
Abstract
Pulmonary lymphangioleiomyomatosis (LAM) is a rare disease that almost exclusively occurs in women of childbearing age, most often appearing as a diffuse cystic lung disease due to proliferations of distinctive smooth-muscle cell-like cells along lymphatic channels progressively involving the pulmonary parenchyma and the pleura [1–3].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Pulmonary lymphangioleiomyomatosis (LAM) is a rare disease that almost exclusively occurs in women of childbearing age, most often appearing as a diffuse cystic lung disease due to proliferations of distinctive smooth-muscle cell-like cells along lymphatic channels progressively involving the pulmonary parenchyma and the pleura [1,2,3].
LAM occurs sporadically in patients with no evidence of genetic disease (sporadic form) and in about one third of women with tuberous sclerosis complex (TSC), an autosomal dominant neurocutaneous syndrome characterized by mutations of TSC-1 (located on chromosome 9q34 and encoding for the protein hamartin) and TSC-2 (located on chromosome 16p13 and encoding for the protein tuberin) genes [4,5,6,7,8,9,10]. TSC occurs in 1 of 5800 live births and leads to hamartoma-like tumor growths in different organs, cerebral calcification, seizures, and mental retardation [5, 6]. Sporadic LAM is a relatively uncommon disease with an estimated prevalence of 2.6 per one million women, while a couple of sporadic LAM were documented in a karyotypically normal men without TS [11,12,13]. The disease tends to present between menarche and menopause, with the mean age at presentation of 34 years [1,2,3, 14].
The clinical features result from the progressive cystic destruction of the lung parenchyma secondary to widespread growth of LAM cells along lymphatics. Cystic abnormalities and airflow obstruction seem to be related to the constrictive effect on airways of LAM growths, but even the imbalance between matrix metalloproteinases and their inhibitors secreted by LAM cells leading to extracellular matrix degradation is involved in the mechanism of cystic modifications [15,16,17,18,19,20,21].
The main pulmonary symptoms consist of progressive dyspnea, recurrent pneumothorax, and chylous pleural effusions [1, 2, 22]. Cough, hemoptysis, and chyloptysis are also reported. Dyspnea is almost always present, while about 60% of patients show concurrent pneumothorax. [1, 2, 5, 22] Abdominal symptoms (nausea, abdominal distension, hematuria, flank pain, abdominal hemorrhage) are related to extrapulmonary manifestations such as abdominal lymphangioleiomyomas (retroperitoneum, abdomen, pelvis), lymphadenopathy, chylous abdominal depositions, and angiomyolipoma (kidney and liver) [23,24,25,26,27,28,29,30]. Around 10% of patients will develop chylous ascites due to lymphatic obstruction.
In early phase of LAM, chest X-ray may be normal even in symptomatic patient [31]. However, increased pulmonary volumes, pneumothorax or chylous pleural effusion, and cystic changes are the main radiological features [3].
The distribution of these lesions is generally bilateral and diffuse with some basilar predominance. LAM cysts have a very thin and regular wall and appear equally distributed. In contrast with centrilobular emphysema, LAM cysts show a well-defined wall lacking the centrilobular artery. Centrilobular emphysema predominates in the upper lobes and is smoking-related [1,2,3, 5]. Cysts of Langerhans cell histiocytosis is another smoking-related interstitial lung disease frequently showing cystic changes. However, cysts are often associated with micronodules and display a thick, irregular wall, also sparing the costophrenic angle [5]. LAM cysts generally have a round shape, do not spare the costophrenic angles, decrease in size on expiration, and tend to progressively increase in number and size. HRCT is also used to evaluate the severity of lung disease. Tiny nodules observed at high-resolution computed tomography consist of small benign proliferation of alveolar type 2 pneumocytes (so-called multifocal nodular type 2 pneumocyte hyperplasia) [3, 5]. Ill-defined ground glass opacities (observed in 25% of the cases) and interlobular septal thickening can be due to alveolar hemorrhage or edema, resulting from the obstruction of pulmonary lymphatic vessels and small veins. In the suspicion of LAM, an abdominal CT scan can provide useful additional findings, namely, renal angiomyolipomas and lymphangioleiomyomas [1,2,3, 5, 30,31,32,33,34,35,36].
