
Abstract
Neurodegenerative diseases share a number of common features with respect to clinical course, pathology, and molecular mechanisms. This chapter outlines the common themes of neurodegeneration, including the concept of selective neuronal vulnerability, genetic susceptibility, aberrant protein structures, disruption in mitochondrial function, altered axonal transport, oxidative stress, and neuroinflammation. A greater understanding of the shared pathophysiologic processes for neurodegenerative diseases will help to develop therapeutic agents that may be beneficial to a range of different clinical phenotypes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
As the population increases in size and life expectancies continue to rise, so do the number of people diagnosed with neurodegenerative diseases. This term refers to age-dependent progressive diseases, caused by degeneration of the central nervous system (CNS). Traditionally, these conditions were characterized clinically, but with advances in imaging, it has become possible to attribute specific clinical manifestations of disease to degeneration in specific anatomical regions of the CNS. Histopathological analysis, genetic studies, and proteomic interrogation have further refined the diagnosis of neurodegenerative diseases.
Neurodegenerative diseases share certain common features including histopathology, clinical course, and molecular mechanisms of pathogenesis. Comparing two different diseases, there may be both overlap with regard to some features and divergence of other aspects. As new categories of disease emerge, some are seen to share common pathogenic features and genetic origins.
The aim of this chapter is to describe the common themes that underlie the major neurodegenerative diseases, and to draw biochemical, histopathological, and molecular genetic parallels across the different disease categories that are outlined in the remainder of this book.
Common Clinical Features of Neurodegenerative Diseases
In 2003, Przedborski and colleagues recognized the clinical and pathological manifestations of neurodegenerative diseases, which:
-
Affect “specific subsets of neurons”
-
Arise without clear explanation and could be either inherited or acquired
-
“Progress relentlessly”
-
Are often age-related, increasing in frequency with advancing age
-
Are often accompanied by microscopic signs of four stages of disorder:
-
Neuronal pathology
-
Neuronal cell death
-
Disappearance of neuronal cell bodies
-
Glial proliferation
-
The following may serve as a brief overview of common clinical features of neurodegenerative diseases:
-
The chronic clinical course is relentlessly progressive until death.
-
The disorder is not reversible by any known therapy although drug therapy or gene therapy may give marginal and temporary improvement.
-
Phenotypic variability is commonly seen.
-
Cognitive impairment and dementia are common manifestations in neurodegenerative disorders but are not seen in all forms. The diagnosis of dementia has been formalized so that cognitive changes in frontotemporal dementia (FTD) and Alzheimer disease can generally be differentiated by formal neuropsychological tests.
-
The major risk factor is advancing age. The term age-related neurodegenerative diseases is commonly used.
-
The condition appears to be heritable in a small percentage of cases.
-
In the familial form of the disease, the onset occurs up to a decade before onset of the sporadic form of the disease.
-
Several different neurodegenerative diseases may appear together within a family.
-
Different clinical manifestations are mediated by dysfunction of different anatomical regions of degeneration.
-
Features of more than one neurodegenerative diseases may appear to coexist in one patient. There is clinical overlap between amyotrophic lateral sclerosis (ALS) and FTD, indeed the two conditions are believed by some to represent a spectrum rather than separate diseases. In the ALS–parkinsonism–dementia complex of Guam, patients have evidence of two motor diseases as well as dementia.
-
Advances in genetics of neurodegenerative diseases have demonstrated that diverse clinical phenotypes may share similar genotypes, and that clinically similar phenotypes may be associated with a wide variety of genotypes.
Classification of Neurodegenerative Diseases
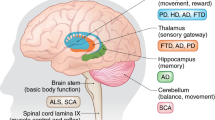
Neurodegenerative diseases are diagnosed primarily on the basis of history and clinical examination (Fig. 1.1). Diagnostic criteria, based primarily on clinical findings, have been generated for most of the common neurodegenerative diseases. The suspected diagnosis is then confirmed by carrying out directed tests in the fields of neurophysiology, neuropsychology, neuroimaging, or genetic analysis. Often, the best diagnostic tool is the observation of the patient over the course of time; antemortem tissue analysis usually does not play a role in diagnosis.

