
Abstract
Pleuroparenchymal fibroelastosis (PPFE) is a rare interstitial lung disease characterized by predominantly upper lobe fibrosis involving the pleura and subpleural lung parenchyma. PPFE has been classified as either idiopathic or secondary. Idiopathic PPFE (iPPFE) is currently defined as a rare but distinct disease entity in the updated classification of idiopathic interstitial pneumonias (IIPs), while secondary PPFE is associated with a variety conditions, such as transplantation, dust exposure, autoimmune diseases, and genetic mutations. Given that diverse conditions cause PPFE, it may represent a pattern of chronic lung injury in response to various stimuli and/or in association with immune dysregulation or genetic predisposition. Nonetheless, the precise pathogenesis remains unclear. Typically, patients with PPFE show a “flattened thoracic cage” with a lean body and restrictive impairments with increased residual volume (RV)/total lung capacity (TLC) on pulmonary function test. The pathologic features of PPFE include parenchymal intra-alveolar fibrosis with marked elastic fiber deposition (fibroelastosis) accompanied by dense visceral pleura thickening. With no effective therapy currently established, its prognosis has remained poor. More recently, reports have shown that this disease is not as rare as previously considered and that its fibrosis is not confined to the upper lobes. Although surgical lung biopsy is essential for a definitive PPFE diagnosis, it is generally discouraged because of poor pulmonary function and post-operative pneumothorax. Thus, several clinical diagnostic criteria have been proposed, albeit not yet extensively validated. Future large-scale studies are therefore needed for further our understanding on the precise clinical behavior and prognosis of PPFE, as well as develop an effective treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Pleuroparenchymal fibroelastosis (PPFE) is a rare interstitial lung disease (ILD) characterized by predominantly upper lobe fibrosis involving the pleura and subpleural lung parenchyma [1,2,3]. In 1992, Amitani et al. had first described a peculiar series of Japanese patients with upper lobe-localized pulmonary fibrosis of unknown etiology [4], which was referred to as “idiopathic pulmonary upper lobe fibrosis.” In 2004, Frankel et al. [1] subsequently introduced the term PPFE. Accordingly, they reported five cases with upper lobe-predominant pulmonary fibrosis of unknown origin similar to what described by Amintani, which they described as a “unique idiopathic pleuroparenchymal lung disease that is characterized by upper lobe radiologic predominance and pathologic findings that do not fit with any currently defined interstitial pneumonia.” Fankel et al. proposed idiopathic PPFE to be a novel clinicopathologic entity of idiopathic interstitial pneumonias (IIPs). Since then, a growing body of literature has continued to report cases of PPFE, while the updated consensus statement for the multidisciplinary diagnosis of IIPs by the American Thoracic Society (ATS)/European Respiratory Society (ERS) has included idiopathic PPFE as a rare but distinct form of IIP [2].
Initially, the fibrotic lesions of PPFE had been thought to be restricted to the upper lobes. However, increasing evidence has demonstrated that quite a few patients with PPFE also had lower-lobe ILD [3, 5,6,7,8,9]. In addition, although PPFE was originally considered idiopathic, many studies have reported that PPFE also occurred in association with several conditions, including bone marrow and lung transplantation [10,11,12], chemotherapy [13, 14], and occupational exposure [15, 16]. To complicate matters, histologic PPFE patterns have also been found among patients with other ILDs, such as idiopathic pulmonary fibrosis (IPF) [17], ILD associated with connective tissue disease (CTD) [18], hypersensitivity pneumonitis [19], and familial interstitial pneumonia [19]. Moreover, recurrent/chronic pulmonary infection can cause PPFE [3, 20, 21]. Thus, although PPFE can be associated with a variety of underlying clinical conditions, the primary etiology and pathogenesis of PPFE have yet to be completely understood. This chapter will focus mainly on idiopathic PPFE (iPPFE) and describe current evidence related thereto, including its clinical, radiologic, and pathologic features. In addition, recently proposed diagnostic criteria, as well as current controversies concerning this disease entity, will be discussed.
Epidemiology
Two distinct forms of PPFE have been recognized: an idiopathic form, which occurs without any specific causes (iPPFE), and a secondary form, which is associated with underlying diseases or conditions (secondary PPFE) [22] (Table 36.1). While the precise incidence rates of each PPFE have yet to be clarified, Nakatani et al. recently reported 12 cases of PPFE (5.9%) out of 205 consecutive ILD cases undergoing surgical lung biopsy [23], among whom 8 (3.9%) were categorized as iPPFE and other 4 (2.0%) as secondary PPFE. In addition, among the patients with IIPs, 10.4% were identified as iPPFE. Shioya et al. showed 29 cases of iPPFE (7.8%) out of 375 consecutive IIP cases [24]. More recently, Fujisawa et al. reported that the out of 444 biopsy-confirmed IIP cases, 4.1% were iPPFE cases diagnosed through multidisciplinary discussion (MDD) [25]. Thus, iPPFE may not be as rare as previously considered with respect to the clinical setting of IIPs. On the other hand, one study showed that secondary PPFE had a prevalence 0.28% and 7.54% in hematopoietic stem cell transplantation and lung transplantation, respectively [25].
Familial forms of PPFE have also been reported [26, 27]. Azoulay et al. had reported three sisters with bilateral isolated apical pleural fibrosis of unknown origin with poor prognosis [26]. Interestingly, mutations of telomere-related genes have been found in patients with PPFE [28]. Indeed, Newton et al. had been the first to demonstrate mutations in the telomere maintenance machinery genes, such as telomerase reverse transcriptase (TERT), telomerase RNA component (TERC), and regulator of telomere elongation helicase 1 (RTEL1), in eight patients with iPPFE. More recently, Nunes et al. identified TERT mutations in 5 out of 10 patients with PPFE, among whom three had iPPFE and two had PPFE associated with Sjogren syndrome [27].
Studies have shown that patients with iPPFE have a median age of 50–70 years with a wide range (13–87 years) [1, 3,4,5, 29, 30]. A systematic review by Thusen et al., which included a total of 78 patients with iPPFE and secondary PPFE, showed a bimodal distribution with an early peak at the third and a later peak at the sixth decade of life [31]. Generally, no gender predominance has been observed. However, PPFE patients with genetic mutations have female predominane [28]. Moreover, despite the absence of an association between smoking habit and iPPFE, 20%–40% of patients with iPPFE have smoking history [3, 5, 29, 30].
