
Abstract
Optogenetics and photopharmacology are two perspective modern methodologies for control and monitoring of biological processes from an isolated cell to complex cell assemblies and organisms. Both methodologies use optically active components that being introduced into the cells of interest allow for optical control or monitoring of different cellular processes. In optogenetics, genetic materials are introduced into the cells to express light-sensitive proteins or protein constructs. In photopharmacology, photochromic compounds are delivered into a cell directly but not produced inside the cell from a genetic material. The development of both optogenetics and photopharmacology is inseparable from the design of improved tools (protein constructs or organic molecules) optimized for specific applications. Herein, we review the main tools that are used in modern optogenetics and photopharmaclogy and describe the types of cellular processes that can be controlled by these tools. Although a large number of different kinds of optogenetic tools exist, their performance can be evaluated with a limited number of metrics that have to be optimized for specific applications. We classify these metrics and describe the ways of their improvement.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Modern photonics provides versatile tools for control and monitoring of biological processes from an isolated cell to complex cell assemblies and organisms. The most straightforward approach is to use intrinsic optical properties of molecules and molecular aggregates that are produced naturally in the cell. Several implementations of this approach have been developed and used for biological and medical applications [1,2,3]. However, the real progress was made by using specially designed tools with desired optical, chemical, and biological properties that, being incorporated into the cellular processes, allow changing them after absorption of a photon at the specific wavelength.
Two main methodologies have been proposed for such optical monitoring and control of cellular processes: optogenetics and photopharmacology. Their key difference is in the tools employed. In optogenetics, light-sensitive proteins or protein constructs are genetically introduced into the cells of interest becoming an integral part of the cellular machinery [4]. Illumination of cells modified in such way allows for the change of various cellular properties and processes, for example, the membrane potential or gene transcription. In photopharmacology, photochromic compounds are delivered into the cell or to its surface [5, 6]. Such compound has to satisfy two requirements: the compound can be transferred from one form to another form by a light stimulus, and physiological activities of these two forms must differ. One of the interesting examples is photochromic ion channel blockers—compounds that block a channel in one form and open in another one, allowing for the change in ion transport from extracellular to intracellular regions.
The development of both optogenetics and photopharmacology is inseparable from the design of improved tools (protein constructs or organic molecules) optimized for specific applications. These improvements regard either to the biological features, such as expression levels, toxicity, an efficiency of localization at the target sites, either to the properties of the light-induced response. In this review, we will cover the main factors determining the functionality of both optogenetic and photopharmacological tools and consider the commonly applied methods for their improvement.
This review is organized as follows. First, we will introduce the types of cellular processes that can be controlled with modern optogenetic and photopharmacological tools. For each target process, the applicable tools will be described. Although a large number of different kinds of optogenetic tools exist, their performance can be evaluated with a limited number of metrics. In the following subsections, we will classify these metrics in the Tables 8.1 and 8.2. For each metric, we will consider challenges and proposed solutions.
2 Optogenetics. Properties to Control or Monitor and Applied Tools
In this section we will consider cellular properties and processes that can be controlled or monitored with optogenetic tools. For each application, we will describe the classes of tools and briefly describe the molecular mechanisms of their functioning.
Control of electrical activity of excitable cells (neurons and cardiomyocytes). In order to achieve this goal, one has to activate ion currents through the cell membrane [7, 8]. Two main protein families are used for this purpose.
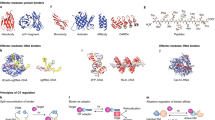
Microbial rhodopsins. Microbial rhodopsins are heptahelical transmembrane proteins naturally found in archaea, algae, fungi, and bacteria (Fig. 8.1a). Working as ion pumps or ion channels, microbial rhodopsins allow effective activation and inhibition of neural electrical activity by changing ion currents through the membrane.


Structure of main classes of optogenetic tools. a Microbial rhodopsin N. pharaonis halorhodopsin (PDB entry 3A7K) with an all-trans retinal chromophore, b Visual rhodopsin from T. Pacificus (PDB entry 2Z73) with an 11-cis retinal chromophore, c–e LOV domain (PDB entry 5EFW) (c), BLUF domain (PDB entry 2IYG) (d) and cryptochrome (PDB entry 2J4D) (g) are optogenetic tools with a flavin-based chromophore, flavin mononucleotide is presented in the figure, f Phytochrome (PDB entry 4OUR) with bilin chromophore, g UVR8 receptor (PDB entry 4DNW) utilizing tryptophan clusters for light absorption
Activation of neural excitability is achieved by intracellular transport of cations, leading to the neural depolarization. Proton-pumping channelrhodopsins are the most commonly used actuators of neural activity [9,10,11,12].
The detailed molecular mechanism of proton pumping in channelrhodopsin has not been determined yet. However, it is known that at some stages of the photocycle the retinal chromophore becomes deprotonated and the opsin converts into an open-gate state, allowing for proton transfer from the chromophore into the cell (neuron). Afterward, the retinal chromophore can be reprotonated with a proton from the intracellular medium and the opsin converts back into a closed-gate state [13]. Several studies demonstrated that channelrhodopsins can function not only as proton pumps but also as cation channels, allowing for cation transfer inside a cell [12]. The pumped protons (or other cations) change the membrane potential of the neuron, leading to its depolarization.
Inhibition of neural activity with other microbial rhodopsins can be achieved via three mechanisms.
-
(1)
Neural hyperpolarization achieved by outward cation transport or inward anion transport. Tools for outward cation transport: proton-pumping archaerhodopsins [14, 15], natural and engineered sodium pumps [16, 17], and potassium pumps [18]. Tools for inward anion transport: chloride-pumping halorhodopsins and cruxhalorhodopsins [19, 20]. Here, the conformational changes in proteins triggered by light absorption lead to the formation of an ion pore, which is highly selective to a certain ion type [18,19,20]. This selectivity is determined by the amino acids forming the pore, however, exact mechanisms are not always clear.
-
(2)
Blocking neural depolarization by triggering “shunting” currents of chloride ions through light-gated chloride channels. Initially, light-activated chloride channels were engineered on the basis of proton-pumping channelrhodopsins [21, 22]. A single replacement of a negative residue on a positive one led to the change of protein ion specificity. Later natural chloride channels with increased conductance were found [23, 24]. While the exact working principle of these anion-conducting channelrhodopsins is not clear yet, recent site-directed mutagenesis studies support the idea that two mechanisms are involved.
The first mechanism is mostly regulated by the protonation of a single Glu68 residue [25]. The slight change of conductance efficiency with pH supports the idea that another pH-dependent group is also involved into the channel dynamics. The second mechanism is fully regulated with a conserved Cys102 residue. Probably, Cys102 residue forms a hydrogen bond with a conserved Asp156 residue in a closed-gate state. Upon light absorption, the hydrogen bond dissociates, and the subsequent conformational changes lead to the formation of an open-gate state [25]. The channel opening leads to the occurrence of “extra” flow of cations, which decreases the local electrical resistance of the cellular system and, subsequently, decreases the excitatory postsynaptic potential.
-
(3)
Excitation of inhibitory interneurons with proton-pumping channelrhodopsins [26]. The activation of interneurons leads to the silencing of local areas of the brain [27].
GPCRs. G protein-coupled receptors constitute a large class of transmembrane proteins found in eukaryotes (Fig. 8.1b). Upon activation with photon absorption or binding to a specific signaling molecule, GPCRs activate signaling transduction by interacting with a specific G protein. GPCRs are most commonly used for the inhibition of neural activity through intrinsic Gi/o and Gs pathways. Usually, visual rhodopsins from vertebrates and invertebrates are used [28, 29].
Specifically, light activation triggers the interaction of visual rhodopsin with G proteins. G proteins are involved in the regulation of G protein inward rectifying potassium (GIRK) channels and presynaptic calcium channels. Thus, the light-activated interaction between rhodopsins and G proteins leads to the light-activated ion flow through GIRK and presynaptic calcium channels that induces neural inhibition [28].
In other studies bi-stable melanopsins, neuropsins, and parapinopsins that can be switched on and off with brief pulses of light with different wavelengths are used [30].
In order to change the G protein-coupling specificity of GPCR, an OptoXR approach is used. In the OptoXR method, rhodopsin intracellular or extracellular loops are replaced with corresponding parts of the ligand-activated GPCR with required specificity [31,32,33]. Alternatively, the ligand-activated GPCR can be attached to the C- or N-terminus of the light-activated rhodopsin [34].
Control of synaptic transmission between neurons. In order to achieve this goal, modern optogenetic tools that can change pH levels in the presynaptic region of neurons are used. Up to date, only proteins from the family of microbial rhodopsins were utilized for this purpose.
Microbial rhodopsins. Microbial rhodopsin archaerhodopsin-3 expressed in presynaptic terminals demonstrated light-activated inhibition of neurotransmitter release via increasing intracellular pH levels [35, 36]. The substantial increase of pH levels at the presynaptic terminals is induced by the light-activated proton pumping activity of archaerhodopsin-3. However, the exact mechanism of neurotransmitter release inhibition via pH increase is not determined yet, even though several speculations have been made [35].
Control of enzyme activity. In order to achieve this goal, modern optogenetic tools mediate a light-induced structural reorganization of a target enzyme. This allows controlling intracellular concentrations of second messengers, such as cyclic adenosine monophosphate (cAMP). Proteins from five protein families are used for this purpose.
LOV proteins. LOV proteins, naturally found in plants, bacteria, algae and fungi, absorb blue light with a flavin mononucleotide (FMN) chromophore (Fig. 8.1c). For optogenetic applications they are attached to specific functional domains. Light activation triggers structural reorganization in the LOV protein.
Specifically, this reorganization starts from the formation of a covalent bond between the FMN chromophore and the conserved cysteine residue [37]. The reorganization is then transmitted via hydrogen bonds and salt bridges to the alpha-helical linker, leading to its rotation. The rotation of the linker in its turn causes the reorientation of the functional domain, triggering its activation or inactivation. The induced effect depends on the number of coils in the alpha-helical linker, but does not critically depend on the linker length. Thus, addition of seven coils conserves the rotational angle of the linker and has a little effect on the signal transmission from the LOV domain to the functional domain.
In one of the examples, LOV domain was attached to histidine kinase (HisK) instead of its natural chemosensory domain, which allowed light activation of the enzyme [38]. Here, LOV-HisK constructs were paired in order to achieve the effective light modulation of HisK activity. It was demonstrated, that alteration of alpha-helical linker length led to three different cases: activation, deactivation or independence of HisK activity upon blue light illumination, supporting the assumption of crucial dependence of LOV-triggered effects on the linker rotational angle.
