRAF mutations, and they remained dependent on upstream Ras-GTPase. The authors proposed that class III BRAF mutations are oncogenic when paired synergistically with upstream activation of Ras-GTPase. Consistent with this hypothesis, the authors found that class III BRAF mutations in melanoma cell lines were often accompanied by activating mutations in RAS or NF1 while class III BRAF mutations in NSCLC and colorectal cancer cell lines had high levels of phosphorylated receptor tyrosine kinases such as EGFR, ERBB2, and MET. As class III BRAF mutations activate the MAPK/ERK pathway in a Ras-dependent manner, the authors also found that patient-derived xenografts of class III BRAF-mutated colorectal cancer were not responsive to BRAF inhibition with vemurafenib. However, they were sensitive to cetuximab (EGFR monoclonal antibody) and, to a lesser degree, trametinib (MEK inhibitor), suggesting dependency on the MAPK/ERK pathway.
The value of the novel class I–III classification system is that it shifts focus away from the structural amino acid changes that characterize different mutations and toward the functional significance of the mutations instead. In the era of targeted therapy, acknowledging the heterogeneous mechanisms by which different BRAF mutations activate the MAPK/ERK pathway is arguably more important and has the potential to shed light on targets beyond BRAF and MEK that may be therapeutically relevant. The distinction between different types of BRAF non-V600 mutations is especially important, given that these mutations had been previously grouped together into a heterogeneous category. Clinical trials for patients with BRAF non-V600 mutations are clearly needed, and drug development as well as clinical trial designs should consider class II and class III BRAF non-V600 mutations separately, given the different functional mechanisms by which these mutations regulate the MAPK/ERK pathway.
BRAF Mutations in Non-small Cell Lung Cancer
BRAF mutations occur most commonly in melanoma, where the incidence of V600E mutations is >50% in metastatic disease [42, 43]. Other cancers in which BRAF is recurrently mutated include papillary thyroid carcinoma, hairy cell leukemia, and microsatellite unstable colorectal cancer [44,45,46]. The biological significance of BRAF mutations in these different tumor types likely varies, which is clear when considering the utility of targeted therapy in different cancers. At one extreme, BRAF inhibition alone with vemurafenib was highly efficacious in small cohorts of relapsed, refractory hairy cell leukemia [47]. At the other extreme, BRAF targeted monotherapy was ineffective when studied in colorectal cancer and has no current role in the disease [48]. These different susceptibilities to BRAF targeted therapy suggest that while the downstream MAPK/ERK pathway is targetable in BRAF-mutated tumors in general, the development of resistance and reliance on other bypass growth pathways likely varies from one tumor type to the other.
In lung cancer, BRAF mutations are relatively uncommon but represent an early, clonal event in cancer development, suggesting that they are an important oncogenic driver in this setting [49]. While they have also been described as secondary resistance mutations arising in previously treated EGFR-mutated NSCLC, it is their role as primary driver mutations that has garnered the most attention [50].
Pathologic and Clinical Characteristics
The overall incidence of BRAF mutations in NSCLC is approximately 2–4% [51,52,53]. Although the distribution of BRAF V600E versus BRAF non-V600E mutations varies between studies, BRAF non-V600E mutations appear to represent nearly half of all BRAF mutations occurring in NSCLC [52, 54,55,56].
Consistent with their role as drivers in NSCLC, BRAF mutations are usually found independently of mutations in EGFR or rearrangements in ALK [57]. Identifying other co-occurring mutations in BRAF-mutated NSCLC has been limited by (1) the relative infrequency with which BRAF mutations occur in lung cancer and (2) sequencing panels that test only for mutations in a select number of driver genes. However, the advent of next-generation sequencing has allowed for broader mutational testing. In one study of 1007 patients with metastatic or recurrent lung adenocarcinoma whose tumors were genotyped using multiplexed assays of up to 10 genes, 2 of the 14 patients whose tumors harbored BRAF V600E mutations also had co-occurring PIK3CA E542K mutations [53]. In another cohort of 174 patients with BRAF-mutated NSCLC, next-generation sequencing of 17 genes identified co-mutations in TP53 in 89 cases (51%) [58]. Findings such as this are potentially consistent with the preclinical studies described previously that suggested that co-occurring mutations contribute to the malignant differentiation of BRAF-mutated tumors.
It remains unclear as to how co-occurring mutations affect survival or response to treatment in patients with BRAF-mutated NSCLC. In our own single-center study of 18 patients with BRAF-mutated NSCLC, we found that those patients with metastatic disease whose tumors harbored co-occurring TP53 mutations (n = 4) had a numerically worse OS compared to patients without TP53 mutations (n = 12) (37.7 vs. 13.7 months; P = 0.23) [59]. Conclusions are limited by the very small sample size, although the suggestion of worse outcomes in patients harboring co-mutations in TP53 is consistent with studies of other driver-mutated lung cancers. For example, shorter OS has been reported in patients with NSCLC whose tumors harbor TP53 mutations alongside driver mutations in EGFR, ALK, or ROS1 [60]. On the other hand, TP53 mutations in KRAS-mutated NSCLC have been associated with increased somatic mutation burden and an enhanced response to immunotherapy [61, 62]. As molecular tumor testing expands and more patients with BRAF-mutated NSCLC are identified, further prognostic and predictive studies of patients with co-occurring TP53 mutations are warranted.
