
Abstract
One of the most important triggers of cell activity is adhesion, a process by which cells and their organelles interact and attach to substrates, internal scaffolds, external interfaces, or other cells. The physiological and pathological significance of cell adhesion is hard to exaggerate, and adhesion is ubiquitous in the living world. Adhesive contacts need to be able to function in widely varying circumstances and must be established in an extremely noisy environment. For these reasons, the control mechanisms of adhesion have had to develop so as to be able to permanently monitor and correct cellular performance. While a lot of effort has been invested into understanding the biochemical aspect of these processes, the underlying physical principles of adhesion regulation have obtained significantly less appreciation. Only in recent years have these two approaches begun to converge in a unified view. Due to the strong coupling of the biochemical reactions to the spatial coordination provided by membranes and the cytoskeleton, biological signaling is subject to a plethora of physical constraints. Indeed, many signaling pathways, particularly those involving the adhesion, involve protein diffusion and aggregation guided by membranes. It is these aspects of adhesion that can be understood in the framework of statistical physics, as we intend to demonstrate in this short review. Here we summarize the developments in understanding cell and membrane adhesion from a theoretical point of view and support it with experiments in model systems as well as with living cells.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Cell adhesion
- Artificial cell
- Ligand–receptor interactions
- Competitive binding
- Multiscale approaches to adhesion
1 Introduction
Biological cells are small-scale machines that permanently process a large amount of input signals in order to perform their normal activities [1]. One of the most important triggers of cell activity is adhesion, a process by which cells and their organelles interact and attach to substrates, internal scaffolds, external interfaces, or other cells. The physiological and pathological significance of cell adhesion is hard to exaggerate. Bacteria and viruses need to adhere and attach themselves to the cell membrane in order to invade it and in turn, dynamic initiation of adhesion intervenes at various points in the immune response of the body. Cell adhesion is essential for the integrity of tissues and needs to be dynamically controlled during embryogenesis. Adhesion is a prerequisite for fusion of, for example, endocytic vesicles to the cell membrane. It is equally important in phagocytosis and is a first step for migration to which it is intimately linked. Importantly, cells communicate in various ways across adhesive junctions, the propagation of nerve signals being a striking example. While adhesion is important for both prokaryotes and eukaryotes, the discussion below will be focused on animal cells, with examples taken mostly from mammalian cells.
Animal cells (Fig. 1) and their organelles are bound by phospholipid membranes [3]. Naturally therefore, the membrane plays a central role in adhesion. Apart from having amazing material properties which can be exploited beyond pure biological aspects, membranes provide a working environment for proteins which are responsible for establishing specific contacts [4]. In the context of adhesion-mediated cell recognition process, the plasma membrane carries specialized receptors called cell adhesion molecules (CAMs) that are usually transmembrane proteins with an extracellular, an intracellular, and a transmembrane domain [3]. The extracellular domain forms specific bonds with the counter-receptor, often called the ligand. Typically, the ligand is either embedded into the extracellular matrix (ECM) and is immobile, or is carried on the surface of another cell in which case it is mobile in the plane of the cellular membrane. On its outer surface, the membrane carries long sugars and proteoglycans, called the glycocalyx, one of whose primary role is to prevent unspecific adhesion and merger of the cells. On the inner surface of the cell, the membrane is connected to a scaffold called the cortex which is made up of a network of the semiflexible polymer actin. When a receptor binds to its ligand, a chain of events are triggered and it is often accompanied by a drastic reorganization of the membrane at a local or cellular scale. This process is partly a result of passive thermodynamics of the membrane and is partly actively modulated by the cell [5,6,7]. Most receptors are sites of cell signaling: upon binding to a ligand on the extracellular side, the receptor transmits a biochemical signal towards the cell interior which then usually leads to a whole cascade of biochemical processes that culminate in a biological action [3]. The importance of the ensemble of these proteins is captured in the concept of “adhesome”—a term used to describe the entirety of proteins and signaling network triggered by a specific adhesion event.

Fluorescence images of specific ligand–receptor-mediated adhesion in living cells. Left: Primary cardiac fibroblasts from embryonic (E19) rat heart. Cells are stained for actin (green) and vinculin (red) and imaged in confocal mode. They clearly show focal adhesions, which are typically associated with integrin-mediated adhesion on immobilized ECM ligands. Image courtesy of R. Merkel and B. Hoffman. Middle: MDCK cell monolayer adhering to collagen I-coated glass. The nuclei (DAPI, blue), the actin cytoskeleton (red), and the focal adhesions (paxillin, green) are stained and imaged in confocal mode [2]. Right: a single T cell adhering to glass coated with ligand against the TCR–complex (anti-CD3) imaged in total internal fluorescence microscopy, with the actin stained and visualized in pseudo-color. No focal adhesions are formed in these cell types. Image courtesy of P. Dillard and L. Limozin
Adhesive contacts need to be able to function in widely varying circumstances and must be established in an extremely noisy environment. For these reasons, the control mechanisms of adhesion have had to develop so as to be able to permanently monitor and correct cellular performance. While a lot of effort has been invested into understanding the biochemical aspect of these processes, the underlying physical principles of adhesion regulation have obtained significantly less appreciation. Only in recent years have these two approaches begun to converge in a unified view. Due to the strong coupling of the biochemical reactions to the spatial coordination provided by membranes and the cytoskeleton, biological signaling is subject to a plethora of physical constraints. Indeed, many signaling pathways, particularly those involving the adhesion, involve protein diffusion and aggregation guided by membranes. It is these aspects of adhesion that can be understood in the framework of statistical physics, as we intend to demonstrate in this short review.
1.1 Establishment of a Minimal System
Many features of cell-to-cell and cell-to-ECM adhesion have been, and continue to be, studied in vitro using single cells interacting with a functionalized surface. However, even with such simplified systems, due to the complexity of the cell and its natural regulation mechanisms, it is often difficult to repeatedly reproduce exactly the same experimental conditions in adhesion experiments. This was the main incentive for the development of a variety of model systems in which “ideal conditions” could be achieved. These ideal conditions comprise a fluid membrane, discernible ligand–receptor interactions, and the ability to mimic the control of the glycocalyx [5, 7, 8]. At least one of the participants in the specific interaction should be mobile and the total number of ligand and receptors controlled.
To achieve these goals, giant unilamellar vesicles (GUVs) with about 30 μm diameter are used to mimic the cell [4, 5, 7,8,9]. GUVs are made of double-chained phospholipids arranged as a bilayer, with additional cholesterol, glycolipids, and/or embedded proteins to capture one or more essential property of the plasma membrane. The second cell can be simulated by another vesicle or a cell as, for example, in the case of experiments with micropipettes. This technique allows for a macroscopic view of the adhering vesicles whereas the information about adhesion events is obtained from the response function of the applied suction pressure [10, 11].
While the experimental methods for studying adhering membranes in three dimensions are quite limited, the variety of surface-sensitive optical techniques and electrical measurements that can be applied to planar systems is vast. Besides confocal microscopy [12], reflection interference contrast microscopy (RICM) is the most commonly used technique in studies of adhesion [13,14,15,16]. This technique allows direct measurement of the average membrane shape and fluctuations and thus provides information on the formation of protein complexes without the use of fluorescent labels [5]. Over the years, the capabilities of RICM were improved and enhanced, including the multiwavelength setups [17,18,19], or dynamic-RICM [1, 13, 20]. The development of these and other surface-sensitive optical imaging techniques is the reason that, flat, functionalized glass interfaces are often used as a cell or ECM mimic, and counterpartner for an adherent GUV (Fig. 2).

Model systems for studying membrane-mediated adhesion. (a) Ligand-decorated GUV adhering to a functionalized substrate, which can expose mobile or immobile binders. (b) Unbinding of a vesicle specifically adhered to a cell, manipulated by the micropipette technique and observed in a differential interference contrast. Image adapted from Prechtel et al. [10]. (c) Interference contrast reflection microscopy can be used when the vesicle is brought in contact to a flat substrate and is more appropriate for detailed studies of the adhesion zone. Due to a very good height resolution of such setups, fluctuations of the membrane in the contact zone can be recorded in real time. This allows the determination of the mean-square amplitude of the fluctuations, and thus the distinction between weakly and strongly adhered (the growing dark patch) parts of the membrane can be obtained [16, 21]. Image courtesy S. Fenz [8]
Functional surfaces rely on anchoring of active molecules, often to supported lipid bilayers (SLB). Flat bilayers can be assembled either by Langmuir–Blodgett lipid deposition or by rupturing of pretensed unilamellar vesicles on hydrophilic surfaces (see reviews by Sackmann and Tanaka [22] or Groves et al. [23] or Richter et al. [24] and references therein). When a bilayer is deposited directly on the surface, a thin film of lubricating water layer (thickness of 1 nm) forms separating the membrane from the support. This layer usually ensures that the lipids remain mobile in the plane of the bilayer. However, large transmembrane proteins such as integrins may become immobile, due to interactions with the surface.
A different strategy for separating the membrane from the substrate involves the inclusion of intercalating water-swelled polymers. This film can be achieved by the chemical grafting or physical absorption of water-soluble polymers or polyelectrolyte multilayers [25,26,27], the reconstitution of lipopolymers covalently bound to lipids in the bilayer [28, 29], or by the deposition of multilayers of amphiphilic molecules such as cellulose with alkyl side chains [30]. Though somewhat more elaborate for preparation and control, these systems ensure that a large proportion of proteins remain mobile and fully functional, and are hence suitable for adhesion assays.
In the context of cells, recently even more sophisticated substrates have been used that are chemically patterned at the nano-/submicron scale [31,32,33,34], or those that form diffusive barriers [35, 36]. Such approaches offer a wealth of possibilities for gaining new insights over the adhesion process by meticulously controlling the microenvironment in which cells or vesicles adhesion takes place. These highly advanced systems are particularly useful for the comparison with theoretical modeling and simulations, which can now be performed in precisely defined conditions that reproduce the experimental situation [37].
Despite the growing appreciation for mimetic approaches, however, a challenge remains to combine insight from these soft matter model systems with molecular specificity and activity found in real cells: The physical mechanisms isolated need to be translated into the realm of biology, and thus, put into the context of active processes and signaling. This is a challenging task already on the level of a single cell, and even more so in vivo. Yet, the prospect of understanding normal and pathological cell function should outweigh the immense work required to take into account the complexity of the system and through a multidisciplinary effort lead ultimately to deep understanding of the cell recognition process.
2 General Mechanisms of Adhesion
2.1 Protein-Mediated Adhesion in Cells and Tissues
Cell adhesion relies on selective binding of receptors which typically belong to one of five major families of cell adhesion proteins, namely cadherins, integrins, selectins, mucines, and immunoglobulins [38]. These proteins either establish homophilic contact as in case of cadherins, or they interact with specific ligands on the opposing surface. Their interactions rely on the so-called lock-and-key principle: the interplay of numerous low-energy interactions is established to produce a binding that is sufficiently strong to be useful in the recognition and adhesion process. The weakness of the individual interactions also conveys the ability to spontaneously dissociate without the need for further control mechanisms to be developed by the cell. These weak intermolecular contacts can be formed by different mechanisms. Specifically, electrostatic interactions between two complementarily charged domains in molecules lead to binding energies of 2–4 k B T. Hydrogen bonds are somewhat weaker and contribute with energies of 1–2 k B T. Furthermore, van der Waals energy can be considerable if the complementarity of protein shapes is large, yielding a large number of low-energy (0.2–0.5 k B T) contacts. Finally, as the proteins are exposed to water, domains in molecules with similar affinity to water will feel attractive forces due to the hydrophobic effect. Overall binding energy measured in solution, i.e. the three-dimensional binding affinity, amounts to 5–10 k B T, where the lower bounds are typical for selectins and cadherins, while larger values are associated with integrins. The affinity measured in solution can be as large as 30 k B T in the case of the unusually strong, avidin–biotin recognition.