In other applications to modulate calcium concentrations, LOV proteins were attached to calcium-binding proteins, altering their calcium-binding capability [39]. The same strategy was used to activate Rac1, regulating cell protrusions [40].
Blue Light Utilizing Flavine adenine dinucleotide (FAD) (BLUF) domains. The action mechanism of BLUF domains is similar to that of LOV proteins (Fig. 8.1d). Photon absorption by the flavin chromophore also triggers structural reorganization of these proteins, which is transferred to distinct functional domains. It is proposed that the first steps of this photoactivated reorganization involve the electron transfer between FAD and the conserved tyrosine amino acid, leading to the formation of two radicals.
Subsequently, the formation of the radicals leads to the alteration of a hydrogen-bonding network of the protein and induces significant conformational alterations. However, the mechanism describing how this structural reorganization is transmitted to the linker between BLUF and functional domains remains is completely unclear [41].
In one of the experiments considering blue light triggered enzyme activation, BLUF domain was coupled to the catalytic domain that produces cAMP second messenger, which allowed blue-light mediated cAMP production [42, 43].
Cryptochromes. Cryptochromes are the third class of optogenetic tools that use the flavine chromophore (FAD) for blue light absorption (Fig. 8.1e). Cryptochromes are naturally found in plants and animals. Their functioning is based on the same principles as the functioning of LOV proteins and BLUF domains. Thus, attachment of cryptochromes to specific enzymes allows for light-induced activation of these proteins. For example, the light-inducible region of Arabidopsis thaliana cryptochrome attached to its binding partner allowed for the activation of the tropomyosin-related kinase, which regulates neurotrophin signaling [44].
Phytochromes. Phytochromes are photoreceptors naturally found in plants. They use bilin chromophore for light absorption and are attached to distinct functional domains in optogenetic experiments (Fig. 8.1f). In a recent study, a red light-activated bacterial phytochrome was linked via an alpha-helical linker to an effector module of Homo sapiens phosphodiesterase 2A. Resulting light-activated phosphodiesterase (LAPD) modulated the hydrolysis of cAMP and cGMP [45]. The mechanism of signal transduction here is similar to that of LOV domains; however, it is not completely described yet.
Microbial rhodopsins. The microbial rhodopsin from fungus Blastocladiella emersonii fused to the guanylyl cyclase catalytic domain allowed green light-activated modulation of cGMP levels [46, 47]. These unique eight alpha-helical rhodopsins allowed for the light-modulated control of cyclic nucleotide gated channels opening, modulating the intracellular cGMP levels.
Recruitment of protein to plasma membrane or organelles. In order to achieve this goal, one has to alter protein binding to specific functional proteins that are attached to the membrane or an organelle in a light-dependent manner. This allows controlling protein availability to its binding partners, modulating its activity. Three main protein families are used for this purpose.
LOV proteins. Light-activated interaction of LOV domain from Arabidopsis thaliana with GIGANTEA protein mediated the recruitment of yellow fluorescent protein to the plasma membrane, which led to the dimerization of the target protein [48]. In other works light-induced protein binding was used for activating scaffold proteins, kinases, and nucleotide-exchange factors [49].
Phytochromes. Plant photoreceptor phytochrome B from Arabidopsis thaliana coupled with phytochrome interaction factor 3 (PIF3) was attached to the cellular membrane. Red-light illumination regulated the binding of PIF3 to target proteins, in this way regulating attachment of target proteins to the membrane. This approach was used to modulate intracellular signaling by changing protein availability to its binding partners and to control nuclear localization of proteins [50,51,52].
Cryptochromes. The activation of Raf kinase occurs when the protein is bound to the cellular membrane. In a recent study, Raf proteins were labeled with cryptochromes. Upon blue-light absorption, cryptochromes attached to their interaction partners, which were recruited to the plasma membrane. Thus, Raf kinase recruitment and activation occurred [53].
Light modulated organelle relocalization. In order to achieve this goal, one has to induce organelle binding to specific functional proteins that are attached to mobile intracellular structures. Control of organelle relocalization allows the modulation of cellular signaling, cell growth, and other vital cellular activities. Up to date, only LOV proteins and cryptochromes were used for this purpose.
Light-oxygen-voltage (LOV) proteins. In order to achieve blue-light induced relocalization of peroxisomes, they were fused to the LOV domain from Avena sativa phototropin 1. Upon blue-light absorption, LOV domain attached to the cytoskeletal motor protein kinesin, which moved the peroxisome [54].
Cryptochromes. In one of the experiments, organelles were labeled with cryptochrome 2. Upon blue-light activation, cryptochrome bound to its interaction partner CIB1. CIB1 in its turn was fused to intracellular molecular motor kinesin. Thus, blue-light activation bound organelles to kinesin, which resulted in their translocation [55].
Control of protein-protein interactions. In order to alter protein-protein interactions, one has to change the position of the target protein relative to its interaction partner. This allows for modulation of protein activation and deactivation, and can be used for light-modulated opening of ion channels. Two main protein classes are used for this purpose.
LOV proteins. In one of the experiments, the photoswitch consisted of a LOV domain bound to a peptide toxin that blocks ion channels. Upon light activation, the photoswitch unfolded, lowering toxin concentration near the cell membrane. The lack of toxin near the cell membrane led to unblocking of ion channels [56]. In another study, target protein was bound to the localized LOV domain. Upon blue-light absorption, target protein detached from LOV domain and moved to the site of action [49]. LOV domains bound to peptides allowed for blue-light mediated control of peptide binding affinity [57].
Phytochromes. Phytochrome B from Arabidopsis thaliana with its interaction partner PIF3 was combined into a conditional protein splicing system. Red light absorption led to the attachment of phytochrome to the specific intein, which triggered protein splicing processes [58].
Control of gene transcription and expression. In order to induce gene transcription or expression in a light-dependent manner, modern optogenetic tools activate specific DNA binding proteins. Four main protein classes are used for this purpose.
LOV domains. Fusion of a LOV domain with DNA-binding protein EL222 prevented the dimerization of the latter protein. Upon blue-light absorption EL222 detached from the LOV domain, underwent dimerization, which led to DNA-binding and triggering of the gene transcription [59]. Light-induced interaction of LOV domain from Arabidopsis thaliana with GIGANTEA protein bound to the DNA-binding domain also allowed to regulate gene transcription [48].
Phytochromes. Red light absorption triggers interaction of phytochrome B with its interaction partner PIF6, reconstituting a factor mediating gene transcription. Upon far-red light absorption, the interaction breaks, silencing gene transcription [60].
Cryptochromes. Plant cryptochrome 2 binds Arabidopsis CIB1 protein upon blue-light absorption. This system allows for light-activated DNA binding and activation of gene expression and DNA transcription [61].
UV Resistance locus 8 (UVR8) receptors. UVR8 receptors are naturally found in plants. These proteins utilize tryptophan clusters for UV-light absorption (Fig. 8.1f). UVR8 receptors from A. thaliana in the dark-adapted state are organized in homo-dimers. Light activation leads to UVR8 monomerization.
As it was demostrated in recent mutational studies supported by dynamic crystallography and quantum chemical calculations, in the dark adapted state the two UVR8 monomers are coupled together with a network of hydrogen bonds and salt bridges [62, 63]. Upon light absorption, the charge separation in the conserved Trp233/Trp285 residues occurs, which leads to the disruption of salt bridges between arginine and aspartic acid residues that connected the two monomers. The molecular dynamics simulations also support the idea that upon light absorption the water molecule located at the interface of two UVR8 monomers is released, which also results in the dissociation of hydrogen bonds between the monomers. However, these assumptions have to be proved experimentally in further investigations [62].
In a construct for UV-activated gene expression, the monomeric form of UVR8 receptor interacts with the E3-ubiquitin ligase COP1 factor activating gene expression [64]. Specifically, upon light absorption the monomeric UVR8 receptor binds to the WD40 domain of the COP1 factor, which activates the target promoter and starts the gene expression [64].
Monitoring the electrical activity of excitable cells (neurons). In order to detect the changes of cellular membrane potential, modern optogenetics uses tools that alter their fluorescent signal upon the change of external electric potential.
Microbial rhodopsins. Several microbial rhodopsins—archaerhodopsin-3, green light-absorbing proteorhodopsin, and Gloebacter violaceus rhodopsin demonstrate the voltage-dependent intensity of fluorescence [65,66,67,68]. They absorb light with the retinal chromophore that can be either in all-trans or 13-cis conformations in the dark-adapted state (corresponding to two coexisting conformations of proteins).
The fluorescence spectroscopy and near-IR resonance Raman confocal microscopy studies of archaerhodopsin-3 suggest that the fluorescent Q-form of the protein is an intermediate that is generated after the photoexcitation of the 13-cis archaerhodopsin-3 conformation [69, 70]. Specifically, during the photocycle of archaerhodopsin-3 the deprotonation of the retinal chromophore occurs (M-state). Subsequently, the chromophore is reprotonated (N-state), and the photoexcitation of the N-state generates the Q-state. In its turn the photoexcitation of the Q-state results in the fluorescent signal.
It is assumed that membrane voltage regulates the equilibrium between the deprotonated M and the protonated N-states, thus regulating the concentration of the Q-form. However, the exact mechanism of potential-dependent fluorescence of archaerhodopsin-3 and other similar proteins is not clear yet [69].
Sensors based on fluorescent proteins. A fluorescent protein or two fluorescent proteins (FPs) are attached to a functional voltage-sensing domain. Membrane voltage triggers structural reorganization in the functional domain, which is transferred to FPs via a connecting linker, altering the fluorescent signal [71,72,73]. For example, in VSFP1 voltage sensor, two fluorescent proteins (CFP and YFP) connected with a flexible linker were fused to the fourth alpha-helix of the Ciona intestinalis voltage-sensing domain [74]. The alteration of membrane voltage induced the rotation of the fourth alpha-helix of the domain and consequently changed the relative orientation of the two fluorescent proteins. In its turn the change of orientation altered the Fluorescence Resonance Energy Transfer (FRET) efficiency between the proteins, and caused the alteration of the output fluorescent signal [74].
In recent studies, GFP was introduced into the neuron in combination with the fluorescent voltage-dependent microbial rhodopsin archaerhodopsin-3. Membrane voltage altered the intensity of the fluorescent signal of archaerhodopsin-3 that was absorbed with the GFP via FRET, which caused a more substantial alteration in the GFP fluorescence [75].