High-resolution computed tomography scan should be performed in all young, non-smoking women with unclear recurrent pneumothorax with/without chylous effusion and/or functional obstruction, since imaging findings are quite specific in the hands of expert radiologists [3, 31]. The diagnosis of LAM is consistently performed on a clinical ground with typical clinical symptoms and HRCT pattern without tissue biopsy in presence of other confirmatory findings, such as chylothorax, angiomyolipoma, lymphangioleiomyoma/lymphangiomyoma, tuberous sclerosis complex, and elevated serum vascular endothelial growth factor—VEGF-D ≥ 800 pg/mL [31].
In the hands of expert pulmonary radiologists, the sensitivity and specificity of HRCT in identifying LAM among several cystic diseases are 87.5% and 97.5%, respectively [3, 31].
Nevertheless, difficult cases showing characteristic cysts at HRCT in absence of additional confirmatory features require lung tissue examination to achieve a definitive diagnosis. Surgical lung biopsy (SLB) is still considered the gold standard biopsy in interstitial lung diseases (ILD), including those with cystic pattern, but less invasive transbronchial lung biopsy (TBLB) may be considered equally effective and safer than SLB in the diagnosis of LAM [31].
Lung biopsy is performed in suspected LAM patients to exclude other cystic lung diseases [32]. The rarity of the disease and the therapeutic and prognostic implications of the diagnosis make tissue sampling recommended in all clinically uncertain cases. Cytology has a little or no role in LAM diagnosis, but the finding of alveolar hemorrhage with hemosiderin-laden macrophages at bronchoalveolar lavage (BAL) fluid analysis may be helpful [31].
LAM is one of diffuse lung diseases that can be successfully diagnosed on TBLB, which is often attempted before submitting the patient to thoracic surgery [32]. Given the random distribution of the lesions, the diagnostic yield of TBLB increases with the number of biopsies. In TBLBs with clinical and CT findings suspicious for LAM and in all biopsies from patients with cystic disease, the use of immunohistochemical stains is strongly recommended to highlight tiny and focally distributed lesions apparently not visible on hematoxylin-eosin-stained slides [33,34,35].
Differential diagnosis mainly includes Langerhans cell histiocytosis, emphysema, lymphoid interstitial pneumonia, and non-specific interstitial pneumonia (NSIP) related to connective tissue diseases (e.g., Sjogren’s disease), Birt-Hogg-Dubé syndrome, amyloidosis, hypersensitivity pneumonia, and lymphangiomatosis.
In the recent years, several authors and the last 2015 World Health Organization (WHO) classification of lung tumors have suggested to consider LAM as a low-grade tumor arising from peculiar cells spreading through lymphatic channels and possibly metastasizing to lymph nodes [36].
Indeed, LAM cells harbor inactivating mutations in tumor suppressor gene TSC1 or TSC2 and activate mammalian target of rapamycin complex 1(mTORC1) leading to cell growth [4]. The 2015 WHO classification has created a group of perivascular epithelioid cell (PEC)-derived tumors including PEComa (formerly clear cell tumor/sugar tumor) and LAM. This view is sustained by the frequent occurrence of extrapulmonary manifestation of LAM, comprising pelvic lymph node infiltration, uterine LAM, and renal angiomyolipoma [36].
In addition, patients with sporadic LAM requiring lung transplantation have developed recurrent LAM in transplanted lungs harboring identical genetic alterations to those originally identified in naïve lungs [37,38,39]. Furthermore, identical TSC2 mutations were detected in LAM cells and angiomyolipomas from the same patients, and LAM cells were isolated from blood and other fluids in patients with sporadic LAM [4, 39].
Although data derive from few and limited case series, the diagnostic yield of TBLB is about 60%, and procedure-related complication rate is about 14% (pneumothorax, hemorrhage, thoracic chest pain, pneumonia) [31]. Nevertheless, the most recent American Thoracic Society (ATS)/Japanese Respiratory Society clinical practice guidelines underline the necessity to submit nondiagnostic TBLB samples to expert pathologists for a second opinion, since a significant quote of cases become entirely diagnostic [31].
Limitations of TBLB are related to the small case series published in literature that preclude a confident estimation of the diagnostic yield in LAM, although the diagnostic bar could mainly depend on the percentage of involved lung. Second, data from previous works give no indication in the correct selection of patients undergoing TBLB.
However, once lung tissue examination is required to have a confident diagnosis of LAM, TBLB should be attempted in light of the significant diagnostic yield (up to 50%) and to prevent unnecessary more invasive surgical lung biopsy. The complication rate of TBLB in LAM is acceptable, ranging from 2 to 14%.