As described previously, PPFE can occur in association with underlying diseases or conditions (secondary PPFE) (Table 36.1). Most importantly, PPFE has been known as a serious late-onset non-infectious pulmonary complication of bone marrow or hematopoietic stem cell transplantation [10, 11, 32, 33] and lung transplantation [12, 34]. Moreover, chronic graft-versus-host disease has been considered a major possible cause of transplantation-associated PPFE. However, cytotoxic agents used for treating hematologic malignancies have also been associated with this disease. Recently, Higo et al. suggested chronic graft-versus-host disease as the main cause of PPFE following allogeneic hematopoietic stem cell transplantation given that majority of patients with PPFE had simultaneous bronchiolitis obliterans, a typical form of chronic graft-versus-host disease [35]. Drugs, especially alkylating agents, can cause PPFE [13, 14]. Beynat-Mouterde et al. described six patients with upper lobe fibrosis suggestive of PPFE who had a history of chemotherapy for malignancy [13]. Among the six patients, five received cyclophosphamide, while one received other alkylating agents. Moreover, several studies have revealed that occupational dust exposure, such as asbestos or aluminum, is associated with PPFE [15, 16, 36, 37]. Interestingly, the response to asbestos in the lung is thought to be more fibroelastic than fibrotic [38]. Chronic pulmonary infection is another condition possibly associated with PPFE [3, 20, 21]. In a series of 12 patients with PPFE, Reddy et al. showed that seven had recurrent pulmonary infections, such as aspergillosis, suggesting that recurrent infections may lead to PPFE [3]. Moreover, Watanabe et al. described patients with rapidly progressive idiopathic pulmonary upper lobe fibrosis who had pulmonary mycobacterium avium complex disease [6]. In addition, PPFE can also develop in patients with autoimmune diseases. Upper lobe fibrosis or apical pulmonary fibrosis, which is suggestive of PPFE, has been observed in patients with ankylosing spondylitis, ulcerative colitis, and psoriasis [39,40,41,42]. Patients with rheumatoid arthritis or Sjögren syndrome have also been found to develop PPFE [23, 27].
Clinical Manifestations
Dry cough and dyspnea on exertion are the most common symptoms among patients with idiopathic or secondary PPFE, while weight loss has been frequently observed among patients with advanced PPFE. Moreover, some patients complain of chest pain due to pneumothorax. A “flattened thoracic cage” or “platythorax,” which is a reduction in the anteroposterior diameter of the chest wall, is often present especially in advanced stages [42]. Harada et al., who assessed the ratio between the anteroposterior and transverse diameter of the thoracic cage using chest computed tomography (CT) [42], found that it was much lower among patients with PPFE than among normal subjects and decreased as the disease progressed with a reduction in force vital capacity (FVC). Clubbing, which is frequently present among patients with IPF, is rare among those with PPFE. Indeed, Ishii et al. reported that only 2 (3.8%) of 52 patients with PPFE exhibited clubbing [7]. Another study showed that less than half of PPFE cases had audible crackles [22]. Particularly, patients with the lower-lobe ILD exhibit bibasilar crackles.
Laboratory Findings
Studies have shown that patients with PPFE had elevated serum levels of Krebs von den Lungen-6 (KL-6), a mucin-like glycoprotein, and surfactant protein D (SP-D), both of which are biomarkers of ILDs [5, 7, 29, 30, 43,44,45]. Generally, KL-6 levels remain around or slightly higher than the upper limit of the normal range, whereas SP-D levels often increase more than twice the upper limit. These observations suggest that serum SP-D elevation is more conspicuous than that of serum KL-6. Indeed, Sato et al. showed that among four consecutive iPPFE cases who had markedly increased serum SP-D levels, three had KL-6 levels within the normal limit [46]. Patients with PPFE who had simultaneous lower-lobe ILD exhibited significantly higher serum KL-6 levels than those without the comorbidity [7]. Moreover, Oyama et al. demonstrated that patients with iPPFE had significantly lower serum KL-6 levels than those with IPF, whereas no difference in serum SP-D levels were observed between them [47]. Some patients with PPFE have serum autoantibodies, such as rheumatologic factor, antinuclear antibody, myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA) [3, 29].
Desmosines are unique amino acids that are derived from the breakdown of mature elastic fibers. Patients with PPFE have considerably increased amounts of elastic fiber in their lungs, which may lead to an elevation of its degradation products, such as desmosines. With this regard, Oyama et al. reported that patients with iPPFE had significantly higher urinary desmosines levels than those with IPF or COPD, suggesting that urinary desmosines may be a useful diagnostic marker for iPPFE [47].
Respiratory Function
Patients with PPFE usually show a restrictive impairment with marked decline in FVC on their pulmonary function test. In addition, total lung capacity (TLC) is also decreased, while the forced expiratory volume in 1 s (FEV1)/FVC ratio is increased. Importantly, the residual volume (RV)/TLC ratio is generally elevated, perhaps due to the compensatory hyperinflation of the lower lobes caused by fibrotic collapse of the upper lobes [30]. Although diffusing capacity for carbon monoxide (DLCO) is usually decreased, the DLCO/VA ratio is relatively preserved [22, 30]. A comparison among patients with iPPFE showed that those with coexisting lower-lobe ILD had significantly lower RV and TLC than those without [8].
During the early stages of PPFE, patients have almost normal partial pressure of oxygen in arterial blood (PaO2), which usually decreases during advanced stages. Importantly, a mild elevation of a partial pressure of carbon monoxide (PaCO2) is noted [30], resulting in a preserved alveolar–arterial gradient of oxygen (AaDO2). Watanabe et al. speculated that the increased PaCO2 in patients with PPFE results from mechanical restriction due to the subpleural parenchyma rather than concomitant obstructive lung disease [30]. This peculiar blood gas analysis profile is distinct from that observed in other ILDs showing a decrease in both PaO2 and PaCO2 together with an increased AaDO2. Moreover, oxygen desaturation on a 6-min walk test (6MWT) is less frequent in PPFE than in other interstitial pneumonias, particularly IPF [48]. Among patients with ILDs registered for lung transplantation, those with PPFE had significantly smaller oxygen desaturation on the 6MWT than those with other ILDs [49].
Collectively, Watanabe et al. reported that the functional characteristics of PPFE include restrictive impairment with high RV/TLC which is often accompanied by a mild elevation of PaCO2 and relatively preserved AaDO2, resulting from subpleural parenchymal fibroelastosis, with a preserved parenchyma distant from the pleura [30].