Monitoring the intracellular pH levels. In order to detect the changes of pH levels, modern optogenetics uses fluorescent proteins that alter their signal properties upon the change of intracellular or extracellular pH level.
Sensors based on fluorescent proteins. Specific mutations in the vicinity of fluorescent protein active site shift the pKa values of the protein chromophore and endow the GFP and its analogs (such as YFP, RFP) with the pH sensitivity [76].
Specifically, the intensities of absorption peaks at two wavelengths, corresponding to the protonated and deprotonated states of the protein chromophore, change with the alteration of the intracellular pH value [77]. Most probably, intracellular pH value regulates the relative concentration of the protonated and deprotonated states of the fluorescent protein.
Optical detection of redox reactions. In order to detect redox reaction, modern optogenetics uses tools that change the properties of their fluorescent signal upon oxidation or reduction.
Sensors based on fluorescent proteins. Redox reactions are indicated by H2O2 concentrations. Fluorescent reporters either consist of a fluorescent protein and a distinct H2O2 sensor, which undergoes structural reorganization upon oxidation, either is represented by mutated fluorescent proteins [78, 79]. In the latter case, additional peptides for the structural stabilization of the fluorescent protein after oxidation are required [79].
Specifically, the first H2O2 sensor consisted of a circularly permutated yellow fluorescent protein (cpYFP) that was inserted into the regulatory domain (RD) of the H2O2 sensitive protein (OxyR). Upon oxydation, the Cys199 converted into a sulfenic acid derivative and formed a disulfide bond with the Cys208 of the RD. The formation of the disulfide bridge dramatically changed the conformation of the RD, leading to the increase of the cpYFP fluorescence [78].
In further investigations, the cpRFP was modified in such a way that the oxydation led to the disulfide bridge formation between cysteines located at the N- and C-termini of the protein, and the induced conformational change led to the alteration of the protein fluorescent signal [79].
Monitoring the intracellular ion concentrations. In order to achieve this goal, modern optogenetics uses fluorescent proteins that change their signal properties upon binding to specific ions.
Sensors based on fluorescent proteins. Specific mutations created fluorescent proteins with conformational changes of their chromophore upon ion binding. This conformational change leads to the change of the fluorescent signal. For example, the chromophore of the GFP S65T/T203Y mutant in chloride-unbound form forms a hydrogen bond with the Tyr203 residue of the apoprotein [80]. The bond dissociation upon chloride binding to Tyr203 leads to the increased distance between the GFP chromophore and the Tyr203 residue and causes the increase of absorbance at 400 nm.
In other constructs, more complex systems involving several fluorescent proteins were developed. For example, in one of the experiments, increase of chloride concentration reduced the excitation efficiency of YFP which was connected with CFP in a FRET system. Thus, the increase of chloride concentration led to the increase of CFP fluorescence relative to the YFP fluorescence [81].
Sensors for a number of ions were developed, including calcium, cadmium, zinc ions [80,81,82]. Here, two general types of sensors exist. In the sensors of first type, ions interact not with fluorescent proteins, but rather with distinct modules. For example, in a calcium sensor the CFP-YFP FRET system was linked to the calmodulin-M13 (CaM-M13) system [81, 83]. In the absence of calcium ions CaM was not bound to M13 and the two fluorescent proteins were located far away from each other, not allowing FRET to occur. Interaction of CaM with calcium ions led to the CaM-M13 binding, decrease of the distance between CFP and YFP and the occurrence of FRET between the two proteins [81].
The second type of sensors does not involve distinct domains for ion binding. For example, in one of zinc sensors, specific mutations were introduced into CFP and YFP, creating zinc-binding sites at their surface. These two fluorescent proteins were linked in a single FRET system. Upon zinc binding, the two proteins stuck together via zinc bridges, which led to the sufficient change of FRET efficiency between the two proteins [84].
Optical monitoring protein expression and protein-protein interactions. In order to achieve this goal, the tool must to change the properties of its fluorescent signal upon binding or interacting with specific proteins.
LOV proteins. LOV proteins have intrinsic dim fluorescence. Rational mutagenesis of the protein active site increased this fluorescence and allowed using LOV proteins as fluorescent reporters of protein-protein interactions [85].
3 Optogenetics. Main Properties to Optimize
In this section, we will consider the main properties that define the performance of optogenetic tools. For each property, we will briefly describe the main directions of improvement and the approaches that are usually used for the optimization of this property to specific experimental conditions. This information is summarized in Table 8.1.
Signal intensity. A lot of studies were aimed at increasing the signal intensity of optogenetic tools. For tools that are aimed at the control of cellular processes, the increase of signal intensity is required to achieve the desired effect upon moderate illumination. For fluorescent reporters of cellular processes or properties, the increase of fluorescent signal intensity allows for more reliable measurements.
Microbial rhodopsins for control of neural activity. Ion pumping microbial rhodopsins usually demonstrate suboptimal currents, because a single ion is conducted upon absorption of a single photon. Weak ion currents cannot trigger strong action potentials or completely inhibit neural electrical activity.
One of the solutions is increasing the conductivity of microbial rhodopsins. For example, proton-pumping channelrhodopsins were converted into light-gated anion channels. In an open-gate state, these constructs allowed chloride ions to move freely through the pore, shunting excitatory ion currents.
Substitution of a single acidic residue with a basic residue in the ion-pumping region led to the halide selectivity of channelrhodopsin-2 [21, 22]. While the first variants demonstrated residual proton currents, additional mutagenesis solved this problem, leading to variants with even more intense photocurrents [119]. Natural anion selective channelrhodopsins were even more efficient [23, 24]. Another approach to obtain large ion currents is shifting the ion selectivity of ion pumps from monovalent to divalent ions [86].
Site-directed mutagenesis combined with screening assays increased the channelrhodopsin-2 photocurrents by introducing T159C point mutation [87]. In other studies, rational mutagenesis in the vicinity of protein active site also led to the increased signal [20, 88].
Microbial rhodopsins as sensors of membrane potential. Wild-type microbial rhodopsins used as fluorescent reporters of membrane potential are very dim and unsuitable for in vivo experiments [66]. Application of directed evolution approach [89], high-throughput screening of mutants in combination with site-directed mutagenesis [90] allowed to increase the fluorescence of archaerhodopsin-3. Red-shifting the absorption spectrum also led to the increase of fluorescence intensity; however, the mechanism of such improvement is unclear [68].
Another approach is combining microbial rhodopsin with bright fluorescent protein in a FRET system, monitoring only the change of FP fluorescence intensity [75]. In future studies, computational methodologies can help to develop a protein with increased signal intensity. Such development can be based on the rational mutagenesis or on application of more efficient artificial chromophores with higher quantum yields [152,153,154,155,156,157,158,159,160,161,162,163].
LOV proteins and cryptochromes. The intensity of LOV signal is defined by the change of the regulatory functions, e.g. of target protein binding affinity, upon light illumination. Rational structure-based mutagenesis of the LOV2 domain from Avena sativa increased the change of binding affinity of the corresponding functional domain to DNA upon light illumination in 14 times. Increased change of binding affinity allowed better control of gene transcription and other DNA-related processes [91].
Structure-guided mutagenesis in combination with screening assays led to LOV domain with increased control of interaction between to peptides [92]. The rational design also increased the light-induced oligomerization of proteins bound to Arabidopsis photoreceptor cryptochrome 2 [93, 94].
UVR8 receptors. The enhancement of UVR8 receptor activity was achieved by coupling two UVR8 receptors via a flexible peptide linker [95].
Sensors based on fluorescent proteins. The signal intensity of fluorescent sensors must also be high enough for reliable monitoring of cellular processes. Finding brighter fluorescent proteins, inserting mutations or circular permutating GFP are possible solutions [96, 98,99,100], along with finding sensors with higher levels of expression and plasma membrane localization [51, 97].
Photoreceptor kinetics. The desired on/off kinetics of an optogenetic tool greatly depends on the application of interest. Generation of frequent neural spikes or monitoring action potentials requires tools with on/off kinetics faster than 1 ms. When the inhibition of neural activity is concerned, long off-kinetics is preferable. Long-living open states of neural silencers allow maintaining neural inhibition with brief pulses of light, reducing the photodamage and increasing the stability of the tool.
Microbial rhodopsins. Natural microbial rhodopsins have millisecond-timescale photocycle kinetics. However, generation of frequent spikes can require additional protein engineering. For example, channelrhodopsin-2 could not stably generate action potentials with frequencies higher than 40 Hz. Rational mutagenesis of the channelrhodopsin-2 active site allowed stable generation of action potentials with frequencies up to 200 Hz [87, 101]. Finding faster natural analogs in another possible approach [103].
Considering microbial rhodopsins used for sensing membrane potential, site-directed mutagenesis of residues involved in the photocycle along with screening assays generated variants with submillisecond kinetics [90, 102]. The mutant of archaerhodopsin-3, QuasAr1, demonstrated response time constants of only 0.05 ms, which is close to the limitations imposed by electronics [90].
Site-directed mutagenesis of C128 and D156 positions of channelrhodopsin-2, which are involved in the protein photocycle, led to the variants with substantially decreased off-kinetics [104, 105, 164]. Thus, D156A mutant had open-gate state lasting for several minutes, compared to about 19 ms of the wild-type protein. Such great elongation of the protein photocycle is supposed to be related to the alteration of hydrogen-bonding network between protein helices [104]. Rhodopsins with long-living open states allowed the engineering of step-function rhodopsins, which are rapidly turned on and off with brief pulses of light [104, 105].
GPCRs. GPCRs have slower kinetics compared to microbial rhodopsins, with photocycle lasting for seconds or minutes. Among them, vertebrate cone opsins have faster responses compared to other types of GPCRs [28, 106]. On the other hand, melanopsins, neuropsins and parapinopsins have very long open states.
Moreover, these three classes of GPCRs are bistable, i.e. can be turned on and off with brief pulses of light [30, 107]. In order to obtain even more stable inhibition of neural activity, sufficient levels of GPCRs were expressed in the target tissue. High expression levels led to the saturation—there always were proteins in active state, which led to constant neural inhibition [108].
LOV and BLUF sensors. Activation kinetics of LOV and BLUF sensors is relatively slow and can last from minutes to hours [48, 165]. The slow off-kinetics of these sensors is related to the long thermal decay of their flavin chromophore to the dark-adapted state [166]. Rational mutagenesis, e.g. altering the protein hydrogen-bonding network in the vicinity of the flavin chromophore, allowed altering the signaling lifetimes of LOV and BLUF sensors by orders of magnitude [109, 110]. It allowed increasing the activation kinetics of protein recruitment to cellular compartments [49, 111], gene expression [112].