Transbronchial fine needle aspiration of enlarged mediastinal lymph nodes could represent an alternative and less invasive approach, as well as the search for LAM cells in chylous effusions using cell block preparation [31].
No studies focused on the efficacy and safety of transbronchial cryobiopsies in LAM have been so far published. Fruchter et al. [40] reported 1 case of LAM among their 75 patients with interstitial lung diseases undergoing histologic examination by transbronchial cryobiopsy. Apparently, the cystic pattern of ILD does not appear a contraindication to transbronchial cryobiopsy.
Survival at 10 years is of 80%, and main treatments include medroxyprogesterone, mTOR inhibitors, and lung transplantation [41,42,43].
1.1 Histology
-
Cysts bilaterally distributed through all the lung fields with involvement of costophrenic angles
-
Proliferation of smooth-muscle-like cells with cytoplasmic clear vacuolization (LAM cells) along the lymphatic routes lining the cystic formation, even leading to small nodules
-
Chronic alveolar hemorrhage around vessels
-
Variable immunohistochemical expression of smooth-muscle markers (smooth-muscle actin, desmin), hormonal receptors, melanocytic markers (HMB45, MART-1), and cathepsin-K
Cystic change and LAM cell growth may be very subtle and quite different in each individual case. The distribution of cysts and LAM cells mainly involves lymphatic routes, such as peribronchiolar areas, vessels, pleura, and lymphatic spaces. LAM cells are typically organized in small clusters at the edges of the cysts and along pulmonary lymphatics. Compared to normal smooth-muscle cells, LAM cells have less eosinophilic cytoplasm and a characteristic clear change/vacuolization. LAM cells may vary from small spindle-shaped to round or oval, to large epithelioid cell growing into the cystic spaces or forming small thickening of the cyst wall.
Since LAM cells involve the vessel walls leading to vascular disruption, a careful observation of chronic alveolar hemorrhage with hemosiderin-laden macrophages may indirectly indicate LAM. Serial sections of each formalin-fixed, paraffin-embedded block and the use of immunostains are mandatory in these cases. Indeed, LAM smooth-muscle-like cells generally express melanocytic markers (HMB45, MART-1), hormonal receptors, smooth-muscle actin, and cathepsin-K [44,45,46,47,48].
Multifocal micronodular pneumocyte hyperplasia is another lesion in LAM appearing as tiny nodular proliferations of type 2 pneumocytes with polygonal or cuboidal appearance.
1.2 Differential Diagnosis
Basically, all lesions leading to pulmonary cystic modifications may enter in differential diagnosis. Identification of LAM cells around cysts using specific immunostains (e.g., HMB45, cathepsin-K) is unique of LAM. Of note, pathologists should be aware that HMB45 expression may be variable, weak, and restricted to few cells. Since histology of LAM is quite peculiar, the disease may be recognizable even in tiny biopsy.
LAM must be distinguished from other more common cystic pulmonary lesions, as emphysema and pulmonary Langerhans cell histiocytosis. These diseases are characterized by distinctive clinical and HRCT findings, such as association with smoke and upper lobe distribution. LCH is characterized by cystic changes and tiny and cavitate nodules depending on its evolution. The wall of cysts consists of fascicle of spindle cells resembling smooth-muscle elements. Metastatic low-grade stromal sarcoma may present with several cysts lined by hormonal and CD10 positive bland spindled tumor cells, but clinical history together with imaging studies and lack of expression for melanocytic markers rule out LAM. Even lung endometriosis with/without catamenial pneumothorax and hemoptysis or smooth-muscle lesions (benign metastasizing leiomyoma) enter in the differential diagnosis with LAM.
2 Case Presentation
-
Clinical Background
-
46-year-old woman
-
Former smoker
-
Unremarkable past medical history
-
-
Onset of Symptoms
-
Progressive dyspnea
-
Recurrent pneumothorax
-
-
Laboratory Findings
-
Routine laboratory tests: unremarkable
-
Autoimmune serum tests: negative
-
-
Pulmonary Function Test
-
FVC 82%; FEV1 61%; FEV1/FVC 58%; RV 88%; DLCO 65%
-
-
Imaging (Figs. 14.1, 14.2, and 14.3)
-
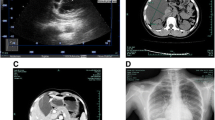
CT scan shows numerous and bilateral thin-walled cysts of variable size, diffusely distributed through all pulmonary fields without a preferential location and involving costophrenic angles.