Radiologic Features
Patients with PPFE have been shown to usually have marked thickening in the bilateral apical portions with an upward shift of hilar structures on chest X-ray (Fig. 36.1a) [1, 22, 50]. Moreover, typical high-resolution computed tomography (HRCT) features include bilateral irregular subpleural dense consolidations and reticulations in the upper lobes and less marked or no involvement of the lower lobes (Fig. 36.1b) (Table 36.2) [1, 3], with consolidations often having traction bronchiectasis with architecture distortion [51]. In advanced stages, consolidations and reticulations extend to the adjacent lobes, while subpleural cysts or bullae are often appreciated. Wedge-shaped pleural-based densities also protrude along parenchymal septa toward hila [3].

(a) Chest radiograph of a patient with idiopathic pleuroparenchymal fibroelastosis (iPPFE) showing bilateral pleural thickening and parenchymal bands in apical portions accompanied by reticular and ground-glass opacities with left-lung predominance. Upper lobe volume loss and trachea deviation are also observed. (b) High-resolution computed tomography (HRCT) showing bilateral subpleural consolidation with pleural thickening
Remarkably, a considerable proportion of patients with PPFE have been shown to have lower-lobe ILD. Although Amitani et al. originally described idiopathic pulmonary upper lobe fibrosis as purely upper lobe-localized fibrosis [4]. recent studies have reported that 42–92% of patients with PPFE have coexisting lower-lobe ILD on HRCT (Table 36.3) [3, 5,6,7,8,9, 23]. The most common HRCT pattern in lower-lobe ILD is the usual interstitial pneumonia (UIP) pattern. Interestingly, Ishii et al. reported that five of eight patients with PPFE who initially had no fibrotic lesions in the lower lung fields developed lower-lobe ILD during the follow-up period, suggesting that the lower-lobe ILD may develop as the disease progresses [7].
Radiological PPFE-like lesions have been reported in a variety of ILDs other than PPFE [17,18,19, 52]. Oda et al. observed radiological PPFE-like lesions, defined as pleural thickening with associated subpleural fibrosis concentrated in the upper lobes, in 11 (10%) of 110 patients with IPF [17]. Moreover, radiologic PPFE-like lesions were found in 21 patients (19%) of 113 patients with CTD-associated ILD [18]. Interestingly, the presence of PPFE-like lesions was associated with poor prognosis. Furthermore, marked PPFE-like lesions on HRCT, which were associated with impaired lung function and increased mortality, were found in 23% of 233 patients with hypersensitivity pneumonitis [19].
Pathologic Features
Originally, Frankel et al. demonstrated markedly dense fibrosis of the pleura and subpleural parenchyma in PPFE [1]. Fibrosis of the subpleural parenchyma is characterized by intra-alveolar fibrosis with prominent deposition of elastic fibers (fibroelastosis) (Fig. 36.2) (Table 36.4). An abrupt border between the fibrotic parenchymal areas and adjacent normal parenchyma is noted. A few fibroblastic foci at the interface of the fibrotic lesions and mild lymphocytic interstitial inflammation are also present [31]. In addition, bronchocentric intra-alveolar fibrosis is occasionally seen [3]. Fibrotic parenchymal lesions in PPFE are very similar to those of the pulmonary apical cap, and distinguishing between both has been histologically difficult. As described previously, although patients with PPFE often have the lower-lobe ILD, a few studies have focused on histologic findings of lower-lobe ILD in PPFE [27, 53, 54]. Accordingly, Nunes et al. reported that among four biopsy-confirmed patients with PPFE who had lower-lobe ILD, three exhibited a UIP pattern in the lower lobes, suggesting that a UIP pattern is a common histologic finding in lower-lobe ILD of PPFE [27].

(a) Surgical lung biopsy specimens from a patient with idiopathic pleuroparenchymal fibroelastosis (iPPFE). A lung section of the left upper lobe stained with hematoxylin and eosin showing subpleural fibrosis with an abrupt transition to normal lung parenchyma and fibroblastic foci. (b) A lung specimen with Elastica van Gieson (EVG) staining demonstrating depositions of dense elastic fibers (elastosis) and intra-alveolar collagen fibers in the subpleural fibrotic lung lesion (b). These features of PPFE on EVG staining are different from those of UIP, which show architectural destruction of normal alveoli. Pleural fibrosis is also observed
One study showed that patients with PPFE have more lymphatic vessels in the lung of compared to those with an apical cap and IPF [55]. Notably, Enomoto et al. tested immunostaining markers that could distinguish PPFE from IPF or apical cap and found that podoplanin-positive myofibroblasts could be a pathologic hallmark of PPFE [56]. They hypothesized that “pleural” mesothelial-to-mesenchymal transition is associated with the fibrosis in PPFE given that podoplanin is usually expressed in mesothelial cells but not myofibroblasts.
Recent studies have shown that histologic PPFE patterns can also be found in other ILDs, such as IPF [17, 18, 52, 54, 57, 58]. Indeed, Oda et al. observed a histologic PPFE pattern in 9 (8.2%) of 110 consecutive patients with biopsy-confirmed IPF [17]. In addition, Kinoshita et al. reported that 11 (22.9%) of 48 patients with biopsy-confirmed IPF exhibited a histologic PPFE pattern [58]. However, the extent of the subpleural parenchymal fibroelastosis in patients with IPF was much smaller than that in patients with PPFE. Moreover, histologic PPFE pattern was identified in 50% of 24 patients with CTD-associated ILD [52]. These observations suggest that the histologic PPFE patterns may indicate chronic lung injury, similar to UIP pattern, and are focally observed in association with a variety of conditions [54].
Diagnosis
Although several radiological and histological criteria for PPFE or PPFE pattern have been proposed [3, 31], no consensus regarding the diagnosis of iPPFE has yet been established. A definitive diagnosis of iPPFE ideally requires histologic confirmation of PPFE features following surgical lung biopsy. However, surgical lung biopsy is usually not feasible considering that patients with iPPFE, especially those with advanced diseases, have severe pulmonary function impairment and often develop persistent post-operative pneumothorax. Recent studies have demonstrated the diagnostic utility and safety of transbronchial lung biopsy (TBLB) and transbronchial lung cryobiopsy (TBLC) for iPPFE [59, 60]. However, given that these studies included only a small number of patients, a larger cohort of patients with iPPFE is needed to validate their findings. Therefore, clinical criteria without surgical lung biopsy have been desired for the diagnosis of iPPFE. Enomoto et al. recently proposed the following clinical criteria for iPPFE: (1) a radiologic PPFE pattern on chest CT (defined as bilateral subpleural dense consolidation with or without pleural thickening in the upper lobes and less marked or absent involvement of the lower lobes, (2) radiologic confirmation of disease progression, and (3) exclusion of other lung diseases with identifiable etiologies [5]. More recently, the Study Group on Diffuse Pulmonary Disorders in Japan has proposed a more comprehensive criteria for iPPFE, which consist of the following four categories: “definite iPPFE,” “radiologically and physiologically probable iPPFE,” “radiologically probable iPPFE,” and “radiologically possible iPPFE” (Box 36.1 and Table 36.5) [61]. “Definite iPPFE” requires surgical lung biopsy, whereas “radiologically and physiologically probable iPPFE,” “radiologically probable iPPFE,” or “radiologically possible iPPFE” do not. Currently, validation studies of the aforementioned criteria are being undertaken.
