
Abstract
Pleuroparenchymal fibroelastosis (PPFE) is a rare and possibly underrecognized idiopathic interstitial pneumonia, as defined by the updated 2013 ATS/ERS Classification. Here we present an emblematic case of pleuroparenchymal fibroelastosis and a detailed description of its clinical, radiological, and histopathological presentation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Pleuroparenchymal fibroelastosis (PPFE) is a rare and possibly underrecognized interstitial pneumonia, as defined by the updated 2013 ATS/ERS Classification [1].
There are two groups of patients, those with a known cause and those without known association, the latter being termed idiopathic PPFE [2].
The first group includes cases of PPFE associated with lung transplantation (late posttransplant complication in 2–7.5% of lung transplant recipients), hematopoietic stem cell transplantation (late complication in 0 and 2% of patients), previous chemotherapy (alkylating agents) and/or radiotherapy, occupational dust exposure (asbestos, aluminum), recurrent pulmonary infections (Aspergillus sp., Mycobacterium avium-intracellulare), autoimmune diseases (ankylosing spondylitis, ulcerative colitis, psoriasis, lupus, rheumatoid arthritis), and familial cases with a possible underlying genetic predisposition (e.g., a short telomere syndrome characterized by telomere-related gene mutations of TERT, TERC, RTEL1, and PARN): the latter are prevalent among female patients [2,3,4,5,6].
When all these causes may be excluded or no specific clinical settings are identified, the disorder is labeled as idiopathic [4].
Curiously PPFE is morphologically similar to a spontaneous syndrome of aged donkeys with a high prevalence (35%) that analogously involves the upper lung zones. PPFE as an effect of aging in humans has also been speculated upon [7].
PPFE is also classified into pure PPFE and PPFE combined with other interstitial pneumonias, such as usual interstitial pneumonia (UIP) and non-specific interstitial pneumonia (NSIP), often involving the lower lung zones [8].
There is a wide age range at diagnosis (13–87 years, mean 53 years) and a bimodal distribution of presentation with an early peak in the third and a later peak in the sixth decade; a striking predominance of female cases in the earlier peak is observed.
A frequent association with a low body mass index (BMI) and with a “platythorax” (reduction in the anterior-posterior diameter of the chest wall) has also been demonstrated [2,3,4].
Recently a possible association between idiopathic PPFE and hypothyroidism (“lung-thyroid syndrome”) and cutaneous manifestations of PPFE that clinically simulate telangiectasia macularis eruptiva perstans, but lacking mast cell infiltrate, have been reported [9, 10].
The most common presenting symptoms are dyspnea, cough, hemoptysis, weight loss, low-grade fever, recurrent infection, pleuritic chest pain, and spontaneous pneumothorax [2,3,4].
Pulmonary function tests typically show a restrictive pattern or a mixed restrictive-obstructive pattern with an increased residual volume/total lung capacity (RV/TLC) ratio, which is a peculiar functional impairment that differs from that seen in IPF [11].
Serum laboratory data show elevation of KL-6 with the disease progression, and also surfactant protein D may be elevated. About a half of patients with PPFE demonstrates increased titers of a variety of serum autoantibodies such as rheumatoid factor, double-stranded DNA, and antinuclear antibody, suggesting a possible role of autoimmune mechanism in the pathogenesis of the disease [3, 4].
Chest radiograph at the early stage of PPFE shows bilateral, apical, irregular thickening of the pleura. Later the elevation of bilateral hila is detected. The lateral view often demonstrates an abnormally narrowed anterior-posterior thoracic dimension [2, 3].
Radiologic criteria for the diagnosis of PPFE are proposed by Reddy as follows: a definite diagnosis of PPFE at HRCT requires upper lobe pleural thickening and subpleural fibrosis (traction bronchiectasis, architectural distortion, upper lobe volume loss, superior hilar retraction), with involvement of the lower lobes being less marked or absent. The presence of a clear demarcation between the affected and the normal lung is a characteristic feature. Pneumothorax, platythorax, parenchymal consolidations, subpleural cysts, and ground glass areas might be present, mainly in the upper zones. A consistent diagnosis of PPFE is considered when there is pleural thickening and subpleural fibrosis that are not concentrated in the upper lobe or there is presence of coexistent disease elsewhere [3].