2.1.1 Focal Adhesion, Integrins, and Mechanotransduction
Receptors of the integrin family are one of the most prevalent adhesion molecules in animal cells [3]. Cells typically use integrins to bind to the extracellular matrix but integrins may occasionally also participate in cell–cell adhesion, the integrin LFA1 in T cells being a prime example [39]. On biding to its ligand, the intracellular part binds to talin which in turn binds to vinculin and other actin-binding proteins. At the same time, integrins cluster in the plane of the membrane. The resulting complex eventually links to actin and is capable of transmitting forces. In many cells, notably fibroblasts, the mature adhesion comprising integrins and the associated proteins takes the form of focal adhesions, linked to actin bundles called stress fibers [3] (see Fig. 1). Integrins are particularly intriguing because of their participation in both inside-out and outside-in signaling [40, 41].
Over the last couple of decades, it was shown that cells sense the mechanics of their surroundings by application of force, along the actin bundles and through the talin–integrin complex in focal adhesions. Intriguingly, the size of a focal adhesion grows with application of physical force [42,43,44,45]. In general, cells tend to reinforce their adhesive contacts under force, and it is now well known that mechanical forces direct a host of cellular and tissue processes. It is believed that cell adhesion complexes may act as force sensors, in concert with the actin cytoskeleton, which also transmits the forces. Cells were shown to spread more on hard and less on soft substrates, and in turn, well spread cells are stiffer than less spread cells [46]. All these considerations link the adhesion state of cells to their mechanics. Today, it is impossible to discuss cell mechanics and cell adhesion separately from each other.
In addition to the actin-mediated mechanosensing, adhesion molecules are themselves force sensitive at the molecular level, often becoming weaker under force [47, 48]. In these “slip” bonds, the bond lifetime is shortened by tensile forces acting on the bond. More recently, integrins have been shown to form “catch bonds”, responding in the opposite manner—in a certain force range, their lifetime increases with tensile force applied to the bond [49, 50]. It is now increasingly clear that integrins are not unique—the mechanosensitivity at single bond level is seen in many other adhesion molecules as well.
2.1.2 Cadherins and Tissue Formation
The cadherin superfamily of receptors is another class of cell adhesion molecules which participate in cell–cell adhesion. They are responsible for the integrity of tissues and are therefore dynamically reorganized during embryogenesis [51,52,53,54,55,56]. Because of this key role, they are also implicated in many forms of cancers [57]. Cadherins exist in different subtypes and form type-specific homodimers, which ensures agglomeration of cells containing only a particular cadherin type [58,59,60]. In addition to forming trans-bonds that connect cells, cadherins are also known to form cis-bonds in the plane of the membrane, which leads to the aggregation of trans-bonds—a phenomenon with clear biological importance [61,62,63,64,65,66]. Interestingly, unlike trans-bonds, cis-interactions have been detected only in membrane-bound cadherins [64], and the origin of cis-interactions remained elusive until recently. Based on computational studies, it has been suggested that the origin of cis- interactions resides in entropic reasons related to mechanics of the monomeric and dimeric states operating at very short distance range [60, 67]. New work, however, points to the intriguing possibility that in addition, long range cis-interactions may in fact be driven purely by membrane entropy related to bending fluctuations [37].
2.1.3 The Immune Synapse and Hybrid Systems
Adhesion of Lymphocytes is an example of a highly dynamic reorganization of the membrane following adhesion. T lymphocytes (also called T cells) undergo repeated adhesion and de-adhesion in order to fulfill their physiological role of recognition of “foreign” peptide fragments displayed on the membrane of specialized cells called antigen presenting cells (APCs) (see, for example, [68] for a review). T cells carry specific receptors called T cell receptors (TCR) on their membrane which bind to their ligand present on the membrane of the APC. This binding or “recognition” triggers a cascade of signals, leading to the initiation of actin polymerization as well as activation of integrins (in this case LFA1 or α L β 2), which subsequently bind to their ligand, ICAM1, also present on the surface of the APC. Adhesion in T cells is rapid and is accompanied by dramatic reorganization of the cell membrane both in terms of molecular distribution and in terms of membrane topography. Upon binding to their ligands, the TCRs first form nano- to micron-size agglomerations [69, 70], the so-called micro-clusters, and then are later actively centralized by actin [36, 70]. Meanwhile, the LFA1 form separate, smaller clusters and ultimately are excluded from the center, forming a concentric ring with the TCR central cluster. Thus, though T cells adhere through a molecule of the integrin family, there is no focal adhesion formation (see Fig. 1).
A particularly successful experimental approach to study T cell activation and reorganization of the receptors has been to use hybrid systems where the APC is mimicked by a functionalized supporting lipid bilayer (SLB) exhibiting mobile ligands [35, 36, 70,71,72]. Such studies have revealed drastic receptor reorganization at the interface, leading to the formation of the immunological synapse, itself formed by coalescence of receptor micro-clusters that are transported along the T cell/APC interface [70]. Experiments connecting adhesive molecules of the substrate within micron-size corrals [35] or submicron patches [34], as much as the systematic comparison between mobile and immobile ligands [72], have emphasized the importance of receptor reorganization in signaling.
2.2 Nonspecific Adhesion
2.2.1 Generic Potentials and Fluctuations
While specific protein interactions were identified a long ago as the key players in cell adhesion, a new understanding has emerged during the past two decades that the cell membrane itself, being a “floppy” sheet, adds another unavoidable, yet not fully understood, interaction with the opposing surface it binds to. Although this interaction does not at all depend on any specific proteins, it can have a major impact on the protein-mediated adhesion and can be viewed as a mechanism that controls the binding affinity to the cell adhesion molecules [73].
A membrane, like any other interface, has different, omnipresent interaction types. Prominent examples include intrinsically continuous potentials of Coulomb, hydration [74], and van der Waals origin [30, 75]. Furthermore, due to their small bending rigidity of 10–100 k B T, membranes experience relatively strong, fluctuations. As first argued by Helfrich, the suppression of these fluctuations by another interface gives rise to a steric repulsion which quadratically diverges close to a wall [76]. In cells, these fluctuations [77,78,79,80,81,82] may be of thermal origin, or may arise as a result of active processes [83,84,85,86,87,88]. Overall, the balance between attractive and repulsive interactions typically results in an effective potential, which has at least one minimum separating two membranes by 5–150 nm [1, 16, 18, 21, 30, 73, 89,90,91].
The difficulty in measuring and modeling this nonspecific potential is that the tension in the cell or vesicle membrane renormalizes the fluctuations and thus the repulsive contribution to the effective potential [92]. In turn, this affects the position of the minimum of the potential and its strength. Consequently, all these parameters should be determined self-consistently [89, 91,92,93], as a function of the membrane stiffness. However, the coupling between the strength of the repulsion, the tension, and the fluctuation spectrum is still not fully understood when the system is of a finite size and away from the unbinding transition [94,95,96,97].
A common way to deal with the effective surface interactions of the membrane is to introduce a harmonic potential, whose strength and position are defined by the curvature γ and the position h 0 of the true minimum [4, 21, 73, 98,99,100]. In this case, the energetics of the membrane of bending stiffness κ and projected area S, put under tension σ, is given by:
Here, the membrane profile is parameterized in the Monge representation, whereby the membrane height h(x) is determined for every vector x residing in the plane of the substrate. The first term in the Hamiltonian in Eq. (1) is the contribution due to the bending of the membrane. The second term accounts for the surface tension, while the last term in Eq. (1) is related to the discussed membrane–substrate interaction potential.
The validity of this model was only recently confirmed [91] in experiments where the vesicle membrane was pinned to square pattern [18, 90], within which the membrane–substrate interaction is purely nonspecific (Fig. 3). In this geometry, the membrane shape and fluctuations could be measured with Dual Wavelength Reflection Interference Contrast Microscopy [91] or with the Dynamic Optical Displacement Spectroscopy [88, 101]. Because the size of the patterned square is much larger than the lateral correlation length of the membrane [21], the membrane in the central part of the square is flat on average, and fluctuates around the minimum of the membrane–substrate interaction potential. These measurements showed that even in this weak interaction limit, the fluctuations are not purely Gaussian (Fig. 3). A holistic description requires abandoning the harmonic approximation, particularly for the reconstruction of the membrane average shape. However, the harmonic approximation for the potential seems sufficient for the description of the power spectral density [102].

Nonspecific potentials and the harmonic approximation. Top left: RICM image of a GUV, partly adhering via a pattern of ligands stamped on the substrate, as is shown schematically just below. The nonadhered parts of the GUV are seen in lighter shade of gray here and are seen to flicker in a dynamic movie. The experimental data can be fitted well with a Mie potential (top right). Bottom: Theoretically generated shapes with either Mie or harmonic potential—it is seen that the nonlinear Mie potential fits the experimental shape. Adapted from [91]
2.2.2 Steric Repulsion of the Glycocalyx
In most cells, the plasma membrane is decorated with a layer of long sugars and proteoglycans, variously called glycocalyx, cell coat, or pericellular matrix (Fig. 4a). This coat may be several microns thick [105,106,107] and can essentially have a repulsive role (see [108] for review). Cells are believed to be able to regulate their adhesion by modulating the glycocalyx, which needs to be expelled from the zone of contact. The glycocalyx has been modeled in GUVs with PEG as a repeller, where variation of the PEG concentration was shown to clearly change the weak nonspecific potential [109]. The concentration of the PEG layer also directly impacts specific adhesion (Fig. 4b–d), especially when the adhesion is mediated by very weak specific interactions [104]. Hyaluronan was used as a more realistic mimic (Fig. 5), but in conjunction with strong nonspecific adhesion [13]. In this case, the system can be switched from nonadhering to weakly or strongly adhering, depending on the thickness of the polymer layer (top vs bottom panels in Fig. 5), as well as the membrane tension [13, 19]. While in the case of membrane-coupled PEG [104], the repellers are thought to be expelled from the adhesion zone, both in case of surface bound hyaluronan [13, 19] and DNA [110, 111] repellers, they are crushed and flattened by the strong membrane interactions. Similar effects are observed in cells on PEG cushions. However, repellers rich in amino acids, which are often mimicked by BSA or HSA very successfully screen adhesion-inducing separation distances between membrane of over 100 nm [91, 104].