-
-
Cryobiopsy (Figs. 14.4, 14.5, 14.6, 14.7, and 14.8)
-
The specimen obtained from the transbronchial cryobiopsy technique consists of three fragments of pulmonary parenchyma ranging from 5 to 8 mm of maximum diameter. Histologically, the lung tissue shows the presence of few cystic spaces characterized by a thin layer of spindled cells with smooth-muscle cell appearance and slightly hyperplastic pneumocytes (Figs. 14.4 and 14.5). Some calcifications are present around spindle cell proliferations. At immunohistochemistry, these cells show a patchy, weak, and focal expression for HMB45 (Fig. 14.6), nuclear positivity with estrogen receptors (Fig. 14.7), and consistent and diffuse staining for cathepsin-K (Fig. 14.8). In addition, a positive staining is observed with smooth-muscle actin and focally with MART-1.
-
-
Diagnosis
-
Pulmonary lymphangioleiomyomatosis with high level of diagnostic confidence.
-

CT scan showing several thin-walled rounded cysts involving bilaterally and diffusely the lung fields without costophrenic angles preservation (upper lobes)

CT scan showing several thin-walled rounded cysts involving bilaterally and diffusely the lung fields without costophrenic angles preservation (upper lobes and middle lobe)

CT scan showing several thin-walled rounded cysts involving bilaterally and diffusely the lung fields without costophrenic angle preservation (lower lobes)

Transbronchial cryobiopsy showing normal lung and subtle aggregates of LAM cells showing smooth-muscle-like appearance and cytoplasmic vacuolization leading to clear changes. Some calcifications are also noted

Transbronchial cryobiopsy showing normal lung and subtle aggregates of LAM cells showing smooth-muscle-like appearance and cytoplasmic vacuolization leading to clear changes

Focal immunohistochemical expression for HMB45 in the cytoplasm of LAM cells

LAM cells showing nuclear staining with estrogen receptor

Consistent immunohistochemical expression for cathepsin-K in LAM cells
3 Discussion
Transbronchial cryobiopsy may be used in patients with suspected LAM. Anecdotic reports also suggest that lung complications related to the procedure have a limited clinical impact. Lung tissue samples so obtained are well preserved, and immunohistochemical analyses or even molecular investigations may be done easily. Recently an increased PD-L1 expression in lung tissue from LAM patients has been proven suggesting new opportunities for therapeutic targeting [49]. The need of lung tissue that is now limited to cases that do not meet all the criteria required for a clinic-radiologic- and laboratory-based diagnosis will probably gain importance.
References
McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133:507–16.
Taylor JR, Ryu J, Colby TV, Raffin TA. Lymphangioleiomyomatosis. Clinical course in 32 patients. N Engl J Med. 1990;323:1254–60.
Abbott GF, Rosado de Christenson ML, Frazier AA, Franks TJ, Pugatch RD, Galvin JR. From the archives of the AFIP: lymphangioleiomyomatosis: radiologic-pathologic correlation. Radiographics. 2005;25:803–28.
Juvet SC, McCormack FX, Kwiatkowski DJ, Downey GP. Molecular pathogenesis of lymphangioleiomyomatosis: lessons learned from orphans. Am J Respir Cell Mol Biol. 2007;36:398–408.
Morbini P, Guddo F, Contini P, Luisetti M, Schiavina M, Zompatori M. Rare diffuse diseases of the lung. Pulmonary alveolar proteinosis, lymphangioleiomyomatosis, amyloidosis. Pathologica. 2010;102:547–56.
Krymskaya VP, McCormack FX. Lymphangioleiomyomatosis: a monogenic model of malignancy. Annu Rev Med. 2017;68:69–83.
Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97:6085–90.
Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998;62:810–5.
Dibble CC, Elis W, Menon S, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell. 2012;47:535–46.
Goncharova EA, Goncharov DA, Eszterhas A, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem. 2002;277:30958–67.
Schiavina M, Di Scioscio V, Contini P, Cavazza A, Fabiani A, Barberis M, et al. Pulmonary lymphangioleiomyomatosis in a karyotypically normal man without tuberous sclerosis complex. Am J Respir Crit Care Med. 2007;176:96–8.
Aubry MC, Myers JL, Ryu JH, et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med. 2000;162:749–52.
Miyake M, Tateishi U, Maeda T, et al. Pulmonary lymphangioleiomyomatosis in a male patient with tuberous sclerosis complex. Radiat Med. 2005;23:525–7.
Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151:527–33.
Finlay G. The LAM cell: what is it, where does it come from, and why does it grow? Am J Physiol Lung Cell Mol Physiol. 2004;286:L690–3.
Crooks DM, Pacheco-Rodriguez G, DeCastro RM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004;101:17462–7.
Chang WY, Cane JL, Blakey JD, Kumaran M, Pointon KS, Johnson SR. Clinical utility of diagnostic guidelines and putative biomarkers in lymphangioleiomyomatosis. Respir Res. 2012;13:34.
Matsui K, Takeda K, Yu ZX, Travis WD, Moss J, Ferrans VJ. Role for activation of matrix metalloproteinases in the pathogenesis of pulmonary lymphangioleiomyomatosis. Arch Pathol Lab Med. 2000;124:267–75.
Hayashi T, Fleming MV, Stetler-Stevenson WG, et al. Immunohistochemical study of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in pulmonary lymphangioleiomyomatosis (LAM). Hum Pathol. 1997;28:1071–8.
Glassberg MK, Elliot SJ, Fritz J, et al. Activation of the estrogen receptor contributes to the progression of pulmonary lymphangioleiomyomatosis via matrix metalloproteinase-induced cell invasiveness. J Clin Endocrinol Metab. 2008;93:1625–33.
Odajima N, Betsuyaku T, Nasuhara Y, Inoue H, Seyama K, Nishimura M. Matrix metalloproteinases in blood from patients with LAM. Respir Med. 2009;103:124–9.
Urban T, Lazor R, Lacronique J, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM“O”P). Medicine (Baltimore). 1999;78:321–37.
Cudzilo CJ, Szczesniak RD, Brody AS, et al. Lymphangioleiomyomatosis screening in women with tuberous sclerosis. Chest. 2013;144:578–85.
Astrinidis A, Khare L, Carsillo T, et al. Mutational analysis of the tuberous sclerosis gene TSC2 in patients with pulmonary lymphangioleiomyomatosis. J Med Genet. 2000;37:55–7.
Gupta R, Kitaichi M, Inoue Y, Kotloff R, McCormack FX. Lymphatic manifestations of lymphangioleiomyomatosis. Lymphology. 2014;47:106–17.
Hayashi T, Kumasaka T, Mitani K, et al. Prevalence of uterine and adnexal involvement in pulmonary lymphangioleiomyomatosis: a clinicopathologic study of 10 patients. Am J Surg Pathol. 2011;35:1776–85.
Matsui K, Tatsuguchi A, Valencia J, et al. Extrapulmonary lymphangioleiomyomatosis (LAM): clinicopathologic features in 22 cases. Hum Pathol. 2000;31:1242–8.
Torres VE, Björnsson J, King BF, et al. Extrapulmonary lymphangioleiomyomatosis and lymphangiomatous cysts in tuberous sclerosis complex. Mayo Clin Proc. 1995;70:641–8.
Kerr LA, Blute ML, Ryu JH, Swensen SJ, Malek RS. Renal angiomyolipoma in association with pulmonary lymphangioleiomyomatosis: forme fruste of tuberous sclerosis? Urology. 1993;41:440–4.
De Pauw RA, Boelaert JR, Haenebalcke CW, Matthys EG, Schurgers MS, De Vriese AS. Renal angiomyolipoma in association with pulmonary lymphangioleiomyomatosis. Am J Kidney Dis. 2003;41:877–83.
Gupta N, Finlay GA, Kotloff RM, Strange C, Wilson KC, Young LR, Taveira-DaSilva AM, Johnson SR, Cottin V, Sahn SA, Ryu JH, Seyama K, Inoue Y, Downey GP, Han MK, Colby TV, Wikenheiser-Brokamp KA, Meyer CA, Smith K, Moss J, McCormack FX, ATS Assembly on Clinical Problems. Lymphangioleiomyomatosis diagnosis and management: high-resolution chest computed tomography, transbronchial lung biopsy, and pleural disease management. An official American Thoracic Society/Japanese Respiratory Society clinical practice guideline. Am J Respir Crit Care Med. 2017;196(10):1337–48.
Leslie KO, Gruden JF, Parish JM, Scholand MB. Transbronchial biopsy interpretation in the patient with diffuse parenchymal lung disease. Arch Pathol Lab Med. 2007;131:407–23.
