
Abstract
X-ray crystallography is a very powerful tool to determine structures of biological macromolecules at the atomic and molecular level and thereby enables insights into their function. Unfortunately, irradiating biological samples with X-rays is an inherently damaging and unavoidable process in conventional X-ray crystallography. The unique properties of X-ray free electron lasers (FELs), with pulse duration in the femtosecond range, allow data collection at time scales shorter than, or equivalent to, the time scales of the X-ray induced radiation damage pathways, offering a plausible way to diminish the ill effects of conventional radiation damage in biological structure determination. However, the high intensities of the X-ray FEL beam necessitated the development of novel techniques for sample preparation, characterization, introduction, data collection, and analysis. Serial femtosecond crystallography (SFX) represents a set of techniques developed to enable X-ray crystallography experiments at X-ray FELs, which encompasses multiple developments in sample introduction and data collection. This chapter summarizes the early experiments that demonstrated the SFX methods along with more recent developments and accomplishments that will be discussed in more detail in the following chapters of this book.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

2.1 Background
Understanding the processes of life at the molecular and atomic levels is a major goal of the biological and life sciences. A challenge in this endeavor is that the length and time scales of interest for studying biological processes span many (10+) orders of magnitude. The components of life are as small as nanometers (size of atoms and molecules) to make a meter-scale living being such as a human. In the temporal domain, molecular dynamics occur on ultrafast time scales (nanoseconds or faster) leading to concerted dynamics on a much slower scale on the order of a second or more at the molecular level, to sustain a human life for up to 100 years, for example. No individual technique has proven to be the panacea to study the processes in biology at these disparate length and time scales. X-ray crystallography has traditionally provided information at high spatial resolution (atomic to molecular resolution) but information of limited temporal resolution, affecting the ability to study the dynamics of biological macromolecules. Since crystallography relies on producing crystals with repeating units of molecules that are (mostly) identical and oriented in a well-ordered manner, there is a pragmatic upper limit to the length scale that can be studied. Growing well-ordered crystals of large macromolecular complexes or complexes with large flexible regions is very difficult. Serial femtosecond crystallography (SFX) is utilizing the unique properties of X-ray free electron lasers (FELs) to expand the reach of crystallography to targets that are more difficult to crystallize. SFX also allows for time-resolved X-ray crystallography studies at room temperature to study the dynamics of biological molecules.
SFX is a growing field with many important experiments done to date. As such, several excellent review articles have been written to date about SFX [1,2,3] and the interested reader is directed to them for different perspectives on the technique.
2.2 The Need for SFX
Crystallizing macromolecules is very different from crystallization of small molecules. Crystallization of larger biomolecules involves slower transport properties in solution. Larger biomolecules are often very fragile and maintain their function and structural integrity only in a narrow window of experimental conditions such as solvent, ionic strength, pH, temperature as well as precipitants—the very parameters that can be used to lower the solubility of the molecule. Membrane protein crystallization is even more complex as they reside in the membrane and are amphiphilic and therefore must be crystallized either in form of protein-detergent micelles or in complex phases of aqueous solvent, such as lipidic cubic phases [4].
Generally, protein crystals have much higher solvent content in the crystal than small molecules, feature a small ratio of crystal contact to total surface area of the molecule and are also grown at much higher levels of supersaturation [5]. Potentially as a direct result of these differences, the growth of large single crystals of proteins can be challenging. Long range disorder effects accumulate and often lead to poor order or even termination of crystal growth. Therefore, the observations of small, nanocrystalline or microcrystalline showers during initial sparse-matrix screening is common [6] and suggests that it may in some cases be easier to generate well-ordered microcrystals than to grow large, well-ordered macrocrystals.
A technique that could make use of microcrystals would be incredibly valuable, as years can be spent optimizing the crystallization conditions for interesting macromolecules such as many of the membrane proteins and macromolecular complexes. Chapter 3 of this book is focused on the growth and characterization of nanocrystals and microcrystals for X-ray FEL studies. The challenge with using microcrystals for high-resolution structure determination is that the very act of collecting data on the samples with X-rays will damage the sample. In essence there is a competition between the X-ray dose required to accumulate high-resolution data and how rapidly the sample decays in the beam. To overcome radiation damage in order to obtain high resolution data sets typically requires spreading the dose to as many molecules as possible, often achieved with large, well-ordered crystals illuminated at a low dose per molecule or shifted during data collection to illuminate fresh spots of the sample. The damage is caused by X-ray induced photoionization followed by diffusion of the radicals causing photoreduction, which leads to rapid reduction of metals, bond breaking and finally destruction of the biomolecule and the crystalline order. In the diffraction data these damage processes will manifest at both a global and local level. Global damage will appear as an overall reduction in scattering power from the sample and can manifest as large-scale destruction of a crystal, whereas local damage will lead to a reduction in interpretability of the electron density maps near certain components of the sample, as shown in Fig. 2.1 [7]. X-ray induced radiation damage will be covered in more depth in Chap. 6.

Ill effects of radiation damage. Specific structural damage inflicted on a cryocooled crystal of apoferritin during sequential data sets collected on beamline ID14-4 at ESRF. (a) 2Fo—Fc map of Glu63 contoured at 0.2 e Å−3 after a dose of 2.5 MGy and (b) after 50 MGy. (c) 2Fo—Fc map of Met96 contoured at 0.2 e Å−3 after a dose of 2.5 MGy and (d) after 50 MGy, showing loss of electron density around the disordered atoms. Figure and caption reproduced from [7]
As discussed in Chap. 1, X-ray FELs have unique properties compared to synchrotron X-ray sources, namely substantially higher peak power and ultrashort pulse durations on the order of tens of femtoseconds (1 fs = 10−15 s). Based on molecular dynamics simulations of the process of Coulomb explosion of a biomolecule irradiated by an X-ray FEL pulse in vacuum, Neutze et al. predicted that if the X-ray FEL pulse duration is shorter than the damage processes in the molecule, the diffraction data from single molecules or crystals could be collected from a damage-free structure, a concept later called diffraction-before-destruction [8]. Early experiments at FLASH (Free-Electron LASer in Hamburg), the soft X-ray FEL at DESY in Hamburg, hinted at the promise of using diffraction-before-destruction for damage-free structure determination by successfully reconstructing an image of a nanofabricated artificial object from a single diffraction pattern measured by exposure to a single FLASH pulse [9]. Further tests of the diffraction-before-destruction concept were performed using FLASH. For example, a time evolution of sample damage was imaged using two illuminations of the sample with a controlled delay time between them. A novel method called time-delay holography was used to reflect the pulse back onto the exploding sample to probe the damage generated by the FLASH pulse as a function of time, confirming some of the theoretical predictions [10].
The excitement quickly built from the early FLASH results and the anticipation of the eventual impact of X-ray FELs on macromolecular crystallography (MX). However, questions remained about the viability of using submicron macromolecular crystals for structure determination, which was considered one of the most promising aspects of using X-ray FELs for MX. During the buildup to the first SFX experiments, powder diffraction measurements using liquid injection of nanocrystals of the integral membrane protein photosystem I (PSI) into the X-ray interaction region were performed at beam line 9.0.1 of the Advanced Light Source at Lawrence Berkeley National Laboratory [11]. PSI is a large membrane protein complex consisting of 36 proteins and 381 cofactors, and is a challenging test case for serial crystallography methods. Ultimately, the experiments were able to measure powder diffraction from PSI crystals that were restricted in size to <220 nm along an edge (very weak diffraction was even recorded from <100 nm PSI crystals) [11, 12]. The experiments demonstrated that submicron crystals of a membrane protein could be effectively delivered in vacuo to the X-ray interaction region and retain their integrity and diffraction power. The results suggested that these submicron crystals might be useful for structure determination, if radiation damage was not a concern, but the idea could not be further tested until MX could be done at an X-ray FEL. These experiments, along with demonstration experiments at FLASH paved the way for SFX at the Linac Coherent Light Source (LCLS), the first high energy X-ray FEL. However, adapting protein crystallography to serial data collection at X-rays FELs proved challenging.
The nature of the X-ray FEL pulses makes conventional crystallography methods challenging and therefore new methods had to be invented, which range from nanocrystal growth and characterization, sample delivery, all the way to new data evaluation algorithms. In crystallography, the reconstructed electron density is a Fourier transform of the structure factors F hkl, where the amplitude of F hkl is correlated to the Bragg peak intensities I hkl in measured diffraction patterns. In order to determine the intensities of the individual Bragg peaks, the reciprocal lattice points associated with the structure factors must meet the Bragg conditions, that is, at a given orientation only a partial subset of the reflections can be measured (for more detailed information please see any of the excellent X-ray crystallography textbooks that are available [13,14,15]). A full data set is typically measured through rotating the crystal during data collection. Typically in conventional crystallography, rotation series from individual (or a small number of) macromolecular crystals are collected in order to measure full reflections from each reciprocal lattice peak [16] and data are collected at cryogenic temperatures to reduce, but not evade, X-ray damage. For large crystals, diffraction data at X-ray FELs can be collected using a highly attenuated beam and moving the crystal between shots [17,18,19]. Generally, the high peak intensity of an X-ray FEL pulse makes multiple measurements on any single particle or microcrystal impossible; thereby diffraction from each microcrystal can only be measured once before being destroyed. Consequently, for microcrystals, new sample introduction methods to the X-ray beam have been developed as well as novel data analysis methods that allow for accurate determination of the structure factor amplitudes from millions of random single crystal diffraction snapshots.
2.2.1 Sample Preparation and Characterization for SFX
As with traditional macromolecular crystallography, sample preparation and characterization are still key to the successful outcome of an experiment. SFX allows microcrystals to be used for high-resolution structure determination but new methods had to be developed to grow microcrystals in sufficient quality and quantity for SFX studies.
While most crystals grown for conventional crystallography are grown by vapor diffusion methods, these are not suitable for most SFX experiments due to the difficulty in scaling them up to meet the high sample consumption requirements of SFX arising from the need for a new crystal on every pulse. Crystallization methods have been adapted and developed for growth of nanocrystals and microcrystals for SFX. In addition, methods have been developed where nanocrystals and small microcrystals are grown by expression of the protein in living insect cells [20, 21] or bacterial cells. Nanocrystals require new methods for detection and characterization and have to be discriminated from non-crystalline aggregates that are not useful for SFX. Unfortunately, nanocrystals are similar in size to the wavelength of visible light and can therefore not be identified with a light microscope. Even for small microcrystals (here defined as between 1 μm and 5 μm in size) the sharp edges typically associated with a crystal are not easily discernible with a light microscope. To make matters worse, checking for birefringence, a powerful method for identifying protein crystals larger than a few micrometers, becomes very challenging, if not impossible, for nanocrystals and small microcrystals [22]. Any additional scattering from the crystallization drop, such as resulting from a PEG (polyethylene glycol) skin, will make optical identification of microcrystals very difficult, and the in meso phases can be notoriously challenging [23]. Fortunately, a lot of knowledge has been gained and new methods have been developed for growth and characterization of the nanocrystals and small microcrystals used for SFX, which are discussed in detail in Chap. 3.
Through the years of rapid development in SFX, numerous techniques have been shown to help in the identification and characterization of microcrystal samples. The crystallinity of submicron particles can be tested using electron microscopy [24, 25] but the throughput is low. Dynamic light scattering and Nanosight tracking (Malvern instruments) can be used to quickly determine size distributions for submicron to micrometer sized proteins, but do not relay information about crystallinity. DLS can identify the size of particles up to about 5 μm (depending on the buffer components), while Nanosight tracking is limited to sizes <500 nm but has the advantage to also allow for identification of the particle density.