Box 36.1 Diagnostic Criteria for Idiopathic Pleuroparenchymal Fibroelastosis (iPPFE) Proposed by the Study Group on Diffuse Pulmonary Disorders in Japan [62]
-
Definite iPPFE (with surgical lung biopsy)
-
1.
Multiple subpleural foci of airspace consolidation with traction bronchiectasis located predominantly in the bilateral upper lobes on high-resolution computed tomography (HRCT) scans.
-
2.
Subpleural zonal or wedge-shaped dense fibrosis consisting of collapsed alveoli and collagen-filled alveoli with septal elastosis, with or without collagenous thickening of visceral pleura in surgical lung biopsy specimens.
-
3.
Exclusion of other diseases with known causes or conditions showing radiological and/or histological PPFE patterns, such as chronic hypersensitivity pneumonia, connective tissue diseases, occupational diseases, and hematopoietic stem cell or lung transplantation-related lung diseases.
-
1.
If all the above three criteria are met, definite iPPFE is diagnosed. If the lower lobes are involved by fibrosis, multidisciplinary discussion is necessary for the final diagnosis.
-
Radiologically and physiologically probable iPPFE (without surgical lung biopsy)
-
1.
Dry cough or exertional dyspnea with insidious onset.
-
2.
Multiple subpleural foci of airspace consolidation with traction bronchiectasis located predominantly in the bilateral upper lobes on HRCT scans.
-
3.
Upward shift of the bilateral hilar structures on chest radiographs and/or volume loss of the upper lobes on HRCT scans.
-
4.
Exclusion of other diseases with known causes or conditions showing radiological and/or histological PPFE patterns, such as chronic hypersensitivity pneumonia, connective tissue diseases, occupational diseases, and hematopoietic stem cell or lung transplantation-related lung diseases.
-
5.
Percentage of predicted values of the ratio between residual volume and total lung capacity (RV/TLC %pred.) ≥ 115%.
-
6.
Body mass index ≤20 kg/m [2] and RV/TLC %pred. ≥ 80%.
If criteria 1, 2, 3, 4, and 5 or 6 are satisfied, radiologically and physiologically probable iPPFE is diagnosed.
-
Radiologically probable iPPFE (without surgical lung biopsy)
-
1.
Dry cough or exertional dyspnea with insidious onset.
-
2.
Multiple subpleural foci of airspace consolidation with traction bronchiectasis located predominantly in the bilateral upper lobes on HRCT scans.
-
3.
Upward shift of the bilateral hilar structures on chest radiographs and/or volume loss of the upper lobes on HRCT scans.
-
4.
Exclusion of other diseases with known causes or conditions showing radiological and/or histological PPFE patterns, such as chronic hypersensitivity pneumonia, connective diseases, occupational diseases, and hematopoietic stem cell or lung transplantation-related lung diseases.
If all aforementioned criteria are satisfied, radiologically probable iPPFE is diagnosed.
-
Radiologically possible iPPFE (without surgical lung biopsy).
-
1.
Multiple subpleural foci of airspace consolidation with traction bronchiectasis located predominantly in the bilateral upper lobes on HRCT scans.
-
2.
Exclusion of other diseases with known causes or conditions showing radiological and/or histological PPFE patterns, such as chronic hypersensitivity pneumonia, connective diseases, occupational diseases, and hematopoietic stem cell or lung transplantation-related lung diseases.
If both criteria are satisfied, radiologically possible iPPFE is diagnosed. Radiologically possible IPPFE includes an apical cap with neither symptoms nor long-term progression in addition to the early and localized stages of iPPFE.
Differential diagnoses include a variety of diseases with upper lobe pleural thickening and/or fibrosis (Table 36.6), with pulmonary apical cap being one of the difficult conditions to differentiate. Both radiologic and histologic features of the apical cap are very similar to those of iPPFE. However, the apical cap usually occurs in older males with a history of smoking, whereas iPPFE affects relatively younger non-smokers with no gender predisposition. Moreover, the apical cap commonly shows a localized upper lobe lesion, whereas iPPFE, despite having upper lobe predominance, often exhibits a more extended distribution beyond the upper lobes. Most importantly, patients with an apical cap usually do not show disease progression over time, unlike those with iPPFE. Other differential diagnoses include chronic hypersensitivity pneumonitis and radiation-induced pneumonitis. Furthermore, to diagnose iPPFE, secondary PPFE, such as drug-induced and post-transplant PPFE (Table 36.1), must be excluded.
Another difficulty encountered when diagnosing iPPFE is distinguishing between this disease and other types of ILDs with radiologic PPFE-like lesions or a histologic PPFE pattern in the upper lobes given that certain patients with other ILDs, such as IPF and CTD-ILD, show radiologic PPFE-like lesions or a histologic PPFE pattern [17,18,19, 51, 52, 55, 58, 59]. In addition, a majority of patients with iPPFE often have lower-lobe ILD [3, 5,6,7,8,9, 24]. Thus, for patients with both upper- and lower-lobe fibrosis, determining upper lobe predominance, which is essential for the diagnosis of iPPFE, is occasionally challenging. Under such circumstances, MDD is necessary.
Treatment
To date, no effective treatments for iPPFE or secondary PPFE have been established.