Sensors based on fluorescent proteins. If a fluorescent protein is attached to a distinct functional, e.g. voltage-sensing, domain, finding faster domains [72, 113,114,115], changing the position of fluorescent protein relative to the functional domain and the cellular membrane [97], and optimizing the linker between fluorescent protein and the functional domain [116] are possible solutions. Different mutation techniques, such as random [100] and rational [98] mutagenesis are also applicable here.
The activation wavelength. A large number of works were aimed at shifting the activation wavelength of optogenetic tools towards the IR region. This direction is related to two issues. First of all, the ability of deep tissue imaging requires the red-shifted activation wavelength, ideally in the IR spectral range, because biological tissues are almost transparent for radiation in the 700–900 nm spectral range [167]. While the modern solutions to this problem include using highly invasive optical fibers or application of a two-photon microscopy technique [168, 169] the availability of deep tissue single-photon activation could be much more effective.
Another reason for obtaining red-shifted optogenetic tools is related to the reduction of phototoxicity—photons with longer wavelength have lower energy and less affect the biological tissues.
Simultaneous applications of two different optogenetic tools also can require tuning of their absorption properties. In this case, it is very important to use variants with substantially different activation wavelengths in order to prevent the spectral cross-talk (see Compatibility of optogenetic tools).
Microbial rhodopsins. Activation wavelengths of microbial rhodopsins alter in a wide range even without additional protein engineering. For example, choosing channelrhodopsin from different bacteria allows varying the activation wavelength from 436 to 590 nm [103, 117, 118]. Rational mutagenesis of the active site and engineering protein chimeras allowed obtaining red-shifted variants of different microbial rhodopsins [89, 120,121,122]. Rational mutagenesis was also used to obtain bi-stable channelrhodopsin with sufficiently different on/off wavelengths (488/600 nm) [119]. Computational methodologies can be a prospective approach for the rational spectral tuning of rhodopsins [153, 160, 170,171,172,173,174].
Directed evolution demonstrated itself as a powerful method for shifting protein absorption maximum. Thus, directed evolution of Gloebacter violaceus rhodopsin generated mutants with absorption maximum red-shifted at 80 nm relative to the wild-type protein [68].
GPCRs. Engineering of GPCRs with required G protein specificity and activation wavelength exploits OptoXR approach. As described above, in the OptoXR approach visual opsin with required absorption maximum wavelength is combined with parts of ligand-activated GPCR of interest. Thus, an independent variation of neural inhibition pathway and activation wavelength is possible [31, 34, 123].
LOV proteins, BLUF, cryptochromes. Color tuning of optogenetic tools with flavin chromophore is impossible. An alternative is using red light absorbing phytochromes that perform the same functions [60].
Phytochromes. Modulation of activation wavelength of phytochromes can be achieved by changing the protein chromophore or finding a natural phytochrome with different absorption [124, 125].
Sensors based on fluorescent proteins. For tools based on fluorescent proteins, choosing FPs with naturally different absorption wavelength or insertion mutations in the current FPs are the two possible options [126, 127].
Stability. There are two important parameters of the stability of optogenetic tools that need to be considered: the stability of the signal during long-lasting experiments and the independence from the cellular environment.
Microbial rhodopsins. Signal intensity of all microbial rhodopsins used for the control of the neural electrical activity, decrease upon prolonged illumination. This instability has two reasons. First, the photocycle of microbial rhodopsins has inactive intermediates with slow recovery kinetics, and these intermediates accumulate with time [175]. Second, because microbial rhodopsins work as intracellular or extracellular ion pumps or ion channels, the change of corresponding ion concentrations leads to the decrease of the current [142].
Finding more stable natural variants or introducing mutations in the existing ones are possible solutions. For example, a channelrhodopsin-1/channelrhodopsin-2 chimera with crossover site at E-F loop exhibited significantly less inactivation upon prolonged light stimulation [176]. Switching to divalent ions also led to the increase of signal stability [86].
Fluorescence of microbial rhodopsins used as sensors of membrane potential also decreases very fast [66, 90]. The speed of photobleaching depends on the illumination intensity and wavelength, and time-varying modulation of the illumination [128]. Less photobleaching variants can be obtained by insertion of specific mutations [90], however, even the most stable mutants available up to date are unsuitable for long-term experiments.
GPCRs. GPCRs demonstrate unstable signal, which adapts under repetitive stimulation [28]. Switching to opsins from invertebrates, e.g. box jellyfish opsin or using specific cone opsins partially solved this problem [106, 123].
LOV proteins. LOV protein can be used as stable fluorescent reporters of protein-protein interactions. LOV proteins have very low photobleaching levels, and their signal is stable over a wide range of pH, temperatures and does not depend on the availability of molecular oxygen, i.e. they can work in the hypoxia conditions [85, 129, 130].
Sensors based on fluorescent proteins. In case of tools based on fluorescent proteins, using brighter FPs and thus decreasing illumination brightness is a possible solution [131].
Sensitivity. The sensitivity of an optogenetic control tool is defined by the change of the signal upon the change of illumination intensity. On the other hand, the sensitivity of fluorescent reporters is defined by the change of fluorescent intensity upon the change of the property of interest. The sensitivity should be maximal in the desired physiological range of the target property [143].
Microbial rhodopsins. The sensitivity of microbial rhodopsins used as actuators/inhibitors of neural activity was achieved by the accumulation of rhodopsins with slow kinetics at the site of interest [87, 105]. Shifting to divalent ions also increased the sensitivity in 70 times compared to the corresponding monovalent ion-pumping rhodopsins [86].
Mutating the residues of fluorescent voltage-dependent microbial rhodopsins, which are involved in the photocycle, increased their sensitivity [90, 102].
GPCRs. GPCRs as a tool for the control of neural activity are on orders of magnitude more sensitive than channelrhodopsins [106, 132].
Sensors based on fluorescent proteins. Considering sensors based on fluorescent proteins the following approaches are applied to increase the sensitivity of the tools.
-
1.
Mutation of fluorescent protein at the periphery of the chromophore or circular permutation of the protein [98, 133, 134].
-
2.
Mutation of the distinct functional domain, e.g. voltage-sensing domain [72, 100].
-
3.
Changing the linker between fluorescent protein and the functional domain [100, 151].
-
4.
Replacing fluorescent protein or functional domain with more sensitive homologs [97, 113, 127].
For example, mutations that alter the chromophore pKa values change the pH sensitivity of the protein [177, 178]. Mutations that alter the affinity of chloride ions to the chromophore change the values of halide sensitivity [80, 179].
Measurement of absolute values. Most of the modern fluorescent sensors are capable of monitoring the changes of the target property, being incapable of measuring the absolute values. A commonly used technique, applied both for measuring absolute pH and membrane potential values, is ratiometric measurements. Here, the relative response of the sensor to illumination with two different wavelengths is measured [135, 136]. However, this approach is quite unstable and has slow signal detection fidelity.
Attempts to measure absolute values of membrane potential using preliminary calibration of sensors were performed, however, such calibration is very unstable and is extremely challenging for in vivo applications [137].
Finally, a complicated reliable approach for measuring absolute values of membrane potential was presented for microbial rhodopsin sensors [138]. The response of archaerhodopsin-3 mutant upon alteration of two wavelengths was measured. Such measurements were possible because of a complex nature of archaerhodopsin-3, which has two dark-adapted conformations.
Compatibility of optogenetic tools. Several studies were aimed at combining optogenetic tools for a more complex interaction with the target cells. Five different combinations are used in the experiments.
Two actuators of neural activity. First of all, two kinds of actuators of neural electrical activity can be simultaneously introduced into different cell types. This allows independent actuation of distinct neural subpopulations and can be applied for studying interactions between these subpopulations [122, 139].
Actuator and inhibitor of neural activity. Second, actuator and inhibitor can be simultaneously introduced into a single neuron. This allows the researchers to easily turn on and off neural activity with the light of different wavelengths as it was demonstrated for the channelrhodopsin/halorhodopsin system [140].
Actuator and sensor of neural activity. The third approach, so-called “closed-loop optogenetics”, combines an optogenetic actuator and a fluorescent voltage sensor in a single system [90, 141]. Ideally, the activity of the actuator must depend on the output signal from the fluorescent sensor.
Several fluorescent sensors. Fourth, several fluorescent sensors can be combined for simultaneous monitoring of different cellular processes [143].
Combination of ion pumps for a complex effect. Fifth, two tools can be combined in order to achieve some complex effect. For example, outward proton-pumping archaerhodopsin was combined with inward chloride pumping halorhodopsin in order to pump chloride ions out from the cell [142].
Requirements for the combined optogenetic tools. The combined optogenetic tools must satisfy certain requirements. First of all, the activation spectrum of the combined tools must be sufficiently different in order to prevent their spectral cross-talk [90, 121]. The only exclusion here is when two tools must be activated simultaneously in order to achieve the desired effect, as in the case of archaerhodopsin/halorhodopsin outward chloride pump [142].
Second, while illumination of a tool with red-shifted activation wavelength cannot trigger the blue-shifted tool, the reverse effect is often observed. Such residual activation can be suppressed by mutations [141].
Finally, when two different tools are expressed into a single cell, the question of using single or separate expression systems occurs. In a recent study, it was demonstrated that a single expression system leads to more stable expression and plasma membrane localization [180].
Biological problems. In this section we will consider four main classes of problems that are related with the functioning of optogenetic tools inside the cell.
-
(a)
Expression levels and plasma membrane localization. High expression levels and excellent plasma localization are of key importance for the efficient and intensive work of the optogenetic tools. Improving these characteristics is an especially acute problem for in vivo applications [114].
Microbial rhodopsins. The first solution is finding natural analogs with higher expression and membrane localization levels. For example, N. pharaonis halorhodopsin has superior expression levels compared to archaerhodopsins from different organisms. Both protein types are used for the inhibition of membrane potential [140, 144]. Between fluorescent sensors of membrane potential, archaerhodopsin-3 demonstrated better membrane localization than green-absorbing proteorhodopsin in eukaryotic cells [66].
The second approach is creating chimeras with highly-expressing proteins. For example, replacing the first two helices of green light-absorbing Volvox carteri rhodopsin with the corresponding part of highly expressing channelrhodopsin-1 resulted in chimeric rhodopsin with superior expression levels compared to wild-type Volvox carteri rhodopsin [122].