Given the fact that other driver mutations are known to be enriched in particular patients with NSCLC, there has been a strong interest in identifying clinicopathologic features that associate with BRAF mutations. Across multiple studies, BRAF mutations have been primarily found in NSCLC with adenocarcinoma histology [52, 55, 56, 59, 63, 64]. As pointed out by Cardarella et al., genomic testing in clinical practice may be more routinely performed for adenocarcinomas compared to other histologic subtypes, which could reflect a source of bias [52]. However, two studies performed genomic sequencing in resected lung cancers from 1046 (37 BRAF-positive) and 2001 (26 BRAF-positive) patients with primarily early stage disease who otherwise might not have undergone routine molecular testing. In both of these studies, BRAF mutations occurred more commonly in adenocarcinoma than in squamous cell cancer, suggesting that histology is closely associated with BRAF mutation status [55, 64].
With respect to demographic characteristics, including age, sex, and smoking history, Cardarella et al. found no differences in 36 patients with BRAF-mutated NSCLC compared to those with wild-type NSCLC as determined by Sanger sequencing [52]. In contrast, it has been difficult to draw conclusions about associations with ethnicity from individual retrospective studies, as many them have been conducted in ethnically homogenous cohorts. In multiple reports consisting primarily of Asian patients, BRAF mutations have been reported in 0.7–1.7% of cases, which is similar to the 0.4% rate reported in an African American cohort but lower than the 4% rate described in a predominantly Caucasian cohort [52, 65,66,67]. It may be tempting to conclude from these numbers that BRAF mutations occur more commonly in certain ethnic groups but comparing results across studies requires a certain degree of caution. In addition, prior studies have suggested that tumor mutation testing may not be performed equally in patients of different ethnic backgrounds, which reflects a potential source of bias [68].
Within the group of patients with BRAF-mutated NSCLC, there may be more significant demographic differences between those with BRAF V600E versus BRAF non-V600E mutations. Sex differences have been reported in two studies that each found BRAF V600E mutations to be more common in females [55, 69]. A more consistent finding has been that patients with BRAF V600E mutations are more likely to have been never or light smokers compared to patients with BRAF non-V600E mutations [52, 54, 55, 59, 69]. Given the potential relationship between smoking, tumor mutation burden, and response to immunotherapy, this difference between patients with BRAF V600E versus BRAF non-V600E mutations may have important therapeutic implications [70]. Of note, fewer studies have evaluated demographic characteristics in patients with BRAF-mutated NSCLC using the newer class I–III classification system. However, one retrospective study that did evaluate BRAF mutations according to this classification system similarly found that patients with class I BRAF V600-mutated NSCLC were more commonly never-smokers than patients with class II or III BRAF non-V600-mutated NSCLC [71].
Outcomes and Prognosis
BRAF-mutated NSCLC was originally thought to have a more aggressive phenotype given its association with papillary and micropapillary histologic features [72]. However, retrospective studies suggest that the outcomes of patients with BRAF-mutated NSCLC compare favorably to the outcomes of patients whose tumors harbor other driver mutations or are wild-type for known drivers. In one of the earlier studies of BRAF-mutated NSCLC, for example, 739 resected adenocarcinomas from patients with primarily early stage disease were sequenced to identify BRAF mutations in exons 11 and 15 [55]. The median OS of 36 patients with BRAF-mutated adenocarcinoma (21 V600E and 15 non-V600E) was not statistically different than that of patients with BRAF wild-type adenocarcinoma (data not reported). In a separate cohort of patients with metastatic or recurrent adenocarcinoma from the Lung Cancer Mutation Consortium, Villaruz et al. reported a median OS of 56 months for 15 patients with BRAF-mutated NSCLC, which was not statistically different than the median OS reported for patients with EGFR mutations (43 months), KRAS mutations (33 months), ALK rearrangements (52 months), or no oncogenic driver (25 months) (P > 0.20) [73]. Notably, only three patients with BRAF-mutated NSCLC in this study had received MEK-directed targeted therapy (selumetinib), and none received BRAF-directed targeted therapy. In a third study of French patients, Tissot et al. similarly found that the median OS of 80 patients with BRAF-mutated NSCLC (42 V600E, 57.5% stage IV) was not statistically different than that of patients whose tumors were wild-type in EGFR, KRAS, HER2, PI3K, and ALK (22.1 vs. 14.5 months; P = 0.095) [56]. Only a small number of patients in this study (n = 9; 11%) received BRAF or combined BRAF-MEK targeted therapy.