Density and thickness of repelling molecules building the cell glycocalyx affect adhesion. (a) The glycocalyx, visualized by the exclusion of red blood cells, significantly changes in the adhesion zone between two cells. Image contributed by J. Curtis. (b) Variation in the surface coverage (denoted below micrographs) of the glycocalyx reconstituted into the vesicle membrane affects the E-selectin-mediated adhesion as seen by a decrease of the size of the black patch in the micrographs. The latter is an adhesion domain built by E-selectin attachments to sLex in the vesicle. The number of bonds, and the spreading pressure of the vesicle decrease with the increased density of the glycocalyx, as predicted by (c) a thermodynamic theory [103] (E r denotes the enthalpic cost per repeller in the contact zone), and observed experimentally [104]. Notably, reconstituted glycocalyx which is shorter than the sLex construct did not affect adhesion

Modeling efforts showed that the effect of the glycocalyx can be integrated as a contribution to the nonspecific adhesion [112], with two competing states of adhesion: initial weak adhesion is followed by slower aggregation of the adhesion molecules into small, tightly bound clusters that coexist with the regions of weak adhesion. If on the other hand cluster of bonds grow extensively, the glycocalyx molecules will be expelled from the zone of contact, with small, nonspecifically bound islands where the molecules of the glycocalyx remain trapped [113]. Naturally, thermodynamics will also play a role, as expulsion of the glycocalyx from the zone of contact will act as a pressure on the adhesion domains. Reversely, after expulsion, competition for space in the nonadherent parts of the vesicle may lead to increase of binders in the contact zone, and strengthening of adhesion sites due to finite size effects [82, 103]. Interestingly, these effects were recently found to play a role in cells [114].
3 Modeling Adhesion
Despite several decades of intensive research [5, 22, 99, 115,116,117,118,119] and the pressing need to find the underlying principles governing the establishment of intracellular contacts in various circumstances [71, 120], the growth of macromolecular structures in membranes is still poorly understood. The complexity emerges from the coupling of molecular diffusion and formation of bonds (occurring with characteristic times of 10−5 − 10−2 s) to fast membrane fluctuations (10−9 − 10−6 s). Moreover, several length scales are involved—from angstrom separations necessary for molecular recognition, cooperative effects between proteins in membranes occurring on the length scale of the membrane correlation length [121], to the micron-sized macromolecular structures that develop over time.
3.1 Nonspecific Adhesion
On the macroscopic scale, adhesion of membranes can be regarded as a wetting phenomenon [9]. A number of features associated with the process of cell spreading were identified in experiments [122,123,124] and could be modeled on this continuous level [124], where a fluid bilayer vesicle is treated as a finite system with, for all practical purposes, a fixed true area (i.e. including residual fluctuations), and enclosed volume [115]. On this scale, it is possible to account for vesicle adhesion by introducing a single parameter W, the effective adhesion strength, which is assumed to be known [125]. Minimizing curvature and adhesion energy subject to the geometrical constraints leads to a two-dimensional phase diagram where regimes of strong adhesion (spherical cap-like shapes), weak adhesion (smooth shapes), and no adhesion can be identified [115, 125]. Similar methodology was used to study the de-adhesion processes induced by an apical force. In this case, depending on the adhesion strength, continuous unbinding through tethering [126] and discontinuous detachment of smoothly deformed shapes [127, 128] were identified and characterized.
In experiments, the emphasis has usually been on screening the nonspecific interactions in order to study specific binding [129,130,131]. Nevertheless, in controlled nonspecific adhesion, salient features could be identified like formation of bubbles [132] or even motion [133] due to electrostatic interactions [12]. Very strong adhesion was used for studies of controlled pore opening [134, 135], as well as the competition between adhesion and hydrodynamic dissipation [19, 136].
3.2 Specific Adhesion: Mechanisms Governing Protein Binding
Early attempts to theoretically model the formation of macromolecular clusters in the adhesion process involved analogies with classical theories of growth [137,138,139,140]. Furthermore, a number of scaling laws were suggested after the analysis of the relationship between various stochastic processes involved [141]. However, only limited experimental confirmation has been obtained to support these arguments [136, 142,143,144].
In experiments, actually, the focus from the earliest time was on specific adhesion in a minimal system. Formation of adhesion plaques was already observed in the earliest experiments with specific linkers [130, 131]. These plaques are domains of closely packed bonds that, in case of strong and numerous bonds eventually, coalesce and fill the entire zone of adhesion. Such behavior was later observed with mammalian adhesion proteins like integrins [20, 142, 145, 146], cadherins [37, 147], and selectins [104]. Interestingly, these plaques can be destroyed by putting competitive antibodies as antagonists for the receptor into the surrounding solution [14].
3.2.1 Mobility and Density of Proteins
Another important aspect of adhesion is the transport of proteins from the regions of a membrane that are not participating in adhesion to regions in contact with another membrane or a cell. This transport is secured by the fluidity of the membrane as well as the membrane-anchoring or transmembrane nature of most cell adhesion proteins that exhibit two-dimensional diffusion. Importantly, the friction coefficient for this motion is dominated by the hydrodynamic coupling between the protein and the fluid bilayer [148]. Further effects arise due to the coupling of the protein motion with the local membrane curvature [149,150,151,152]. However, in the context of adhesion so far, these effects have been considered as small, and it is typically assumed that proteins perform a random walk with a rescaled diffusion coefficient [151, 153].
While it is intuitively clear that the diffusion of proteins will affect the dynamics of adhesion, it is perhaps less appreciated that both the equilibrium and steady-state adhesion depend on the density (Fig. 6) and mobility (Fig. 7) of binders. Namely, immobilization prior to adhesion, strong frictional coupling upon the formation of a bond, and trapping effects induced by preexisting bonds may immobilize newly formed complexes [102], the consequence of which is an entropic cost for binding. This entropic cost depends on the density of binders and has a different impact if one or both binding partners are mobile [20]. The loss of entropy is compensated by the enthalpic gain associated with the binding affinity of the pair [116]. The balance between these two contributions to the free energy [116], which naturally depends on the actual number of proteins available (Figs. 6 and 7e), in turn defines the final concentration of formed bonds [34, 104, 154, 155], and the spreading pressure of the cell or the vesicle (Fig. 6b). This highlights the importance of the correct choice of the thermodynamic ensemble—to explain the experimentally observed behavior, a cell or a vesicle must be treated as a finite reservoir of binders, while mobile receptors on the SLB should be coupled to a reservoir of a constant chemical potential.

Role of the density of receptors immobilized on the passivated glass support in GUV and cell adhesion. (a) Vesicle adhesion mediated by the binding of a lipid-anchored sialyl-LewisX (sLeX) motive to E-selectin receptors. As the concentration of E-selectin on the surface increases, the size of the domains grows linearly until saturation, as seen in the graph and in the associated RICM snapshots above the graph [104]. No adhesion was observed on substrates with less than 800 E-selectins per μm2. Inset: Fraction of bound ligands as a function of receptor density for increasing binding affinity, as calculated from the entropy–enthalpy balance, shows the same linear increase and saturation [116]. (b) T-cell adhesion on substrates which are decorated with pMHC-functionalized nanoparticles with well-defined spacing (top left micrograph). Coupling of the density to the spreading pressure is evident from the change in the contact area as a function of spacing between ligand-coated beads (top right). Furthermore, adhesion versus particle density shown in the graph displays a rapid quasi-linear increase until the density reaches 300 particles per μm2. As suggested by theory, and experiments with vesicles, this regime is followed by the saturation at higher particle densities. At low densities (spacing greater than 150 nm), most cells failed to adhere (micrographs in the inset). Image adapted from [154]. Similar dependence on ligand density is also reported in [34]

Role of ligand/receptor mobility in GUV and cell spreading. Top: GUVs carrying the tripeptide Arg-Gly-Asp motive (RGD) spread on SLB carrying integrins. (a) When the integrins are immobile and abundant, the GUVs adhere tightly with a large adhesion zone [137]. (b) At low concentrations of integrins, the vesicles are not visibly adhered but nevertheless they resist being pulled off at the points shown with arrows [20, 145]. (c) On mobile integrins at comparably low integrin concentrations, dark, compact adhesions (black domains) are formed (black arrows). (d) If the contact zone is made smaller, compact adhesions grow (red arrow). In addition, dilute domains (white arrows in (c) and (d)), which densify under force could be identified by mapping membrane fluctuations [1, 20]. (e) The difference in baseline increased density between mobile and immobile, as well as the increase of enrichment for decreased contact can be captured by a thermodynamic theory. [116]. (f)–(h) T cells spreading on SLBs carrying anti-CD3 ligands. Image adopted from [72]. (f) Similar enrichment is seen between cells binding to immobile and mobile receptors, reminiscent to observations in vesicle adhesion. (g) Immobilized ligands can sustain traction-inducing cell spreading. No local increase in ligand/receptor density is observed. (h) When the ligands are mobile, the substrate is unable to sustain traction forces, and the cells are unable to spread. As in the case of GUVs, ligand–receptor pairs are pulled towards the cell interior, thus increasing their local density (see [72] for images of receptor distribution)
As a consequence of the interplay between enthalpy and entropy of binding, complex thermodynamic response can be recovered, including the passive growth of adhesion domains subject to retracting force [20]. This increase in adhesion, which is usually reflected in the increase of the cell or vesicle spreading pressure (Fig. 6b, top right panel), couples with the deformation of the macroscopic membrane shape [128, 156]. Furthermore, even in the absence of any signaling or cytoskeleton, repeated pulling on a vesicle strengthens adhesion by compaction of dilute and growth dense agglomerates of bonds, which is reminiscent of mechanotransduction. Furthermore, if both, the ligand and the receptor, are mobile, a disjointing force acting on the membranes results in displacement of intact ligand–receptor bonds between a vesicle and the SLB, without breaking. This mimics similar observations in T cell adhesion on mobile ligands of T cell receptor complex [72] and explains the observation that cells have significant difficulties to produce traction on supported bilayers if both binders are mobile. The role of ligand mobility has also been emphasized in integrin-mediated adhesion of fibroblasts where the formed adhesion structures on mobile or immobilized tripeptide Arg-Gly-Asp motive (RGD) spread on were very different [157].
Another aspect of the interplay between different contributions to the overall free energy is the accumulation of ligand–receptor pairs in the contact zone between two adherent membranes (Fig. 7) that leads to reorganization of the membrane molecular components [103, 113, 116, 156]. Combined with steric hindrance from molecules, this accumulation can lead to both the formation of unexpected patterns [1], and the self-assembly of adhesion corals, as observed first in vesicles [102, 158] and than in cells [66]. Recently, it was shown that not only proteins but also associated lipids phase separates upon adhesion [159].
From the point of view of dynamics, if the time to find a binding partner exceeds the time to bind, the growth is considered diffusion limited, and in the opposite case, reaction limited. In the context of radial growth, when there is no unbinding from immobilized receptors, these two regimes are associated with universal power laws describing the growth of the adhesion area as a function of time. The exponents adopt values of one or two for diffusion and reaction limited aggregation, respectively [137,138,139,140]. The transition from the reaction- to diffusion-dominated regime, induced by the depletion of binders, was demonstrated in vesicles [104, 137] and cell spreading experiments [124]. More recently, the change of regime could be exploited to estimate the reaction rate for RGD–integrin, sLex-E-selectin, and biotin–neutravidin binding [121]. More complex growth exponents are, however, expected in the presence of unbinding [160], and when both partners are mobile [144]. This was demonstrated experimentally when some nonuniversal growth exponents were measured [136], showing that membrane adhesion is an ideal playground for studying the rich growth phase space in the presence of weak to strong fluctuations and short- to long-range interactions.
3.2.2 Stochastic Binding Rates and Explicit Membrane Simulations of Adhesion
Understanding adhesion requires dealing with local stochastic interactions associated with ligand–receptor binding. A common way to model these interactions is to consider the proteins as thermalized springs with stiffness λ and rest length l 0, contributing to the energy \(\mathcal {H}_B\) associated with the membrane profile h(r) by:
Here, N b is the number of formed bonds, 𝜖 b accounts for the bond enthalpy gain for forming a bond, and δ(r) is the Dirac-delta function for a bond at the position r.