Corticosteroids, immunosuppressants, or N-acetyl cysteine have been shown to achieve no improvement or, if any, transient response [48, 62, 63]. Nonetheless, very preliminary observations have suggested that anti-fibrotic agents, pirfenidone and nintedanib, might have some benefit [64, 65]. Sato et al. reported that pirfenidone treatment was followed by FVC stabilization in a patient with iPPFE [64]. Moreover, Nasser et al. showed that nintedanib treatment in five patients with PPFE, among whom three had iPPFE and two had PPFE secondary to chemotherapy, was followed by reduced FVC decline in all patients [65]. However, given that these studies included only a small number of patients, prospective studies including a larger cohort are needed to confirm their findings. Although several reports have recently demonstrated that lung transplantation may be an effective treatment option for iPPFE and secondary PPFE [62, 66,67,68,69,70,71], evidence has still been limited. Owing to reports of recurrent infections among patients with PPFE, such as aspergillosis and non-tuberculosis mycobacterial infection [3], infection control should be taken into consideration in such cases.
Prognosis
The prognosis of PPFE is heterogeneous. Yoshida et al. reported two patterns of disease progression: rapid FVC decline over a short time period and slow decline over a long period [72]. However, clinical differences at the baseline had not been described in detail between the two patterns. They speculated that, in patients with PPFE, FVC decline gradually to a certain point in time, after which FVC begins to drop rapidly. To date, several studies have performed survival analysis among patients with PPFE [5, 8, 9, 25, 72, 73], subsequently revealing 5-year survival rates and median survival durations ranging from 29% to 58% and 2.0–8.0 years, respectively, with wide variability (Table 36.7). Although a few studies have directly compared the prognosis between PPFE and other ILDs, such as IPF, Fujisawa et al. reported that patients with iPPFE had significantly shorter survival than those with IPF [25]. Collectively, these observations suggest that PPFE seems to have a poor prognosis.
Several prognostic factors have been identified. Indeed, Suzuki. et al. identified male gender and low erector spinae muscle attenuation (ESMMA) determined through CT imaging as independent factors for poor prognosis among patients with iPPFE [73]. Similarly, Khiroya et al. found that male sex was a predictor of increased risk of mortality among patients with PPFE [53]. Histologically, the coexistence of granulomas was associated with a significant decrease in mortality. Moreover, multivariate analysis by Kono et al. and Kato et al. identified low %FVC and high fibrosis score assessed through HRCT as factors for poor prognosis in iPPFE, respectively [8, 9]. Recently, Ishii et al. reported that serum KL-6 levels were significantly associated with outcomes in 52 patients with PPFE (48 iPPFE, 4 secondary PPFE) such that patients with KL-6 levels >600 U/mL showed significantly shorter survival than those with KL-6 levels <600 U/mL [7]. Given that patients with PPFE usually have normal upper limits or slightly higher serum KL-6 levels, the increase in KL-6 levels might have been attributed to the coexistence of lower-lobe ILDs. This suggest that patients with PPFE who develop lower-lobe ILDs may have poor prognosis. Similarly, Kono et al. recently demonstrated that patients with iPPFE who had lower-lobe ILD exhibited significantly worse survival with higher serum KL-6 levels than those without the lower-lobe ILD [8]. More importantly, patients with a lower-lobe UIP pattern had significantly shorter survival than those with a lower-lobe non-UIP pattern, suggesting that a radiologic UIP pattern in the lower-lobe ILD is an important prognostic determinant. Accordingly, Kato et al. also described that a lower-lobe UIP pattern was an independent factor for poor prognosis among patients with iPPFE [9]. Moreover, patients with iPPFE who had a lower-lobe UIP pattern tended to exhibit poorer prognosis than those with IPF [17].
During the course of PPFE, two particular conditions should be considered: pneumothorax and acute exacerbation. Recurrent pneumothorax often occurs especially in advanced diseases with multiple bullae, with pneumothorax incidence rates ranging from 17% to 75% [3, 5, 6, 8, 9, 23, 72]. The pneumothorax is often intractable and occasionally accompanied by pneumomediastinum. Recently, it has become evident that patients with PPFE develop acute exacerbation, as seen in those with IPF [7, 17, 24, 73, 74]. Suzuki et al. reported that acute exacerbation occurred in 8 (18.6%) of 43 patients with iPPFE over a median observation period of 31.1 months [73], while Ishi et al. found acute exacerbation in 7 (13%) of 52 patients with PPFE [7]. Outcomes following acute exacerbation of PPFE have generally been poor and often fatal. Moreover, precise annual incidence rates or risk factors of acute exacerbation have yet to be fully determined.
Conclusions
PPFE has been recognized as a distinct pattern of pulmonary fibrosis with characteristic clinical, radiologic, and histologic features. PPFE has been classified as either idiopathic or secondary. iPPFE is currently characterized as a rare IIP, while secondary PPFE is associated with diverse conditions, such as transplantation, dust exposure, autoimmune diseases, and genetic mutations. Given that a variety of conditions cause PPFE, it may represent a pattern of chronic lung injury in response to various stimuli and/or in association with immune dysregulation and genetic predisposition. However, the precise pathogenesis has remained undetermined. While no effective therapy for PPFE has been established, its prognosis remains poor. Recently, awareness for PPFE has considerably increased, leading to studies showing that this disease is not rare as previously considered and that fibrosis is not confined to the upper lobes. Practically, validated clinical diagnostic criteria without surgical lung biopsy have been developed considering that performing lung biopsy in patients with PPFE is generally discouraged. Future large-scale studies will be required to further understand the precise clinical behavior and prognosis of PPFE, as well as develop effective treatments.
Clinical Vignette
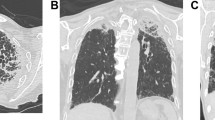
A 73-year-old man, non-smoker, with no occupational exposure was referred to our hospital due to dry cough, progressive dyspnea on exertion, and weight loss. His physical examination revealed low body mass index (BMI), a flattened thorax, and bilateral fine crackles but no clubbed fingers or skin lesions. Serum KL-6 was slightly increased (563 IU/mL) (normal range: ≤500 IU/mL), while serum SP-D was remarkably elevated (372 ng/mL) (normal range: ≤110 ng/mL). Serological tests revealed no autoantibodies. Pulmonary function test showed low forced vital capacity (FVC) (39.9% predict.), low forced expiratory volume in 1 s (FEV1) (49.8% predict.), normal FEV1/FVC 100%, increased residual volume (RV) (160.5% predict.), and high RV/total lung capacity (TLC) (157.7%). Diffusing capacity for carbon monoxide (DLCO) was decreased (70.4%). Blood gas analysis at room air showed decreased partial pressure of oxygen in arterial blood (PaO2) (68 Torr) and slightly increased partial pressure of carbon monoxide (PaCO2) (51 Torr). Chest radiography and computed tomography exhibited bilateral apical pleural thickening with parenchymal bands and reticulations in the upper lobes (Fig. 36.3a and b). Some reticular opacities were also found in the lower lobes. Based on these findings, idiopathic pleuroparenchymal fibroelastosis (iPPFE) was suspected, after which a video-associated thoracoscopic surgical lung biopsy was conducted to confirm the diagnosis. The biopsy revealed marked thickening of the visceral pleura and subpleural parenchymal intra-alveolar fibrosis with massive deposition of elastic fibers, which were consistent with the histologic pattern of PPFE. Thus, a diagnosis of iPPFE was established, and the patient had been followed up without treatment. During observation, he had developed intractable pneumothorax three times and eventually died of chronic respiratory failure 3 years after diagnosis.