Although all BRAF mutations were analyzed together in the above studies, subgroup analyses from these and other studies suggest that not all BRAF mutations behave the same. Marchetti et al. found that 21 patients with BRAF V600E-mutated early stage disease undergoing resection had a shorter median disease-free survival (DFS) (15.2 vs. 52.1 months; P < 0.001) and OS (29.3 vs. 72.4 months; P < 0.001) compared to those with BRAF V600E wild-type disease [55]. In contrast, 15 patients with BRAF non-V600E mutations had similar DFS (42.8 vs. 43.2 months; P = 0.84) and OS (56.4 vs. 65.1 months; P = 0.42) compared to those with BRAF non-V600E wild-type disease. In a cohort of patients with stage IV NSCLC, Cardarella et al. similarly reported a non-significantly lower response rate to platinum-based chemotherapy (29% vs. 71%; P = 0.286) and shorter progression-free survival (PFS) (4.1 vs. 8.9 months; P = 0.297) in patients with metastatic NSCLC harboring BRAF V600E (n = 7) versus BRAF non-V600E (n = 7) mutations [52]. A few additional patients with metastatic disease were available for the survival analysis, which revealed non-significantly shortened OS (10.8 vs. 15.2 months; P = 0.726) in patients with BRAF V600E-mutated NSCLC (n = 12) versus BRAF non-V600E-mutated NSCLC (n = 12). In contrast to these studies, Litvak et al. found that the 3-year OS rate for patients with stage IIIB/IV BRAF V600E-mutated NSCLC (n = 20) was 24% compared to 0% for patients with non-V600E mutations (n = 9) (P < 0.001) [54]. The 3-year OS rate of patients with BRAF V600E-mutated NSCLC was not statistically different than that of patients with unresectable EGFR-mutated NSCLC (38%). However, 94% of patients with EGFR-mutated NSCLC in this study received EGFR-directed targeted therapy compared to only 50% of patients with BRAF-mutated NSCLC receiving BRAF-directed targeted therapy.
In conjunction with these conflicting findings are multiple case reports that have described long-term survival lasting several years in select patients with metastatic BRAF V600E-mutated NSCLC [74, 75]. However, the small patient numbers in these reports as well as in the retrospective studies described above have limited conclusions regarding the differential effect of BRAF V600E versus BRAF non-V600E mutations on outcomes. A recent study from Dagogo-Jack et al. begins to overcome this limitation by describing one of the largest cohorts of BRAF-mutated NSCLC to date [71]. Using the newer class I–III classification system, the authors identified 236 patients at their center whose tumors had a BRAF mutation detected by either next-generation sequencing or multiplex polymerase chain reaction. This included 107 patients with class I BRAF V600 mutations and 129 patients with class II or III BRAF non-V600 mutations. Among the 139 patients with metastatic disease at diagnosis, 62 of them received first-line carboplatin plus pemetrexed. The median PFS was longer in patients with class I BRAF mutations compared to patients with class II or III BRAF mutations (6.2 vs. 3.3 months, P = 0.069 for class I vs. class II and 6.2 vs. 4.9 months, P = 0.034 for class I vs. class III). Overall survival for the entire group of 139 patients with metastatic disease was also longer in those with class I BRAF mutations compared to class II or III BRAF mutations (40.1 vs. 13.9 months, P < 0.001 for class I vs. class II and 40.1 vs. 15.6 months, P = 0.023 for class I vs. class III).
Interestingly, the authors found that patients with class I BRAF mutations were less likely to have brain metastases at diagnosis than patients with class II or III BRAF mutations. They were also more likely to have intrathoracic metastases alone than patients with class II BRAF mutations. When survival analyses were limited to patients with extra-thoracic metastases who did not receive targeted therapy, median OS between the mutation classes was not significantly different (9.4 months for class I vs. 7.9 months for class II vs. 9.7 months for class III), suggesting that extent of disease and response to targeted therapy are prognostically important. Although additional studies are needed for confirmation, these results suggest that favorable outcomes may be achieved in a subset of patients with BRAF-mutated NSCLC and that patients with class I BRAF mutations in particular may have a better prognosis due to the different clinical behavior of these mutations compared to class II or III BRAF non-V600E mutations.
A Changing Therapeutic Landscape
Efficacy of Targeted Therapy
Despite long-term survival in some cases of BRAF-mutated NSCLC, additional advances are required to improve outcomes. Furthermore, prior to the approval of targeted therapies for BRAF-mutated NSCLC, more than half of patients with these tumors received only best supportive care in the second-line setting, suggesting a need for additional therapeutic options for this patient population [76].
Targeted therapies inhibiting the MAPK/ERK signaling pathway have been most extensively studied in melanoma, due to the frequency of BRAF mutations in this disease. Initial front-line trials in melanoma evaluated reversible BRAF kinase inhibitors (dabrafenib or vemurafenib) as monotherapy in the treatment of patients with metastatic or unresectable BRAF V600E-mutated disease. In this setting, dabrafenib was associated with an overall response rate (ORR) of 50% (95% CI 42.4–57.1) compared to 6% (95% CI 1.8–15.5) with dacarbazine [77]. Single-agent vemurafenib was similarly effective with an ORR of 57% (95% CI NR) versus 9% (95% CI NR) for dacarbazine [78]. Vemurafenib was also associated with an OS benefit of 13.6 months versus 9.7 months (HR 0.70, 95% CI 0.57–0.87, P = 0.0008). As resistance to BRAF targeted therapies developed quickly in these patients, subsequent studies focused on the combination of reversible MEK kinase inhibitors (trametinib or cometinib) with BRAF kinase inhibitors in untreated metastatic melanoma. Combined therapy in this setting was associated with significantly improved response rates and longer PFS and OS compared to BRAF kinase inhibitors alone, thus establishing dual inhibition as the standard of care [79,80,81,82].