Clinical classification of neurodegenerative diseases
Eponymous classifications (e.g., Alzheimer disease, Parkinson disease, and Huntington disease) remain useful in a clinical setting, as the diagnosis generates a framework for clinical discussion, prognostication, and disease management. However, it is increasingly recognized that neurodegenerative diseases can also be subdivided into categories based on pathological or genetic characteristics, as outlined in Fig 1.1, and as the field advances, diagnostic categories for some diseases will accordingly require some adjustment.
A number of common themes have emerged in the pathogenesis of various neurodegenerative diseases. Whether these commonalities are simply secondary processes, which reflect the fact that neurons have a limited repertoire by which to die, or whether they reveal important initiating upstream mechanisms remains unclear.
In the following section, some putative common pathogenic molecular mechanisms will be discussed, but it is worth bearing in mind that it is not currently possible to distinguish between mechanisms that initiate disease and those that contribute to disease progression. Both may yield targets suitable for therapeutic intervention, but only the former can yield knowledge that will lead to disease prevention.
Common Themes in Genetics
Familial aggregation of specific neurodegenerative diseases is well recognized. Excepting the trinucleotide repeat disorders, which exhibit Mendelian inheritance with full penetrance and anticipation, neurodegenerative disorders have a small percentage of familial cases and a large percentage of apparently sporadic cases. Sporadic and familial cases are usually phenotypically and histologically indistinguishable, although the onset of familial cases tends be earlier. This suggests that genetic mutations accelerate the molecular processes that lead to late-onset sporadic disease.
A number of causative genes have been discovered for specific neurodegenerative diseases. An understanding of gene function has helped to elaborate mechanisms of disease, as well as providing a platform for research into similar mechanisms in other neurodegenerative diseases. For example, mutations in APP, Presenilin 1, and Presenilin 2, which occur in early onset Alzheimer disease (AD), cause altered protein production and increased aggregation of β-amyloid protein (Aβ). Similarly, PARK 1, the first gene to be identified in Parkinson disease (PD), alters the production of the protein α-synuclein. Mutations in genes associated with oxidative stress pathways, SOD1 and DJ1, have been implicated in familial amyotrophic lateral sclerosis (ALS) and PD, respectively. The discovery of TAR-DP and FUS in ALS and frontotemporal dementia (FTD)-ALS has pointed to the likely role of altered RNA regulation in some neurodegenerative diseases.
Genome-wide association studies and high-throughput sequencing have identified susceptibility genes in many neurodegenerative conditions. Moreover, overlap between susceptibility genes has been reported. APOE4 is well established as a risk factor for late-onset AD. However, meta-analysis has shown that presence of the allele is also linked to PD and FTD. Although the incidence of APOE4 is the same in patients with ALS as the general population, the presence of the APOE4 allele is associated with earlier age of disease onset.
Common Themes in Neuropathology
Thorough pathological diagnosis depends on the pathologist having access to accurate clinical information as well as tissue analysis. Pathological diagnostic criteria presume that phenomena involved in degeneration tend to occur together, but invariably they are evident at different time points during the disease. Thus, interpretation of the neuropathological data must take into account when in the clinical course of the disease the pathological assessment has been performed.
Both gross and microscopic tissue examination can help to identify pathological processes and can also differentiate features that are purely degenerative, and those that emerge from the innate responses to protect and repair. Finding the site of the earliest visible alteration helps in establishing the diagnosis. Degeneration in the hippocampal and frontal lobe pyramidal neurons is associated with AD, in the dopaminergic neurons of the substantia nigra with PD, in the upper and lower motor neurons of the pyramidal system with ALS and in the medium-sized spiny GABAergic neurons of the striatum with HD.
Dementia coupled with mainly limbic atrophy suggests Alzheimer disease (AD), while mild atrophy implies Lewy body disease. Moderate cognition decline in the setting of asymmetric, motor and sensory impairment, with reduced metabolic activities, and atrophy prevailing around the central sulcus is indicative of corticobasal degeneration. However, if these changes involve the lateral half of the putamen, caudal to the mammillary body, multiple system atrophy (MSA) is the most likely diagnosis. Despite the topographical differences in neurodegenerative diseases, gross histopathological findings are similar – there is regional atrophy with gliosis and neuronal loss as well as abnormal accumulation of protein.