If one assumes that the structural fluctuations of free receptors occur on faster timescales than the membrane dynamics (femtoseconds compared to nanoseconds), each bond should fulfill a local detailed balance condition for the transitions between the bound and unbound states, given by the instantaneous rates \(k_{\text{off}}\left (h(\mathbf {r},t)\right )\) and \(k_{\text{on}}\left (h(\mathbf {r},t)\right )\):
Here, α is the range of the interaction potential of the ligand–receptor bond.
Condition (3) naturally includes the stretching energy associated with the slow structural changes of the protein due to binding (first term in the exponent) and the intrinsic binding affinity (second term) [161]. The last term accounts for the entropic cost associated with the suppression of structural fluctuations of a protein upon binding[140, 162]. Following such defined detailed balance, each bond is locally in thermal equilibrium with the instantaneous membrane shape.
Starting from (3), one can use arguments by Dembo et al. [49] and weigh in the intrinsic reaction rate k 0 with the Boltzmann factor that depends on the energy of protein structural fluctuations allowing ligands and receptors to come in the binding range [49, 149, 163]:
From this, the local off-rate can be determined readily using Eq. (3):
This off-rate recovers the dependence of the unbinding rate on the force \(\lambda \left (h(\mathbf {r},t)-l_0\right )\) acting on a bond, which was suggested by the Bell in one of the first models for protein recognition occurring between membranes [161]. While it is easy to physically motivate the rates equations (4) and (5) from the physics perspective, their development from a minimal model is still missing. Nevertheless, the simplicity of these rates secured their broad usage in modeling the dynamics of binding, in the framework of adhesion as discussed in the next section, but also in a much broader context [108, 164].
3.2.3 Simulating Adhesion with High Accuracy
The development of accurate mesoscopic schemes within which the membrane and the proteins are explicitly treated became possible with the increase in computing power. An example are kinetic Monte Carlo approaches [165,166,167,168]. An alternative are Langevin simulations [99, 149, 169,170,171], which have the advantage of coupling the complexation rates with the instantaneous membrane shape. Specifically, the membrane described by Eqs. (1) and (2) is propagated in time by means of the Langevin equation in the Fourier space spanned by modes k [99, 140, 149]:
Here, the tension is set to zero, Λ(k) = (4ηk)−1 is the Oseen tensor, describing the hydrodynamic interaction between membrane and surrounding fluid of a viscosity η, and A is the area of the membrane. The stochastic force ξ(k) is set by the temperature of the surrounding fluid by the fluctuation–dissipation theorem:
This, and similar schemes, allowed for the direct comparison of the simulated shape of a pinned membrane with the experiment [99]. Furthermore, they were instrumental for the understanding that the affinity of the protein binding depends on the membrane fluctuations [149]. Furthermore, these simulations showed that the instantaneous roughness of the membrane reflects the instantaneous density of bonds, independently on their strength (bottom left panel in Fig. 8) [171]. Since the roughness is a non-monotonous function of the bond density, nucleation of an adhesion domain is accompanied by the appearance of a hot spot on the membrane, a fact first noticed in the context of cells [82], and rationalized by the comparison of experiments in GUVs and Langevin simulations [172]. Since the bonds are the main contributor to the roughness, spatially and temporally averaged roughness is directly proportional to the overall variance in the number of bonds (right panel in Fig. 8), both for unstable and stable adhesion.

Phase behavior of the adhered membrane and the relation between membrane roughness and bond density as obtained from explicit membrane simulations. Top left: Snapshot from a Langevin simulations of a membrane (blue), adhered to a substrate (gray) by elastic ligand–receptor bonds (yellow springs and red beads). Bottom left: Instantaneous roughness ζ ⊥ (mean-square deviation from a flat shape in a particular moment of time), as a function of the instantaneous bond density ϕ, is found to be non-monotonous and independent of the bond strength (symbols as shown in the legend). Right: Mean fluctuation amplitude of a membrane segment \(\bar \Xi \) obtained after temporal and spatial averaging (left axis, dots) is directly proportional to the fluctuation in bond concentration Δϕ (right axis, crosses). Results are presented as a function of the mean bond density \(\bar \phi \). A branch of stable adhesion (red dots and black crosses, high mean bond density) is separated from a branch of unstable adhesion (blue dots and yellow crosses at low mean density) by a dashed perpendicular line denoting a first-order-like phase transition. Figure adopted from [171]
While these insights contribute to deeper understanding of the adhesion process, only relatively small membrane segments (sizes of a single focal adhesion) can be explored for relatively short times. Consequently, long-timescale dynamics associated with the formation of adhesion structures and diffusion-limited processes remained out of reach with these techniques, which prompted the development of coarse-grained simulation methods based on mapping of adhesion to a lattice gas or Ising-like systems of bonds [100, 173,174,175]. While providing valuable understanding of cooperative dynamics in membranes, these approaches are, however, accurate only in a limited range of parameters.
3.2.4 Implicit Membrane Adhesion Dynamics: From Nucleation to Growth Patterns
A breakthrough in modeling the adhesion process was the realization that there is a clear separation of timescales between protein binding and diffusion on one hand, and membrane fluctuations on the other hand [73]. Accordingly, the mean membrane shape can be regarded as fixed, as long as the configuration of bonds pulling on the membrane remains unchanged. During this time, the membrane, and with it, the proteins sample the entire probability distribution of distances between ligands and receptors by means of membrane fluctuations.
3.2.4.1 Coarse Graining the Complexation Rates
The separation of timescales permits the integration of the effects of the membrane into effective rates for the (de)complexation of proteins. Consequently, the explicit treatment of the membrane can be fully circumvented with a negligible loss of accuracy. The averaging of the instantaneous rates equations (3)–(5) should be performed over all possible, and appropriately weighted configurations of the membrane at the position of the receptor, as height distribution {p}. Naturally, {p} is sensitive to a configuration of bonds in the neighborhood of the receptor of interest. Accordingly,
where h b and h r signify the height of the membrane at the position of a bond, and above a free receptor, respectively. The distribution {p} can be calculated analytically for an arbitrary configuration of bonds in the surrounding [162], which permits to obtain effective rates (Eq. (8)) as a function on the average \(\overline {h}\) (local mean membrane height) and the variance σ (local fluctuation amplitude) of the relevant distribution {p} [73].
The analysis of the dependence of rates on properties of the membrane shows that typically membrane fluctuations and deformation increase the binding rate. This is because the probability for the encounter between a ligand and a receptor increases in average. Since stronger fluctuations can exert stronger stochastic forces, the unbinding rate is also increased, albeit to a lesser extent than the binding rate (Fig. 9a). Somewhat surprisingly, the overall effect is the effective stabilization of bonds by fluctuations.

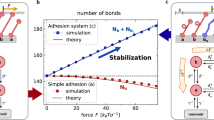
Complexation rates and stability of bonds strongly depend on membrane-mediated, cooperative effects. (a) Binding (green) and unbinding (red) rates for a ligand receptor pair at distance x from a preexisting bond, showing appreciable changes up to 400-nm separations. Inset: Deformation of a bond (blue), and the affinity of a second bond as a function of the separation the bond inducing the deformation. At short distances (positive affinity), the second bond is stable, unlike at larger distances when the formation of a bond is associated with a free energy loss (negative affinity). Graph contributed by J. Vlajcevic. (b) Fluctuations of the membrane destabilize isolated bonds, thus leading to stabilization of bond clusters as shown in the cartoon. (c) Snapshots acquired at identical time from simulations run with cooperative effects switched off (top) and on (bottom). (d) Association rate determined from the radial growth of a domain [121]. In the case of biotin–avidin recognition, increasing the residual fluctuations increases the binding rate for a factor of two. Similarly, for RGD–integrin binding, decrease in density of the glycocalyx (weakening of the nonspecific potential) may increase the binding rate for over an order of magnitude. Sketch contributed by T. Bihr
The validity of the concept of average membrane-dependent rates could be independently verified by an alternative approach. Specifically, the binding rate \(K_{\mathrm{on}}^{1}\) to form a bond in a neighborhood of an already existing bond can be extracted by fitting the time evolution of the area of a radially growing domain with a growth law obtained from a solution of a modified Stefan’s equation [121]. The associated experiments show clearly that membrane fluctuations can change the binding rate by an order of magnitude in the case of RGD–integrin, or biotin–avidin pairing (Fig. 9d). Similar stabilization of biotin–neutravidin bonds was observed in switching from ultra-weak to strong adhesion, which takes place with the suppression of fluctuations within mature adhesion domains [172].
An important consequence of such a description is the strong sensitivity of the effective rates (Eq. (6)) to the neighboring distribution of bonds, which is equivalent to cooperative effects that may enhance or prevent further (de)complexation (Fig. 9b–c). This is best demonstrated for a scaffold comprising a domain of densely packed receptors. Deep within the domain, where each bonded receptor is surrounded by other bonds, the unbinding rate is nearly zero. The bonds at the smooth edge of the domain unbind with a probability that is several times larger, while fully exposed bonds are even more unstable. Likewise, compared to receptors far away from the cluster, where the binding probability is very low, free receptors at the edge of the cluster have a significantly larger probability to form a bond [162].
3.2.4.2 Binding Affinity
As a consequence of the above argument, the binding affinity E b is also a function of the local membrane environment (Fig. 9a). Such a finding is consistent with the significant spread in the values measured for binding energies of membrane-embedded proteins, where the membrane environment could not be strictly controlled in cells [176,177,178]. This spread, on the other hand, may be relevant physiologically, since it implies a subtle means of controlling binding affinity either by thermal or by active fluctuations, which have already been found to be important in the case of cadherin junction formation in cells [179] and in model membranes [37].
At a level of an individual bond, membrane fluctuations affect the affinity, which is, in principle, quadratic [162] and in units of k B T takes the form:
with the effective elastic stiffness of the bond-membrane construct being:
Here, the stiffness of the receptor–ligand bond is denoted by λ, and the fluctuation amplitude of the unbound membrane σ f and 𝜖 b signifies the (3D) binding affinity in solution.
The first term on the right-hand side of Eq. (9) is the deformation energy stored in the bond-membrane construct and is a quadratic function of the average vertical distance between the ligand and receptor \((\overline {h}-l_0)\). Here, \(\overline {h}\) is the distance between the two membranes before the bond is formed and l 0 is the typical size of the protein assembly. The second term on the right-hand side of Eq. (9) is the entropic penalty associated with the suppression of the membrane fluctuations and the structural conformational space of the ligand–receptor pair [162]. It is important to notice that this 2D affinity is always smaller than its 3D counterpart, but also that the affinity of an ensemble of several bonds is larger (Fig. 9a). This is because a group of bonds which are separated by less than a couple of membrane correlation lengths cooperatively share the cost of bending the membrane and suppressing its fluctuations.
More detailed analysis of the free energy for the bond formation [162] shows that the affinity depends on the distance between bonds. In principle, there is a global minimum at zero distance between bonds, which suggests that densely packed domains should be most commonly observed, and that an effective attractive force acts between bonds in the membrane. However, depending on the details of the system, an additional minimum appears in the free energy at finite distance between bonds, suggesting that a sparse configuration of bonds may be meta-stable, as observed experimentally in several experiments [8, 20].
The conclusion which arises from the present discussion is that because of strong cooperative effects induced by the membrane, the mass action law is inapplicable, except in some particular limits where cooperative effects play no role. The affinity (free energy gain per bond) is, in principle, nonadditive with the bond number, due to the nonadditive effect of fluctuations. The affinity can be evaluated for fixed distribution of bonds. Affinity is, hence, a property of the steady state, and it adopts no universal value. Instead, it is a direct function of bond density, membrane elasticity, glycocalyx thickness, and the mobility of binders.