Support for using BRAF targeted therapy in lung cancer initially came from case reports that described responses in individual patients [83,84,85]. This was supported by a retrospective study of 35 patients with BRAF-mutated NSCLC (29 V600E, 6 non-V600E) who were treated with a BRAF kinase inhibitor (dabrafenib, vemurafenib, or sorafenib) outside of the clinical trial setting. The response rate among all patients, the majority of whom received targeted therapy in the later-line setting, was 53% (95% CI 35–70) [86].
The earliest prospective studies evaluating BRAF targeted therapy in lung cancer were basket trials that included not only NSCLC but also multiple other cancers harboring BRAF V600E mutations as well. The first was a phase 1 trial of dabrafenib that enrolled one patient with BRAF V600E-mutated NSCLC who achieved a partial response with an 83% reduction in tumor size [87]. In a phase 2 trial enrolling 20 patients with BRAF V600E-mutated NSCLC, most of whom had received prior systemic therapy, the response rate to vemurafenib was 42% (95% CI 20–67), consisting entirely of partial responses [88].
Based on these encouraging results, Planchard et al. conducted three phase 2 multicenter, open-label trials evaluating the use of targeted therapy in patients with metastatic BRAF V600E-mutated NSCLC (Table 2) [89,90,91]. In the first trial, previously treated patients received BRAF targeted therapy alone with dabrafenib 150 mg twice daily. Treatment was active in a subset of patients, with an overall response rate of 33% (95% CI 23–45) and a median PFS of 5.5 months (95% CI 3.4–7.3). In the second trial enrolling another cohort of previously treated patients, combination BRAF and MEK targeted therapy was administered with dabrafenib 150 mg twice daily and trametinib 2 mg once daily, respectively. Outcomes in response to combination therapy were better than those reported in the dabrafenib monotherapy cohort, although the two groups were enrolled separately and not intended for direct comparison. Nonetheless, the response rate to combination therapy was 63.2% (95% CI 49.3–75.6) with a median PFS of 9.7 months (95% CI 6.9–19.6). This led to a third study evaluating combination dabrafenib plus trametinib in patients with previously untreated metastatic BRAF V600E-mutated NSCLC. Combination therapy in the frontline setting was active, with an overall response rate of 64% (95% CI 46–79) that included two complete responses and a median PFS of 10.9 months (95% CI 7.0–16.6). Only 5 patients (14%) in this cohort had a best response of progressive disease.
The most common grade 3 or higher adverse events in patients receiving dabrafenib monotherapy were asthenia, squamous cell carcinoma of the skin, and basal cell carcinoma. The side effect profile of dabrafenib plus trametinib was slightly different with fever and cytopenias occurring more commonly but basal and squamous cell carcinoma of the skin occurring less commonly. In patients with previously treated NSCLC, dabrafenib plus trametinib was also associated with higher rates of treatment discontinuation (12% vs. 6%), treatment interruption (61% vs. 43%), and dose reduction (35% vs. 18%) compared to patients receiving dabrafenib alone. However, side effects in both groups were ultimately felt to be manageable overall. Based on the acceptable safety profile and potential benefit associated with targeted therapy, dabrafenib 150 mg twice daily plus trametinib 2 mg once daily is now approved as of 2017 for patients with untreated or previously treated, advanced BRAF V600E-mutated NSCLC, making it the only combination targeted therapy regimen that is approved in lung cancer [92].
As described above, NSCLC is unique in that approximately half of BRAF mutations are non-V600E. However, clinical trials of BRAF targeted therapies have excluded patients with BRAF non-V600E mutations. It has been reasonable to assume that BRAF non-V600E mutations may be less responsive to BRAF-directed targeted therapy given that the mechanisms by which they activate MAPK/ERK signaling are different than those of V600E mutations [40, 41]. However, further studies are needed for confirmation. The hope is that by further exploring the signaling mechanisms that underlie different BRAF mutations, new targets for the treatment of BRAF non-V600E-mutated NSCLC will emerge that can be tested in the clinical trial setting.
Mechanisms of Resistance to Targeted Therapy
Although BRAF-directed targeted therapies are effective, the development of resistance and eventual disease progression are inevitable. Much more is known about secondary resistance in BRAF-mutated melanoma given that targeted therapy for metastatic BRAF V600E-mutated NSCLC was only recently approved. As a result, our current understanding of secondary resistance in NSCLC is limited primarily to case reports.