Using additional sequences in an expression system, e.g. Golgi export sequences or plasma membrane targeting motifs, can also increase the membrane localization levels [145, 146].
Sensors based on fluorescent proteins. In case of optogenetic sensors based on fluorescent proteins, which utilize distinct functional domains, using smaller domains [72, 73], optimization of coupling between the functional domain and the fluorescent protein [97], or replacing the fluorescent protein with analog that do not lead to accumulation of intracellular aggregates [147] are possible solutions.
-
(b)
Availability of the chromophore. Except for UVR8 receptors, which absorb light via intrinsic tryptophan clusters, optogenetic tools require a chromophore cofactor for functioning. This cofactor cannot be encoded in the expression system of the protein-based tool and binds to the protein inside the cells. For this reason, the availability of chromophore molecules in the cells of interest is of critical importance for the tool functioning.
The retinal chromophore, which is required for the functioning of microbial rhodopsins and GPCRs, and the flavin chromophore required for LOV-based tools and cryptochromes, are available in animal tissues or can be easily delivered into their organisms with food [10, 130].
On the other hand, phytochromes, one of the common tools for altering protein-protein interactions, exploit bilin chromophores, which are absent in animals and require additional two-stage synthesis [148, 149]. Because of this obstacle, phytochromes were rarely used for the modulation of protein-protein interactions in animals, giving the preference to LOV-based protein constructs and cryptochromes.
-
(c)
Undesirable altering the cellular physiology. Another challenge is related to minimizing the side effects imposed by optogenetic tool functioning on the cell.
Microbial rhodopsins. Proton and chloride pumping microbial rhodopsins, such as archaerhodopsins and halorhodopsins, alter intracellular pH level, proton, and chloride concentrations during functioning [14, 15]. Changing of ion specificity to sodium or potassium is a possible solution [16].
Anion conducting channelrhodopsins, which inhibit neural excitations via shunting (decreasing ionic flux) rather than hyperpolarization of the cell membrane, also do not cause chloride concentration changes [119, 181].
Light illumination of microbial rhodopsins which are used as fluorescent reporters of membrane potential can cause the emergence of residual photocurrents. These photocurrents can be blocked by mutating a single residue involved in the photocycle of the rhodopsin [66].
LOV, BLUF, cryptochromes. Optogenetic tools with a flavin chromophore, i.e. LOV, BLUF domains, and cryptochromes, demonstrate significant dark activity [43]. Using alternative BLUF domains with reduced dark activity [46, 47] and truncated versions of cryptochromes [94] can help to solve the problem. Another approach is expressing these sensors with additional constructs which suppress dark activity [150].
Sensors based on fluorescent proteins. Fluorescent sensors of membrane voltage increase the capacitance of cellular membrane and alter the properties of the cellular electrical activity [182]. Solving this problem can be achieved by reducing the expression levels of the tool, which should be compensated by increased fluorescence intensity.
-
(d)
Phototoxicity. Another source of cellular damage during optogenetic experiments is light. Phototoxicity involves altering cell morphology and physiology, DNA damage [183, 184]. Light photons of longer wavelength contain less energy, and for this reason, cause less damage [185]. Unfortunately, red-shifting the activation wavelength is not applicable to all kinds of tools, but only to microbial rhodopsins, GPCRs, phytochromes, and tools based on fluorescent proteins.
Other approaches for decreasing phototoxicity include the decrease of illumination time and the decrease of illumination intensity [87, 105].
4 Photopharmacology. Some Tools and Ways for Their Improvement
Photopharmacology is a new and fast developing area of medicine [6]. The workhorses of photopharmacology are different types of organic molecules, which change their conformation upon light illumination, such as azobenzenes, diarylethenes, azonaphthalenes [186,187,188]. In order to control specific functions in a light-dependent manner, these compounds are attached to different pharmaceuticals. Two types of constructs are most commonly used. In the first variant, the photoswitch is used as a linker between two distinct parts of the drug. Upon light absorption, the photoswitch isomerizes, and the distance between the pharmacores change leading to drug activation [187]. For example, in several experiments, azobenzene-based switches were inserted between acrylamide moiety and quaternary ammonium ions (AAQ) (Fig. 8.2). In the trans-conformation of the photoswitch, the AAQ conformation allows the quaternary ammonium ions to effectively block neural potassium channels. Upon light illumination, trans-to-cis conversion of azobenzene takes place, leading to the unblocking of potassium channels [189, 190]. Such optical control of neurons can be used for the treatment of vision loss caused by neural degradation [190,191,192,193]. In the second type of constructs, molecular photoswitch is bound to the one-piece drug, altering its affinity to the receptor upon light illumination. For example, this strategy was used for the control of drugs acting on glutamate and GABAA receptors [194, 195]. The variation of molecular photoswitches is obtained by chemical modification of various molecules, which have already proved their effectiveness. These modifications are aimed at optimization of key characteristics of the photoswitches for different experimental requirements. Below we will consider these characteristics and run over the standard methods used for their optimization (Table 8.2).

Structural formula of a commonly used in photopharmacology azobenzene derivative, AAQ
The efficiency of the photopharmaceutical. The efficiency of the photopharmaceutical depends on two key parameters: the efficiency of the photoswitch isomerization and the change in the drug activity upon light activation. The efficiency of isomerization is defined by the difference between relative concentrations of active/inactive states of the molecular photoswitch in the dark and illuminated conditions. Ideally, only non-active form must be present in the dark-adapted state, and only active form in the light-adapted state. However, such situation is prevented by the laws of thermodynamics. The presence of a small fraction of the active state in the darkness is not critical if this fraction is below a certain biological threshold [5].
Studies are aimed at increasing the isomerization efficiency of molecular photoswitches because this will allow increasing the activity of the drug along with decreasing the required illumination intensity. Up to date, chemical modification of azobenzene photoswitches allowed obtaining more than 95% cis-isomer upon light activation [196]. The magnitude of activity change of the drug can be varied by modification of the photochromic compound. For example, in case of AAQ and the similar switches used for blocking of potassium channels, their efficiency can be altered by increasing the geometry change upon trans-to-cis isomerization. This alteration can be obtained by adding proper moieties into azobenzene linkers [197].
Activation wavelength. As in the case of optogenetic tools, numerous works were aimed at red-shifting the absorption maximum of photochromic switches, which is related to less phototoxicity and the possibility of illuminating deeper tissues. Unfortunately, most of the switches used nowadays, such as azobenzenes, are activated by UV light that is extremely harmful to cells [167].
Activation of the drug prior injecting it into the organism is one of the possible solutions. However, this methodology is not always applicable. Extending π-conjugated system of the molecule is one of the mechanisms that leads to the red-shift of the absorption [198]. Thus, azobenzenes para-substituted with phenylacetylene absorb in the visible spectrum range. Another mechanism, which is commonly applied to azobenzenes, is introducing electron-donating (ED) or electron-withdrawing (EW) groups at ortho or para positions relative to the double N-N bond [199]. Two variants of azobenzene derivatives are generated with these methods—“amino” azobenzenes with one or both rings functionalized with ED groups or “push-pull” azobenzenes with AD group at one ring and AW group at another one. Both modifications lead to substantial red-shift of absorption spectrum to the visible region [200]. Another approach is providing an additional bridge between the ring moieties of the molecule [201, 202]. Finally, ortho-methoxy and ortho-fluoro azobenzenes also demonstrated absorption in the visible spectrum range [203].
Red-shifting the absorption spectrum is usually related to alteration of other characteristics. Thus, an extension of a π-conjugated system or adding ED and EW groups leads to faster thermal relaxation times [200]. On the other hand, adding a ring strain into the molecule led to the inverse thermal stability of the isomers [201]. Finally, ortho-fluorinating azobenzenes led to both isomers with approximately equal stability [203].
Kinetics of thermal relaxation. The optimal kinetics of the thermal relaxation of molecular photoswitches depends on the application of interest. For example, control of neural excitability, e.g. applied for the vision restoration, requires very fast (of millisecond timescale) thermal relaxation time constants [204]. On the other hand, obtaining active conformations with very long relaxation kinetics allows creating bi-stable molecular photoswitches, which can be turned on and off with the light of different wavelengths [161, 201, 205].
The kinetics of thermal relaxation is directly related to the energy difference between the two conformations of the molecular photoswitch. Decreasing the energy gap leads to slower relaxation kinetics. If the energy of two conformations is equal, both forms remain stable. The variation of thermal isomerization time constants can be controlled by adding different functional moieties, which alter electronic and steric nature of the molecules. For example, adding electron donating and electron withdrawing groups, creating “push-pull” azobenzenes, led to a much faster kinetics of thermal relaxation [199, 200]. Modern quantum chemical methodologies allow for high quality investigation of different chemical reactions [206,207,208] and can greatly facilitate the development of molecular photoswitches with altered kinetics.
Other issues. Other issues related to the use of synthetic photochromic compounds as light-activated drugs are the stability of their work in living organisms and their toxicity. Studying molecular mechanisms of the photoswitch degradation allows finding modifications that prevent the very first steps in the degradation process. For example, the mechanism of azobenzene degradation under the influence of different enzymes and glutathione were thoroughly investigated. Subsequently, insertion of specific electron-donating moieties prevented the degradation initiation reactions [209,210,211].
If the application of interest requires long-term work of the photopharmaceutical, the intrinsic stability of the photoswitch must be considered. In this case, the molecule should not lose the efficiency of conversion even after billions of transitions. For example, in visual restoration single photocycle of a photoswitch corresponds to the single action of photon acquisition by the eye retina [69]. In this case, millisecond timescale of the photocycle is required and efficient work of the drug for at least several days is desirable.
The toxicity of various azobenzene photoswitches was tested, and the main sources of their toxicity were defined [212]. In order to prevent the toxicity of azobenzenes, different chemical modifications are performed.
5 Conclusions
Large progress of optogenetics and photopharmacology achieved since the beginning of the XXI century is inseparably related with the development of improved tools. This improvement, guided by the experiment requirements, applied numerous approaches for achieving the goal. Unfortunately, in most cases these were solely experimental techniques, usually not based on solid surface of understanding the molecular mechanisms, which define the certain property of the tool. For example, in a number of experiments structure-guided rational design of channelrhodopsin-2, which applied as an actuator of membrane potential, was based on the crystallographic structure of channelrhodopsin-1/channelrhodopsin-2 chimeric protein [160]. However, FTIR and electrophysiological experiments demonstrated that this chimeric protein has different light-induced responses and biophysical properties [12]. Luckily, in November 2017 the high-quality crystallographic structure of channelrhodopsin-2 was published, and its analysis has already shed light on the molecular mechanisms of channelhrodopsin-2 functioning [213].