At the advanced stage, fibrotic shadows extend to lower lung fields, and the diaphragm is elevated with the loss of bilateral lung volume. Multiple bullae and large cysts often appear in the upper lung fields [3].
The radiological differential diagnosis includes familial pulmonary fibrosis, connective tissue disease (particularly ankylosing spondylitis), fibrotic sarcoidosis, and chronic hypersensitivity pneumonitis (HP) [3, 11].
Once PPFE becomes symptomatic, patients may remain stable for a long period of time or progress rapidly (60% of cases) to hypoxemic and hypercapnic respiratory failure [1,2,3,4].
Urinary desmosines (degradation product of mature elastin) have been proposed as a novel, noninvasive diagnostic biomarker, since urinary levels are significantly higher in patients with PPFE than those in patients with IPF or healthy controls [12].
At present, there are no established therapeutic options for PPFE, except for transplantation. Patients have been treated empirically with corticosteroids, N-acetylcysteine, prophylactic antibiotics, and immunosuppressant (cyclophosphamide, azathioprine) although none have demonstrated clear evidence of efficacy except for supportive care (oxygen therapy). A potential efficacy of pirfenidone in preventing lung function decline has been suggested in single cases of PPFE combined with UIP/IPF, leading to the consideration that a subset of PPFE may potentially benefit from anti-fibrotic drugs, especially in the setting of PPFE combined with UIP/IPF [13].
1.1 Histology
-
Upper lobes and peripheral predominant distribution.
-
Collagenous fibrosis of the visceral pleura with haphazardly arranged elastic fibers.
-
Subpleural intra-alveolar elastotic fibrosis.
-
Alveolar septal elastosis.
-
Increase in subpleural network of elastin fibers best appreciated at histochemistry with elastic tissue stains.
-
Abrupt transition between the areas of fibroelastosis and the surrounding unaffected parenchyma with occasional fibroblastic foci at the leading edge of fibrosis (i.e., interface of PPFE and adjacent lung).
-
Mild and patchy chronic inflammation within areas of fibrosis.
-
Vascular fibrointimal thickening with partial stenosis in arteries and/or vascular microthrombi.
-
Absence of classic honeycombing.
-
Possible coexistence of other histologic features and/or patterns: granulomas, UIP/IPF, HP, NSIP.
-
Diffuse alveolar damage, alveolar hemorrhage, and obliterative bronchiolitis are possible concomitant findings in lung or hematopoietic stem cell transplant recipients.
1.2 Differential Diagnosis
The main histologic differential diagnosis is between PPFE and UIP, either in its idiopathic form (IPF) or secondary to other known causes such as chronic HP, connective tissue disease-related interstitial lung disease (CTD-related ILD), drug reaction, asbestosis, or chronic fibrosing sarcoidosis. In the pure forms, PPFE and UIP are readily distinguishable. As the name implies, PPFE is a morphologically descriptive term denoting intense elastotic fibrosis of the visceral pleura and adjacent lung parenchyma. Elastosis is a distinctive form of chronic scarring in the lung that differs from the common scarring associated with extensive remodeling of the lung parenchyma and honeycomb changes which are appreciable in UIP pattern. Usually, PPFE lungs contain twice as much elastin fibers compared to UIP/IPF appreciable both on hematoxylin and eosin and using elastic stains in the upper lobes. The distinction between PPFE and UIP/IPF relies above all in the different predominant involvement of the lung parenchyma as follows: PPFE is an upper lobe-dominant elastotic fibrosis, whereas IPF is a lower lobe-dominant collagenous fibrosis. Both PPFE and UIP/IPF are characterized by a similar abrupt transition between fibrotic areas and spared juxtaposed lung, but PPFE lacks honeycombing and shows a smaller number of fibroblastic foci. In addition PPFE is prevalent among non-smokers (62–85%), while the majority of patients with UIP/IPF are smokers [2,3,4].
Since UIP/IPF and PPFE can coexist in the same patient, the histologic interpretation becomes more difficult. These cases represent a disease entity distinct from UIP/IPF. Compared with patients with IPF, patients with PPFE and UIP/IPF had (1) higher complication rate of pneumothorax or pneumomediastinum, (2) lower BMI, (3) flattened chest, and (4) distinct pulmonary function with restrictive pattern, alveolar hypoventilation, and increased RV/TLC ratio and PaCO2. The prognosis is worse, and the survival time is shorter in patients with PPFE and UIP/IPF than in patients with IPF [8]. The histologic problem centers on whether a PPFE-like change or UIP-like regions are significant or not, and the issue is best addressed with clinical and radiologic correlation.