Koba T, Arai T, Kitaichi M, Kasai T, Hirose M, Tachibana K, Sugimoto C, Akira M, Hayashi S, Inoue Y. Efficacy and safety of transbronchial lung biopsy for the diagnosis of lymphangioleiomyomatosis: a report of 24 consecutive patients. Respirology. 2018;23(3):331–8.
Meraj R, Wikenheiser-Brokamp KA, Young LR, Byrnes S, McCormack FX. Utility of transbronchial biopsy in the diagnosis of lymphangioleiomyomatosis. Front Med. 2012;6(4):395–405.
Harari S, Torre O, Cassandro R, Taveira-DaSilva AM, Moss J. Bronchoscopic diagnosis of Langerhans cell histiocytosis and lymphangioleiomyomatosis. Respir Med. 2012;106(9):1286–92.
Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–60.
Karbowniczek M, Astrinidis A, Balsara BR, et al. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167:976–82.
Bittmann I, Rolf B, Amann G, Löhrs U. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insights into pathogenesis. Hum Pathol. 2003;34:95–8.
Steagall WK, Zhang L, Cai X, Pacheco-Rodriguez G, Moss J. Genetic heterogeneity of circulating cells from patients with lymphangioleiomyomatosis with and without lung transplantation. Am J Respir Crit Care Med. 2015;191:854–6.
Fruchter O, Fridel L, El Raouf BA, Abdel-Rahman N, Rosengarten D, Kramer MR. Histological diagnosis of interstitial lung diseases by cryo-transbronchial biopsy. Respirology. 2014;19(5):683–8.
Rossi GA, Balbi B, Oddera S, Lantero S, Ravazzoni C. Response to treatment with an analog of the luteinizing-hormone-releasing hormone in a patient with pulmonary lymphangioleiomyomatosis. Am Rev Respir Dis. 1991;143:174–6.
Taveira-DaSilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126:1867–74.
McCormack FX, Gupta N, Finlay GR, Young LR, Taveira-DaSilva AM, Glasgow CG, Steagall WK, Johnson SR, Sahn SA, Ryu JH, Strange C, Seyama K, Sullivan EJ, Kotloff RM, Downey GP, Chapman JT, Han MK, D’Armiento JM, Inoue Y, Henske EP, Bissler JJ, Colby TV, Kinder BW, Wikenheiser-Brokamp KA, Brown KK, Cordier JF, Meyer C, Cottin V, Brozek JL, Smith K, Wilson KC, Moss J, ATS/JRS Committee on Lymphangioleiomyomatosis. Official American Thoracic Society/Japanese Respiratory Society clinical practice guidelines: lymphangioleiomyomatosis diagnosis and management. Am J Respir Crit Care Med. 2016;194(6):748–61.
Nijmeh J, El-Chemaly S, Henske EP. Emerging biomarkers of lymphangioleiomyomatosis. Expert Rev Respir Med. 2018;12(2):95–102.
Tanaka H, Imada A, Morikawa T, et al. Diagnosis of pulmonary lymphangioleiomyomatosis by HMB45 in surgically treated spontaneous pneumothorax. Eur Respir J. 1995;8:1879–82.
Fetsch PA, Fetsch JF, Marincola FM, Travis W, Batts KP, Abati A. Comparison of melanoma antigen recognized by T cells (MART-1) to HMB-45: additional evidence to support a common lineage for angiomyolipoma, lymphangiomyomatosis, and clear cell sugar tumor. Mod Pathol. 1998;11:699–703.
Chilosi M, Pea M, Martignoni G, Brunelli M, Gobbo S, Poletti V, et al. Cathepsin-k expression in pulmonary lymphangioleiomyomatosis. Mod Pathol. 2009;22:161–6.
Martignoni G, Pea M, Reghellin D, et al. Molecular pathology of lymphangioleiomyomatosis and other perivascular epithelioid cell tumors. Arch Pathol Lab Med. 2010;134:33–40.
Maisel K, Merrilees M, Atochina-Vassermaqn EN, et al. Immune checkpoint ligand PD-L1 is upregulated in pulmonary lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2018;59(6):723–32.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Rossi, G., Valli, M., Dubini, A., Spagnolo, P. (2019). Lymphangioleiomyomatosis. In: Poletti, V. (eds) Transbronchial cryobiopsy in diffuse parenchymal lung disease. Springer, Cham. https://doi.org/10.1007/978-3-030-14891-1_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-14891-1_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14890-4
Online ISBN: 978-3-030-14891-1
eBook Packages: MedicineMedicine (R0)