A set of methods has been developed and is now available to identify nanocrystals and microcrystals from other protein-rich or crystalline phases. Second-order nonlinear imaging of chiral crystals (SONICC) [26, 27] is a two photon microscopy technique [28] and shows strong signal for protein crystals down to hundreds of nanometers, and can be greatly enhanced by chromophores present in the protein [29]. SONICC signal depends on the symmetry of the sample and the orientation of crystals in the field of view, and can give false positives for crystals of certain chiral inorganic and organic molecules and ligands [30]. UV-fluorescence, achieved from two-photon or standard methods, can be used to determine the presence of tryptophan in the small aggregates, allowing protein-rich phases to be discriminated from salt crystals, PEG skins, etc., but the contrast between the fluorescence signal of crystals and other aggregates may make unambiguous identification of macromolecular crystals difficult [31]. Testing for reversibility of precipitation conditions could also be used to identify potential nanocrystallization and microcrystallization conditions [6]. X-ray powder diffraction can assess the difference in diffraction quality between crystal batches, and can easily distinguish macromolecular crystals from other aggregates and salt crystals, and can be used for all space groups, but the resolution will generally be limited compared to diffraction results from an X-ray FEL [2].
2.2.2 Sample Introduction for SFX
If a microcrystal can only be measured once with an X-ray FEL beam before being destroyed, new and effective methods had to be developed to deliver the sample to the X-ray interaction region. We provide here an overview, while Chap. 5 describes these sample delivery methods in more detail.
The primary method developed for crystallography at X-ray FELs has been the serial crystallography approach, in which the crystals are delivered to the X-ray interaction region in a serial way. In the serial approach, the sample is moved through the interaction region during data collection and the data are taken from many copies of similar samples. SFX was developed for X-ray FEL sources but the techniques have recently been applied to serial crystallography data collection at room temperature with millisecond X-ray exposures of larger crystals (10–50 μm) at synchrotron sources [32,33,34]. While primary X-ray damage cannot be outrun at synchrotron sources, the millisecond exposure time reduces secondary damage effects.
The sample must be delivered and replenished at faster rates than the X-ray FEL frequency to avoid X-ray damage from the previous pulse. While current sample delivery systems have been optimized for the current X-ray FELs that operate at 10–120 Hz high repetition rates, the European X-ray FEL (XFEL.EU), which began user operation in the fall 2017, and LCLS-II (under construction) will approach or exceed MHz peak rates by 2020, creating a new challenge for both sample delivery and detector read out rates.
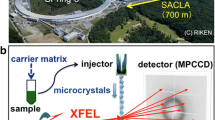
The high solvent content typical of macromolecular crystals means that the crystals need to be treated delicately in order to avoid mechanical damage. One compelling option to deliver the sample is to use a liquid stream or droplets to transport a suspension of microcrystals in their mother liquor to the X-ray interaction region, with the stream allowing replenishment of the sample while removing “used” sample. During initial development, several requirements needed to be met by liquid jets in order to be considered suitable: (1) avoid damage or alteration of the sample during deliver, (2) be compatible with in vacuo work due to soft X-ray operations and the constraints of existing X-ray FEL instruments, (3) produce thin jets in order to minimize the background from the crystallization mother liquors, and (4) use minimal amounts of sample due to the low abundance of many of the samples.
Rayleigh jets are easy to make and use, but were found to be unsuitable for sample injection because they use a large amount of sample, are prone to ice formation when running protein buffers in vacuum, and smaller, <10 μm diameter Rayleigh nozzles tend to clog quickly [35]. Gañán-Calvo showed that by replacing the glass walls of a Rayleigh nozzle with a laminar accelerating gas stream a thin liquid jet can be formed [36]. The gas dynamic virtual nozzle (GDVN) [37], shown in Fig. 2.2, was developed for SFX sample injection based upon laminar gas acceleration through an aperture and delivered samples into vacuum with significantly reduced frequency of clogging or freezing compared to the Rayleigh jet. The GDVN was a major breakthrough because (1) it produces thin jets, producing low background from the mother liquor, (2) it facilitates sample injection in vacuum because the accelerating sheath gas reduces the evaporative cooling of the injected liquid, decreasing the likelihood of ice formation on the nozzle, and (3) the large inner capillaries that deliver the sample are less likely to get clogged than the <10 μm Rayleigh nozzles needed for comparably thin jets [38]. The GDVN injection is generally driven by a high-performance liquid chromatography (HPLC) pump, allowing for a large variety of samples to be effectively injected, from soluble proteins to membrane proteins grown in certain in meso phases, such as lipidic sponge phase. The major limitation to the GDVN is relatively high sample consumption, so many new sample introduction methods have been developed to compliment the GDVN, such as the lipidic cubic phase (LCP) injector [39], double flow focusing injectors [40], microfluidic electrokinetic sample holder (MESH) [41, 42], and fixed target approaches [17, 19, 43, 44]. GDVN, as a fast flowing jet, remains a key tool of sample injection, especially for time-resolved SFX, and is discussed in detail, along with alternate injection methods, in Chap. 5.

Light Microscope image of a nozzle with liquid jet. A 360 μm OD × 40 μm ID capillary tube (a) with external taper injects a liquid stream into a convergent gas flow in a 1.2 mm OD capillary nozzle (b). This yields a rapidly tapering liquid jet (c) surrounded by a co-flowing gas. Figure and caption reproduced from [38]
2.2.3 Data Analysis for SFX
One of the major topics of discussion directly before the initial SFX experiments (and to this day!) was data processing. Unlike conventional crystallography at synchrotrons, where intensities of full reflections are recorded in a rotation series, in SFX a data set consists of tens of thousands of diffraction patterns. Each SFX diffraction pattern is essentially a “still” image (i.e., a very thin “slice” through the reciprocal lattice) and is thereby comprised of partial reflections. The integrated intensity for a reflection that is associated with the structure factors cannot be determined from a single SFX diffraction pattern [45]. To conquer the challenge, Kirian et al. [45] developed a new data evaluation method that uses Monte Carlo methods to determine accurate structure factors by averaging over the separate intensity measurements made from different crystals (in separately recorded diffraction patterns). An “average” structure factor could be calculated and used to determine the underlying structure. This “Monte Carlo” processing of the diffraction data has proven to be the workhorse for SFX data analysis, even as more sophisticated post refinement methods, such as determining the partiality of the measured Bragg peak [46, 47], are implemented.
In practice, SFX data analysis is more than simply indexing and merging of the data. After dark and gain corrections for each recorded diffraction pattern, significant effort goes into finding diffraction patterns that contain Bragg peaks (and disregard blanks), known as “hit finding.” An iterative process of hit finding and indexing is usually done until a sufficiently large number of indexable diffraction patterns are identified. SFX data evaluation thereby requires assessment of data quality and quantity in real time. Furthermore intensity variations of the individual X-ray FEL pulses as well as variation of the diffraction volume of the individual crystals require data sets with high multiplicity. The amount of data required for the determination of accurate structure factors for a complete data set is a controversial point of current discussion and also depends on the resolution of the data sets, the methods used for phasing, as well as the data evaluation programs. The average SFX data set consists of more than 10,000 patterns, while large data sets over 100,000 patterns have been reported and may be required for certain time-resolved studies [48] and for experimental phasing [49]. Consequently, much work has gone into software to facilitate SFX data analysis. The major SFX analysis programs are Cheetah [50] for hitfinding, CrystFEL [51] for indexing, merging, and mtz generation, and cctbx.xfel [52] for hit finding through mtz generation. The detailed considerations for SFX data analysis are featured in Chap. 7.
2.3 Early Experiments of Serial Femtosecond Crystallography (SFX)
The first SFX experiments took place in 2009 and many advances and milestones that made SFX possible were contributed by a large and active research community. Details of the initial experiment will be discussed in the coming section but the interested reader is directed to read the original publications (cited throughout the chapter) for detailed descriptions.
2.3.1 The “Birth” of SFX
The first demonstration of SFX methods took place at LCLS simply by the nature of LCLS being the first FEL source in the X-ray regime available to users. LCLS commissioning and early user operations started in early 2009 [53]. The first protein crystallography experiments [54] were performed in December 2009 using microcrystals of Photosystem I (PSI) at the Atomic Molecular and Optical (AMO) [55] instrument in the CAMP endstation [56] using a photon energy of 1.8 keV (6.9 Å wavelength) with a 7 μm FWHM focus. The experiments also investigated X-ray induced radiation damage by measuring diffraction data at 20, 70, and 200 fs pulse duration, with a peak power density of 1016 W/cm2 and an estimated absorbed dose for the protein crystals of 700 MGy, well above the conventional X-ray dose limit of 30 MGy [57]. Two sets of detectors were used to measure the diffraction data, a “front” detector at a working distance of 68 mm that collected high spatial resolution data and a “back” detector that collected high angular resolution data.
Diffraction data from PSI nanocrystals were collected to 8.5-Å resolution at the edges of the detector, which corresponded to the resolution limit for the given experimental geometry and photon energy. In total, over 3,000,000 diffraction patterns were collected from the sample of PSI crystals between 200 and 2000 nm in size (many were blank). A total of 112,725 were identified as having over 10 Bragg peaks of which 15,445 patterns were successfully indexed and merged for the final data set and phasing was performed by molecular replacement [54, 58]. Figure 2.3 shows diffraction patterns and the electron densities derived from the SFX diffraction pattern from the PSI nanocrystals.

Diffraction intensities and electron density of photosystem I. (a) Diffraction pattern recorded on the front pnCCDs with a single 70-fs pulse after background subtraction and correction of saturated pixels. Some peaks are labelled with their Miller indices. The resolution in the lower detector corner is 8.5 Å. (b) Precession-style pattern of the [001] zone for photosystem I, obtained from merging femtosecond nanocrystal data from over 15,000 nanocrystal patterns, displayed on the linear color scale shown on the right. (c, d) Region of the 2mFo—DFc electron density map at 1.0σ (purple mesh), calculated from the 70-fs data (c) and from conventional synchrotron data truncated at a resolution of 8.5 Å and collected at a temperature of 100 K (d) (Methods). The refined model is depicted in yellow. Figure and caption reproduced from [54]
The 8.5-Å data and electron density maps generated from these initial SFX experiments were the first breakthrough for SFX. However, several other interesting results were obtained in these first experiments. Data collected on the back detector had high angular resolution, and interference fringes between the Bragg peaks were detected for some of the smaller crystals, as shown in Fig. 2.4. In 1952, Sayre had proposed that if one were ever able to collect X-ray diffraction data from crystals with a small number of unit cells one would see intensity between the Bragg peaks. The intensity between the peaks arises from non-delta function reciprocal lattice peaks from the finite sum of unit cells in the crystal, while interference fringes from the small crystals result from the high coherence of the beam and the faceting of the crystals, essentially representing the Fourier transform of the size and shape of the object. This was one of the first observations of intensity between the Bragg peaks in protein crystallography data. These data can be used to reconstruct the size of the object as shown in the insets of Fig. 2.4 and these particular reconstructions were accomplished using the shrinkwrap [59] algorithm. The added information could potentially be used to solve the crystallographic phase problem [60], as will be discussed in Chap. 8.

Coherent crystal diffraction. Low-angle diffraction patterns recorded on the rear pnCCDs, revealing coherent diffraction from the structure of the photosystem I nanocrystals, shown using a logarithmic, false-color scale. The Miller indices of the peaks in (a) were identified from the corresponding high-angle pattern. In (c) we count seven fringes in the b* direction, corresponding to nine unit cells, or 250 nm. Insets, real-space images of the nanocrystal, determined by phase retrieval (using the Shrinkwrap algorithm [59]) of the circled coherent Bragg shape transform. Figure and caption reproduced from [54]
Although the electron density maps did not contain any evidence of conventional X-ray induced radiation damage, the impact of pulse duration on crystallographic data quality was explored using pulse durations of 20, 70, and 200 fs. The collected data showed a distinct reduction in (normalized) scattered intensity at larger scattering angles for the longer pulses indicating that alterations of the sample due to its interaction with the X-rays occur at longer pulse duration. The result was an important initial finding but research into radiation damage in SFX is even more complex and still ongoing. However, during the early operation of LCLS the reported X-ray pulse durations were derived from the electron bunch durations without direct measurement of the X-ray pulse duration itself. An experiment measuring photoionization of Ne atoms suggested that the X-ray pulse durations were approximately half the electron bunch duration [61]. Experiments have been dedicated to better understand this important aspect of SFX and X-ray FEL data collection in general, as discussed in later sections of this chapter and Chap. 6.