Studies of melanoma have identified mechanisms of both innate and acquired resistance to BRAF-directed targeted therapy. With respect to innate resistance, the tumor microenvironment is thought to play an important role. In a study by Straussman et al., extracellular hepatocyte growth factor (HGF) secreted by stromal cells was shown to activate the MAPK and PIK3CA pathways in melanoma cells via binding to the MET receptor [93]. In a series of experiments, the authors showed that while PLX4720, a BRAF kinase inhibitor, effectively inhibited proliferation of melanoma cells in vitro, the addition of increasing concentrations of HGF resulted in resistance and sustained melanoma cell growth. Notably, when HGF expression was then measured by immunohistochemistry in 34 melanoma biopsy samples derived from patients enrolled in clinical trials, HGF positivity pretreatment was associated with a lower response to BRAF inhibition with or without dual inhibition of MEK.
On the other hand, acquired resistance in melanoma has been attributed to multiple secondary mutations that induce either upregulation of the MAPK/ERK signaling pathway or activation of bypass pathways. In the case of other driver-mutated cancers, acquired gatekeeper mutations occurring in the same driver genes can confer resistance and pathway reactivation. In the classic example of EGFR-mutated NSCLC, for example, a gatekeeper T790 M mutation arises in a subset of patients treated with erlotinib or gefitinib. The T790 M mutation renders the EGFR kinase resistant to binding by first- and second-generation EGFR tyrosine kinase inhibitors, thus restoring EGFR-mediated growth signaling and cancer progression [94]. In the case of BRAF-mutated melanoma, gatekeeper mutations producing an amino acid substitution for threonine at residue 529 within the BRAF kinase have been successfully introduced into cell lines and confer resistance in vitro to PLX4720 [95]. However, the spontaneous occurrence of gatekeeper mutations in BRAF-mutated melanoma cell lines or patient tumor samples has not been described, with multiple studies demonstrating instead that secondary BRAF mutations are uncommon and that V600E mutations are preserved [96,97,98]. Retention of the original BRAF mutation suggests that treatment after the development of resistance might benefit not only from additional therapy to target the mechanism of resistance but also ongoing inhibition of the MAPK/ERK pathway since the mutant BRAF kinase could be a source of continued signaling.
The MAPK/ERK pathway is commonly reactivated in BRAF-mutated melanoma by secondary mutations in upstream Ras-GTPases. Across multiple studies of melanoma resistant to BRAF inhibition alone, mutations have been identified in both NRAS (G12D/R, G13R, Q61K/R/L) and KRAS (G12C/R, Q61H, K117 N) [96,97,98]. Reactivation of the MAPK/ERK pathway may also occur secondary to BRAF amplification, increased expression of the CRAF kinase, and activating C121S mutations in MEK1 [97, 99, 100]. Resistance mutations have also been identified in the PI3K pathway, including AKT (E17K and Q79K), PIK3CA, PIK3CG, and PTEN, suggesting that PI3K may act as a bypass pathway [97].
A certain number of these resistance mutations occurring secondary to BRAF inhibitor monotherapy might be expected to be less effective at conferring resistance to combination BRAF plus MEK inhibition. RAS mutations, for example, have been shown to mediate resistance to BRAF inhibition by activating CRAF, which initiates signaling through the MAPK/ERK pathway at the level of MEK [96, 98]. In one study using a resistant KRAS K117 N-mutated melanoma xenograft, tumor growth was effectively inhibited by the combination of vemurafenib plus a MEK inhibitor, confirming that the effectiveness of MEK inhibition was maintained in the setting of an upstream RAS activating mutation. Nonetheless, NRAS G12D and Q61K mutations have been identified in melanoma samples derived from patients who had progressed on combination dabrafenib plus trametinib, which raises the possibility that upstream Ras activation might confer resistance by mechanisms other than CRAF-mediated MEK activation alone [101]. Interestingly, BRAF amplification has also been observed in melanoma samples resistant to combination targeted therapy, similar to what has been described for melanomas resistant to BRAF inhibition alone. However, the increase in BRAF copy number has been noted to be significantly higher in those cells that are resistant to dabrafenib plus trametinib compared to cells resistant to BRAF inhibition alone. Thus, while there is clearly some degree of overlap with respect to the targets of resistance in melanomas treated with BRAF inhibition alone compared to those treated with combination BRAF plus MEK inhibition, the mechanisms by which these targets confer resistance likely vary.
Furthermore, downstream mutations in the MAPK/ERK pathway appear to be more common in melanomas treated with combination dabrafenib plus trametinib. In addition to identifying NRAS mutations and BRAF amplification, Long et al. discovered MEK1/2 mutations in 27% of patient melanoma samples with acquired resistance to dabrafenib plus trametinib, which is higher than the 3–5% incidence reported in studies of melanomas treated with BRAF inhibitor therapy alone [101]. Mutations in MEK2 were more common than mutations in MEK1, and only the MEK2 C125S mutation was associated with increased colony growth in the presence of dabrafenib plus trametinib. Together, these findings suggest that particular isoforms of MEK may be more capable of mediating resistance to targeted therapy combinations than others [100].