Extensive computational techniques can also facilitate the progress in this area. Thus, modern homology modeling techniques can be applied for the prediction of the proteins of interest if their crystallographic structure is not available, and for prediction of structural changes caused by mutations. On the other hand, techniques for the accurate calculation of protein absorption properties also exist. These two methodologies can be combined for in silico search for new protein-based tools with altered activation wavelength. Moreover, complex computational simulations, including molecular dynamics and hybrid quantum-mechanics/molecular-mechanics calculations can help to understand the molecular mechanisms of protein functioning and greatly facilitate the rational design approach.
References
A.B. Ghisaidoobe, S.J. Chung, Int. J. Mol. Sci. 15, 22518 (2014)
M. Monici, Biotechnol. Ann. Rev. 11, 227 (2005)
S. Schmitz-Valckenberg, F.G. Holz, A.C. Bird, R.F. Spaide, Retina 28, 385 (2008)
B.R. Rost, F. Schneider-Warme, D. Schmitz, P. Hegemann, Neuron 96, 572 (2017)
W.A. Velema, W. Szymanski, B.L. Feringa, J. Am. Chem. Soc. 136, 2178 (2014)
M.M. Lerch, M.J. Hansen, G.M. van Dam, W. Szymanski, B.L. Feringa, Angew. Chem. Int. Ed. 55, 10978 (2016)
J.S. Wiegert, M. Mahn, M. Prigge, Y. Printz, O. Yizhar, Neuron 95, 504 (2017)
A.B. Arrenberg, D.Y. Stainier, H. Baier, J. Huisken, Science 330, 971 (2010)
F. Zhang, L.-P. Wang, E.S. Boyden, K. Deisseroth, Nat. Methods 3, 785 (2006)
G. Nagel, M. Brauner, J.F. Liewald, N. Adeishvili, E. Bamberg, A. Gottschalk, Curr. Biol. 15, 2279 (2005)
E.S. Boyden, F. Zhang, E. Bamberg, G. Nagel, K. Deisseroth, Nat. Neurosci. 8, 1263 (2005)
F. Schneider, C. Grimm, P. Hegemann, Annu. Rev. Biophys. 44, 167 (2015)
V.A. Lórenz-Fonfría, J. Heberle, Biochim. Biophys. Acta (BBA)-Bioenerg. 1837, 626 (2014)
B.Y. Chow et al., Nature 463, 98 (2010)
X. Han et al., Front Syst. Neurosci. 5, 18 (2011)
H.E. Kato et al., Nature 521, 48 (2015)
M.R. Hoque, T. Ishizuka, K. Inoue, R. Abe-Yoshizumi, H. Igarashi, T. Mishima, H. Kandori, H. Yawo, PLoS ONE 11, e0166820 (2016)
I. Gushchin et al., Nat. Struct. Mol. Biol. 22, 390 (2015)
V. Gradinaru, K.R. Thompson, K. Deisseroth, Brain Cell Biol. 36, 129 (2008)
A.S. Chuong et al., Nat. Neurosci. 17, 1123 (2014)
J. Wietek et al., Science 344, 409 (2014)
A. Berndt, S.Y. Lee, C. Ramakrishnan, K. Deisseroth, Science 344, 420 (2014)
E.G. Govorunova, O.A. Sineshchekov, R. Janz, X. Liu, J.L. Spudich, Science, aaa7484 (2015)
E.G. Govorunova, O.A. Sineshchekov, J.L. Spudich, Photochem. Photobiol. 92, 257 (2016)
O.A. Sineshchekov, E.G. Govorunova, H. Li, J.L. Spudich, Proc. Natl. Acad. Sci. 112, 14236 (2015)
L. Madisen et al., Nat. Neurosci. 15, 793 (2012)
J.-Z. Guo et al., Elife 4 (2015)
X. Li, D.V. Gutierrez, M.G. Hanson, J. Han, M.D. Mark, H. Chiel, P. Hegemann, L.T. Landmesser, S. Herlitze, Proc. Natl. Acad. Sci. U.S.A. 102, 17816 (2005)
H.J. Bailes, L.-Y. Zhuang, R.J. Lucas, PLoS ONE 7, e30774 (2012)
K. Spoida et al., Curr. Biol. 26, 1206 (2016)
R.D. Airan, K.R. Thompson, L.E. Fenno, H. Bernstein, K. Deisseroth, Nature 458, 1025 (2009)
P.A. Barish, Y. Xu, J. Li, J. Sun, Y.P. Jarajapu, W.O. Ogle, Eur. J. Pharmacol. 705, 42 (2013)
E.R. Siuda et al., Nat. Commun. 6, 8480 (2015)
E. Oh, T. Maejima, C. Liu, E. Deneris, S. Herlitze, J. Biol. Chem. 285, 30825 (2010)
M. El-Gaby, Y. Zhang, K. Wolf, C.J. Schwiening, O. Paulsen, O.A. Shipton, Cell Rep. 16, 2259 (2016)
M. Mahn, M. Prigge, S. Ron, R. Levy, O. Yizhar, Nat. Neurosci. 19, 554 (2016)
E. Peter, B. Dick, S.A. Baeurle, Nat. Commun. 1, 122 (2010)
A. Möglich, R.A. Ayers, K. Moffat, J. Mol. Biol. 385, 1433 (2009)
N. Fukuda, T. Matsuda, T. Nagai, ACS Chem. Biol. 9, 1197 (2014)
Y.I. Wu, D. Frey, O.I. Lungu, A. Jaehrig, I. Schlichting, B. Kuhlman, K.M. Hahn, Nature 461, 104 (2009)
S. Masuda, Plant Cell Physiol. 54, 171 (2012)
T. Nagahama, T. Suzuki, S. Yoshikawa, M. Iseki, Neurosci. Res. 59, 81 (2007)
S. Schröder-Lang et al., Nat. Methods 4, 39 (2007)
K.-Y. Chang et al., Nat. Commun. 5, 4057 (2014)
C. Gasser, S. Taiber, C.-M. Yeh, C.H. Wittig, P. Hegemann, S. Ryu, F. Wunder, A. Möglich, Proc. Natl. Acad. Sci. U.S.A. 111, 8803 (2014)
G.M. Avelar, R.I. Schumacher, P.A. Zaini, G. Leonard, T.A. Richards, S.L. Gomes, Curr. Biol. 24, 1234 (2014)
S. Gao, J. Nagpal, M.W. Schneider, V. Kozjak-Pavlovic, G. Nagel, A. Gottschalk, Nat. Commun. 6, 8046 (2015)
M. Yazawa, A.M. Sadaghiani, B. Hsueh, R.E. Dolmetsch, Nat. Biotechnol. 27, 941 (2009)
H. Wang et al., Nat. Methods 13, 755 (2016)
A. Levskaya, O.D. Weiner, W.A. Lim, C.A. Voigt, Nature 461, 997 (2009)
X. Yang, A.P.-T. Jost, O.D. Weiner, C. Tang, Mol. Biol. Cell 24, 2419 (2013)
H.M. Beyer et al., ACS Synth. Biol. 4, 951 (2015)
K. Zhang, L. Duan, Q. Ong, Z. Lin, P.M. Varman, K. Sung, B. Cui, PLoS ONE 9, e92917 (2014)
P. Van Bergeijk, M. Adrian, C.C. Hoogenraad, L.C. Kapitein, Nature 518, 111 (2015)
L. Duan, D. Che, K. Zhang, Q. Ong, S. Guo, B. Cui, Chem. Biol. 22, 671 (2015)
D. Schmidt, P.W. Tillberg, F. Chen, E.S. Boyden, Nat. Commun. 5, 3019 (2014)
O.I. Lungu, R.A. Hallett, E.J. Choi, M.J. Aiken, K.M. Hahn, B. Kuhlman, Chem. Biol. 19, 507 (2012)
A.B. Tyszkiewicz, T.W. Muir, Nat. Methods 5, 303 (2008)
B.D. Zoltowski, L.B. Motta-Mena, K.H. Gardner, Biochemistry 52, 6653 (2013)
K. Müller et al., Nucleic Acids Res. 41, e77 (2013)
M.J. Kennedy, R.M. Hughes, L.A. Peteya, J.W. Schwartz, M.D. Ehlers, C.L. Tucker, Nat. Methods 7, 973 (2010)
X. Yang, S. Montano, Z. Ren, Photochem. Photobiol. 91, 993 (2015)
K. Tilbrook, A.B. Arongaus, M. Binkert, M. Heijde, R. Yin, R. Ulm, The Arabidopsis book. Am. Soc. Plant Biol. 11 (2013)
K. Müller et al., Nucleic Acids Res. 41, e124 (2013)
J.M. Kralj, D.R. Hochbaum, A.D. Douglass, A.E. Cohen, Science 333, 345 (2011)
J.M. Kralj, A.D. Douglass, D.R. Hochbaum, D. Maclaurin, A.E. Cohen, Nat. Methods 9, 90 (2012)
N.C. Flytzanis, C.N. Bedbrook, H. Chiu, M.K. Engqvist, C. Xiao, K.Y. Chan, P.W. Sternberg, F.H. Arnold, V. Gradinaru, Nat. Commun. 5, 4894 (2014)
M.K. Engqvist, R.S. McIsaac, P. Dollinger, N.C. Flytzanis, M. Abrams, S. Schor, F.H. Arnold, J. Mol. Biol. 427, 205 (2015)
D. Maclaurin, V. Venkatachalam, H. Lee, A.E. Cohen, Proc. Natl. Acad. Sci. U. S. A. 110, 5939 (2013)
E.C. Saint Clair, J.I. Ogren, S. Mamaev, D. Russano, J.M. Kralj, K.J. Rothschild, J. Phys. Chem. B 116, 14592 (2012)
M.S. Siegel, E.Y. Isacoff, Neuron 19, 735 (1997)
D. Dimitrov, Y. He, H. Mutoh, B.J. Baker, L. Cohen, W. Akemann, T. Knöpfel, PLoS ONE 2, e440 (2007)
B.E. Kang, B.J. Baker, Sci. Rep. 6, 23865 (2016)
R. Sakai, V. Repunte-Canonigo, C.D. Raj, T. Knöpfel, Eur. J. Neurosci. 13, 2314 (2001)