While the design and construction of the hard X-ray instruments at LCLS and the SPring-8 Angstrom Compact free electron Laser (SACLA) were still ongoing, several additional SFX experiments were done at the AMO instrument. Crystals of a photosynthetic reaction center grown in lipidic sponge phase (LSP) were introduced to the interaction point using a GDVN injector. Data from the <10 μm crystals were collected to the geometric limit of 7.4 Å for the experiment with the electron density reconstructed to 8.2-Å resolution [62].
Additional experiments were performed to better understand the impact of radiation damage, as the initial experiments showed a pulse duration dependence on the quality of the collected data. Detailed analysis showed that X-ray damage mechanism is different in SFX compared to synchrotron-based data collection. In synchrotron-based damage studies, the total intensity remains constant while reflections become broader and background increases until they “merge” with the background. In contrast, the first SFX damage studies show that with increased pulse duration “termination” of Bragg peaks occurs. Barty et al. [63] showed that the crystalline order of the samples would decrease during the plasma formation and onset of nuclear motion. The reduction of crystalline order will cause the Bragg diffraction to cease in a process termed “self-terminating diffraction” and shown in Fig. 2.5 [63]. Self-termination of the Bragg peaks could be interpreted as meaning that the pulse duration in SFX is inconsequential, as the Bragg diffraction terminates and the scattering of the destroyed crystals only leads to an increase in the homogenous diffuse background. However, this picture is likely overly simplistic. Ionization of the atoms that results in an even distribution throughout the unit cell will lead to an overall decrease in scattered intensity in the Bragg peaks; uncorrelated ionization would lead to a background increase caused by homogenous diffuse scattering. Lomb et al. found a clear trend that the R-factors between data sets increase as the pulse duration increases [64]. At this point, it was clear that pulse duration had an impact on the quality of SFX data collected but the limited resolution by 2 keV X-rays was a limitation on the further exploration of the topic. These questions were further addressed as higher energy X-rays became available and are discussed in detail in Chap. 6.

Bragg termination observed at approximately constant X-ray pulse fluence I0T. (a) “Virtual powder pattern” formed by summing 3792 single-pulse patterns obtained with X-ray pulses with a duration of 300 fs. The spots in the pattern are Bragg peaks, which are visible out to the corners of the detector, corresponding to a resolution of d 1/4 0.76 nm. Because of the large unit cell size of the crystal, Debye–Scherrer rings overlap and are not resolved at q > 0.5 nm−1. (b) Bragg signal I(q; T) of Photosystem I nanocrystals averaged over q shells of virtual powder patterns for nominal pulse durations T varying between 70 fs and 300 fs. (c) Bragg signal relative to the shortest pulses, plotted as solid lines. Dashed lines give the computed ratios of I(q; T)/I(q; T 1/4 40 fs) from the Cretin simulations. Previous experiments at LCLS indicate that the nominal “70 fs” pulses are shorter than indicated [61]. We achieve a best fit assuming these pulses have a duration of 40 fs. (d) Comparison of the calculated dynamic disorder factor g(q; T) (solid lines) compared to a Debye–Waller factor best-fit to the same data (dashed lines). Figure and caption reproduced from [63]
2.3.2 SFX at High Resolution
The early SFX experiments were limited in resolution due to the photon energy usable by the available instruments at LCLS. However, in mid-to-late 2010, the X-ray pump–probe (XPP) [65] and coherent X-ray imaging (CXI) [66] instruments came online and experiments could be done using hard X-rays, with the potential to extend to atomic resolution for the first time.
In February 2011, the first SFX experiments were performed at CXI using 9.4 keV (1.33 Å wavelength) X-rays with 40 fs pulse duration using lysozyme microcrystals [67]. Data were collected to 1.9 Å resolution and the calculated difference-density maps between structures derived from SFX and low-dose synchrotron experiments showed no evident signs of radiation damage at the molecular level, as shown in Fig. 2.6. This result provided further evidence for the diffraction-before-destruction idea. The result showed the first proof-of-principle that structures can be determined by SFX at near atomic resolution without significant structural damage at room temperature from crystals that are typically too small to be used for synchrotron-based crystallography. This result has major implications for the study of membrane proteins, macromolecular complexes, and formed the basis for the first time serial time-resolved studies at X-ray FELs. The impact of high resolution SFX was felt immediately with a major increase in demand for beamtime by the user community, still seen today with structural biology representing a large fraction of X-ray FEL use.

High resolution lysozyme structure. (a) Final, refined 2mFobs—DFcalc (1.5σ) electron density map of lysozyme at 1.9 Å resolution calculated from 40-fs pulse data. (b) Fobs(40 fs)—Fobs (synchrotron) difference Fourier map, contoured at +3 σ (green) and −3 σ (red). No interpretable features are apparent. The synchrotron data set was collected with a radiation dose of 24 kGy. Figure and caption reproduced from [67]
2.3.3 SFX of G-Protein Coupled Receptors
G-coupled protein receptors (GPCRs) are the largest class of integral membrane proteins in the human genome (>more than 600 genes code for GPRCs). They are receptors that are involved in many biochemical pathways and act as key players in signal transduction, neuronal function, hormone action, vision, sensing, mood control, pain sensing and relief and are major drug targets against diseases, with >50% of all current drugs being targeted to GPCRs [68]. However, membrane proteins are notoriously difficult to crystallize. While over 100,000 structures of proteins have been determined to date, structures for <800 unique membrane proteins have been solved to date. The intrinsic amphiphilic nature of membrane integral proteins makes them difficult to express, isolate intact from the membrane, purify and crystallize. They often form only small, weakly diffracting crystals that deteriorate rapidly when exposed to synchrotron radiation. Despite their high medicinal impact, leading to huge efforts to determine structures of GPCRs, they resisted crystallization and structure determination until a breakthrough was achieved in the field with crystallization of GPCRs in lipidic phases that mimic the native membrane [4, 69]. As discussed in Chap. 4, crystallizing GPCRs using these in meso media, such as lipidic cubic phase (LCP) or lipidic sponge phase (LSP), has been very successful to date and led to the first high-resolution GPCR structure in 2007 [70]. However, many of the crystallization trials lead to small crystals that are insufficient for high-resolution data collection at a synchrotron due to rapid deterioration in the X-ray beam even at cryogenic temperatures. Consequently, the development of new methods to enable the use of SFX for LCP-grown GPCR crystals was a very important goal. This was greatly enabled by a new sample injector capable of effectively delivering the high viscosity media (which have the consistency of tooth paste) to the X-ray FEL interaction region. These newly developed injectors are called high viscosity extruders (HVE) [32] or LCP injectors [39] and are discussed in depth in Chap. 5.
The first SFX experiments on LCP-grown GPCR crystals were done in 2012 [71]. Microcrystals of the human serotonin receptor 5-HT2B bound to the agonist ergotamine were delivered using the LCP injector and a full data set was collected to 2.8-Å resolution at room temperature. The crystals used in the SFX experiments were 100× smaller in volume than the crystals required for the synchrotron experiments. This was the first X-ray structure determined from a GPCR at room temperature. The SFX-derived electron density maps were compared to synchrotron-derived electron density maps, and although the overall structures were very similar (rmsd = 0.45 Å for the backbone) significant differences were identified. Several side chains were much better resolved in the X-ray FEL structure compared to the synchrotron-based structure, and structural changes were detected in the loop regions of the proteins, side chain conformations and even part of the backbone of one of the helices was altered (it was kinked in the synchrotron structure but straight in the SFX structure). These structural differences may represent the more native conformations of the GPCR as the SFX data were collected at room temperature whereas the synchrotron-based structures are derived from data at cryogenic temperatures. The crystal size reduction is a major development for protein crystallography, as growth of large well-ordered crystals of GPCRs and other membrane protein samples can take years to finish, if ever. SFX of membrane protein targets, and especially GPCRs, represent a prominent area of current research at X-ray FELs [72, 73] and is covered in detail in Chap. 10.
The high viscosity of the LCP-grown crystal samples limits the velocity of the sample moving through the X-ray interaction region. This results in much lower sample consumption compared to GDVN (Liu et al. [71] reported 300 μg of sample used versus 10s of mg for GDVN) as the flow rate of the jet can be tightly controlled from 1 to 500 nl/min, and the jet speed is adjusted so that the sample is just replenished between the X-ray pulses. The viscous injectors have therefore been highly desired for sample delivery of crystals grown conventionally in solution by embedding crystals in LCP [74] or other viscous media. Alternate viscous media include agarose [75] or hydroxyethyl cellulose [76], grease [77], as well as high molecular weight PEG [33] matrices. The development of viscous injection methods for X-ray FELs has also recently enabled serial microsecond crystallography studies at RT at synchrotron sources [34], which highlights the strong synergistic effects of X-ray FEL developments on conventional crystallography.
2.3.4 Phasing of SFX Data
The first structures determined by SFX were all solved by use of the method of molecular replacement (in which the phases from a structurally-similar molecule are used [78]), however experimental phasing is required to solve novel structures for which no homologous structure exists. In principle the same phasing methods that are established for conventional crystallography (Single and Multiple Isomorphous Replacement (SIR and MIR), as well as Single and Multiple Anomalous Diffraction/Dispersion (SAD and MAD) can be applied to SFX. However these phasing methods are more challenging for SFX than for conventional crystallography, as the isomorphous or anomalous differences are generally small and require very accurate structure factor determination. During the Self-Amplified Spontaneous Emission (SASE) process that generates the normal lasing of an X-ray laser (see Chap. 1 for details), the X-rays are delivered with variations in pulse energy, wavelength, and pulse duration, which decrease the accuracy of individual measurements and complicate experimental phasing. Consequently, experimental phasing is easier to achieve with conventional crystallography where data can be collected on one crystal and there are smaller fluctuations of the delivered X-rays between images. These challenges can be met by a high multiplicity of the SFX data sets and several proof-of-principle studies have shown successful de novo phasing of SFX data [79, 80]. Since the initial successes using SAD phasing, SAD has been demonstrated using several the elements, such as copper [18], mercury [81], iodine [82], and. SAD phasing accounted for ∼70% of all novel macromolecular structures in 2013 with the most commonly used element being selenium [83] (because selenomethionine, a derivative of methionine in which the sulfur is replaced by selenium, is easily incorporated into many proteins during expression); experiments done at LCLS [84, 85] and SACLA [86] have demonstrated Se-SAD. Additionally, native SAD using sulfur atoms has been demonstrated using lysozyme [87], thaumatin [49], and human A2A adenosine receptor [80].
The X-ray FEL equivalent of MAD phasing experiments has been successfully demonstrated at SACLA [88]. In this example, two color or split beam experiments in which crystals are hit by two pulses of different color are used to record two diffraction patterns with different anomalous content for each pulse and each crystal, and can in principle improve phasing efficiency by comparing data from the same crystal. The demonstration of experimentally determining the phases of novel structures with SFX was a major milestone for the technique, both showing the accuracy of the measurements and the progress made in the analysis of the data.
It deserves mentioning that the SFX SAD/MAD phasing cases to date have been done using standard proteins, that is, proteins that would not need to have the phases determined experimentally and generally scatter very strongly (important for SAD/MAD since the relative difference signal increases with resolution). However, in the case of the mosquito larvicide protein BinAB, experimental phasing from SFX data was needed because the crystals are limited to <500 nm in size, not affording a structure from conventional crystallography. The novel structure was phased using the Multiple Isomorphous Replacement and Anomalous Scatter (MIRAS) approach, collecting data on iodine, gadolinium, and mercury derivatives of in vivo grown nanocrystals [89].