The secondary UIP pattern seen in chronic HP should be differentiated from PPFE for its bronchiolocentric/centrilobular involvement with frequent sparing of the pleural surfaces and presence of scattered interstitial granulomas and/or giant cells. In addition a diagnosis of PPFE should only be made in absence of a relevant history of exposure to inhaling antigens.
The fibrotic stage of Langerhans cell histiocytosis may present with upper lobe fibrosis, and the distinction with PPFE depends on the identification of CD1a-positive Langerhans cells within fibrotic areas, on the stellate bronchiolocentric scars, and/or on the coexistent smoking-related damage typical of Langerhans cell histiocytosis.
The differential diagnosis out of PPFE also includes the residua of infections (previous history of tuberculosis and tuberculosis pneumothorax treatment, aspergillosis), as well as asbestos exposure, advanced chronic fibrotic sarcoidosis, or connective tissue disease.
The histological pattern of asbestos-related fibrosis tends to display more prominent parietal pleural thickening and more advanced remodeling and architectural distortion than PPFE.
Nonetheless, a confident diagnosis of PPFE should only be made in absence of a relevant occupational exposure history to asbestos as well as the lack of asbestos bodies, sarcoid-like granulomas, microorganisms at special stains, dense lymphoplasmacytic infiltrates, and/or numerous follicles with germinal centers suggestive of a CTD-related ILD.
The incidental lesion that morphologically and radiologically closely mimics PPFE is apical cap.
Apical cap is a localized lesion of lung apices in the form of subpleural pyramidal fibroelastotic scar, often resected for a radiologically suspected carcinoma. Characteristically, apical caps occur in older asymptomatic males, mostly smokers and are stable in time, while PPFE also affects younger, non-smoker patients, who present with symptoms and poor clinical outcome. Radiologically, apical caps do not involve the pleura circumferentially like PPFE. Histologically, apical caps consist of dense collagenous fibrosis and are often associated with pleural plaques, extensive alveolar collapse, and smoking-related damage of the adjacent parenchyma, which are not typically present in PPFE [2,3,4].
Finally, an upper lobe pulmonary fibrosis, radiologically consistent with PPFE but limited to unilateral lung, has been observed as an iatrogenic reaction in patients with a history of thoracotomy for resecting lung or esophageal cancers. In these cases the lesions are limited to the operated side [14].
1.3 Role of Cryobiopsy in the Diagnosis of PPFE
The pathologic diagnostic criteria of PPFE on cryobiopsy are the same as those on surgical lung biopsies: “Definite PPFE” is assigned when there are upper zone pleural fibrosis with subjacent intra-alveolar fibrosis accompanied by alveolar septal elastosis and sparing of the parenchyma distant from the pleura, at most mild patchy lymphoplasmacytic infiltrates and at most small numbers of fibroblastic foci; “Consistent with PPFE” is considered when intra-alveolar fibroelastosis is present, but it is not (1) associated with significant pleural fibrosis, (2) not predominantly beneath the pleura, or (3) not in an upper lobe biopsy; and “Inconsistent with PPFE” is the definition for cases lacking the requisite features above described [3].
Due to the transbronchial method of sampling, the pleura is included in cryobiopsies in about a third of cases; as a consequence the multidisciplinary discussion team including pulmonologists, radiologists, and pathologists with careful revision of imaging findings and knowledge of the clinical background of the patient (drug reaction, infection, inhalation exposures, lung/bone marrow transplantation, etc.) is always necessary to reach the proper diagnosis of PPFE [2,3,4]. However, the morphologic evidence of a prominent interstitial and/or intra-alveolar fibroelastosis in a cryobiopsy of symptomatic patient with a predominant upper lobe disease at imaging could be highly suggestive of PPFE.