Case reports of patients with BRAF-mutated NSCLC progressing on targeted therapy suggest that upstream activation of Ras-GTPase may be a mediator of resistance in lung cancer as well. In two reports of single patients with BRAF V600E-mutated NSCLC who progressed on third-line dabrafenib plus trametinib and later-line dabrafenib monotherapy, respectively, re-biopsy at the time of progression identified a new NRAS Q61K mutation in the first patient and a new KRAS G12D mutation in the second patient [102, 103]. In both cases, the original BRAF V600E mutation was maintained at the time of progression, which is consistent with studies from melanoma. In a third report, the tumor of a patient with BRAF V600E NSCLC receiving vemurafenib did not have a RAS mutation at the time of progression but did have increased chromosomal instability, suggesting that multiple pathways of resistance are possible [104].
Future Directions
Overcoming resistance to targeted therapy represents just one of the ongoing challenges in the treatment of BRAF V600E-mutated NSCLC. Another unanswered question is how best to sequence therapies now that multiple treatment options exist for metastatic BRAF V600E-mutated NSCLC, including targeted therapy, chemotherapy, and immunotherapy. Even in melanoma, for which BRAF targeted therapy has been approved for longer, the optimal sequence is not clear, with recommendations drawn primarily from retrospective studies. Previous reviews have suggested that OS may be better in patients with metastatic melanoma who are treated with ipilimumab prior to BRAF targeted therapy [105]. However, BRAF and MEK inhibitors may increase the expression of PD-1 on melanoma cells, which raises the question as to whether outcomes may be improved by using targeted therapy followed by PD-L1 axis-directed therapy [106]. In the case of NSCLC, it is also currently unclear as to how well BRAF-mutated NSCLC responds to immunotherapy. In a recent study, high PD-L1 expression, defined as ≥50%, was seen in nearly half of patients with either BRAF V600E or non-V600E mutations [107]. Tumor mutation burden, on the other hand, was low to intermediate in both groups, and the overall response rate to immunotherapy in primarily the later-line setting was 25% in patients with BRAF V600E mutations and 33% in patients with BRAF non-V600E mutations. As pointed out by the authors, these response rates are comparable to those of second-line immunotherapy in unselected patients with NSCLC. While this suggests that immunotherapy may be more effective in BRAF-mutated NSCLC than it is in NSCLC harboring other driver mutations (e.g., EGFR, ALK), larger studies are needed.
Conclusion
Although BRAF mutations occur infrequently in NSCLC, their ability to constitutively activate the MAPK/ERK signaling pathway has proven to be an important mechanism of cancer cell survival. Understanding this molecular link between BRAF mutations and malignancy has led to the approval of targeted therapy (dabrafenib plus trametinib) in both untreated and relapsed, refractory BRAF V600E-mutated metastatic NSCLC. While BRAF mutations are distinguished from one another in part based on their susceptibility to targeted therapy, it is becoming increasingly clear that other inherent biological differences likely contribute to variability in clinical behavior and prognosis. In addition, much remains unknown regarding the effect of co-occurring mutations on prognosis or response to therapy. More routine use of next-generation sequencing and collaboration between centers specializing in thoracic malignancies should be encouraged, with the goal of addressing ongoing gaps in our knowledge and improving the management of patients with BRAF-mutated NSCLC.
References
Flaherty KT, McArthur G. BRAF, a target in melanoma: implications for solid tumor drug development. Cancer. 2010;116(21):4902–13.
Baik CS, Myall NJ, Wakelee HA. Targeting BRAF-mutant non-small cell lung cancer: from molecular profiling to rationally designed therapy. Oncologist. 2017;22(7):786–96.
Odogwu L, et al. FDA approval summary: dabrafenib and trametinib for the treatment of metastatic non-small cell lung cancers harboring BRAF V600E mutations. Oncologist. 2018;23(6):740–5.
Santarpia L, Lippman SL, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–19.
Yang SH, Sharrocks AD, Whitmarsh AJ. MAP kinase signalling cascades and transcriptional regulation. Gene. 2013;513(1):1–13.
Lee JC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372(6508):739–46.
Lu HT, et al. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999;18(7):1845–57.
Yang D, et al. Targeted disruption of the MKK4 gene causes embryonic death, inhibition of c-Jun NH2-terminal kinase activation, and defects in AP-1 transcriptional activity. Proc Natl Acad Sci U S A. 1997;94(7):3004–9.
Burotto M, et al. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–56.
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83.
McCubrey JA, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012;3(9):954–87.
Chang F, et al. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. 2003;17(7):1263–93.
Klemke RL, et al. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137(2):481–92.
Le Gallic L, et al. Transcriptional repressor ERF is a Ras/mitogen-activated protein kinase target that regulates cellular proliferation. Mol Cell Biol. 1999;19(6):4121–33.
Mischak H, et al. Negative regulation of Raf-1 by phosphorylation of serine 621. Mol Cell Biol. 1996;16(10):5409–18.
Sasaki A, et al. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat Cell Biol. 2003;5(5):427–32.
Dankner M, et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 2018;37(24):3183–99.
Roskoski R Jr. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399(3):313–7.