In addition to being able to continue the experimental phasing approaches developed at synchrotrons, SFX opens new avenues for phasing including phasing based on shape transforms from finite crystals [60] and direct phasing based on continuous diffraction (see below). These novel methods are discussed in Chaps. 8 and 9 in more detail.
2.3.5 Time-Resolved SFX
Time-Resolved crystallography (TR-MX) studies have been pioneered at synchrotron sources utilizing Laue diffraction methods and generally optical lasers as pumps [90,91,92]. However, TR-Laue-MX data collection is challenging as it requires very large crystals, which makes homogeneously pumping the sample challenging, X-ray damage can be severe and time-resolution is limited to ∼100 ps due to the pulse duration at synchrotrons [90, 93].
The ultrashort time duration of X-ray FEL pulses enables data collection at room temperature from very small nanocrystals and microcrystals, and is ideally suited to further develop the method of time-resolved crystallography. The theoretical temporal resolving power of an X-ray FEL is limited by the pulse duration (although the ultimate temporal resolution will be dependent on the sample and experimental setup) and therefore Time-Resolved SFX (TR-SFX) can explore shorter timescale phenomenon in biology than is possible using synchrotrons.
The development of the first TR-SFX experiments at X-ray FELs was driven by the motivation to study the light-driven processes in photosynthesis. The first experiments were performed using soft X-rays at the AMO beamline using a complex of Photosystem I with its electron acceptor ferredoxin [94]. With the increase in the usable X-ray FEL energy, the study of the water splitting mechanism of Photosystem II (PSII) became a very active field of X-ray FEL research at both LCLS and SACLA. The full photocycle studies can only be done at X-ray FELs as the core of the oxygen-evolving complex consists of a Mn4O5Ca cluster which cycles through five oxidation states during the catalytic cycles. X-ray damage of the cluster by reduction of the metals is severe, but can be outrun by X-ray FEL pulses as shown by the first undamaged dark structure of PSII determined from large frozen (1 mm) crystals, which are shifted after each X-ray FEL shot [19]. TR-SFX studies on PSII are very complex, as they require multiple laser excitations to cycle PSII through the five stages of the catalytic cycle. The resolution for TR-SFX studies has been improved from 5 Å for the first TR-SFX studies on PSII [95, 96] to 2.2–2.5 Å resolution for recent TR-SFX studies [48, 97].
Besides the ongoing PSII work, many experiments have been done establishing the ability to measure interpretable difference signals between the excited and ground states of photoactive proteins to high spatial and temporal resolution. Difference data from TR-SFX experiments using photoactive yellow protein (PYP) extended to 1.6 Å resolution and showed excellent quality and strong difference density features, as shown in Fig. 2.7 [99]. The 1-μs Difference Electron Density (DED) maps showed similar, but much stronger, features compared to 1-μs DED maps produced from synchrotron data collected using Laue diffraction.

Comparison of electron density and DED maps in the chromophore pocket obtained by TR-SFX and the Laue method. The dark state is shown in yellow in all maps. (a, d) Electron density maps for the PYP dark state obtained with TR-SFX and Laue, respectively (contour level 1.1 s, 1.6 Å resolution).The PCA chromophore and nearby residues are marked in (a). Arrow: Double bond in the chromophore about which isomerization occurs. (b) TR-SFX DED map at 10 ns. Light green structure: ICT intermediate. Features marked by dotted arrows belong to additional intermediates not shown. (c) TR-SFX DED map at 1 ms. Pink and red structures: structures of pR1 and pR2 intermediates, respectively. (e) Laue 32-ns DED map correlates best to the TR-SFX 10-ns map. (f) Laue 1-ms DED map. Contour levels of the DED maps: red/white −3 s/−4 s, blue/cyan +3 s/+5 s, except for (c) where cyan is +7 s. Figure and caption reproduced from [98]
The proof-of-principle PYP TR-SFX experiments demonstrated several substantial benefits of SFX for laser-excited time resolved experiments. For the time-resolved Laue experiments previously done using synchrotron sources, large PYP crystals were needed to achieve high resolution and several pumping cycles were needed to collect a full data set. When a ns laser is used to initiate the photocycle, the laser beam size is usually smaller than the crystal and does not penetrate fully or uniformly through the PYP crystal—the percentage of molecules photoactivated in the TR-SFX experiment was calculated at ∼40%, whereas 10–15% is maximally achieved in the synchrotron experiments, leading to decreased map quality. The repeated pumping of the same crystal in the Laue experiment leads to strain in the crystal, which sets an upper limit to the laser fluence that can be used for the experiment. These issues are avoided with TR-SFX, as the crystals used were smaller than the absorption length of the laser in the PYP crystals and each crystal was at most used for one diffraction pattern.
Several experiments have explored TR-SFX to investigate the sub-picosecond dynamics of proteins (and the chromophores typically absorbing the light energy), such as CO dissociation from myoglobin [100], cis/trans isomerization of PYP chromophore [98], and chromophore twisting of a photoswitchable fluorescent protein [101]. These experiments have shown the potential for TR-SFX to reach time resolutions below 200 fs [98], a previously inaccessible time scale. Each of these pioneering experiments required careful experimental design in order to accurately measure small differences in diffraction data. For an in-depth discussion of TR-SFX the reader is referred to Chap. 11.
The ability to use microcrystals in SFX to achieve atomic and molecular resolution also allows for reactions to be triggered by mixing, in which case a substrate/ligand is mixed with the crystals of interest [102]. The substrate diffuses into the crystal and the progression of the time-dependent changes is measured at specific time points after the mixing. This method allows for snapshots of the reaction to be recorded, which paves the way for molecular movies to be recorded in the future, when higher repetition rate X-ray FELs become operational. Reactions of biomolecules (e.g., enzymatic catalysis) could be recorded on a microsecond to second timescale. Proof-of-principle experiments on X-ray FEL-based Mix and Inject Serial Crystallography (MISC) include the study of the complex of β-lactamase and the antibiotic ceftriaxone at 2.4 Å resolution [103], and the first observation of conformational changes in riboswitches at 2.8-Å resolution [104]. An overall schematic picture of the β-lactamase experiment [103] is shown in Chap. 12. The overall design of the riboswitch MISC experiment, in which a T-junction was used for time points >10 min, is shown in Fig. 2.8a. The large differences in the riboswitch samples in the apo and ligand-bound structure are shown in Fig. 2.8b.

The experimental setup of 3DX and conversion of the structure and crystal lattice after ligand mixing. (a) Schematic illustration of the SFX ligand-mixing experiment. For details, see the Methods. (b) The unit cells of the crystals of apo (P21) and ligand-bound (P21212), whose structure was converted in crystallo from the apo structures after >10 min mixing with adenine ligand. Figure and caption reproduced from [105]
MISC is an exciting and novel way for X-ray FELs to impact structural biology beyond the limited field of photoactive proteins and is discussed in greater detail in Chap. 12. The high spatio-temporal resolution offered by X-ray FELs combined with the higher repetition rate machines, such as EuXFEL and (potentially) the high energy extension of LCLS-II (LCLS-II-HE, which would enable LCLS-II to produce hard X-rays) may offer a paradigm shift for structural enzymology, changing the experiments from studying a few time points that underdetermine the time course of the enzyme to a sufficiently temporally determined system. Future X-ray FEL sources and the types of experiments they will facilitate are discussed in Chap. 16.
2.3.6 Imperfect Crystals and the Push Toward Crystal-Free Imaging
Continuously modulated diffraction intensities have frequently been encountered in MX [105,106,107,108,109,110] and were generally considered a detrimental feature, although the data could be used for understanding the defects present in a crystal and potentially to infer protein dynamics within the unit cell [111]. Smaller macromolecular crystals may have fewer defects than larger crystals, and may have smaller unit cell parameter variability and mosaicity throughout [112]. In large crystals, long range order defects accumulate where disturbances during growth leads to shifts of the crystal lattice indicated by an increase in mosaicity. While these long-range growth defects can be diminished in nanocrystals and small microcrystals, other defects like dislocations caused by thermal motions in crystals with weak crystal contacts or defects caused by heterogeneities of the molecules will still be present.
SFX data that contain continuously modulated diffraction intensities from crystals of the large membrane protein complex Photosystem II (PSII) were collected at LCLS. While the Bragg peaks terminated at 4.5 Å resolution, diffuse scattering extended beyond the Bragg peaks, as shown in Fig. 2.9. While the cause of diffuse scattering was previously discussed to be arising from flexibility within the molecule, Ayyer et al. proposed a model in which continuous diffraction is based on diffraction from rigid units of molecules within the unit cell (the PSII-dimer in this case) that are translationally displaced within the unit cell (not rotated). An rmsd displacement of the molecules within the unit cells by 0.8 Å could account for the Bragg peaks terminating at 5 Å resolution [114]; this displacement is smaller than the distance between a carbon–carbon bond. The diffraction from the displaced molecules would lead to a diffraction pattern that corresponds to single particle diffraction pattern of the content of the unit cell (here four PSII dimer molecules). This is the case because all unit cells are assumed in the same orientation but with no constructive interference from repeating positions, leading to an incoherent sum of the intensities from all unit cells.

Molecular coherent diffraction. An XFEL snapshot “still” diffraction pattern of a PSII microcrystal shows a weak speckle structure beyond the extent of Bragg peaks, which is enhanced in this figure by limiting the displayed pixel values. Figure and caption reproduced from [113]
A hybrid approach where Bragg and continuous diffraction data were treated as separate data sets was applied to the analysis of the diffraction patterns. The orientation of each pattern in the reciprocal space was determined by indexing the Bragg data. Then the structure was first solved based on evaluation of the Bragg data, which were phased by molecular replacement. The pixels that contain Bragg peaks were then masked to generate the continuous diffraction reciprocal space. The continuous diffraction data were then phased directly by iterative methods, where a low-resolution 9 Å molecular envelope of the complete PSII-dimer was the only input from the Bragg-diffraction-based X-ray structure. The iterative phasing of the continuous diffraction data extended the resolution range to 3.5 Å, and after convergence of the solution, the Fourier amplitudes and phases from the Bragg peaks (terminating at 4.5 Å) and the continuous diffraction data were combined to generate a 3.5 Å structure. Notably, the new electron density map showed better definition of the side chains as well as the features of the chlorophyll-cofactors when compared to the electron density maps produced solely from the Bragg data. This result has been received with much interest and controversy in the overall community. Data that were once considered reason to discard a macromolecular crystal might be useful for high-resolution structure determination, if the disorder is mainly caused by translation of the molecules in the crystal. This has sparked renewed discussion about how to interpret diffuse diffraction patterns measured in crystallography experiments as well as which metrics can be used to reflect data quality. The topic will be discussed in depth in Chap. 9.
2.3.7 Towards Single Particle X-ray Diffraction
The use of the continuous diffraction data along with the Bragg data is a hybrid approach combining methods from crystallography with techniques from coherent diffractive imaging (CDI). Many fields utilize CDI to study non-periodic objects, such as inorganic nanoparticles [115], frozen cells [113, 116], or even integrated circuits [117]. In fact, one of the great promises of the highly coherent, ultrashort-pulse X-ray FELs was that they would allow for Single Particle Imaging (SPI) of biological specimens, such as viruses and protein complexes, without the need to crystallize the samples to get high-resolution information.