Y. Gong, M.J. Wagner, J.Z. Li, M.J. Schnitzer, Nat. Commun. 5, 3674 (2014)
G.H. Patterson, S.M. Knobel, W.D. Sharif, S.R. Kain, D.W. Piston, Biophys. J. 73, 2782 (1997)
M. Kneen, J. Farinas, Y. Li, A. Verkman, Biophys. J. 74, 1591 (1998)
V.V. Belousov, A.F. Fradkov, K.A. Lukyanov, D.B. Staroverov, K.S. Shakhbazov, A.V. Terskikh, S. Lukyanov, Nat. Methods 3, 281 (2006)
Y. Fan, Z. Chen, H.-W. Ai, Anal. Chem. 87, 2802 (2015)
R.M. Wachter, S.J. Remington, Curr. Biol. 9, R628 (1999)
A. Germond, H. Fujita, T. Ichimura, T.M. Watanabe, Biophys. Rev. 8, 121 (2016)
A. Miyawaki, J. Llopis, R. Heim, J.M. McCaffery, J.A. Adams, M. Ikura, R.Y. Tsien, Nature 388, 882 (1997)
M. Ikura, G.M. Clore, A.M. Gronenborn, G. Zhu, C.B. Klee, A. Bax, Science 256, 632 (1992)
K.K. Jensen, L. Martini, T.W. Schwartz, Biochemistry (Moscow) 40, 938 (2001)
S. Chapman, C. Faulkner, E. Kaiserli, C. Garcia-Mata, E.I. Savenkov, A.G. Roberts, K.J. Oparka, J.M. Christie, Proc. Natl. Acad. Sci. U.S.A. 105, 20038 (2008)
F. Schneider, D. Gradmann, P. Hegemann, Biophys. J. 105, 91 (2013)
A. Berndt, P. Schoenenberger, J. Mattis, K.M. Tye, K. Deisseroth, P. Hegemann, T.G. Oertner, Proc. Natl. Acad. Sci. U.S.A. 108, 7595 (2011)
Y. Sudo et al., J. Biol. Chem. 288, 20624 (2013)
R.S. McIsaac et al., Proc. Natl. Acad. Sci. U.S.A. 111, 13034 (2014)
D.R. Hochbaum et al., Nat. Methods 11, 825 (2014)
D. Strickland, X. Yao, G. Gawlak, M.K. Rosen, K.H. Gardner, T.R. Sosnick, Nat. Methods 7, 623 (2010)
G. Guntas, R.A. Hallett, S.P. Zimmerman, T. Williams, H. Yumerefendi, J.E. Bear, B. Kuhlman, Proc. Natl. Acad. Sci. U.S.A. 112, 112 (2015)
A. Taslimi, J.D. Vrana, D. Chen, S. Borinskaya, B.J. Mayer, M.J. Kennedy, C.L. Tucker, Nat. Commun. 5, 4925 (2014)
A. Taslimi, B. Zoltowski, J.G. Miranda, G.P. Pathak, R.M. Hughes, C.L. Tucker, Nat. Chem. Biol. 12, 425 (2016)
R.P. Crefcoeur, R. Yin, R. Ulm, T.D. Halazonetis, Nat. Commun. 4, 1779 (2013)
H. Tsutsui, S. Karasawa, Y. Okamura, A. Miyawaki, Nat. Methods 5, 683 (2008)
F. St-Pierre, J.D. Marshall, Y. Yang, Y. Gong, M.J. Schnitzer, M.Z. Lin, Nat. Neurosci. 17, 884 (2014)
L. Jin, Z. Han, J. Platisa, J.R. Wooltorton, L.B. Cohen, V.A. Pieribone, Neuron 75, 779 (2012)
G.S. Baird, D.A. Zacharias, R.Y. Tsien, Proc. Natl. Acad. Sci. U.S.A. 96, 11241 (1999)
A.S. Abdelfattah et al., J. Neurosci. 36, 2458 (2016)
L.A. Gunaydin, O. Yizhar, A. Berndt, V.S. Sohal, K. Deisseroth, P. Hegemann, Nat. Neurosci. 13, 387 (2010)
Y. Gong, J.Z. Li, M.J. Schnitzer, PLoS ONE 8, e66959 (2013)
N.C. Klapoetke et al., Nat. Methods 11, 338 (2014)
C. Bamann, R. Gueta, S. Kleinlogel, G. Nagel, E. Bamberg, Biochemistry 49, 267 (2009)
A. Berndt, O. Yizhar, L.A. Gunaydin, P. Hegemann, K. Deisseroth, Nat. Neurosci. 12, 229 (2009)
O.A. Masseck, K. Spoida, D. Dalkara, T. Maejima, J.M. Rubelowski, L. Wallhorn, E.S. Deneris, S. Herlitze, Neuron 81, 1263 (2014)
T. Yamashita et al., J. Biol. Chem. 289, 3991 (2014)
B.L. Roth, Neuron 89, 683 (2016)
J.M. Christie, Annu. Rev. Plant Biol. 58, 21 (2007)
S. Raffelberg, M. Mansurova, W. Gärtner, A. Losi, J. Am. Chem. Soc. 133, 5346 (2011)
D. Strickland, Y. Lin, E. Wagner, C.M. Hope, J. Zayner, C. Antoniou, T.R. Sosnick, E.L. Weiss, M. Glotzer, Nat. Methods 9, 379 (2012)
L.B. Motta-Mena, A. Reade, M.J. Mallory, S. Glantz, O.D. Weiner, K.W. Lynch, K.H. Gardner, Nat. Chem. Biol. 10, 196 (2014)
B.J. Baker, L. Jin, Z. Han, L.B. Cohen, M. Popovic, J. Platisa, V. Pieribone, J. Neurosci. Methods 208, 190 (2012)
Y. Gong, C. Huang, J. Z. Li, B.F. Grewe, Y. Zhang, S. Eismann, M.J. Schnitzer, Science, aab0810 (2015)
Y. Mishina, H. Mutoh, C. Song, T. Knöpfel, Front. Mol. Neurosci. 7, 78 (2014)
U. Sung, M. Sepehri-Rad, H.H. Piao, L. Jin, T. Hughes, L.B. Cohen, B.J. Baker, PLoS ONE 10, e0141585 (2015)
E.G. Govorunova, O.A. Sineshchekov, H. Li, R. Janz, J.L. Spudich, J. Biol. Chem. 288, 29911 (2013)
O. Yizhar et al., Nature 477, 171 (2011)
A. Berndt et al., Proc. Natl. Acad. Sci. U.S.A. 113, 822 (2016)
J.Y. Lin, P.M. Knutsen, A. Muller, D. Kleinfeld, R.Y. Tsien, Nat. Neurosci. 16, 1499 (2013)
K. Erbguth, M. Prigge, F. Schneider, P. Hegemann, A. Gottschalk, PLoS ONE 7, e46827 (2012)
M. Prigge, F. Schneider, S.P. Tsunoda, C. Shilyansky, J. Wietek, K. Deisseroth, P. Hegemann, J. Biol. Chem. 287, 31804 (2012)
W.A. Karunarathne, L. Giri, V. Kalyanaraman, N. Gautam, Proc. Natl. Acad. Sci. U.S.A. 110, E1565 (2013)
N.C. Rockwell, D. Duanmu, S.S. Martin, C. Bachy, D.C. Price, D. Bhattacharya, A.Z. Worden, J.C. Lagarias, Proc. Natl. Acad. Sci. U.S.A. 111, 3871 (2014)
K.D. Piatkevich, F.V. Subach, V.V. Verkhusha, Chem. Soc. Rev. 42, 3441 (2013)
M. Ormö, A.B. Cubitt, K. Kallio, L.A. Gross, R.Y. Tsien, S.J. Remington, Science 273, 1392 (1996)
N.C. Shaner, R.E. Campbell, P.A. Steinbach, B.N. Giepmans, A.E. Palmer, R.Y. Tsien, Nat. Biotechnol. 22, 1567 (2004)
K.M. Dean, J.L. Lubbeck, J.K. Binder, L.R. Schwall, R. Jimenez, A.E. Palmer, Biophys. J. 101, 961 (2011)
T.E. Swartz, S.B. Corchnoy, J.M. Christie, J.W. Lewis, I. Szundi, W.R. Briggs, R.A. Bogomolni, J. Biol. Chem. 276, 36493 (2001)
J.M. Christie, J. Gawthorne, G. Young, N.J. Fraser, A.J. Roe, Mol. Plant 5, 533 (2012)
N.C. Shaner et al., Nat. Methods 10, 407 (2013)
S. Kleinlogel, K. Feldbauer, R.E. Dempski, H. Fotis, P.G. Wood, C. Bamann, E. Bamberg, Nat. Neurosci. 14, 513 (2011)
S. Jayaraman, P. Haggie, R.M. Wachter, S.J. Remington, A. Verkman, J. Biol. Chem. 275, 6047 (2000)
M.J. Mahon, Adv. Biosci. Biotechnol. (Print) 2, 132 (2011)
T. Kuner, G.J. Augustine, Neuron 27, 447 (2000)
B.A. Wilt, J.E. Fitzgerald, M.J. Schnitzer, Biophys. J. 104, 51 (2013)
M.B. Hoppa, G. Gouzer, M. Armbruster, T.A. Ryan, Neuron 84, 778 (2014)
J.H. Hou, V. Venkatachalam, A.E. Cohen, Biophys. J. 106, 639 (2014)
F. Zhang, M. Prigge, F. Beyrière, S.P. Tsunoda, J. Mattis, O. Yizhar, P. Hegemann, K. Deisseroth, Nat. Neurosci. 11, 631 (2008)
X. Han, E.S. Boyden, PLoS ONE 2, e299 (2007)
S. Lou et al., J. Neurosci. 36, 11059 (2016)
H. Alfonsa, E.M. Merricks, N.K. Codadu, M.O. Cunningham, K. Deisseroth, C. Racca, A.J. Trevelyan, J. Neurosci. 35, 7715 (2015)
M. Tantama, Y.P. Hung, G. Yellen, Progress in Brain Research (Elsevier, 2012), p. 235
F. Zhang et al., Nature 446, 633 (2007)
V. Gradinaru, F. Zhang, C. Ramakrishnan, J. Mattis, R. Prakash, I. Diester, I. Goshen, K.R. Thompson, K. Deisseroth, Cell 141, 154 (2010)
W. Akemann, H. Mutoh, A. Perron, Y.K. Park, Y. Iwamoto, T. Knöpfel, J. Neurophysiol. 108, 2323 (2012)
K. Tsutsui, T. Ubuka, G.E. Bentley, L. Kriegsfeld, Front. Neurosci. 7, 60 (2013)
K. Müller, R. Engesser, J. Timmer, F. Nagy, M.D. Zurbriggen, W. Weber, Chem. Commun. 49, 8970 (2013)
C.E. Buckley, R.E. Moore, A. Reade, A.R. Goldberg, O.D. Weiner, J.D. Clarke, Dev. Cell 36, 117 (2016)