3.2.4.3 Nucleation Dynamics
It is natural to expect that the sensitivity of rates to membrane parameters reflects in the nucleation dynamics of adhesion domains. The process of nucleation is typically characterized by two parameters—the number of bonds forming the smallest stable adhesion domain N c, and its characteristic formation time \(\bar \tau \) (Fig. 10a, b). For the membrane associated nucleation [73], N c is calculated within the capillary approximation:
where ξ || is the lateral correlation length of the membrane and ρ b is the bond density within the domain. The analysis of Eq. (11) shows that the minimal number of bonds within a stable adhesion site is typically small. Importantly, N c increases with the fourth power of the separation between the unbound ligand and receptor, and decreases with the fourth power of the membrane fluctuation amplitude.

Characteristics of the nucleation process in adhering membranes. (a) Dependence of the characteristic nucleation time \(\bar \tau \) and the number of bonds forming a stable seed of the adhesion domain N c, as a function of the distance between receptors. Strong sensitivity is observed with respect to the flexibility of the receptors, and small changes in receptor density may lead to large changes in the nucleation time. Results of an analytic approach (solid lines) are shown together with explicit membrane simulations (symbols). Panel reproduced from [73]. (b) Similar level of responsiveness is observed in the phase diagram for the nucleation time (colored background) and the number of bonds forming the seed (symbols), presented as a function of the membrane fluctuation amplitude and the initial separation between membranes (from [37]). (c) Dynamics of nucleation as observed by RICM height micrographs (left column), and Dy-RICM imaging the fluctuation amplitude of the membrane (right column). As the membrane gradually transitions from the nonspecific minimum to the bound state, the fluctuations show a non-monotonous behavior. Specifically, the site of nucleation appears initially as a hot spot in fluctuations as predicted by explicit membrane simulations presented in Fig. 8. Panel adopted from [172]
The average nucleation time \(\bar \tau \) is directly related to effective association and dissociation rates of the first (\(K^0_{\text{on}}\), \(K^1_{\text{off}}\)) and the second bond (\(K^1_{\text{off}}\) and \(K^2_{\text{on}}\)), even if N c > 2 [73], giving rise to:
This expression, extracted as an analytic solution to the master equation was successfully compared to explicit membrane Langevin dynamics in which the bond formation is governed by instantaneous rates (Eqs. (3)–(5)), as well as to implicit membrane Monte Carlo simulations, where the formation of bonds is driven by coarse-grained rates (Eq. (6)) [140].
From the functional form of the effective reaction rates [73], one can conclude that in the regime of moderate fluctuations, the nucleation time is a decreasing exponential function of the square of the fluctuation amplitude. On the other hand, \(\bar \tau \) is an increasing exponential function of the square of the initial separation between ligands and receptors. This extreme sensitivity of both, the critical size and the characteristic nucleation time, is fully consistent with the experimentally observed variability of the number and size of growing adhesion junctions due to very small variations in the vesicle membrane fluctuation amplitude as well as in the initial height separation between the vesicle and the substrate [37].
In the model-membrane systems, the fluctuations are of thermal origin. Cells may, of course, locally regulate their activity, which is then reflected in the dynamic roughness of the membrane. Interestingly, locally increased fluctuations have been observed in the early stages of the formation of adhesion contacts in vesicles [172] and in cells [82]. Moreover, a typical source of fluctuations are protrusions and retractions of filopodia which have been found necessary for the initiation of cell spreading through integrin-related adhesion, but also for the nucleation of cadherin junctions [66]. As expected, disruption of actin polymerization prevented adhesion [180], even though the association of cadherins with actin is not important for initial cadherin recruitment [179]. However, it is known that E-cadherin accumulation depends on transient activation of phosphatidylinositol 3-kinase and Rac1, the latter intensifying membrane fluctuations [179]. Upon the formation of tightly packed contacts equivalent to a nucleation site, fluctuations are suppressed within the domain, followed by a drop in the Rac1 activity. These findings, put in context of the theory of nucleation, suggest that there is a coupling between membrane mechanics and the signaling pathways already in the early stages of adhesion, prior to maturation of adhesion domain and the formation of the complete mechanosome.
3.2.4.4 Dynamics of Growth
Clearly, effective rates (Eq. (6)) can be also used within a Monte Carlo approach [140], in which systems of the size of a cell can be simulated for tens of seconds, because the membrane is no longer resolved explicitly. The simulation relies on determining the bond configuration around each binder (free or engaged) in each step. Because the binding rates decay very fast with increasing distance between the bonds, the current implementation explicitly checks only for first and second neighbors. Their configuration is used to determine the local mean shape and fluctuation amplitude for each binder. These rates are then applied in attempting to associate or dissociate each binder. After updating the bond configuration, all free proteins perform a diffusion step.
Even though the membrane is no longer explicitly treated, the described MC simulation operates without loss of accuracy, as shown by comparison to the 106 times more expensive Langevin simulations [171], which themselves were shown to agree very well with experiments in the context of the nucleation [172] and the morphology of adhesion domains [149]. As mentioned before, in this higher-level approach, the membrane deformations and fluctuations are explicitly simulated through a Langevin equation with the hydrodynamics fully resolved [140]. Furthermore, the scheme explicitly accounts for the diffusion and the complexation of binders, the latter modeled directly by Bell–Dembo’s rates (Eq. (4)) [149]. This agreement fully validates the concept of the effective rates and enables studies of the early stages of the adhesion process in the regimes that are either not accessible to analytic modeling or are extremely demanding from the computational point of view. Examples of such regimes are fast nucleation, competitive growth of multiple seeds, or the diffusion-limited nucleation and growth. Actually, detailed analysis of the phase space shows a rich phase diagram that emerges from the competition between diffusivity, binding, cooperativity, and molecular crowding [37, 102] (Figs. 11 and 12). Furthermore, the spreading velocity of the membrane and diffusivity was found to strongly affect the extent of binder accumulation (Fig. 11a), where large enrichment factors are obtained at fast spreading (Fig. 11b, c), both in simulations and experiments [102]. While this effect was demonstrated on relatively large scale, the mechanism applies also for small areas of contact. This would allow cells to regulate the density of binders in the forming adhesions by controlling the protruding and retracting speed of lamellipodia and filopodia that make the first adhesive contact with the environment.

Enrichment of mobile receptors in the adhesion zone. Figure adapted from [102]. (a) Enrichment factor as a function of the diffusion constant. At high temperature (fast diffusion, black dots), the adhesion zone is fully filled with receptors (blue dashed line), while for low diffusion constant enrichment factors are smaller in simulations (cyan full line) and in associated experiments (red squares), due to the buildup of corals at the edge of the adhesion zone. (b) Bond surface coverage (fraction of the adhesion zone occupied by bonds) as a function of the inverse of the velocity of spreading v s, at various concentrations (surface coverage) of receptors. At small spreading velocities, migration of binders into the contact zone can lead to full filling, and the formation of the corals can be circumvented [8]. (c) As expected from simulations, at identical receptor densities, higher initial concentrations of ligands (top panel) lead to higher concentrations of bonds, compared to the system with small ligand concentration (bottom panel). In both systems, full filling is achieved due to small spreading velocities and corals are avoided. However, the enrichment factors differ considerably

Formation of ring-shaped coral in vesicles and cells. (a) Left panel shows an experimental fluorescent image of a ring formed by adhesion of a biotinylated GUV to an SLB decorated with mobile neutravidin as receptors. The sequence on the right shows the dynamics of the ring closure as obtained from implicit membrane simulations for the equivalent set of parameters. Stable ring was found to occur only at relatively small values of the parameters ι (Eq. (13)). Image adopted from [102]. (b) The formation of the ring was found to take place in cells only when the diffusion of binders is strongly suppressed relative to their characteristic binding time (decreasing ι). Panel reproduced from [66]. (c) Implicit membrane simulations of cadherin-mediated adhesion for various parameters ι. At high ι (fast diffusion, slow binding), full filling in a steady state is obtained an order of magnitude slower than a ring-like morphology at low ι (slow diffusion, fast binding)
Another interesting problem that this simulation can access is the formation of corals and patterns in the contact zone of vesicles and cells. Depending on the density of receptors, the adhesions may develop into peripheral ring-like structures [6]. The latter were found to be caused by the jamming of bulky proteins at the edge of the contact zone (Fig. 12a), and stabilized by membrane-transmitted correlations between bonds [102]. The simulations were able to recover the dependence of the ring thickness on the density of binders, as measured in experiments performed on biotinylated GUVs interacting with neutravidin-enriched SLBs, and show that the properties of the ring depend on the interplay between characteristic length of the path that protein makes upon entering the contact zone before forming a bond, and the mean free path of simple diffusion. Consequently, fast proteins reach the center of the contact zone prior to forming a bond, having a large dimensionless parameter:
The slow proteins (small ι) get recruited to adhesions at the edge of the contact zone, gradually building a coral. Similar effects have been observed in cell adhesion (Fig. 12b), where the fluidity (diffusivity) was found to affect the macroscopic organization of cadherins in cells [66]. At high cadherin diffusivity (high fluidity), the adhesions formed uniformly over the entire contact zone. At low diffusivity, a ring of cadherin junctions appeared spontaneously between two adherent cells [55], in cells binding to the substrate [66], and in analogous simulations as shown in Fig. 12c.
Another interesting system where the experiments and simulations showed a very good agreement is the cadherin-mediated GUV–SLB adhesion [37]. At small fluctuations and initial large separations between the GUV and the SLB, the cadherin agglomerate was a single, radially expanding domain with tightly packed bonds, consistently with strong, membrane-induced cooperative effects, recognized as cis interactions (bottom panels in Fig. 13a). Only 4-nm larger mean fluctuation amplitude of the GUV membrane resulted in a decrease of the characteristic nucleation time for three orders of magnitude. As a result, many domains developed simultaneously, producing a gas-like distribution of bonds (top panels in Fig. 13a). This is consistent with the finding that the critical number of bonds for stable nucleation has dropped to unity, even further decreasing the nucleation time (Fig. 10b), and the appearance of different patterns in the adhesion zone (Fig. 13b). At the same time, the affinity in the steady state for cadherin trans-binding grew by about 1–2 k B Ts (background in Fig. 13b) with increasing the fluctuations, as expected from the previous discussion.

Adhesion of GUV to SLBs via E-cadherin fragments. (a) An exceptionally good comparison of experiments and implicit membrane simulations demonstrates a particular sensitivity of the adhesion process to the fluctuation amplitude of the GUV membrane. Changes of only few nanometers can drive the system from growth via gas like distribution of bonds (top rows), where each individual bond is stable, to radial growth dominated by strong cooperative effects (bottom rows). (b) This sensitivity is captured in the phase diagram of growth patterns. Coarsening of the adhesion process occurs with the decrease of the effective binding affinity (background), e.g., the increase of the initial separation between the membranes and the decrease in the fluctuation amplitude. Parameters in this plot are identical to the ones used to plot the phase diagram for nucleation in Fig. 10b. (c) Topological parameter reflecting the edge roughness of adhesions, as obtained both in simulations and experiments. Image adopted from [37]
These results together strongly challenge the paradigmatic idea that specific protein molecules embedded in the cell wall (or membrane) are alone responsible for cell adhesion. Instead, a new realization is emerging that the cell membrane itself, being a floppy and deformable, adds another unavoidable, but not yet understood interaction. Although this interaction does not depend on any specific proteins at all, it can have a major impact on the protein-mediated interactions and the organization on the membrane, which will have both dynamic and structural consequences.