The first SPI experiments at LCLS were performed in December 2009 collecting single particle X-ray diffraction data from mimivirus samples at the AMO [55] instrument in the CAMP endstation [56] using a photon energy of 1.8 keV (6.9 Å wavelength) with a 10 μm focus at FWHM [118]. Reconstructed single particle images from the collected data yielded 32-nm full-period resolution. Subsequent work on mimivirus data collected at LCLS allowed a 3D reconstruction to 125 nm, based upon the phase-retrieval transfer-function [119].
In early 2014, a meeting was held to discuss a roadmap for SPI to explore routes to reach atomic resolution, 3D imaging at X-ray FELs and more specifically LCLS. In response to the roadmap provided by the scientific community, LCLS started the SPI initiative [120]. The initiative, where more than 100 researchers from different groups work to explore the potential of SPI and seek solutions to the technical challenges, has proven very useful to perform highly technical proof-of-principle studies on model single particles of biological ensembles. The current status and exciting potential of SPI is discussed in greater detail in Chap. 14.
2.3.8 Structure Determination Based on fs Small and Wide Angle Solution X-ray Scattering
Small and Wide-Angle X-ray Scattering (SAXS/WAXS) are important tools for studying large-scale morphology and conformational changes of biomolecules in solution. The increasing use and utility of SAXS and WAXS for bioscience is indicated by dedicated beamlines being developed at most synchrotron facilities [121]. Collecting SAXS/WAXS data at an X-ray FEL allows for the method to be extended to the ultrafast time scales and, potentially for mixing experiments, analogously to MISC.
Time-Resolved SAXS/WAXS (TR-SAXS/WAXS) can be used to study important questions in light-driven biochemical processes such as how these systems cope with the energy that is absorbed by the chromophores, since the absorbed energy is similar in magnitude to the folding energy for the protein molecules themselves! In order to better understand the response of photo-activated molecules upon light excitation, the response of bacterial reaction center (RCvir), a light-activated membrane protein, was studied using TR-WAXS at LCLS and showed that RCvir relieved the strain induced via the photon absorption by structural deformation that propagates throughout the protein structure [122]. The propagating structural changes are called a protein quake and the changes were observed to continue for several picoseconds. The analysis indicated an ultrafast protein conformational mode that decayed with a half-life of ∼44 ps and reached the half-maximum value in 1.4 ps after photoexcitation with the 800-nm pump laser. Although the study was done with higher than physiologically relevant pump energies, the data and analysis show that strain induced by the absorbed light at the RCvir, is relaxed via protein structural changes that propagate more rapidly than heat.
Including SAXS measurements in the analysis of structural changes to molecules after light excitation allows structural parameters such as the radius of gyration and molecular volume to be followed in the time course, giving additional information to TR-WAXS. The TR-SAXS/WAXS method has been used to measure ultrafast dynamics in a complex of myoglobin and carbon monoxide (MbCO) by photoexciting the solution with a 538-nm pump laser and probing the sample at several time delays up to 100 ps, with a time resolution of ∼500 fs [123]. The results demonstrated that Mb undergoes significant global structure rearrangement after the photolysis of the Mb-CO bond. Similar to what was shown for RCvir, the light-induced structural changes occurring at the Mb-CO bond propagate through the protein as a quake-like intramolecular rearrangement, and the structural parameters of the molecules (radius of gyration and protein volume) oscillate about their equilibrium values for several picoseconds after the initial excitation. The maximum of the ultrafast change in the radius of gyration occurred in 1 ps, and with a radius of gyration of ∼17 Å this would put the speed of the strain relief at approximately 20 Å/ps, consistent with the expected speed of sound in the protein.
The MbCO complex is a model system for studying ultrafast dynamics in proteins. Using TR-SAXS/WAXS at X-ray FELs is an active area of research on photo-activated proteins and appears likely to contribute novel information about protein dynamics in the future.
Another method for structure determination of non-periodic objects is through Fluctuation X-ray Scattering (FXS). The basic idea behind FXS is that if X-ray scattering data are collected from randomly oriented molecules in solution in a time frame shorter than the rotational diffusion times of the molecules, more detailed information can be extracted from the pattern than from a traditional SAXS/WAXS pattern [124]. X-ray FELs have pulse durations shorter than the rotational diffusion times of biomolecules and could thereby open a new era for FXS structure determination of biomolecules in physiological conditions. The theory and practical considerations of FXS are discussed in Chap. 15.
2.4 Pushing SFX Forward
2.4.1 Serial Operations at Beamlines for SFX at Current X-ray FELs
Access to beamtime at X-ray FELs has been a hindrance to the SFX community and increased access to the X-rays would be very beneficial. In addition to multiplexing instruments using upstream monochromators to remove small portions of the SASE spectrum [125], another method to increase access is through refocusing the unused beam from SFX experiments to a downstream, serial experiment. The CXI instrument of LCLS is already conducting serial SFX operations [84], and the SPB/SFX instrument of XFEL.EU has designs for serial operations starting in the early 2020s.
2.4.2 New Experimental Endstations for X-ray FEL Experiments
All initial SFX experiments were performed in vacuum, which set strict limitations to the way samples could be introduced into the X-ray FEL. These sample delivery methods are the workhorse for SFX, however the throughput achieved strongly depends on the crystal density, which is restricted by the clogging challenge. The DAPHNIS (Diverse Application Platform for Hard X-ray diffraction in SACLA) [126] endstation at SACLA and the MFX beamline at LCLS, which operate in air or in a He atmosphere (which can be humidified) have paved the way for the development of new sample delivery methods for SFX based on the fixed target approach.
Two very promising new systems have been developed for measurements at atmospheric pressure: one novel system is based in acoustic droplet generation onto a conveyer belt that delivers the crystals within the droplet one-by one to the X-ray beam [127]. As clogging is not an issue with the tape drive, high hit rates can be achieved and sample consumption is minimized. However, the system is currently only used at 10 Hz but in principle the system could also be adjusted to 120 Hz data collection rates in the future. This tape drive system also allows for combination of X-ray emission spectroscopy (XES) with X-ray diffraction and has been successfully used for combined TR-SFX and TR-XES experiments at LCLS [97]. Chapter 13 will discuss XES spectroscopy using this setup.
The second system, known as the roadrunner system, is a goniometer-based system for fixed target measurements [44]. This system features a silicon chip with 120,000 windows that allows for data collection with 120 Hz at up to 50% hit rate, where >50% of the windows are covered by crystals. In this setup the windows of the chips are aligned to the X-ray interaction region using an inline microscope. This system has recently been used for the high-resolution structure determination of a virus [44]. Further developments are on the way to enable TR-SFX experiment as well as combination with spectroscopy in parallel to X-ray diffraction on the chip using the roadrunner. These represent just two examples of sample delivery methods that were developed specifically for X-ray FELs and other sample delivery methods in use at SACLA and LCLS will also be discussed in Chap. 5.
2.4.3 Higher Repetition Rate X-ray FELs
One very promising way to increase the number of experiments and enable new types of experiments that can be conducted using biological samples at X-ray FELs is the increase of the repetition rate of the X-ray FELs. Two new X-ray FELs, the European XFEL and LCLS-II will deliver X-ray pulses at much higher repetition rates than the current maximum of 120 Hz at LCLS. The European XFEL differs from current X-ray FELs as it delivers pulses as ten bunch trains per second. For the first operation cycle, the bunch train length will be 60 μs with 1–60 equally spaced pulses per bunch. In the future, the train length can be increased to 600 μs and it is planned that eventually up to 27,000 pulses can be delivered in 1 s.
While this increase in repetition rate can in principle dramatically increase the amount of data collected per time, it poses new challenges for the application of the SFX method, mainly with respect to sample delivery and data collection and evaluation. Sample replenishment and destruction of the flow in the jet will be an increasing challenge as pulses in the pulse train will come with a very rapid rate with spacing of only 1 μs between the pulses (for comparison the LCLS pulses are spaced 8.3 ms between pulses). Imaging of jet explosions have shown the impact of the focused beam on the flowing jet and raise a key concern about the ultimate limitation from sample delivery at high repetition rates [128]. Fixed target approaches as well as viscous jets will not be able to achieve MHz scale sample replenishment and likely only very rapid jets such as the GDVN can currently meet these goals. New experimental approaches using droplet or jet on demand techniques may prove useful for the higher repetition rate machines, although the continuous waveform machines above ∼100 kHz, such as LCLS-II, will favor jets. However, the pulse train structure also has advantages, especially for time resolved studies as reactions can be induced in the time between pulses and then up to 60 (and later 2700) images of the reaction can be recorded in the pulse train, thereby getting close to the goal of molecular movies of biomolecules at work.
A further step in this direction will be LCLS-II, which is currently under construction and will start operating in 2020. The unique feature of LCLS-II is that it will reach extremely high repetition rates of close to one million pulses/s. While LCLS-II will operate in the soft to tender X-ray energy regime (up to 5 keV for the first harmonic), which limits the achievable resolution, a proposal for a high energy upgrade (to >12 keV) has already been developed, which will allow for high resolution data to be collected at ultrahigh repetition rates. With the maximal rate of one million pulses/s, the pulses will come with a frequency of ∼1 pulse per microsecond and therefore at a similar rate to the ∼1 pulse per 230 ns pulse train at XFEL.EU. Therefore, sample delivery systems that are being developed to meet the speed requirement at the XFEL.EU can directly be used for the data collection at LCLS-II. As LCLS-II will deliver the pulses equally spaced the sample would not run to waste. Huge multiplicities of data could be achieved, which would allow for extremely accurate structure factor determination for SFX. In addition, the high repetition rate will allow a high number of images for single particle diffraction, which may significantly increase the resolution limit.
With high repetition rate machines X-ray spectroscopy and solution scattering experiments could take only few seconds instead of hours, which would bring a huge increase in data quality and quantity and allow for more time points to be measured for time resolved experiments for on single crystals and by solution scattering.
However, the very high repetition rates of the XFEL.EU and LCLS-II also lead to formidable novel challenges concerning detectors, data acquisition, data evaluation and data storage. An overview of the ongoing developments of upcoming X-ray FEL facilities will be given in Chap. 16.
2.5 Outlook
Since the start of user operations at X-ray FELs, new methods have been developed to study structure and dynamics in biological samples and systems, with SFX being a major contributor. SFX results to date have shown that the hope of structures being determined while avoiding the ill effects of conventional radiation damage were not overly optimistic. The unique capabilities of X-ray FELs have allowed new frontiers of structural biology to be pursued, pushing further into the ultrafast processes that drive photobiology and studying biological reactions of enzymes with an unprecedented combination of spatial and temporal resolutions. Although the results to date have been exciting, the high-repetition rate X-ray FELs will further enable bioscience at high spatial and temporal resolutions. The new instruments and techniques may continue a revolution in structural biology, transforming high-resolution information from (mostly) static structures into information about the structural dynamics and, ultimately, to a combined view containing structural information as it evolves in time.
References
Aquila, A., Barty, A., Bostedt, C., Boutet, S., Carini, G., dePonte, D., et al. (2015). The linac coherent light source single particle imaging road map. Structural Dynamics, 2(4), 041701. https://doi.org/10.1063/1.4918726.
Aquila, A., Hunter, M. S., Doak, R. B., Kirian, R. A., Fromme, P., White, T. A., et al. (2012). Time-resolved protein nanocrystallography using an X-ray free-electron laser. Optics Express, 20(3), 2706–2716. https://doi.org/10.1364/OE.20.002706.
Arnlund, D., Johansson, L. C., Wickstrand, C., Barty, A., Williams, G. J., Malmerberg, E., & Neutze, R. (2014). Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nature Methods, 11(9), 923–926. https://doi.org/10.1038/nmeth.3067 https://www.nature.com/articles/nmeth.3067#supplementary-information.
Ayyer, K., Yefanov, O. M., Oberthür, D., Roy-Chowdhury, S., Galli, L., Mariani, V., et al. (2016). Macromolecular diffractive imaging using imperfect crystals. Nature, 530(7589), 202–206. https://doi.org/10.1038/nature16949.