2 Case Presentation
-
Clinical Background
-
55-year-old female
-
Non-smoker
-
Family history: no family history of ILD
-
Past medical history: Hashimoto thyroiditis on therapy with L-thyroxine; negative for prior chemotherapy or transplant
-
-
Onset of Symptoms
-
Exertional dyspnea for a year and half
-
Cough, fever, weight loss, and fatigue for 6 months
-
-
Laboratory Findings
-
Laboratory findings: unremarkable
-
Autoimmune status: negative
-
Quantiferon: negative
-
-
Pulmonary Function Test
-
Slight reduction of DLCO
-
-
-
High-resolution computed tomography (HRCT) showed pleural and subpleural irregular fibrosis with traction bronchiectasis and tiny subpleural cysts in the apices and middle zones of both lungs.
-
-
Bronchoscopy with Cryobiopsy (Figs. 15.3, 15.4, 15.5, and 15.6)
-
Cryobiopsies of the right upper lobe were obtained: two fragments of tissue 7.5 and 6.9 mm in maximum diameter, respectively (Fig. 15.3).
-
The biopsies showed a subpleural and bronchiolocentric fibroelastotic process characterized by interstitial fibroelastosis with focal/mild inflammatory infiltrate (Fig. 15.4). Abrupt transition to the normal parenchyma was present, and scattered fibroblastic foci were also appreciated (Fig. 15.5).
-
Histochemistry with Elastic van Gieson staining evidenced the prominent network of elastic fibers (Fig. 15.6).
-
-
Diagnosis
-
Idiopathic pleuroparenchymal fibroelastosis.
-

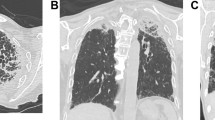
HRCT of upper lung lobes evidenced pleural and subpleural thickening, fibrotic changes in the marginal parenchyma, traction bronchiectasis, architectural distortion, and fine subpleural cyst

A sharp demarcation between the affected and the normal lung was appreciated as well as an apical-caudal gradient

Low-power view of the two cryobiopsies of 7.5 and 6.9 mm of maximum diameter. The sharp demarcation between the affected and the normal lung parenchyma was appreciable also at low-power

Prominent septal fibroelastosis. Haphazardly arranged elastic fibers were detected at hematoxylin and eosin stains as short, curled, intensely eosinophilic fragments within a loose edematous fibroelastotic interstitium. A mild and patchy lymphoplasmacytic infiltrate coexisted

The parenchyma adjacent to the fibroelastotic area was unremarkable, and a small fibroblastic focus was visible

Histochemistry with elastic stain showed short, curled, and randomly oriented elastic fibers within alveolar septa
3 Discussion
It has been recently showed that transbronchial cryobiopsy may be a valid method to obtain a histopathologic diagnosis in patients with suspected idiopathic PPFE and even airway-centered FE and that it can be obtained with an acceptable complication rate. In fact, no bleeding (mild, moderate, or severe) was observed, and pneumothorax was documented in three out of eight cases, none of them requiring drainage or lasting more than 3 days. No death, acute lung injury, persistent fever, prolonged air leak, pneumonia/empyema, or other adverse events occurred after the procedure in any of the cases [15]. The critical point, mainly in cases of idiopathic PPFE, is to stay very peripheral with the probe (around 1 cm from the pleura) in order to retrieve tissue containing pleural-subpleural structures. The use of smaller cryoprobes (1.9 instead of 2.4) is recommended to reach the upper portion of the lungs mainly in smaller subjects or in patients with bronchial malacia.
References
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, Ryerson CJ, Ryu JH, Selman M, Wells AU, Behr J, Bouros D, Brown KK, Colby TV, Collard HR, Cordeiro CR, Cottin V, Crestani B, Drent M, Dudden RF, Egan J, Flaherty K, Hogaboam C, Inoue Y, Johkoh T, Kim DS, Kitaichi M, Loyd J, Martinez FJ, Myers J, Protzko S, Raghu G, Richeldi L, Sverzellati N, Swigris J, Valeyre D. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Camus P, von der Thüsen J, Hansell DM, Colby TV. Pleuroparenchymal fibroelastosis: one more walk on the wild side of drugs? Eur Respir J. 2014;44(2):289–96.