3.3 Conclusion
Here, we have presented a framework for understanding GUV adhesion that has been constructed from experimental data as well as simulation and analytical modeling. To summarize, for nonspecific adhesion as well as abundant or strong linkers, the adhesion dynamics is determined by hydrodynamics and the final state from the competition between elastic deformation and adhesion; for specific adhesion with weak or sparse linkers, the dynamics can be reaction dominated, diffusion dominated or may show a transition between the two. The final state is usually a competition between adhesion enthalpy and entropy of different sort—positional entropy of the linkers, that of the repellers and the configurational entropy of the membrane expressed as fluctuations. The fluctuations can subtly control both the equilibrium state and the dynamics.
The model systems illustrated here are particularly adept at capturing specific aspects of cell adhesion. For example, the regulation of adhesion by the presence of polymers was modeled experimentally as well as theoretically, and has been long observed in cells, including in cancer invasion. Recently, this was quantified, and a very similar model was proposed for cells. The role of ligand mobility too was modeled in GUVs and using the theory, and its relevance was demonstrated later for cells. Crowding-induced pattern formation is another aspect that was first observed in GUV/SLB system and modeled as such and later also seen in the cellular context. Regulation of both in-plane bond clustering and out-of-plane bond formation by membrane fluctuations was demonstrated in model membranes, in analytical calculations as well as coarse-grained simulations using cadherins as example. These observations link up very well with recent reports on early stages of adhesion of cells, where the fluctuations are however active rather than thermal.
We hope to have demonstrated that while model membranes cannot be expected to simultaneously mimic all facets of cell adhesion, they can indeed capture and reproduce specific aspects, which can then be studied in detail and in isolation. This in turn facilitates clean and clear mathematical modeling that in turn can feed back into our understanding of the biology of cell adhesion.
References
Smith A-S, Fenz SF, Sengupta K (2010) Inferring spatial organization of bonds within adhesion clusters by exploiting fluctuations of soft interfaces. EPL 89:28003:1–6
Kaliman S, Jayachandran C, Rehfeldt F, Smith A-S (2014) Novel growth regime of MDCK II model tissues on soft substrates. Biophys J 106(7):L25–L28
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008) Molecular biology of the cell. Garland Science, New York, 1392 pp
Lipowsky R (1995) Generic interactions of flexible membranes. In: Lipowsky R, Sackmann, E (eds) Structure and dynamics of membranes, Chapter 11. Elsevier, Amsterdam, pp 521–602
Smith A-S, Sackmann E (2009) Progress in mimetic studies of cell adhesion and the mechanosensing. ChemPhysChem 10(1):66–78
Fenz SF, Sengupta, K (2012) Giant vesicles as cell models. Integr Biol 4(9):982–995
Sackmann E, Smith A-S (2014) Physics of cell adhesion: some lessons from cell-mimetic systems. Soft Matter 10(11):1644–1659
Fenz SF, Smith A-S, Merkel R, Sengupta K (2011) Inter-membrane adhesion mediated by mobile linkers: effect of receptor shortage. Soft Matter 7(3):952
Bruinsma R, Sackmann E (2001) Bioadhesion and the dewetting transition. C R Acad Sci 2:803–815
Prechtel K, Bausch AR, Marchi-Artzner V, Kantlehner M, Kessler H, Merkel R (2002) Dynamic force spectroscopy to probe adhesion strength of living cells. Phys Rev Lett 89(2):028101
Evans EA (1980) Analysis of adhesion of large vesicles to surfaces. Biophys J 31(3):425
Steinkühler J, Agudo-Canalejo J, Lipowsky R, Dimova R (2016) Modulating vesicle adhesion by electric fields. Biophys J 111(7):1454–1464
Limozin L, Sengupta K (2007) Modulation of vesicle adhesion and spreading kinetics by hyaluronan cushions. Biophys J 93(9):3300–3313
Smith A-S, Lorz BG, Seifert U, Sackmann E (2006) Antagonist-induced deadhesion of specifically adhered vesicles. Biophys J 90:1064–1080
Rädler JO, Feder TJ, Strey HH, Sackmann E (1995) Fluctuation analysis of tension-controlled undulation forces between giant vesicles and solid substrates. Phys Rev E 51:4526–4536
Limozin L, Sengupta K (2009) Quantitative reflection interference contrast microscopy (RICM) in soft matter and cell adhesion. ChemPhysChem 10(16):2752–2768
Schilling J, Sengupta K, Goennenwein S, Bausch AR, Sackmann E (2004) Absolute interfacial distance measurements by dual-wavelength reflection interference contrast microscopy. Phys Rev E 69:021901
Monzel C, Fenz SF, Merkel R, Sengupta K (2009) Probing biomembrane dynamics by dual-wavelength reflection interference contrast microscopy. ChemPhysChem 10:2828–2838
Sengupta K, Limozin L (2010) Adhesion of soft membranes controlled by tension and interfacial polymers. Phys Rev Lett 104(8):088101
Smith A-S, Sengupta K, Goennenwein S, Seifert U, Sackmann E (2008) Force-induced growth of adhesion domains is controlled by receptor mobility. Proc Natl Acad Sci U S A 105(19):6906–6911
Rädler JO, Feder TJ, Strey HH, Sackmann E (1995) Fluctuation analysis of tension-controlled undulation forces between giant vesicles and solid substrates. Phys Rev E 51:4526–4536
Sackmann E (1996) Supported membranes: scientific and practical applications. Science 271(5245):43
Groves JT, Ulman N, Boxer SG (1997) Micropatterning fluid lipid bilayers on solid supports. Science 275(5300):651–653
Richter RP, Bérat R, Brisson AR (2006) Formation of solid-supported lipid bilayers: an integrated view. Langmuir 22(8):3497–3505
Elender G, Kühner M, Sackmann E (1996) Functionalisation of si/sio 2 and glass surfaces with ultrathin dextran films and deposition of lipid bilayers. Biosens Bioelectron 11(6):565–577
Fischlechner M, Zaulig M, Meyer S, Estrela-Lopis I, Cuéllar L, Irigoyen J, Pescador P, Brumen M, Messner P, Moya S, et al (2008) Lipid layers on polyelectrolyte multilayer supports. Soft Matter 4(11):2245–2258
Mulligan K, Jakubek ZJ, Linda JJ (2011) Supported lipid bilayers on biocompatible polysaccharide multilayers. Langmuir 27(23):14352–14359
Wagner ML, Tamm LK (2000) Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys J 79(3):1400–1414
Naumann CA, Prucker O, Lehmann T, Rühe J, Knoll W, Frank CW (2002) The polymer-supported phospholipid bilayer: tethering as a new approach to substrate-membrane stabilization. Biomacromolecules 3(1):27–35
Tanaka M, Sackmann E (2005) Polymer-supported membranes as models of the cell surface. Nature 437(7059):656–663
Ada Cavalcanti-Adam E, Volberg T, Micoulet A, Kessler H, Geiger B, Spatz JP (2007) Cell spreading and focal adhesion dynamics are regulated by spacing of integrin ligands. Biophys J 92(8):2964–2974
Spatz JP, Geiger B (2007) Molecular engineering of cellular environments: cell adhesion to nano-digital surfaces. Methods Cell Biol 83:89–111
Pi F, Dillard P, Alameddine R, Benard E, Wahl A, Ozerov I, Charrier A, Limozin L, Sengupta K (2015) Size-tunable organic nanodot arrays: a versatile platform for manipulating and imaging cells. Nano Lett 15(8):5178–5184. PMID: 26161675
Dillard P, Pi F, Lellouch AC, Limozin L, Sengupta K (2016) Nano-clustering of ligands on surrogate antigen presenting cells modulates t cell membrane adhesion and organization. Integr Biol 8(3):287–301
Mossman KD, Campi G, Groves JT, Dustin ML (2005) Altered TCR signaling from geometrically repatterned immunological synapses. Science 310(5751):1191–1193
Hartman NC, Nyeb JA, Groves JT (2009) Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci U S A 106:12729–12734
Fenz S, Bihr T, Schmidt D, Merkel R, Seifert U, Sengupta K, Smith A-S (2017) Membrane fluctuations mediate lateral interactions between cadherin bonds. Nat Phys 13:906–913
Lodish H, Zipursky SL (2001) Molecular cell biology. Biochem Mol Biol Educ 29:126–133
Janeway C, Murphy KP, Travers P, Walport M (2008) Janeway’s immunobiology. Garland Science, New York
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110(6):673–687
Kim M, Carman CV, Springer TA (2003) Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science 301(5640):1720–1725
Bershadsky A, Kozlov M, Geiger B (2006) Adhesion-mediated mechanosensitivity: a time to experiment, and a time to theorize. Curr Opin Cell Biol 18:472–481
Vogel V, Sheetz M (2006) Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 7(4):265–275
Geiger B, Yamada KM (2011) Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol 3(5):a005033
Iskratsch T, Wolfenson H, Sheetz MP (2014) Appreciating force and shape the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol 15(12):825–833
Solon J, Levental I, Sengupta K, Georges PC, Janmey PA (2007) Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J 93(12):4453–4461
Merkel R, Nassoy P, Leung A, Ritchie K, Evans E (1999) Energy landscapes of receptor–ligand bonds explored with dynamic force spectroscopy. Nature 397(6714):50–53
Evans EA, Calderwood DA (2007) Forces and bond dynamics in cell adhesion. Science 316(5828):1148–1153
Dembo M, Torney DC, Saxman K, Hammer D (1988) The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc R Soc Lond B Biol Sci 234(1274):55–83
Kong F, García AJ, Paul Mould A, Humphries MJ, Zhu C (2009) Demonstration of catch bonds between an integrin and its ligand. J Cell Biol 185(7):1275–1284
Leckband DE, de Rooij J (2014) Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol 30:291–315
Lecuit T, Yap AS (2012) E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol 17(5):533–539
Volk T, Cohen O, Geiger B (1987) Formation of heterotypic adherens-type junctions between l-cam-containing liver cells and a-cam-containing lens cells. Cell 50(6):987–994
Ayalon O, Sabanai H, Lampugnani MG, Dejana E, Geiger B (1994) Spatial and temporal relationships between cadherins and pecam-1 in cell-cell junctions of human endothelial cells. J Cell Biol 126(1):247–258
Engl W, Arasi B, Yap LL, Thiery JP, Viasnoff V (2014) Actin dynamics modulate mechanosensitive immobilization of E-cadherin at adherens junctions. Nat Cell Biol 16(6):587–594
Gumbiner BM (2005) Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol 6(8):622–634