Chest X-ray (a) and coronal section of a chest CT (b) of a patient with idiopathic pleuroparenchymal fibroelastosis. Upward shift of the bilateral hilar structures on chest x-ray and volume loss of the upper lobes on HRCT scan
References
Frankel SK, Cool CD, Lynch DA, Brown KK. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004;126(6):2007–13.
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Reddy TL, Tominaga M, Hansell DM, von der Thusen J, Rassl D, Parfrey H, et al. Pleuroparenchymal fibroelastosis: a spectrum of histopathological and imaging phenotypes. Eur Respir J. 2012;40(2):377–85.
Amitani R, Niimi A, Kuse F. Idiopathic pulmonary upper lobe fibrosis (IPUF). Kokyuu. 1992;11:693–9.
Enomoto Y, Nakamura Y, Satake Y, Sumikawa H, Johkoh T, Colby TV, et al. Clinical diagnosis of idiopathic pleuroparenchymal fibroelastosis: a retrospective multicenter study. Respir Med. 2017;133:1–5.
Watanabe K, Nagata N, Kitasato Y, Wakamatsu K, Nabeshima K, Harada T, et al. Rapid decrease in forced vital capacity in patients with idiopathic pulmonary upper lobe fibrosis. Respir Investig. 2012;50(3):88–97.
Ishii H, Watanabe K, Kushima H, Baba T, Watanabe S, Yamada Y, et al. Pleuroparenchymal fibroelastosis diagnosed by multidisciplinary discussions in Japan. Respir Med. 2018;141:190–7.
Kono M, Fujita Y, Takeda K, Miyashita K, Tsutsumi A, Kobayashi T, et al. Clinical significance of lower-lobe interstitial lung disease on high-resolution computed tomography in patients with idiopathic pleuroparenchymal fibroelastosis. Respir Med. 2019;154:122–6.
Kato M, Sasaki S, Kurokawa K, Nakamura T, Yamada T, Sasano H, et al. Usual interstitial pneumonia pattern in the lower lung lobes as a prognostic factor in idiopathic Pleuroparenchymal fibroelastosis. Respiration. 2019;97(4):319–28.
von der Thusen JH, Hansell DM, Tominaga M, Veys PA, Ashworth MT, Owens CM, et al. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol. 2011;24(12):1633–9.
Mariani F, Gatti B, Rocca A, Bonifazi F, Cavazza A, Fanti S, et al. Pleuroparenchymal fibroelastosis: the prevalence of secondary forms in hematopoietic stem cell and lung transplantation recipients. Diagn Interv Radiol. 2016;22(5):400–6.
Ofek E, Sato M, Saito T, Wagnetz U, Roberts HC, Chaparro C, et al. Restrictive allograft syndrome post lung transplantation is characterized by pleuroparenchymal fibroelastosis. Modern pathology : an official journal of the United States and Canadian academy of pathology. Inc. 2013;26(3):350–6.
Beynat-Mouterde C, Beltramo G, Lezmi G, Pernet D, Camus C, Fanton A, et al. Pleuroparenchymal fibroelastosis as a late complication of chemotherapy agents. Eur Respir J. 2014;44(2):523–7.
Baroke E, Heussel CP, Warth A, Eichinger M, Oltmanns U, Palmowski K, et al. Pleuroparenchymal fibroelastosis in association with carcinomas. Respirology. 2016;21(1):191–4.
Huang Z, Li S, Zhu Y, Zhu H, Yi X. Pleuroparenchymal fibroelastosis associated with aluminosilicate dust: a case report. Int J Clin Exp Pathol. 2015;8(7):8676–9.
Xu L, Rassaei N, Caruso C. Pleuroparenchymal fibroelastosis with long history of Asbestos and silicon exposure. Int J Surg Pathol. 2018;26(2):190–3.
Oda T, Ogura T, Kitamura H, Hagiwara E, Baba T, Enomoto Y, et al. Distinct characteristics of pleuroparenchymal fibroelastosis with usual interstitial pneumonia compared with idiopathic pulmonary fibrosis. Chest. 2014;146(5):1248–55.
Enomoto Y, Nakamura Y, Colby TV, Johkoh T, Sumikawa H, Nishimoto K, et al. Radiologic pleuroparenchymal fibroelastosis-like lesion in connective tissue disease-related interstitial lung disease. PLoS One. 2017;12(6):e0180283.
Jacob J, Odink A, Brun AL, Macaluso C, de Lauretis A, Kokosi M, et al. Functional associations of pleuroparenchymal fibroelastosis and emphysema with hypersensitivity pneumonitis. Respir Med. 2018;138:95–101.
Cha YJ, Han J, Chung MP, Kim TJ, Shin S. Pleuroparenchymal fibroelastosis in heterogeneous clinical conditions: clinicopathologic analysis of 7 cases. Clin Respir J. 2018;12(4):1495–502.
Kushima H, Ishii H, Kinoshita Y, Fujita M, Watanabe K. Chronic pulmonary aspergillosis with Pleuroparenchymal fibroelastosis-like features. Intern Med. 2019;58(8):1137–40.
Watanabe K. Pleuroparenchymal fibroelastosis: its clinical characteristics. Curr Respir Med Rev. 2013;9:299–37.
Nakatani T, Arai T, Kitaichi M, Akira M, Tachibana K, Sugimoto C, et al. Pleuroparenchymal fibroelastosis from a consecutive database: a rare disease entity? Eur Respir J. 2015;45(4):1183–6.
Shioya M, Otsuka M, Yamada G, Umeda Y, Ikeda K, Nishikiori H, et al. Poorer prognosis of idiopathic Pleuroparenchymal fibroelastosis compared with idiopathic pulmonary fibrosis in advanced stage. Can Respir J. 2018;2018:6043053.