Reddy TL, Tominaga M, Hansell DM, von der Thusen J, Rassl D, Parfrey H, Guy S, Twentyman O, Rice A, Maher TM, Renzoni EA, Wells AU, Nicholson AG. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012;40(2):377–85.
von der Thüsen JH. Pleuroparenchymal fibroelastosis: its pathological characteristics. Curr Respir Med Rev. 2013;9(4):238–47.
Mariani F, Gatti B, Rocca A, Bonifazi F, Cavazza A, Fanti S, Tomassetti S, Piciucchi S, Poletti V, Zompatori M. Pleuroparenchymal fibroelastosis: the prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn Interv Radiol. 2016;22(5):400–6.
Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, Meyer K, Raghu G, Collard HR, Garcia CK. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710–20.
Miele A, Dhaliwal K, Du Toit N, Murchison JT, Dhaliwal C, Brooks H, Smith SH, Hirani N, Schwarz T, Haslett C, Wallace WA, McGorum BC. Chronic pleuropulmonary fibrosis and elastosis of aged donkeys: similarities to human pleuroparenchymal fibroelastosis. Chest. 2014;145(6):1325–32.
Oda T, Ogura T, Kitamura H, Hagiwara E, Baba T, Enomoto Y, Iwasawa T, Okudela K, Takemura T, Sakai F, Hasegawa Y. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest. 2014;146(5):1248–55.
Awano N, Izumo T, Fukuda K, Tone M, Yamada D, Takemura T, Ikushima S, Kumasaka T. Is hypothyroidism in idiopathic pleuroparenchymal fibroelastosis a novel lung-thyroid syndrome? Respir Investig. 2018;56(1):48–56.
Lowther CM, Morrison AO, Candelario NM, Khalafbeigi S, Cockerell CJ. Novel cutaneous manifestations of pleuroparenchymal fibroelastosis. Am J Dermatopathol. 2016;38(10):e140–3.
Enomoto Y, Nakamura Y, Satake Y, Sumikawa H, Johkoh T, Colby TV, Yasui H, Hozumi H, Karayama M, Suzuki Y, Furuhashi K, Fujisawa T, Enomoto N, Inui N, Iwashita T, Kuroishi S, Yokomura K, Koshimizu N, Toyoshima M, Imokawa S, Yamada T, Shirai T, Hayakawa H, Suda T. Clinical diagnosis of idiopathic pleuroparenchymal fibroelastosis: a retrospective multicenter study. Respir Med. 2017;133:1–5.
Oyama Y, Enomoto N, Suzuki Y, Kono M, Fujisawa T, Inui N, Nakamura Y, Kuroishi S, Yokomura K, Toyoshima M, Imokawa S, Oishi K, Watanabe S, Kasahara K, Baba T, Ogura T, Ishii H, Watanabe K, Nishioka Y, Suda T. Evaluation of urinary desmosines as a noninvasive diagnostic biomarker in patients with idiopathic pleuroparenchymal fibroelastosis (PPFE). Respir Med. 2017;123:63–70.
Sato S, Hanibuchi M, Takahashi M, Fukuda Y, Morizumi S, Toyoda Y, Goto H, Nishioka Y. A patient with idiopathic pleuroparenchymal fibroelastosis showing a sustained pulmonary function due to treatment with pirfenidone. Intern Med. 2016;55(5):497–501.
Sekine A, Satoh H, Iwasawa T, Matsui K, Ikeya E, Ikeda S, Yamakawa H, Okuda R, Kitamura H, Shinohara T, Baba T, Komatsu S, Kato T, Hagiwara E, Ogura T. Unilateral upper lung field pulmonary fibrosis radiologically consistent with pleuroparenchymal fibroelastosis after thoracotomy: a new disease entity related to thoracotomy. Respiration. 2017;94(5):431–41.
Kronborg-White S, Ravaglia C, Dubini A, et al. Cryobiopsies are diagnostic in pleuroparenchymal and airway-centered fibroelastosis. Respir Res. 2018;19:135.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Mengoli, M.C., Colby, T., Dubini, A., Rossi, G., Cavazza, A. (2019). Pleuroparenchymal Fibroelastosis. In: Poletti, V. (eds) Transbronchial cryobiopsy in diffuse parenchymal lung disease. Springer, Cham. https://doi.org/10.1007/978-3-030-14891-1_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-14891-1_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14890-4
Online ISBN: 978-3-030-14891-1
eBook Packages: MedicineMedicine (R0)