Jeanes A, Gottardi CJ, Yap AS (2008) Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27(55):6920–6929
Bello SM, Millo H, Rajebhosale M, Price SR (2012) Catenin-dependent cadherin function drives divisional segregation of spinal motor neurons. J Neurosci 32(2):490–505
Katsamba P, Carroll K, Ahlsen G, Bahna F, Vendome J, Posy S, Rajebhosale M, Price S, Jessell TM, Ben-Shaul A, Shapiro L, Honig BH (2009) Linking molecular affinity and cellular specificity in cadherin-mediated adhesion. Proc Natl Acad Sci U S A 106(28):11594–11599
Wu Y, Vendome J, Shapiro L, Ben-Shaul A, Honig B (2011) Transforming binding affinities from three dimensions to two with application to cadherin clustering. Nature 475(7357):510–513
Hong S, Troyanovsky RB, Troyanovsky SM (2010) Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc Natl Acad Sci U S A 107(8):3528–3533
Taveau J-C, Dubois M, Le Bihan O, Trépout S, Almagro S, Hewat E, Durmort C, Heyraud S, Gulino-Debrac D, Lambert O (2008) Structure of artificial and natural ve-cadherinbased adherens junctions. Biochem Soc Trans 36(2):189–193
Ozaki C, Obata S, Yamanaka H, Tominaga S, Suzuki ST (2010) The extracellular domains of e- and n-cadherin determine the scattered punctate localization in epithelial cells and the cytoplasmic domains modulate the localization. J Biochem 147(3):415
Harrison OJ, Jin X, Hong S, Bahna F, Ahlsen G, Brasch J, Wu Y, Vendome J, Felsovalyi K, Hampton CM, Troyanovsky RB, Ben-Shaul A, Frank J, Troyanovsky SM, Shapiro L, Honig B (2011) The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure 19(2):244–256
Hong S, Troyanovsky RB, Troyanovsky SM (2013) Binding to f-actin guides cadherin cluster assembly, stability, and movement. J Cell Biol 201(1):131–143
Biswas KH, Hartman KL, Yu C, Harrison OJ, Song H, Smith AW, Huang WYC, Lin W, Guo Z, Padmanabhan A, Troyanovsky SM, Dustin ML, Shapiro L, Honig B, Zaidel-Bara R, Groves JT (2015) E-cadherin junction formation involves an active kinetic nucleation process. Proc Natl Acad Sci U S A 112(35):10932–10937
Brasch J, Harrison OJ, Honig B, Shapiro L (2012) Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol 22(6):299–310
Dustin ML, Groves JT (2012) Receptor signaling clusters in the immune synapse. Ann Rev Biophys 41:543
Varma R, Campi G, Yokosuka T, Saito T, Dustin ML (2006) T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25:117–127
Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD (2007) Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in jurkat T cells. Proc Natl Acad Sci U S A 104(51):20296–20301
Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML (1999) The immunological synapse: a molecular machine controlling t cell activation. Science 285(5425):221–227
Dillard P, Varma R, Sengupta K, Limozin L (2014) Ligand-mediated friction determines morphodynamics of spreading t cells. Biophys J 107(11):2629–2638
Bihr T, Seifert U, Smith A-S (2012) Nucleation of ligand-receptor domains in membrane adhesion. Phys Rev Lett 109:258101
Pincus P, Joanny J-F, Andelman D (1990) Electrostatic interactions, curvature elasticity, and steric repulsion in multimembrane systems. Europhys Lett 11:763
Safinya CR, Roux D, Smith GS, Sinha SK, Dimon P, Clark NA, Bellocq AM (1986) Steric interactions in a model multimembrane system: a synchrotron X-ray study. Phys Rev Lett 57:2718
Helfrich W (1973) Elastic properties of lipid bilayers: theory and possible experiments. Z Naturforsch C J Biosci 28:693–703
Brochard F, Lennon JF (1975) Frequency spectrum of the flicker phenomenon in erythrocytes. J Phys Fr 36:11
Evans E, Rawicz W (1990) Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys Rev Lett 64:2094–2097
Zidovska A, Sackmann E (2006) Brownian motion of nucleated cell envelopes impedes adhesion. Phys Rev Lett 96(4):048103
Pelling AE, Veraitch FS, Chu CP-K, Nicholls BM, Hemsley AL, Mason C, Horton MA (2007) Mapping correlated membrane pulsations and fluctuations in human cells. J Mol Recognit 20:467
Auth T, Safran SA, Gov NS (2007) Fluctuations of coupled fluid and solid membranes with application to red blood cells. Phys Rev E 76:051910
Pierres A, Benoliel A-M, Touchard D, Bongrand P (2008) How cells tiptoe on adhesive surfaces before sticking. Biophys J 94:4114
Safran SA, Gov N, Nicolas A, Schwarz US, Tlusty T (2005) Physics of cell elasticity, shape and adhesion. Physica A 352:171
Betz T, Lenz M, Joanny J-F, Sykes C (2009) ATP-dependent mechanics of red blood cells. Proc Natl Acad Sci U S A 106(36):15320–15325
Loubet B, Seifert U, Lomholt MA (2012) Effective tension and fluctuations in active membranes. Phys Rev E 85:031913
Turlier H, Fedosov DA, Audoly B, Auth T, Gov NS, Sykes C, Joanny J-F, Gompper G, Betz T (2016) Equilibrium physics breakdown reveals the active nature of red blood cell flickering. Nat Phys 12:513–519
Smith A-S (2016) Biophysics: alive and twitching. Nat Phys 12(5):378–379
Monzel C, Schmidt D, Seifert U, Smith A-S, Merkel R, Sengupta K (2016) Nanometric thermal fluctuations of weakly confined biomembranes measured with microsecond time-resolution. Soft Matter 12(21):4755–4768
Netz RR, Lipowsky R (1995) Stacks of fluid membranes under pressure and tension. Europhys Lett 29:345–350
Monzel C, Fenz SF, Giesen M, Merkel R, Sengupta K (2012) Mapping fluctuations in biomembranes adhered to micropatterns. Soft Matter 8(22):6128
Schmidt D, Monzel C, Bihr T, Merkel R, Seifert U, Sengupta K, Smith A-S (2014) Signature of a nonharmonic potential as revealed from a consistent shape and fluctuation analysis of an adherent membrane. Phys Rev X 4(2):021023
Seifert U (1995) Self-consistent theory of bound vesicles. Phys Rev Lett 74:5060–5063
Mecke KR, Charitat T, Graner F (2003) Fluctuating lipid bilayer in an arbitrary potential: theory and experimental determination of bending rigidity. Langmuir 19:2080–2087
Lipowsky R, Leibler S (1986) Unbinding transitions of interacting membranes. Phys Rev Lett 56:2541–2544
Netz RR (1995) Complete unbinding of fluid membranes in the presence of short-ranged forces. Phys Rev E 51(3):2286–2294
Hategan A, Sengupta K, Kahn S, Sackmann E, Discher DE (2004) Topographical pattern dynamics in passive adhesion of cell membranes. Biophys J 87(5):3547–3560
Manghi M, Destainville N (2010) Statistical mechanics and dynamics of two supported stacked lipid bilayers. Langmuir 26:4057–4068
Bruinsma R, Goulian M, Pincus P (1994) Self-assembly of membrane junctions. Biophys J 67:746–750
Brown FLH (2008) Elastic modeling of biomembranes and lipid bilayers. Annu Rev Phys Chem 59:685–712
Speck T (2011) Effective free energy for pinned membranes. Phys Rev E 83:050901
Monzel C, Schmidt D, Kleusch C, Kirchenbüchler D, Seifert U, Smith A-S, Sengupta K, Merkel R (2015) Measuring fast stochastic displacements of bio-membranes with dynamic optical displacement spectroscopy. Nat Commun 6:8162
Schmidt D, Bihr T, Fenz S, Merkel R, Seifert U, Sengupta K, Smith A-S (2015) Crowding of receptors induces ring-like adhesions in model membranes. Biochim Biophys Acta Mol Cell Res 1853:2984–2991
Smith A-S, Seifert U (2007) Vesicles as a model for controlled (de-)adhesion of cells: a thermodynamic approach. Soft Matter 3:275–289
Lorz BG, Smith A-S, Gege C, Sackmann E (2007) Adhesion of giant vesicles mediated by weak binding of sialyl-LewisX to E-selectin in the presence of repelling poly(ethylene glycol) molecules. Langmuir 23:12293–12300
Boehm H, Mundinger TA, Boehm CHJ, Hagel V, Rauch U, Spatz JP, Curtis JE (2009) Mapping the mechanics and macromolecular organization of hyaluronan-rich cell coats. Soft Matter 5:4331–4337
McLane LT, Chang P, Granqvist A, Boehm H, Kramer A, Scrimgeour J, Curtis JE (2013) Spatial organization and mechanical properties of the pericellular matrix on chondrocytes. Biophys J 104(5):986–996
Chang PS, McLane LT, Fogg R, Scrimgeour J, Temenoff JS, Granqvist A, Curtis JE (2016) Cell surface access is modulated by tethered bottlebrush proteoglycans. Biophys J 110(12):2739–2750
Robert P, Limozin L, Benoliel A-M, Bongrand P (2006) Glycocalyx regulation of cell adhesion. Principles of cellular engineering: understanding the biomolecular interface. Academic, Boston, p 143
Marx S, Schilling J, Sackmann E, Bruinsma R (2002) Helfrich repulsion and dynamical phase separation of multicomponent lipid bilayers. Phys Rev Lett 88:138102
Hisette M-L, Haddad P, Gisler T, Marques CM, Schröder AP (2008) Spreading of bio-adhesive vesicles on DNA carpets. Soft Matter 4(4):828–832
Nam G, Hisette ML, Sun YL, Gisler T, Johner A, Thalmann F, Schröder AP, Marques CM, Lee N-K (2010) Scraping and stapling of end-grafted DNA chains by a bioadhesive spreading vesicle to reveal chain internal friction and topological complexity. Phys Rev Lett 105(8):088101
Bruinsma R, Behrisch A, Sackmann E (2016) Adhesive switching of membranes: experiment and theory. Phys Rev E 61:4253–4267
Schmid EM, Bakalar MH, Choudhuri K, Weichsel J, Ann HS, Geissler PL, Dustin ML, Fletcher DA (2000) Size-dependent protein segregation at membrane interfaces. Nat Phys 12:704–711
Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, Rubashkin MG, Magbanua MJ, Thorn KS, Davidson MW, Rugo HS, Park JW, Hammer DA, Giannone G, Bertozzi CR, Weaver VM (2014) The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511:319–325
Seifert U (1997) Configurations of fluid membranes and vesicles. Adv Phys 46:13–137
Smith A-S, Seifert U (2005) Effective adhesion strength of specifically bound vesicles. Phys Rev E 71:061902
Groves JT (2007) Bending mechanics and molecular organization in biological membranes. Annu Rev Phys Chem 58:697–717
Brown FLH (2011) Continuumsimulations of biomembrane dynamics and the importance of hydrodynamic effects. Q Rev Biophys 44:391–432
Schwarz US, Safran SA (2013) Physics of adherent cells. Rev Mod Phys 85:1327–1381
Dustin ML, Chakraborty AK, Shaw AS (2010) Understanding the structure and function of the immunological synapse. Cold Spring Harb Perspect Biol 2(10):a002311
Bihr T, Fenz S, Sackmann E, Merkel R, Seifert U, Sengupta K, Smith A-S (2014) Association rates of membrane-coupled cell adhesion molecules. Biophys J 107(11):L33–L36
Döbereiner H-G, Dubin-Thaler BJ, Hofman JM, Xenias HS, Sims TN, Giannone G, Dustin ML, Wiggins CH, Sheetz MP (2006) Lateral membrane waves constitute a universal dynamic pattern of motile cells. Phys Rev Lett 97(3):038102