Barends, T. R., Foucar, L., Botha, S., Doak, R. B., Shoeman, R. L., Nass, K., et al. (2014). De novo protein crystal structure determination from X-ray free-electron laser data. Nature, 505(7482), 244–247. https://doi.org/10.1038/nature12773.
Barends, T. R. M., Foucar, L., Ardevol, A., Nass, K., Aquila, A., Botha, S., et al. (2015). Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science. https://doi.org/10.1126/science.aac5492.
Barty, A., Caleman, C., Aquila, A., Timneanu, N., Lomb, L., White, T. A., et al. (2012). Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nature Photonics, 6(1), 35–40. http://www.nature.com/nphoton/journal/v6/n1/abs/nphoton.2011.297.html#supplementary-information.
Barty, A., Kirian, R. A., Maia, F. R., Hantke, M., Yoon, C. H., White, T. A., & Chapman, H. (2014). Cheetah : Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. Journal of Applied Crystallography, 47(Pt 3), 1118–1131. https://doi.org/10.1107/S1600576714007626.
Batyuk, A., Galli, L., Ishchenko, A., Han, G. W., Gati, C., Popov, P. A., et al. (2016). Native phasing of x-ray free-electron laser data for a G protein–coupled receptor. Science Advances, 2(9). https://doi.org/10.1126/sciadv.1600292.
Botha, S., Nass, K., Barends, T. R. M., Kabsch, W., Latz, B., Dworkowski, F., et al. (2015). Room-temperature serial crystallography at synchrotron X-ray sources using slowly flowing free-standing high-viscosity microstreams. Acta Crystallographica Section D, 71(2), 387–397. https://doi.org/10.1107/S1399004714026327.
Boutet, S., Lomb, L., Williams, G. J., Barends, T. R., Aquila, A., Doak, R. B., et al. (2012). High-resolution protein structure determination by serial femtosecond crystallography. Science, 337(6092), 362–364. https://doi.org/10.1126/science.1217737.
Caffrey, M. (2003). Membrane protein crystallization. Journal of Structural Biology, 142(1), 108–132. https://doi.org/10.1016/S1047-8477(03)00043-1.
Caspar, D. L. D., Clarage, J., Salunke, D. M., & Clarage, M. (1988). Liquid-like movements in crystalline insulin. Nature, 332(6165), 659–662.
Chapman, H. N., Barty, A., Bogan, M. J., Boutet, S., Frank, M., Hau-Riege, S. P., et al. (2006). Femtosecond diffractive imaging with a soft-X-ray free-electron laser. Nature Physics, 2(12), 839–843.
Chapman, H. N., Fromme, P., Barty, A., White, T. A., Kirian, R. A., Aquila, A., et al. (2011). Femtosecond X-ray protein nanocrystallography. Nature, 470(7332), 73–77. https://doi.org/10.1038/nature09750.
Chapman, H. N., Hau-Riege, S. P., Bogan, M. J., Bajt, S., Barty, A., Boutet, S., et al. (2007). Femtosecond time-delay X-ray holography. Nature, 448(7154), 676–679.
Cho, H. S., Dashdorj, N., Schotte, F., Graber, T., Henning, R., & Anfinrud, P. (2010). Protein structural dynamics in solution unveiled via 100-ps time-resolved x-ray scattering. Proceedings of the National Academy of Sciences, 107(16), 7281–7286. https://doi.org/10.1073/pnas.1002951107.
Chollet, M., Alonso-Mori, R., Cammarata, M., Damiani, D., Defever, J., Delor, J. T., et al. (2015). The X-ray pump–probe instrument at the Linac coherent light source. Journal of Synchrotron Radiation, 22(3), 503–507. https://doi.org/10.1107/S1600577515005135.
Cohen, A. E., Soltis, S. M., González, A., Aguila, L., Alonso-Mori, R., Barnes, C. O., et al. (2014). Goniometer-based femtosecond crystallography with X-ray free electron lasers. Proceedings of the National Academy of Sciences, 111(48), 17122–17127. https://doi.org/10.1073/pnas.1418733111.
Colletier, J.-P., Sawaya, M. R., Gingery, M., Rodriguez, J. A., Cascio, D., Brewster, A. S., & Eisenberg, D. S. (2016). De novo phasing with X-ray laser reveals mosquito larvicide BinAB structure. Nature, 539, 43. https://doi.org/10.1038/nature19825 https://www.nature.com/articles/nature19825#supplementary-information.
Colletier, J. P. (n.d.). Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nature Chemistry.
Conrad, C. E., Basu, S., James, D., Wang, D., Schaffer, A., Roy-Chowdhury, S., et al. (2015). A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ, 2(Pt 4), 421–430. https://doi.org/10.1107/S2052252515009811.
DePonte, D. P., Doak, R. B., Hunter, M., Liu, Z., Weierstall, U., & Spence, J. C. H. (2009). SEM imaging of liquid jets. Micron, 40(4), 507–509. https://doi.org/10.1016/j.micron.2008.12.009.
DePonte, D. P., Weierstall, U., Schmidt, K., Warner, J., Starodub, D., Spence, J. C. H., & Doak, R. B. (2008). Gas dynamic virtual nozzle for generation of microscopic droplet streams. Journal of Physics D: Applied Physics, 41(19), 195505.
Dörner, K., Martin-Garcia, J. M., Kupitz, C., Gong, Z., Mallet, T. C., Chen, L., et al. (2016). Characterization of protein Nanocrystals based on the reversibility of crystallization. Crystal Growth & Design, 16(7), 3838–3845. https://doi.org/10.1021/acs.cgd.6b00384.
Doucet, J., & Benoit, J. P. (1987). Molecular dynamics studied by analysis of the X-ray diffuse scattering from lysozyme crystals. Nature, 325(6105), 643–646.
Drenth, J. (2007). Principles of protein X-ray crystallography (3rd ed.). New York: Springer-Verlag.
Echalier, A., Glazer, R. L., Fulop, V., & Geday, M. A. (2004). Assessing crystallization droplets using birefringence. Acta Crystallographica Section D, 60(4), 696–702. https://doi.org/10.1107/S0907444904003154.
Ekeberg, T., Svenda, M., Abergel, C., Maia, F. R. N. C., Seltzer, V., Claverie, J.-M., et al. (2015). Three-dimensional reconstruction of the Giant Mimivirus particle with an X-ray free-Electron laser. Physical Review Letters, 114(9), 098102.
Emma, P., Akre, R., Arthur, J., Bionta, R., Bostedt, C., Bozek, J., et al. (2010). First lasing and operation of an ångstrom-wavelength free-electron laser. Nature Photonics, 4(9), 641–647. https://doi.org/10.1038/nphoton.2010.176.
Faure, P., Micu, A., Perahia, D., Doucet, J., Smith, J. C., & Benoit, J. P. (1994). Correlated intramolecular motions and diffuse x-ray scattering in lysozyme. Nature Structural & Molecular Biology, 1(2), 124–128.
Ferguson, K. R., Bucher, M., Bozek, J. D., Carron, S., Castagna, J.-C., Coffee, R., et al. (2015). The atomic, molecular and optical science instrument at the Linac coherent light source. Journal of Synchrotron Radiation, 22(3), 492–497. https://doi.org/10.1107/S1600577515004646.
Foadi, J., Aller, P., Alguel, Y., Cameron, A., Axford, D., Owen, R. L., et al. (2013). Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography, 69(Pt 8), 1617–1632. https://doi.org/10.1107/S0907444913012274.
Frank, M., Carlson, D. B., Hunter, M. S., Williams, G. J., Messerschmidt, M., Zatsepin, N. A., et al. (2014). Femtosecond X-ray diffraction from two-dimensional protein crystals. IUCrJ, 1(2), 95–100. https://doi.org/10.1107/S2052252514001444.
Fromme, R., Ishchenko, A., Metz, M., Chowdhury, S. R., Basu, S., Boutet, S., et al. (2015). Serial femtosecond crystallography of soluble proteins in lipidic cubic phase. IUCrJ, 2(5), 545–551. https://doi.org/10.1107/S2052252515013160.
Fukuda, Y., Tse, K. M., Nakane, T., Nakatsu, T., Suzuki, M., Sugahara, M., et al. (2016). Redox-coupled proton transfer mechanism in nitrite reductase revealed by femtosecond crystallography. Proceedings of the National Academy of Sciences, 113(11), 2928–2933. https://doi.org/10.1073/pnas.1517770113.
Fuller, F. D., Gul, S., Chatterjee, R., Burgie, E. S., Young, I. D., Lebrette, H., & Yano, J. (2017). Drop-on-demand sample delivery for studying biocatalysts in action at X-ray free-electron lasers. Nature Methods, 14(4), 443–449. https://doi.org/10.1038/nmeth.4195 http://www.nature.com/nmeth/journal/v14/n4/abs/nmeth.4195.html#supplementary-information.
Gañán-Calvo, A. M. (1998). Generation of steady liquid microthreads and Micron-sized Monodisperse sprays in gas streams. Physical Review Letters, 80(2), 285–288.
Garman, E. (2010). Radiation damage in macromolecular crystallography: What is it and why should we care? Acta Crystallographica Section D, 66(4), 339–351. https://doi.org/10.1107/S0907444910008656.
Ginn, H. M., Brewster, A. S., Hattne, J., Evans, G., Wagner, A., Grimes, J. M., et al. (2015). A revised partiality model and post-refinement algorithm for X-ray free-electron laser data. Acta Crystallographica Section D, 71(6), 1400–1410. https://doi.org/10.1107/S1399004715006902.
Gorel, A., Motomura, K., Fukuzawa, H., Doak, R. B., Grünbein, M. L., Hilpert, M., et al. (2017). Multi-wavelength anomalous diffraction de novo phasing using a two-colour X-ray free-electron laser with wide tunability. Nature Communications, 8(1), 1170. https://doi.org/10.1038/s41467-017-00754-7.
Graewert, M. A., & Svergun, D. I. (2013). Impact and progress in small and wide angle X-ray scattering (SAXS and WAXS). Current Opinion in Structural Biology, 23(5), 748–754. https://doi.org/10.1016/j.sbi.2013.06.007.
Gualtieri, E. J., Guo, F., Kissick, D. J., Jose, J., Kuhn, R. J., Jiang, W., & Simpson, G. J. (2011). Detection of membrane protein two-dimensional crystals in living cells. Biophysical Journal, 100(1), 207–214. https://doi.org/10.1016/j.bpj.2010.10.051.
Guizar-Sicairos, M., Johnson, I., Diaz, A., Holler, M., Karvinen, P., Stadler, H.-C., et al. (2014). High-throughput ptychography using Eiger: Scanning X-ray nano-imaging of extended regions. Optics Express, 22(12), 14859–14870. https://doi.org/10.1364/OE.22.014859.
Hendrickson, W. A. (2014). Anomalous diffraction in crystallographic phase evaluation. Quarterly Reviews of Biophysics, 47(1), 49–93. https://doi.org/10.1017/S0033583514000018.
Hunter, M. S., DePonte, D. P., Shapiro, D. A., Kirian, R. A., Wang, X., Starodub, D., et al. (2011). X-ray diffraction from membrane protein Nanocrystals. Biophysical Journal, 100(1), 198–206. https://doi.org/10.1016/j.bpj.2010.10.049.
Hunter, M. S., Yoon, C. H., DeMirci, H., Sierra, R. G., Dao, E. H., Ahmadi, R., & Boutet, S. (2016). Selenium single-wavelength anomalous diffraction de novo phasing using an X-ray-free electron laser. Nature Communications, 7, 13388. https://doi.org/10.1038/ncomms13388 http://www.nature.com/articles/ncomms13388#supplementary-information.