Fujisawa T, Mori K, Mikamo M, Ohno T, Kataoka K, Sugimoto C, et al. Nationwide cloud-based integrated database of idiopathic interstitial pneumonias for multidisciplinary discussion. Eur Respir J. 2019;53(5):1802243.
Azoulay E, Paugam B, Heymann MF, Kambouchner M, Haloun A, Valeyre D, et al. Familial extensive idiopathic bilateral pleural fibrosis. Eur Respir J. 1999;14(4):971–3.
Nunes H, Jeny F, Bouvry D, Picard C, Bernaudin JF, Menard C, et al. Pleuroparenchymal fibroelastosis associated with telomerase reverse transcriptase mutations. Eur Respir J. 2017;49(5):1700696.
Newton CA, Batra K, Torrealba J, Meyer K, Raghu G, Garcia CK. Pleuroparenchymal fibroelastosis associated with telomerase reverse transcriptase mutations. Eur Respir J. 2017;49(5):1700696.
Kusagaya H, Fujisawa T, Enomoto N, Inui N, Nakamura Y, Suda T. Co-occurrence of pneumoperitoneum and pneumothorax in a patient with Pleuroparenchymal fibroelastosis. Am J Respir Crit Care Med. 2015;191(10):1200–1.
Watanabe S, Waseda Y, Takato H, Matsunuma R, Johkoh T, Egashira R, et al. Pleuroparenchymal fibroelastosis: distinct pulmonary physiological features in nine patients. Respir Investig. 2015;53(4):149–55.
von der Thusen JH. Pleuroparenchymal fibroelastosis: its pathological characteristics. Curr Respir Med Rev. 2013;9(4):238–47.
Meignin V, Thivolet-Bejui F, Kambouchner M, Hussenet C, Bondeelle L, Mitchell A, et al. Lung histopathology of non-infectious pulmonary complications after allogeneic haematopoietic stem cell transplantation. Histopathology. 2018;73(5):832–42.
Okimoto T, Tsubata Y, Hamaguchi M, Sutani A, Hamaguchi S, Isobe T. Pleuroparenchymal fibroelastosis after haematopoietic stem cell transplantation without graft-versus-host disease findings. Respirol Case Rep. 2018;6(3):e00298.
Hirota T, Fujita M, Matsumoto T, Higuchi T, Shiraishi T, Minami M, et al. Pleuroparenchymal fibroelastosis as a manifestation of chronic lung rejection? Eur Respir J. 2013;41(1):243–5.
Higo H, Miyahara N, Taniguchi A, Maeda Y, Kiura K. Cause of pleuroparenchymal fibroelastosis following allogeneic hematopoietic stem cell transplantation. Respir Investig. 2019;57(4):321–4.
Becker CD, Gil J, Padilla ML. Idiopathic pleuroparenchymal fibroelastosis: an unrecognized or misdiagnosed entity? Mod Pathol. 2008;21(6):784–7.
Piciucchi S, Tomassetti S, Casoni G, Sverzellati N, Carloni A, Dubini A, et al. High resolution CT and histological findings in idiopathic pleuroparenchymal fibroelastosis: features and differential diagnosis. Respir Res. 2011;12:111.
Wick MR, Kendall TJ, Ritter JH. Asbestosis: demonstration of distinctive interstitial fibroelastosis: a pilot study. Ann Diagn Pathol. 2009;13(5):297–302.
Davies D. Ankylosing spondylitis and lung fibrosis. Q J Med. 1972;41(164):395–417.
Singh R, Sundaram P, Joshi JM. Upper lobe fibrosis in ulcerative colitis. J Assoc Physicians India. 2003;51:515–7.
Bourke S, Campbell J, Henderson AF, Stevenson RD. Apical pulmonary fibrosis in psoriasis. Br J Dis Chest. 1988;82(4):444–6.
Harada T, Yoshida Y, Kitasato Y, Tsuruta N, Wakamatsu K, Hirota T, et al. The thoracic cage becomes flattened in the progression of pleuroparenchymal fibroelastosis. Eur Respir Rev. 2014;23(132):263–6.
Kohno N, Kyoizumi S, Awaya Y, Fukuhara H, Yamakido M, Akiyama M. New serum indicator of interstitial pneumonitis activity. Sialylated carbohydrate antigen KL-6. Chest. 1989;96(1):68–73.
Kuroki Y, Takahashi H, Chiba H, Akino T. Surfactant proteins A and D: disease markers. Biochim Biophys Acta. 1998;1408(2–3):334–45.
Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, et al. Serum surfactant proteins a and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1109–14.
Sato S, Hanibuchi M, Fukuya A, Yabuki Y, Bando H, Yoshijima T, et al. Idiopathic pleuroparenchymal fibroelastosis is characterized by an elevated serum level of surfactant protein-D, but not Krebs von den Lungen-6. Lung. 2014;192(5):711–7.
Oyama Y, Enomoto N, Suzuki Y, Kono M, Fujisawa T, Inui N, et al. Evaluation of urinary desmosines as a noninvasive diagnostic biomarker in patients with idiopathic pleuroparenchymal fibroelastosis (PPFE). Respir Med. 2017;123:63–70.
Bonifazi M, Montero MA, Renzoni EA. Idiopathic Pleuroparenchymal fibroelastosis. Curr Pulmonol Rep. 2017;6(1):9–15.
Tanizawa K, Handa T, Kubo T, Chen-Yoshikawa TF, Aoyama A, Motoyama H, et al. Clinical significance of radiological pleuroparenchymal fibroelastosis pattern in interstitial lung disease patients registered for lung transplantation: a retrospective cohort study. Respir Res. 2018;19(1):162.
Ishii H, Kinoshita Y, Kushima H, Ogura T, Watanabe K. The upward shift of hilar structures and tracheal deviation in pleuroparenchymal fibroelastosis. Multidiscip Respir Med. 2019;14:10.
Johkoh T, Fukuoka J, Tanaka T. Rare idiopathic intestinal pneumonias (IIPs) and histologic patterns in new ATS/ERS multidisciplinary classification of the IIPs. Eur J Radiol. 2015;84(3):542–6.
Kinoshita Y, Watanabe K, Ishii H, Kushima H, Hamasaki M, Fujita M, et al. Pleuroparenchymal fibroelastosis as a histological background of autoimmune diseases. Virchows Arch. 2019;474(1):97–104.
Khiroya R, Macaluso C, Montero MA, Wells AU, Chua F, Kokosi M, et al. Pleuroparenchymal fibroelastosis: a review of histopathologic features and the relationship between histologic parameters and survival. Am J Surg Pathol. 2017;41(12):1683–9.