Eychene A, et al. Chromosomal assignment of two human B-raf(Rmil) proto-oncogene loci: B-raf-1 encoding the p94Braf/Rmil and B-raf-2, a processed pseudogene. Oncogene. 1992;7(8):1657–60.
Fagerberg L, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406.
Sithanandam G, et al. Complete coding sequence of a human B-raf cDNA and detection of B-raf protein kinase with isozyme specific antibodies. Oncogene. 1990;5(12):1775–80.
Ghosh S, et al. The cysteine-rich region of raf-1 kinase contains zinc, translocates to liposomes, and is adjacent to a segment that binds GTP-ras. J Biol Chem. 1994;269(13):10000–7.
Mott HR, et al. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc Natl Acad Sci U S A. 1996;93(16):8312–7.
Chong H, Guan KL. Regulation of Raf through phosphorylation and N terminus-C terminus interaction. J Biol Chem. 2003;278(38):36269–76.
Cutler REJ, et al. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A. 1998;95(16):9214–9.
Tran NH, Wu X, Frost JA. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J Biol Chem. 2005;280(16):16244–53.
Zhang BH, Guan KL. Activation of B-Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J. 2000;19(20):5429–39.
Desideri E, Cavallo AL, Baccarini M. Alike but different: RAF paralogs and their signaling outputs. Cell. 2015;161(5):967–70.
Ritt DA, et al. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30(3):806–19.
Davies H, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–54.
Brummer T, et al. Functional analysis of the regulatory requirements of B-Raf and the B-Raf(V600E) oncoprotein. Oncogene. 2006;25(47):6262–76.
Pratilas CA, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res. 2008;68(22):9375–83.
Wan PT, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116(6):855–67.
Ji H, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67(10):4933–9.
Dankort D, et al. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21(4):379–84.
Trejo CL, et al. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res. 2013;73(21):6448–61.
Chudnovsky Y, et al. Use of human tissue to assess the oncogenic activity of melanoma-associated mutations. Nat Genet. 2005;37(7):745–9.
Hoeflich KP, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69(7):3042–51.
Micel LN, et al. Antitumor activity of the MEK inhibitor TAK-733 against melanoma cell lines and patient-derived tumor explants. Mol Cancer Ther. 2015;14(2):317–25.
Yao Z, et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacologic inhibition. Cancer Cell. 2015;28(3):370–83.
Yao Z, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548(7666):234–8.
Kumar R, et al. BRAF mutations in metastatic melanoma: a possible association with clinical outcome. Clin Cancer Res. 2003;9(9):3362–8.
Shinozaki M, et al. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004;10(5):1753–7.
Tiacci E, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364(24):2305–15.
Nikiforova MN, et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J Clin Endocrinol Metab. 2003;88(11):5399–404.
Saridaki Z, et al. BRAF mutations, microsatellite instability status and cyclin D1 expression predict metastatic colorectal patients’ outcome. Br J Cancer. 2010;102(12):1762–8.
Tiacci E, et al. Targeting mutant BRAF in relapsed or refractory hairy-cell leukemia. N Engl J Med. 2015;373(18):1733–47.
Kopetz S, et al. Phase II pilot study of vemurafenib in patients with metastatic BRAF-mutated colorectal cancer. J Clin Oncol. 2015;33(34):4032–8.
Jamal-Hanjani M, et al. Tracking the evolution of non-small-cell lung cancer. N Engl J Med. 2017;376(22):2109–21.
Ohashi K, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109(31):E2127–33.
Brose MS, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62(23):6997–7000.
Cardarella S, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res. 2013;19(16):4532–40.
Kris MG, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311(19):1998–2006.
Litvak AM, et al. Clinical characteristics and course of 63 patients with BRAF mutant lung cancers. J Thorac Oncol. 2014;9(11):1669–74.
Marchetti A, et al. Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol. 2011;29(26):3574–9.
Tissot C, et al. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer. 2016;91:23–8.
Dearden S, et al. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013;24(9):2371–6.
Kron A, et al. Impact of co-occurring genomic alterations on overall survival of BRAF V600E and non-V600E mutated NSCLC patients: results of the network genomic medicine. Ann Oncol. 2017;28(Suppl_5):mdx380.003.
Myall NJ, et al. Natural disease history, outcomes, and co-mutations in a series of patients with BRAF-mutated non-small cell lung cancer. Clin Lung Cancer. 2018;20(2):e208–17.
Aisner DL, et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations-the Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res. 2018;24(5):1038–47.
Dong ZY, et al. Potential predictive vaue of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res. 2017;23(12):3012–24.
Skoulidis F, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5(8):860–77.
Brustugun OT, et al. BRAF-mutations in non-small cell lung cancer. Lung Cancer. 2014;84(1):36–8.
Kinno T, et al. Clinicopathological features of nonsmall cell lung carcinomas with BRAF mutations. Ann Oncol. 2014;25(1):138–42.
Araujo LH, et al. Somatic mutation spectrum of non-small-cell lung cancer in African Americans: a pooled analysis. J Thorac Oncol. 2015;10(10):1430–6.