Johansson, L. C., Arnlund, D., White, T. A., Katona, G., DePonte, D. P., Weierstall, U., & Neutze, R. (2012). Lipidic phase membrane protein serial femtosecond crystallography. Nature Methods, 9(3), 263–265. http://www.nature.com/nmeth/journal/v9/n3/abs/nmeth.1867.html#supplementary-information .
Jung, Y. O., Lee, J. H., Kim, J., Schmidt, M., Moffat, K., Šrajer, V., & Ihee, H. (2013). Volume-conserving trans–cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nature Chemistry, 5(3), 212–220. http://www.nature.com/nchem/journal/v5/n3/abs/nchem.1565.html#supplementary-information .
Kam, Z., Koch, M. H. J., & Bordas, J. (1981). Fluctuation X-ray scattering from biological particles in frozen solution by using synchrotron radiation. Proceedings of the National Academy of Sciences of the United States of America, 78(6), 3559–3562.
Kern, J., Tran, R., Alonso-Mori, R., Koroidov, S., Echols, N., Hattne, J., & Yachandra, V. K. (2014). Taking snapshots of photosynthetic water oxidation using femtosecond X-ray diffraction and spectroscopy. Nature Communications, 5, 4371. https://doi.org/10.1038/ncomms5371 https://www.nature.com/articles/ncomms5371#supplementary-information.
Kirian, R. A., Wang, X., Weierstall, U., Schmidt, K. E., Spence, J. C. H., Hunter, M., et al. (2010). Femtosecond protein nanocrystallography—Data analysis methods. Optics Express, 18(6), 5713–5723. https://doi.org/10.1364/OE.18.005713.
Kirian, R. A., White, T. A., Holton, J. M., Chapman, H. N., Fromme, P., Barty, A., et al. (2011). Structure-factor analysis of femtosecond microdiffraction patterns from protein nanocrystals. Acta Crystallographica Section A, 67(2), 131–140. https://doi.org/10.1107/S0108767310050981.
Kissick, D. J., Gualtieri, E. J., Simpson, G. J., & Cherezov, V. (2010). Nonlinear optical imaging of integral membrane protein crystals in Lipidic Mesophases. Analytical Chemistry, 82(2), 491–497. https://doi.org/10.1021/ac902139w.
Kissick, D. J., Wanapun, D., & Simpson, G. J. (2011). Second-order nonlinear optical imaging of Chiral crystals. Annual Review of Analytical Chemistry (Palo Alto, Calif.), 4, 419–437. https://doi.org/10.1146/annurev.anchem.111808.073722.
Koopmann, R., Cupelli, K., Redecke, L., Nass, K., DePonte, D. P., White, T. A., & Duszenko, M. (2012). In vivo protein crystallization opens new routes in structural biology. Natural Methods, 9(3), 259–262. http://www.nature.com/nmeth/journal/v9/n3/abs/nmeth.1859.html#supplementary-information .
Kupitz, C., Basu, S., Grotjohann, I., Fromme, R., Zatsepin, N. A., Rendek, K. N., & Fromme, P. (2014). Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature, 513(7517), 261–265. https://doi.org/10.1038/nature13453 http://www.nature.com/nature/journal/v513/n7517/abs/nature13453.html#supplementary-information.
Kupitz, C., Olmos, J. L., Holl, M., Tremblay, L., Pande, K., Pandey, S., & Schmidt, M. (2017). Structural enzymology using X-ray free electron lasers. Structural Dynamics, 4(4), 044003. https://doi.org/10.1063/1.4972069.
Landau, E. M., & Rosenbusch, J. P. (1996). Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proceedings of the National Academy of Sciences, 93(25), 14532–14535.
Levantino, M., Schirò, G., Lemke, H. T., Cottone, G., Glownia, J. M., Zhu, D., & Cammarata, M. (2015). Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nature Communications, 6, 6772. https://doi.org/10.1038/ncomms7772 https://www.nature.com/articles/ncomms7772#supplementary-information.
Liang, M., Williams, G. J., Messerschmidt, M., Seibert, M. M., Montanez, P. A., Hayes, M., et al. (2015). The coherent X-ray imaging instrument at the Linac coherent light source. Journal of Synchrotron Radiation, 22(Pt 3), 514–519. https://doi.org/10.1107/S160057751500449X.
Liu, W., Wacker, D., Gati, C., Han, G. W., James, D., Wang, D., et al. (2013). Serial femtosecond crystallography of G protein-coupled receptors. Science, 342(6165), 1521–1524. https://doi.org/10.1126/science.1244142.
Lomb, L., Barends, T. R. M., Kassemeyer, S., Aquila, A., Epp, S. W., Erk, B., et al. (2011). Radiation damage in protein serial femtosecond crystallography using an x-ray free-electron laser. Physical Review B, 84(21), 214111.
Madden, J. T., DeWalt, E. L., & Simpson, G. J. (2011). Two-photon excited UV fluorescence for protein crystal detection. Acta Crystallographica Section D, 67(10), 839–846. https://doi.org/10.1107/S0907444911028253.
Malkin, A. J., Kuznetsov, Y. G., Land, T. A., DeYoreo, J. J., & McPherson, A. (1995). Mechanisms of growth for protein and virus crystals. Nature Structural & Molecular Biology, 2(11), 956–959.
Marchesini, S., He, H., Chapman, H. N., Hau-Riege, S. P., Noy, A., Howells, M. R., et al. (2003). X-ray image reconstruction from a diffraction pattern alone. Physical Review B, 68(14), 140101.
Martin-Garcia, J. M., Conrad, C. E., Nelson, G., Stander, N., Zatsepin, N. A., Zook, J., et al. (2017). Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ, 4(4), 439–454. https://doi.org/10.1107/S205225251700570X.
Moffat, K. (2001). Time-resolved biochemical crystallography: A mechanistic perspective. Chemical Reviews, 101(6), 1569–1582. https://doi.org/10.1021/cr990039q.
Nakane, T., Hanashima, S., Suzuki, M., Saiki, H., Hayashi, T., Kakinouchi, K., et al. (2016). Membrane protein structure determination by SAD, SIR, or SIRAS phasing in serial femtosecond crystallography using an iododetergent. Proceedings of the National Academy of Sciences of the United States of America, 113(46), 13039–13044. https://doi.org/10.1073/pnas.1602531113.
Nakane, T., Song, C., Suzuki, M., Nango, E., Kobayashi, J., Masuda, T., et al. (2015). Native sulfur/chlorine SAD phasing for serial femtosecond crystallography. Acta Crystallographica Section D, 71(12), 2519–2525. https://doi.org/10.1107/S139900471501857X.
Nass, K., Meinhart, A., Barends, T., Foucar, L., Gorel, A., Aquila, A., et al. (2016a). Protein structure determination by single-wavelength anomalous diffraction phasing of X-ray free-electron laser data. IUCrJ, 3, 1–12.
Nass, K., Meinhart, A., Barends, T. R. M., Foucar, L., Gorel, A., Aquila, A., et al. (2016b). Protein structure determination by single-wavelength anomalous diffraction phasing of X-ray free-electron laser data. IUCrJ, 3(3), 180–191. https://doi.org/10.1107/S2052252516002980.
Nave, C. (1998). A description of imperfections in protein crystals. Acta Crystallographica Section D, 54(5), 848–853. https://doi.org/10.1107/S0907444998001875.
Neutze, R., Wouts, R., van der Spoel, D., Weckert, E., & Hajdu, J. (2000). Potential for biomolecular imaging with femtosecond X-ray pulses. Nature, 406(6797), 752–757. https://doi.org/10.1038/35021099.
Nogly, P., James, D., Wang, D., White, T. A., Zatsepin, N., Shilova, A., et al. (2015). Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. IUCrJ, 2(2), 168–176. https://doi.org/10.1107/S2052252514026487.
Oberthuer, D., Knoška, J., Wiedorn, M. O., Beyerlein, K. R., Bushnell, D. A., Kovaleva, E. G., et al. (2017). Double-flow focused liquid injector for efficient serial femtosecond crystallography. 7, 44628. https://doi.org/10.1038/srep44628 http://dharmasastra.live.cf.private.springer.com/articles/srep44628#supplementary-information
Owen, R. L., Rudiño-Piñera, E., & Garman, E. F. (2006). Experimental determination of the radiation dose limit for cryocooled protein crystals. Proceedings of the National Academy of Sciences of the United States of America, 103(13), 4912–4917. https://doi.org/10.1073/pnas.0600973103.
Padayatti, P., Palczewska, G., Sun, W., Palczewski, K., & Salom, D. (2012). Imaging of protein crystals with two-photon microscopy. Biochemistry, 51(8), 1625–1637. https://doi.org/10.1021/bi201682q.
Pande, K., Hutchison, C. D. M., Groenhof, G., Aquila, A., Robinson, J. S., Tenboer, J., et al. (2016). Femtosecond structural dynamics drives the trans/cis isomerization in photoactive yellow protein. Science, 352(6286), 725–729. https://doi.org/10.1126/science.aad5081.
Perez, J., Faure, P., & Benoit, J.-P. (1996). Molecular rigid-body displacements in a tetragonal Lysozyme crystal confirmed by X-ray diffuse scattering. Acta Crystallographica Section D, 52(4), 722–729. https://doi.org/10.1107/S0907444996002594.
Rasmussen, S. G. F., Choi, H.-J., Rosenbaum, D. M., Kobilka, T. S., Thian, F. S., Edwards, P. C., & Kobilka, B. K. (2007). Crystal structure of the human [bgr]2 adrenergic G-protein-coupled receptor. Nature, 450(7168), 383–387. http://www.nature.com/nature/journal/v450/n7168/suppinfo/nature06325_S1.html .
Redecke, L., Nass, K., DePonte, D. P., White, T. A., Rehders, D., Barty, A., et al. (2013). Natively inhibited Trypanosoma brucei Cathepsin B structure determined by using an X-ray laser. Science, 339(6116), 227–230. https://doi.org/10.1126/science.1229663.
Roedig, P., Ginn, H. M., Pakendorf, T., Sutton, G., Harlos, K., Walter, T. S., et al. (2017). High-speed fixed-target serial virus crystallography. Natural Methods., advance online publication, 14, 805–810. https://doi.org/10.1038/nmeth.4335 http://www.nature.com/nmeth/journal/vaop/ncurrent/abs/nmeth.4335.html#supplementary-information.
Rossmann, M. G. (1990). The molecular replacement method. Acta Crystallographica. Section A, 46(Pt 2), 73–82.
Rupp, B. (2013). Macromolecular crystallography: Overview. In G. C. K. Roberts (Ed.), Encyclopedia of biophysics (pp. 1346–1353). Berlin, Heidelberg: Springer Berlin Heidelberg.
Schlichting, I. (2015). Serial femtosecond crystallography: The first five years. IUCrJ, 2(Pt 2), 246–255. https://doi.org/10.1107/S205225251402702X.
Schlichting, I., & Miao, J. (2012). Emerging opportunities in structural biology with X-ray free-electron lasers. Current Opinion in Structural Biology, 22(5), 613–626. https://doi.org/10.1016/j.sbi.2012.07.015.
Schmidt, M. (2013). Mix and inject: Reaction initiation by diffusion for time-resolved macromolecular crystallography. Advances in Condensed Matter Physics, 2013, 10. https://doi.org/10.1155/2013/167276.
Schotte, F., Soman, J., Olson, J. S., Wulff, M., & Anfinrud, P. A. (2004). Picosecond time-resolved X-ray crystallography: Probing protein function in real time. Journal of Structural Biology, 147(3), 235–246. https://doi.org/10.1016/j.jsb.2004.06.009.
Seibert, M. M., Ekeberg, T., Maia, F. R. N. C., Svenda, M., Andreasson, J., Jonsson, O., et al. (2011). Single mimivirus particles intercepted and imaged with an X-ray laser. Nature, 470(7332), 78–81.