Rosenbaum JN, Butt YM, Johnson KA, Meyer K, Batra K, Kanne JP, et al. Pleuroparenchymal fibroelastosis: a pattern of chronic lung injury. Hum Pathol. 2015;46(1):137–46.
Kinoshita Y, Watanabe K, Ishii H, Kushima H, Fujita M, Nabeshima K. Significant increases in the density and number of lymphatic vessels in pleuroparenchymal fibroelastosis. Histopathology. 2018;73(3):417–27.
Enomoto Y, Matsushima S, Meguro S, Kawasaki H, Kosugi I, Fujisawa T, et al. Podoplanin-positive myofibroblasts: a pathological hallmark of pleuroparenchymal fibroelastosis. Histopathology. 2018;72(7):1209–15.
Enomoto N, Kusagaya H, Oyama Y, Kono M, Kaida Y, Kuroishi S, et al. Quantitative analysis of lung elastic fibers in idiopathic pleuroparenchymal fibroelastosis (IPPFE): comparison of clinical, radiological, and pathological findings with those of idiopathic pulmonary fibrosis (IPF). BMC Pulm Med. 2014;14:91.
Kinoshita Y, Watanabe K, Ishii H, Kushima H, Fujita M, Nabeshima K. Proliferation of elastic fibres in idiopathic pulmonary fibrosis: a whole-slide image analysis and comparison with pleuroparenchymal fibroelastosis. Histopathology. 2017;71(6):934–42.
Kushima H, Hidaka K, Ishii H, Nakao A, On R, Kinoshita Y, et al. Two cases of pleuroparenchymal fibroelastosis diagnosed with transbronchial lung biopsy. Respir Med Case Rep. 2016;19:71–3.
Kronborg-White S, Ravaglia C, Dubini A, Piciucchi S, Tomassetti S, Bendstrup E, et al. Cryobiopsies are diagnostic in Pleuroparenchymal and airway-centered fibroelastosis. Respir Res. 2018;19(1):135.
Watanabe K, Ishii H, Kiyomi F, Terasaki Y, Hebisawa A, Kawabata Y, et al. Criteria for the diagnosis of idiopathic pleuroparenchymal fibroelastosis: a proposal. Respir Investig. 2019;57(4):312–20.
Huang H, Feng R, Li S, Wu B, Xu K, Xu Z, et al. A CARE-compliant case report: lung transplantation for a Chinese young man with idiopathic pleuroparenchymal fibroelastosis. Medicine (Baltimore). 2017;96(19):e6900.
Shimada A, Terada J, Tsushima K, Tateishi Y, Abe R, Oda S, et al. Veno-venous extracorporeal membrane oxygenation bridged living-donor lung transplantation for rapid progressive respiratory failure with pleuroparenchymal fibroelastosis after allogeneic hematopoietic stem cell transplantation. Respir Investig. 2018;56(3):258–62.
Sato S, Hanibuchi M, Takahashi M, Fukuda Y, Morizumi S, Toyoda Y, et al. A patient with idiopathic Pleuroparenchymal fibroelastosis showing a sustained pulmonary function due to treatment with pirfenidone. Intern Med. 2016;55(5):497–501.
Nasser M, Chebib N, Philit F, Senechal A, Traclet J, Zarza V, et al. Treatment with nintedanib in patients with pleuroparenchymal fibroelastosis. Eur Respir J. 2017;50:PA4876.
Portillo K, Guasch I, Becker C, Andreo F, Fernandez-Figueras MT, Ramirez Ruz J, et al. Pleuroparenchymal fibroelastosis: a new entity within the Spectrum of rare idiopathic interstitial pneumonias. Case Rep Pulmonol. 2015;2015:810515.
Yanagiya M, Sato M, Kawashima S, Kuwano H, Nagayama K, Nitadori J, et al. Flat chest of Pleuroparenchymal fibroelastosis reversed by lung transplantation. Ann Thorac Surg. 2016;102(4):e347–9.
Hata A, Nakajima T, Yoshida S, Kinoshita T, Terada J, Tatsumi K, et al. Living donor lung transplantation for Pleuroparenchymal fibroelastosis. Ann Thorac Surg. 2016;101(5):1970–2.
Chen F, Matsubara K, Miyagawa-Hayashino A, Tada K, Handa T, Yamada T, et al. Lung transplantation for pleuroparenchymal fibroelastosis after chemotherapy. Ann Thorac Surg. 2014;98(5):e115–7.
Righi I, Morlacchi L, Rossetti V, Mendogni P, Palleschi A, Tosi D, et al. Lung transplantation as successful treatment of end-stage idiopathic Pleuroparenchymal fibroelastosis: a case report. Transplant Proc. 2019;51(1):235–8.
Ali MS, Ramalingam VS, Haasler G, Presberg K. Pleuroparenchymal fibroelastosis (PPFE) treated with lung transplantation and review of the literature. BMJ Case Rep. 2019;12(4):e229402.
Yoshida Y, Nagata N, Tsuruta N, Kitasato Y, Wakamatsu K, Yoshimi M, et al. Heterogeneous clinical features in patients with pulmonary fibrosis showing histology of pleuroparenchymal fibroelastosis. Respir Investig. 2016;54(3):162–9.
Suzuki Y, Yoshimura K, Enomoto Y, Yasui H, Hozumi H, Karayama M, et al. Distinct profile and prognostic impact of body composition changes in idiopathic pulmonary fibrosis and idiopathic pleuroparenchymal fibroelastosis. Sci Rep. 2018;8(1):14074.
Murakami Y, Sakamoto K, Okumura Y, Suzuki A, Mii S, Sato M, Yokoi T, Hashimoto N, Hasegawa Y. Acute Exacerbation of Pleuroparenchymal Fibroelastosis Secondary to Allogenic Hematopoietic Stem Cell Transplantation. Intern Med. 2020;59(21):2737–43. https://doi.org/10.2169/internalmedicine.4995-20. Epub 2020 Jul 14.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Suda, T. (2023). Pleuroparenchymal Fibroelastosis. In: Cottin, V., Richeldi, L., Brown, K., McCormack, F.X. (eds) Orphan Lung Diseases. Springer, Cham. https://doi.org/10.1007/978-3-031-12950-6_36
Download citation
DOI: https://doi.org/10.1007/978-3-031-12950-6_36
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12949-0
Online ISBN: 978-3-031-12950-6
eBook Packages: MedicineMedicine (R0)