Ding X, et al. Clinicopathologic characteristics and outcomes of Chinese patients with non-small-cell lung cancer and BRAF mutation. Cancer Med. 2017;6(3):555–62.
Serizawa M, et al. Assessment of mutational profile of Japanese lung adenocarcinoma patients by multitarget assays: a prospective, single-institute study. Cancer. 2014;120(10):1471–81.
Lynch JA, et al. Underutilization and disparities in access to EGFR testing among Medicare patients with lung cancer from 2010 - 2013. BMC Cancer. 2018;18(1):306.
Chen D, et al. BRAF mutations in patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS One. 2014;9(6):e101354.
Davis AA, et al. Association of tumor mutational burden with smoking and mutation status in non-small cell lung cancer (NSCLC). J Clin Oncol. 2017;35(7_suppl):24–24.
Dagogo-Jack I, et al. Impact of BRAF mutation class on disease characteristics and clinical outcomes in BRAF-mutant lung cancer. Clin Cancer Res. 2019;25(1):158–65.
Yousem SA, Nikiforova M, Nikiforov Y. The histopathology of BRAF-V600E-mutated lung adenocarcinoma. Am J Surg Pathol. 2008;32(9):1317–21.
Villaruz LC, et al. Clinicopathologic features and outcomes of patients with lung adenocarcinomas harboring BRAF mutations in the Lung Cancer Mutation Consortium. Cancer. 2015;121(3):448–56.
Myall NJ, et al. Long-term survival of a patient with non-small-cell lung cancer harboring a V600E mutation in the BRAF oncogene. Clin Lung Cancer. 2016;17(2):e17–21.
Nakanishi Y, et al. Favorable outcome with pemetrexed treatment for advanced BRAF-V600E-positive lung adenocarcinoma in a patient followed up over 8 years. J Thorac Oncol. 2018;13(10):e199–202.
Barlesi F, et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet. 2016;387(10026):1415–26.
Hauschild A, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65.
McArthur GA, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15(3):323–32.
Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–703.
Larkin J, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371(20):1867–76.
Long GV, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88.
Robert C, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372(1):30–9.
Gautschi O, et al. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol. 2012;7(10):e23–4.
Peters S, Micheielin O, Zimmermann S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J Clin Oncol. 2013;31(20):e341–4.
Robinson SD, et al. BRAF V600E-mutated lung adenocarcinoma with metastases to the brain responding to treatment with vemurafenib. Lung Cancer. 2014;85(2):326–30.
Gautschi O, et al. Targeted therapy for patients with BRAF-mutant lung cancer: results from the European EURAF cohort. J Thorac Oncol. 2015;10(10):1451–7.
Falchook GS, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–901.
Hyman DM, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015;373(8):726–36.
Planchard D, et al. Dabrafenib plus trametinib in patients with previously treated BRAF V600E -mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial. Lancet Oncol. 2016;17(7):984–93.
Planchard D, et al. Dabrafenib in patients with BRAFV600E-positive advanced non-small-cell lung cancer: a single-arm, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(5):642–50.
Planchard D, et al. Dabrafenib plus trametinib in patients with previously untreated BRAF V600E -mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol. 2017;18(10):1307–16.
National Comprehensive Cancer Network. Non-small cell lung cancer (Version 6.2018). https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed 22 Oct 2018.
Straussman R, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4.
Pao W, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73.
Whittaker S, et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci Transl Med. 2010;2(35):35–41.
Nazarian R, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7.
Shi H, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93.
Su F, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72(4):969–78.
Montagut C, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68(12):4853–61.
Wagle N, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29(22):3085–96.
Long GV, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5:5694.
Abravanel DL, et al. An acquired NRAS Q61K mutation in BRAF V600E-mutant lung adenocarcinoma resistant to dabrafenib plus trametinib. J Thorac Oncol. 2018;13(8):e131–3.
Rudin CM, Hong K, Streit M. Molecular characterization of acquired resistance to the BRAF inhibitor dabrafenib in a patient with BRAF-mutant non-small-cell lung cancer. J Thorac Oncol. 2013;8(5):e41–2.
Lucchesi C, et al. Molecular determinants of acquired resistance to BRAF inhibition in human lung cancer. Lung Cancer. 2018;126:227.
Atkins MB, Larkin J. Immunotherapy combined or sequenced with targeted therapy in the treatment of solid tumors: current perspectives. J Natl Cancer Inst. 2016;108(6):djv414.
Sanlorenzo M, et al. BRAF and MEK inhibitors increase PD-1-positive melanoma cells leading to a potential lymphocyte-independent synergism with anti-PD-1 antibody. Clin Cancer Res. 2018;24(14):3377–85.
Dudnik E, et al. BRAF mutant lung cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol. 2018;13(8):1128–37.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Myall, N.J., Padda, S.K. (2019). BRAF: Novel Therapies for an Emerging Target. In: Salgia, R. (eds) Targeted Therapies for Lung Cancer. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-030-17832-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-17832-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-17831-4
Online ISBN: 978-3-030-17832-1
eBook Packages: MedicineMedicine (R0)