Sengupta K, Aranda-Espinoza H, Smith L, Janmey P, Hammer D (2006) Spreading of neutrophils: from activation to migration. Biophys J 91:4638–4648
Cuvelier D, Théry M, Chu Y-S, Dufour S, Thiéry J-P, Bornens M, Nassoy P, Mahadevan L (2007) The universal dynamics of cell spreading. Curr Biol 17(8):694–699
Seifert U, Lipowsky R (1990) Adhesion of vesicles. Phys Rev A 42:4768–4771
Smith A-S, Sackmann E, Seifert U (2004) Pulling tethers from adhered vesicles. Phys Rev Lett 92:208101
Smith A-S, Sackmann E, Seifert U (2003) Effects of a pulling force on the shape of a bound vesicle. Europhys Lett 64:281–287
Smith A-S, Lorz BG, Goennenwein S, Sackmann E (2006) Force-controlled equilibria of specific vesicle-substrate adhesion. Biophys J 90:L52–L54
Feder TJ, Weissmüller G, Žekš B, Sackmann E (1995) Spreading of giant vesicles on moderately adhesive substrates by fingering: a reflection interference contrast microscopy study. Phys Rev E 51(4):3427
Albersdörfer A, Feder T, Sackmann E (1997) Adhesion-induced domain formation by interplay of long-range repulsion and short-range attraction force: a model membrane study. Biophys J 73:245–257
Kloboucek A, Behrisch A, Faix J, Sackmann E (1999) Adhesion-induced receptor segregation and adhesion plaque formation: a model membrane study. Biophys J 77(4):2311–2328
Nardi J, Bruinsma R, Sackmann E (1998) Adhesion-induced reorganization of charged fluid membranes. Phys Rev E 58(5):6340
Solon J, Streicher P, Richter R, Brochard-Wyart F, Bassereau P (2006) Vesicles surfing on a lipid bilayer: self-induced haptotactic motion. Proc Natl Acad Sci U S A 103(33):12382–12387
Sandre O, Moreaux L, Brochard-Wyart F (1999) Dynamics of transient pores in stretched vesicles. Proc Natl Acad Sci U S A 96(19):10591–10596
Bernard A-L, Guedeau-Boudeville M-A, Sandre O, Palacin S, di Meglio J-M, Jullien L (2000) Permeation through lipid bilayers by adhesion of giant vesicles on decorated surfaces. Langmuir 16(17):6801–6808
Puech P-H, Askovic V, De Gennes P-G, Brochard-Wyart F (2006) Dynamics of vesicle adhesion: spreading versus dewetting coupled to binder diffusion. Biophys Rev Lett 01(01):85–95
Boulbitch A, Guttenberg Z, Sackmann E (2001) Kinetics of membrane adhesion mediated by ligand–receptor interaction studied with a biomimetic system. Biophys J 81:2743–2751
Shenoy VB, Freund LB (2005) Growth and shape stability of a biological membrane adhesion complex in the diffusion-mediated regime. Proc Natl Acad Sci U S A 102:9
Gao H, Shi W, Freund LB (2005) Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A 102:27
Bihr T, Seifert U, Smith A-S (2015) Multiscale approaches to protein-mediated interactions between membranes—relating microscopic and macroscopic dynamics in radially growing adhesions. New J Phys 17(8):083016
Brochard-Wyart F, De Gennes PG (2002) Adhesion induced by mobile binders: dynamics. Proc Natl Acad Sci U S A 99(12):7854–7859
Guttenberg Z, Lorz B, Sackmann E, Boulbitch A (2001) First-order transition between adhesion states in a system mimicking cell-tissue interaction. Europhys Lett 54(6):826
Cuvelier D, Nassoy P (2004) Hidden dynamics of vesicle adhesion induced by specific stickers. Phys Rev Lett 93(22):228101
De Gennes P-G, Puech P-H, Brochard-Wyart F (2003) Adhesion induced by mobile stickers: a list of scenarios. Langmuir 19:7112–7119
Goennenwein S, Tanaka M, Hu B, Moroder L, Sackmann E (2003) Functional incorporation of integrins into solid supported membranes on ultrathin films of cellulose: impact on adhesion. Biophys J 85(1):646–655
Streicher P, Nassoy P, Bärmann M, Dif A, Marchi-Artzner V, Brochard-Wyart F, Spatz J, Bassereau P (2009) Integrin reconstituted in guvs: a biomimetic system to study initial steps of cell spreading. Biochim Biophys Acta Biomembr 1788(10):2291–2300
Puech P-H, Feracci H, Brochard-Wyart F (2004) Adhesion between giant vesicles and supported bilayers decorated with chelated e-cadherin fragments. Langmuir 20(22):9763–9768
Saffman PG, Delbrück M (1975) Brownian motion in biological membranes. Proc Natl Acad Sci U S A 72:3111–3113
Reister-Gottfried E, Sengupta K, Lorz B, Sackmann E, Seifert U, Smith A-S (2008) Dynamics of specific vesicle-substrate adhesion: from local events to global dynamics. Phys Rev Lett 101(20):208103:1–4
Naji A, Atzberger PJ, Brown FLH (2009) Hybrid elastic and discrete-particle approach to biomembrane dynamics with application to the mobility of curved integral membrane proteins. Phys Rev Lett 102:138102
Reister-Gottfried E, Leitenberger SM, Seifert U (2010) Diffusing proteins on a fluctuating membrane: analytical theory and simulations. Phys Rev E 81:031903
Quemeneur F, Sigurdsson JK, Renner M, Atzberger PJ, Bassereau P, Lacoste D (2014) Shape matters in protein mobility within membranes. Proc Natl Acad Sci U S A 111(14):5083–5087
Bartossek T, Jones NG, Schäfer C, Cvitkovic M, Glogger M, Mott HR, Kuper J, Brennich M, Carrington M, Smith A-S, Fenz S, Kisker C, Engstler M (2017) Structural basis for the shielding function of the dynamic trypanosome variant surface glycoprotein coat. Nat Microbiol 2:1523–1532
Deeg J, Axmann M, Matic J, Liapis A, Depoil D, Afrose J, Curado S, Dustin ML, Spatz JP (2013) T cell activation is determined by the number of presented antigens. Nano Lett 13(11):5619–5626. PMID: 24117051
Deeg JA, Louban I, Aydin D, Selhuber-Unkel C, Kessler H, Spatz JP (2011) Impact of local versus global ligand density on cellular adhesion. Nano Lett 11(4):1469–1476. PMID: 21425841
Smith A-S, Seifert U (2005) Force-induced de-adhesion of specifically bound vesicles: strong adhesion in competition with tether extraction. Langmuir 21(24):11357–11367. PMID: 16285811.
Yu C-h, Rafiq NBM, Krishnasamy A, Hartman KL, Jones GE, Bershadsky AD, Sheetz MP (2013) Integrin-matrix clusters form podosome-like adhesions in the absence of traction forces. Cell Rep 5(5):1456–1468
Fenz SF, Smith A-S, Merkel R, Sengupta K (2011) Inter-membrane adhesion mediated by mobile linkers: effect of receptor shortage. Soft Matter 7(3):952–962
Shindell O, Mica N, Ritzer M, Gordon VD (2015) Specific adhesion of membranes simultaneously supports dual heterogeneities in lipids and proteins. Phys Chem Chem Phys 17:15598–15607
Bihr T, Sadafi F-Z, Seifert U, Taylor RK, Smith A-S (2017) Radial growth in 2d revisited: the effect of finite density, binding affinity, reaction rates, and diffusion. Adv Mater Int 4(1600310):1–7
Bell GI (1978) Models for the specific adhesion of cells to cells. Science 200:618–627
Schmidt D, Bihr T, Seifert U, Smith A-S (2012) Coexistence of dilute and densely packed domains of ligand-receptor bonds in membrane adhesion. Europhys Lett 99:38003
Erdmann T, Schwarz US (2006) Bistability of cell-matrix adhesions resulting from nonlinear receptor-ligand dynamics. Biophys J 91(6):L60–L62
Evans E, Leung A, Heinrich V, Zhu C (2004) Mechanical switching and coupling between two dissociation pathways in a p-selectin adhesion bond. Proc Natl Acad Sci U S A 101(31):11281–11286
Weikl TR (2001) Fluctuation-induced aggregation of rigid membrane inclusions. Europhys Lett 54:547–553
Weikl TR, Andelman D, Komura S, Lipowsky R (2002) Adhesion of membranes with competing specific and generic interactions. Eur Phys J E 8:59–66
Weikl TR, Lipowsky R (2004) Pattern formation during t-cell adhesion. Biophys J 87(6):3665–3678
Weikl TR, Asfaw M, Krobath H, Rózycki B, Lipowsky R (2009) Adhesion of membranes via receptor–ligand complexes: domain formation, binding cooperativity, and active processes. Soft Matter 5:3213–3224
Lin LC-L, Groves JT, Brown FLH (2006) Analysis of shape, fluctuations, and dynamics in intermembrane junctions. Biophys J 91:3600–3606
Lin LC-L, Gov N, Brown FLH (2006) Nonequilibrium membrane fluctuations driven by active proteins. J Chem Phys 124(7):074903
Reister E, Bihr T, Seifert U, Smith A-S (2011) Two intertwined facets of adherent membranes: membrane roughness and correlations between ligand–receptors bonds. New J Phys 13:025003:1–15
Fenz SF, Bihr T, Merkel R, Seifert U, Sengupta K, Smith A-S (2011) Switching from ultraweak to strong adhesion. Adv Mater 27:2622–2626
Farago O (2008) Membrane fluctuations near a plane rigid surface. Phys Rev E 78:051919
Farago O (2010) Fluctuation-induced attraction between adhesion sites of supported membranes. Phys Rev E 81(5):050902
Speck T, Reister E, Seifert U (2010) Specific adhesion of membranes: mapping to an effective bond lattice gas. Phys Rev E 82:021923
Dustin M, Bromley SK, Davis MM, Zhu C (2001) Identification of self through two-dimensional chemistry and synapses. Annu Rev Cell Dev Biol 17(3):133–157
Zhu D-M, Dustin ML, Cairo CW, Golan DE (2007) Analysis of two-dimensional dissociation constant of laterally mobile cell adhesion molecules. Biophys J 92(3):1022–1034
Hu J, Lipowsky R, Weikl TR (2013) Binding constants of membrane-anchored receptors and ligands depend strongly on the nanoscale roughness of membranes. Proc Natl Acad Sci U S A 110:15283–15288
Perez TD, Tamada M, Sheetz MP, Nelson WJ (2008) Immediate-early signaling induced by E-cadherin engagement and adhesion. J Biol Chem 283(8):5014–5022
Bazellières E, Conte V, Elosegui-Artola A, Serra-Picamal X, Bintanel-Morcillo M, Roca-Cusachs P, Muñoz JJ, Sales-Pardo M, Guimerà R, Trepat X (2015) Control of cell–cell forces and collective cell dynamics by the intercellular adhesome. Nat Cell Biol 17(4):409–420
Acknowledgements
We are grateful to our mentors Erich Sackmann, Udo Seifert, and Rudolf Merkel for their insights and ongoing collaborations. We are thankful to our teams and colleagues who were instrumental in realizing the work summarized herein, especially Susanne Fenz, Cornelia Monzel, Daniel Smith, Timo Bihr, and Laurent Limozin. We thank Josip Augustin Janeš for the help with formatting and proofreading this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sengupta, K., Smith, AS. (2018). Adhesion of Biological Membranes. In: Bassereau, P., Sens, P. (eds) Physics of Biological Membranes. Springer, Cham. https://doi.org/10.1007/978-3-030-00630-3_18
Download citation
DOI: https://doi.org/10.1007/978-3-030-00630-3_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00628-0
Online ISBN: 978-3-030-00630-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)