Y.I. Wu, X. Wang, L. He, D. Montell, K.M. Hahn, Methods in Enzymology (Elsevier, 2011), p. 393
H.H. Yang, F. St-Pierre, J. Neurosci. 36, 9977 (2016)
L. Herwig et al., Cell Chem. Biol. 24, 415 (2017)
I. Schapiro, M.N. Ryazantsev, L.M. Frutos, N. Ferré, R. Lindh, M. Olivucci, J. Am. Chem. Soc. 133, 3354 (2011)
A. Melloni et al., J. Am. Chem. Soc. 132, 9310 (2010)
A. Sinicropi et al., Proc. Natl. Acad. Sci. U.S.A. 105, 17642 (2008)
I.L. Zheldakov, M.N. Ryazantsev, A.N. Tarnovsky, J. Phys. Chem. Lett. 2, 1540 (2011)
P.Z. El-Khoury, A.N. Tarnovsky, I. Schapiro, M.N. Ryazantsev, M. Olivucci, J. Phys. Chem. A 113, 10767 (2009)
J.C. Williams, J. Xu, Z. Lu, A. Klimas, X. Chen, C.M. Ambrosi, I.S. Cohen, E. Entcheva, PLoS Comp. Biol. 9, e1003220 (2013)
J. Kuhne, K. Eisenhauer, E. Ritter, P. Hegemann, K. Gerwert, F. Bartl, Angew. Chem. Int. Ed. 54, 4953 (2015)
Y. Hontani, M. Marazzi, K. Stehfest, T. Mathes, I.H. Stokkum, M. Elstner, P. Hegemann, J.T. Kennis, Sci. Rep. 7, 7217 (2017)
M. Sumita, M.N. Ryazantsev, K. Saito, PCCP 11, 6406 (2009)
M.S. Panov, V.D. Voskresenska, M.N. Ryazantsev, A.N. Tarnovsky, R.M. Wilson, J. Am. Chem. Soc. 135, 19167 (2013)
A. Filatov, N. Knyazev, M. Ryazantsev, V. Suslonov, A. Larina, A. Molchanov, R. Kostikov, V. Boitsov, A. Stepakov, Org. Chem. Front. 5, 595 (2018)
A. Dawydow et al., Proc. Natl. Acad. Sci. U.S.A. 111, 13972 (2014)
R.E. Dixon, C. Yuan, E.P. Cheng, M.F. Navedo, L.F. Santana, Proc. Natl. Acad. Sci. U.S.A. 109, 1749 (2012)
A. Losi, W. Gärtner, Photochem. Photobiol. 87, 491 (2011)
I. Yoon, J.Z. Li, Y.K. Shim, Clin. Endosc. 46, 7 (2013)
B.A. Wilt, L.D. Burns, E.T. Wei Ho, K.K. Ghosh, E.A. Mukamel, M.J. Schnitzer, Annu. Rev. Neurosci. 32, 435 (2009)
K. Svoboda, R. Yasuda, Neuron 50, 823 (2006)
K. Welke, J.S. Frähmcke, H.C. Watanabe, P. Hegemann, M. Elstner, J. Phys. Chem. B 115, 15119 (2011)
M.N. Ryazantsev, A. Altun, K. Morokuma, J. Am. Chem. Soc. 134, 5520 (2012)
D.M. Nikolaev, A. Emelyanov, V.M. Boitsov, M.S. Panov, M.N. Ryazantsev, F1000Research 6 (2017)
I. Schapiro, M.N. Ryazantsev, W.J. Ding, M.M. Huntress, F. Melaccio, T. Andruniow, M. Olivucci, Aust. J. Chem. 63, 413 (2010)
D.M. Nikolaev, A.A. Shtyrov, M.S. Panov, A. Jamal, O.B. Chakchir, V.A. Kochemirovsky, M. Olivucci, M.N. Ryazantsev, ACS Omega 3, 7555 (2018)
J. Mattis et al., Nat. Methods 9, 159 (2012)
J.Y. Lin, M.Z. Lin, P. Steinbach, R.Y. Tsien, Biophys. J. 96, 1803 (2009)
N.C. Shaner, M.Z. Lin, M.R. McKeown, P.A. Steinbach, K.L. Hazelwood, M.W. Davidson, R.Y. Tsien, Nat. Methods 5, 545 (2008)
D.E. Johnson, H.-W. Ai, P. Wong, J.D. Young, R.E. Campbell, J.R. Casey, J. Biol. Chem. 284, 20499 (2009)
L.J. Galietta, P.M. Haggie, A. Verkman, FEBS Lett. 499, 220 (2001)
S. Kleinlogel, U. Terpitz, B. Legrum, D. Gökbuget, E.S. Boyden, C. Bamann, P.G. Wood, E. Bamberg, Nat. Methods 8, 1083 (2011)
S.M. Iyer et al., Sci. Rep. 6, 30570 (2016)
W. Akemann, A. Lundby, H. Mutoh, T. Knöpfel, Biophys. J. 96, 3959 (2009)
M. Pflaum, C. Kielbassa, M. Garmyn, B. Epe, Mutat. Res. DNA Repair 408, 137 (1998)
R. Dixit, R. Cyr, PlJ 36, 280 (2003)
V. Magidson, A. Khodjakov, Methods in Cell Biology (Elsevier, 2013), p. 545
M. Irie, T. Fukaminato, K. Matsuda, S. Kobatake, Chem. Rev. 114, 12174 (2014)
C. Chittasupho, Ther. Deliv. 3, 1171 (2012)
M. Irie, Chem. Rev. 100, 1685 (2000)
R.O. Blaustein, P.A. Cole, C. Williams, C. Miller, Nat. Struct. Mol. Biol. 7, 309 (2000)
L. Laprell et al., J. Clin. Invest. 127, 2598 (2017)
I. Tochitsky, J. Trautman, N. Gallerani, J.G. Malis, R.H. Kramer, Sci. Rep. 7, 45487 (2017)
A. Polosukhina et al., Neuron 75, 271 (2012)
A.Y. Rotov, L.A. Astakhova, V.S. Sitnikova, A.A. Evdokimov, V.M. Boitsov, M.V. Dubina, M.N. Ryazantsev, M.L. Firsov, Acta Naturae 10, 75 (2018)
M. Volgraf, P. Gorostiza, R. Numano, R.H. Kramer, E.Y. Isacoff, D. Trauner, Nat. Chem. Biol. 2, 47 (2006)
M. Stein, S.J. Middendorp, V. Carta, E. Pejo, D.E. Raines, S.A. Forman, E. Sigel, D. Trauner, Angew. Chem. Int. Ed. 51, 10500 (2012)
W. Szymański, B. Wu, C. Poloni, D.B. Janssen, B.L. Feringa, Angew. Chem. Int. Ed. 52, 2068 (2013)
J. Broichhagen, J.A. Frank, D. Trauner, Acc. Chem. Res. 48, 1947 (2015)
G.M. Tsivgoulis, J.M. Lehn, Adv. Mater. 9, 627 (1997)
S. Samanta, A.A. Beharry, O. Sadovski, T.M. McCormick, A. Babalhavaeji, V. Tropepe, G.A. Woolley, J. Am. Chem. Soc. 135, 9777 (2013)
J. Garcia-Amorós, A. Bučinskas, M. Reig, S. Nonell, D. Velasco, J. Mater. Chem. C 2, 474 (2014)
S. Samanta, C. Qin, A.J. Lough, G.A. Woolley, Angew. Chem. Int. Ed. 51, 6452 (2012)
A.A. Beharry, O. Sadovski, G.A. Woolley, J. Am. Chem. Soc. 133, 19684 (2011)
C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A.M. Brouwer, P. Saalfrank, S. Hecht, D. Bléger, Chem. Eur. J. 20, 16492 (2014)
R.H. Kramer, A. Mourot, H. Adesnik, Nat. Neurosci. 16, 816 (2013)
D. Bléger, J. Schwarz, A.M. Brouwer, S. Hecht, J. Am. Chem. Soc. 134, 20597 (2012)
M.N. Ryazantsev, A. Jamal, S. Maeda, K. Morokuma, PCCP 17, 27789 (2015)
D.S. Parker, B.B. Dangi, R.I. Kaiser, A. Jamal, M. Ryazantsev, K. Morokuma, J. Phys. Chem. A 118, 12111 (2014)
L.G. Muzangwa, T. Yang, D.S. Parker, R.I. Kaiser, A.M. Mebel, A. Jamal, M. Ryazantsev, K. Morokuma, PCCP 17, 7699 (2015)
C. Boulegue, M. Löweneck, C. Renner, L. Moroder, ChemBioChem 8, 591 (2007)
E.M. Kosower, H. Kanety-Londner, J. Am. Chem. Soc. 98, 3001 (1976)
S. Zbaida, Drug Metab. Rev. 27, 497 (1995)
M.A. Brown, S.C. De Vito, Crit. Rev. Environ. Sci. Technol. 23, 249 (1993)
O. Volkov et al., Science 358, eaan8862 (2017)
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant numbers 15-29-03872 ofi_m and 16-04-00494 A) and by the program No. 32 of the Basic Research of Presidium RAS “Nanostructures: physics, chemistry, biology, basics of technology”. The work was supported by the Skolkovo Foundation (grant agreement for Russian educational and scientific organization No. 7 dd 19.12.2017) and the Skolkovo Institute of Science and Technology (General agreement No. 3663-MRA dd. 25.12.2017). The work was also supported by Ministry of Education and Science of Russian Federation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nikolaev, D.M. et al. (2019). Perspective Tools for Optogenetics and Photopharmacology: From Design to Implementation. In: Yamanouchi, K., Tunik, S., Makarov, V. (eds) Progress in Photon Science. Springer Series in Chemical Physics, vol 119. Springer, Cham. https://doi.org/10.1007/978-3-030-05974-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-05974-3_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-05973-6
Online ISBN: 978-3-030-05974-3
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)