Shapiro, D., Thibault, P., Beetz, T., Elser, V., Howells, M., Jacobsen, C., et al. (2005). Biological imaging by soft x-ray diffraction microscopy. Proceedings of the National Academy of Sciences, 102(43), 15343–15346. https://doi.org/10.1073/pnas.0503305102.
Shapiro, D. A., Chapman, H. N., DePonte, D., Doak, R. B., Fromme, P., Hembree, G., et al. (2008). Powder diffraction from a continuous microjet of submicrometer protein crystals. Journal of Synchrotron Radiation, 15(6), 593–599. https://doi.org/10.1107/S0909049508024151.
Sierra, R. G., Gati, C., Laksmono, H., Dao, E. H., Gul, S., Fuller, F., et al. (2016). Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nature Methods, 13(1), 59–62. https://doi.org/10.1038/nmeth.3667.
Sierra, R. G., Laksmono, H., Kern, J., Tran, R., Hattne, J., Alonso-Mori, R., et al. (2012). Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallographica Section D, 68(11), 1584–1587. https://doi.org/10.1107/S0907444912038152.
Spence, J. C. H., & Chapman, H. N. (2014). The birth of a new field. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1647). https://doi.org/10.1098/rstb.2013.0309.
Spence, J. C. H., Kirian, R. A., Wang, X., Weierstall, U., Schmidt, K. E., White, T., et al. (2011). Phasing of coherent femtosecond X-ray diffraction from size-varying nanocrystals. Optics Express, 19(4), 2866–2873. https://doi.org/10.1364/OE.19.002866.
Stagno, J. R., Liu, Y., Bhandari, Y. R., Conrad, C. E., Panja, S., Swain, M., & Wang, Y. X. (2016). Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature., advance online publication, 541(7636), 242–246. https://doi.org/10.1038/nature20599 http://www.nature.com/nature/journal/vaap/ncurrent/abs/nature20599.html#supplementary-information.
Stan, C. A., Milathianaki, D., Laksmono, H., Sierra, R. G., McQueen, T. A., Messerschmidt, M., & Boutet, S. (2016). Liquid explosions induced by X-ray laser pulses. Nature Physics, 12(10), 966–971. https://doi.org/10.1038/nphys3779 http://www.nature.com/nphys/journal/v12/n10/abs/nphys3779.html#supplementary-information.
Stevens, R. C., Cherezov, V., Katritch, V., Abagyan, R., Kuhn, P., Rosen, H., & Wuthrich, K. (2013). The GPCR network: A large-scale collaboration to determine human GPCR structure and function. Nature Reviews. Drug Discovery, 12(1), 25–34.
Stevenson, H. P., DePonte, D. P., Makhov, A. M., Conway, J. F., Zeldin, O. B., Boutet, S., & Cohen, A. E. (2014a). Transmission electron microscopy as a tool for nanocrystal characterization pre- and post-injector. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1647). https://doi.org/10.1098/rstb.2013.0322.
Stevenson, H. P., Makhov, A. M., Calero, M., Edwards, A. L., Zeldin, O. B., Mathews, I. I., et al. (2014b). Use of transmission electron microscopy to identify nanocrystals of challenging protein targets. Proceedings of the National Academy of Sciences, 111(23), 8470–8475. https://doi.org/10.1073/pnas.1400240111.
Strüder, L., Epp, S., Rolles, D., Hartmann, R., Holl, P., Lutz, G., et al. (2010). Large-format, high-speed, X-ray pnCCDs combined with electron and ion imaging spectrometers in a multipurpose chamber for experiments at 4th generation light sources. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment, 614(3), 483–496. https://doi.org/10.1016/j.nima.2009.12.053.
Suga, M., Akita, F., Hirata, K., Ueno, G., Murakami, H., Nakajima, Y., et al. (2015). Native structure of photosystem II at 1.95 a resolution viewed by femtosecond X-ray pulses. Nature, 517(7532), 99–103. https://doi.org/10.1038/nature13991.
Suga, M., Akita, F., Sugahara, M., Kubo, M., Nakajima, Y., Nakane, T., et al. (2017). Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature, 543(7643), 131–135. https://doi.org/10.1038/nature21400.
Sugahara, M., Mizohata, E., Nango, E., Suzuki, M., Tanaka, T., Masuda, T., & Iwata, S. (2015). Grease matrix as a versatile carrier of proteins for serial crystallography. Natural Methods, 12(1), 61–63. https://doi.org/10.1038/nmeth.3172 http://www.nature.com/nmeth/journal/v12/n1/abs/nmeth.3172.html#supplementary-information.
Sugahara, M., Nakane, T., Masuda, T., Suzuki, M., Inoue, S., Song, C., et al. (2017). Hydroxyethyl cellulose matrix applied to serial crystallography. Scientific Reports, 7, 703. https://doi.org/10.1038/s41598-017-00761-0.
Tenboer, J., Basu, S., Zatsepin, N., Pande, K., Milathianaki, D., Frank, M., et al. (2014). Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science, 346(6214), 1242–1246. https://doi.org/10.1126/science.1259357.
Thibault, P., Elser, V., Jacobsen, C., Shapiro, D., & Sayre, D. (2006). Reconstruction of a yeast cell from X-ray diffraction data. Acta Crystallogr. Sect. A, 62(4), 248–261. https://doi.org/10.1107/S0108767306016515.
Tono, K., Nango, E., Sugahara, M., Song, C., Park, J., Tanaka, T., et al. (2015). Diverse application platform for hard X-ray diffraction in SACLA (DAPHNIS): Application to serial protein crystallography using an X-ray free-electron laser. Journal of Synchrotron Radiation, 22(3), 532–537. https://doi.org/10.1107/S1600577515004464.
Van Benschoten, A. H., Liu, L., Gonzalez, A., Brewster, A. S., Sauter, N. K., Fraser, J. S., & Wall, M. E. (2016). Measuring and modeling diffuse scattering in protein X-ray crystallography. Proceedings of the National Academy of Sciences, 113(15), 4069–4074. https://doi.org/10.1073/pnas.1524048113.
Wall, M. E., Ealick, S. E., & Gruner, S. M. (1997). Three-dimensional diffuse x-ray scattering from crystals of staphylococcal nuclease. Proceedings of the National Academy of Sciences, 94(12), 6180–6184.
Wampler, R. D., Kissick, D. J., Dehen, C. J., Gualtieri, E. J., Grey, J. L., Wang, H.-F., et al. (2008). Selective detection of protein crystals by second harmonic microscopy. Journal of the American Chemical Society, 130(43), 14076–14077. https://doi.org/10.1021/ja805983b.
Wanapun, D., Kestur, U. S., Kissick, D. J., Simpson, G. J., & Taylor, L. S. (2010). Selective detection and Quantitation of organic molecule crystallization by second harmonic generation microscopy. Analytical Chemistry, 82(13), 5425–5432. https://doi.org/10.1021/ac100564f.
Weierstall, U., Doak, R. B., Spence, J. C. H., Starodub, D., Shapiro, D., Kennedy, P., et al. (2008). Droplet streams for serial crystallography of proteins. Experiments in Fluids, 44(5), 675–689. https://doi.org/10.1007/s00348-007-0426-8.
Weierstall, U., James, D., Wang, C., White, T. A., Wang, D., Liu, W., et al. (2014). Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nature Communications, 5, 3309–3309. https://doi.org/10.1038/ncomms4309.
Welberry, T. R., Heerdegen, A. P., Goldstone, D. C., & Taylor, I. A. (2011). Diffuse scattering resulting from macromolecular frustration. Acta Crystallographica Section B, 67(6), 516–524. https://doi.org/10.1107/S0108768111037542.
White, T. A. (2014). Post-refinement method for snapshot serial crystallography. Philosophical Transactions of the Royal Society B: Biological Sciences, 369(1647). https://doi.org/10.1098/rstb.2013.0330.
White, T. A., Kirian, R. A., Martin, A. V., Aquila, A., Nass, K., Barty, A., & Chapman, H. N. (2012). CrystFEL: A software suite for snapshot serial crystallography. Journal of Applied Crystallography, 45(2), 335–341. https://doi.org/10.1107/s0021889812002312.
Woolfson, M. M. (1997). An introduction to X-ray crystallography (2nd ed.). Cambridge: Cambridge University Press.
Xiong, G., Moutanabbir, O., Reiche, M., Harder, R., & Robinson, I. (2014). Coherent X-ray diffraction imaging and characterization of strain in silicon-on-insulator nanostructures. Advanced Materials, 26(46), 7747–7763. https://doi.org/10.1002/adma.201304511.
Yamashita, K., Kuwabara, N., Nakane, T., Murai, T., Mizohata, E., Sugahara, M., et al. (2017). Experimental phase determination with selenomethionine or mercury-derivatization in serial femtosecond crystallography. IUCrJ, 4(5), 639–647. https://doi.org/10.1107/S2052252517008557.
Yamashita, K., Pan, D., Okuda, T., Sugahara, M., Kodan, A., Yamaguchi, T., et al. (2015). An isomorphous replacement method for efficient de novo phasing for serial femtosecond crystallography. Scientific Reports, 5, 14017. https://doi.org/10.1038/srep14017.
Yoon, C. H., DeMirci, H., Sierra, R. G., Dao, E. H., Ahmadi, R., Aksit, F., et al. (2017). Se-SAD serial femtosecond crystallography datasets from selenobiotinyl-streptavidin. Scientific Data, 4, 170055. https://doi.org/10.1038/sdata.2017.55.
Young, I. D., Ibrahim, M., Chatterjee, R., Gul, S., Fuller, F. D., Koroidov, S., & Yano, J. (2016). Structure of photosystem II and substrate binding at room temperature. Nature, 540(7633), 453–457. https://doi.org/10.1038/nature20161 http://www.nature.com/nature/journal/v540/n7633/abs/nature20161.html#supplementary-information.
Young, L., Kanter, E. P., Krässig, B., Li, Y., March, A. M., Pratt, S. T., et al. (2010). Femtosecond electronic response of atoms to ultra-intense X-rays. Nature, 466, 56. https://doi.org/10.1038/nature09177.
Zeldin, O. B., Brewster, A. S., Hattne, J., Uervirojnangkoorn, M., Lyubimov, A. Y., Zhou, Q., & Brunger, A. T. (2015). Data exploration toolkit for serial diffraction experiments. Acta Crystallographica Section D, 71(2), 352–356. https://doi.org/10.1107/S1399004714025875.
Zhang, H., Han, G. W., Batyuk, A., Ishchenko, A., White, K. L., Patel, N., & Cherezov, V. (2017a). Structural basis for selectivity and diversity in angiotensin II receptors. Nature., advance online publication, 544(7650), 327–332. https://doi.org/10.1038/nature22035 http://www.nature.com/nature/journal/vaop/ncurrent/abs/nature22035.html#supplementary-information.
Zhang, X., Zhao, F., Wu, Y., Yang, J., Han, G. W., Zhao, S., & Xu, F. (2017b). Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nature Communications, 8, 15383. https://doi.org/10.1038/ncomms15383 https://www.nature.com/articles/ncomms15383#supplementary-information.
Zhu, D., Feng, Y., Stoupin, S., Terentyev, S. A., Lemke, H. T., Fritz, D. M., et al. (2014). Performance of a beam-multiplexing diamond crystal monochromator at the Linac coherent light source. Review of Scientific Instruments, 85(6), 063106. https://doi.org/10.1063/1.4880724.
Acknowledgements
This chapter describes the work of a very large number of people at SLAC National Accelerator Laboratory, the users of LCLS, and the wider community. Use of the LCLS is supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hunter, M.S., Fromme, P. (2018). Serial Femtosecond Crystallography (SFX): An Overview. In: Boutet, S., Fromme, P., Hunter, M. (eds) X-ray Free Electron Lasers. Springer, Cham. https://doi.org/10.1007/978-3-030-00551-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-00551-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00550-4
Online ISBN: 978-3-030-00551-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)