A large subset of neurodegenerative diseases display protein aggregates. Ubiquitinated neuronal nuclear inclusions occur in polyglutaminopathies including Huntington disease (HD). Either parenchymal or vascular accumulation of Aβ occurs with aging, as well as in the occurrence of AD in children with Down syndrome. Neostriatal large neurons are rather resistant compared to medium size neurons in HD, but they degenerate in progressive supranuclear palsy and in AD. Loss of spinal motor neurons is typical in amyotrophic lateral sclerosis (ALS), whereas glial cells degenerate in MSA. Lewy bodies are a hallmark of Parkinson disease with or without dementia and involve many classes of neurons.
In summary, the three practical steps that are useful while appraising the pathologic phenotypes of neurodegenerative diseases are:
-
1.
Identifying the sites or systems where the brunt of the tissue loss occurs, which might be revealed on neuroimaging at some time points during the disease, or eventually on postmortem examination.
-
2.
Cataloging the cells undergoing degeneration.
-
3.
Identifying the abnormal aggregates, their cellular (neuronal vs. glial cells) and topographic propensities (extracellular, cytoplasmic, or nuclear).
General experience confirms that these steps are crucial for assigning the most appropriate diagnosis to most of the currently classifiable neurodegenerative diseases.
Selective Vulnerabilities
Cells are constantly placed under stress because of intrinsic metabolic processes and also because of a number of extrinsic factors. Neurons are even more vulnerable as they facilitate neurotransmission and maintain the metabolic needs required for long axonal projections. Larger neurons with myelinated axons extending long distances appear to be most vulnerable. These neurons have high energy requirements, are especially reliant on axonal transport, and have a larger surface area for exposure to environmental toxins. Coupled with the fact that once damaged they cannot regenerate, neurons in general represent a vulnerable group of cells.
So why then are certain subgroups of neurons more susceptible than others? A number of hypotheses have been proposed to account for specific selective susceptibility of certain neurons to specific pathological processes (Table 1.1). Neuroblasts arise from the neuroectoderm at 8 weeks of fetal life and then proceed to differentiate into highly specialized neurons. Even though all neurons of an individual have the same genes, it is the gene expression profile that determines the highly specialized function of a specific neuron. This degree of specialization is thought to render individual neurons more susceptible to anoxia and oxidative stress, and factors that lead to selective vulnerability include the type of neuron and the neuronal microenvironment.
The dopaminergic neurons located in the substantia nigra in PD are particularly prone to reactive oxygen species (ROS)-induced injury. This selective vulnerability is believed to derive from the fact that neurons in the substantia nigra have very high levels of iron and copper, both of which are capable of catalyzing ROS formation. Additional evidence suggests that the substantia nigra may have low stores of antioxidant molecules such as glutathione, thereby increasing neuronal susceptibility to the damaging effects of ROS. However, this does not explain the vulnerability of neurons associated with PD in other regions of the brain such as autonomic ganglia, brainstem, and spinal cord.
In ALS, motor neurons are affected primarily, although it is now recognized that non-motor neurons are involved in cognitive dysfunction. Motor neurons are large cells and often have long axonal fibers. In lower motor neurons, these fibers may stretch over a meter in order to supply distal muscles. They require a strong cytoskeleton, neurofilament network, and efficient axonal transport system. Motor neurons are highly metabolic and exquisitely sensitive to energy demands. Any one or a multitude of these factors makes motor neurons vulnerable. As is the case in the substantia nigra, deficiencies of protective agents such as glutathione and cytosolic calcium-binding proteins can add to neuronal stress.
The different phenotypic characteristics of individual diseases remain difficult to explain. In Mendelian diseases, differences can be evident within families with the same mutation. For example, a proband with familial ALS may present with flail arm and slow progression, while another family member may present with rapidly progressive bulbar onset disease. Similarly, some patients with AD may have executive or visuospatial deficits, while others have an amnestic syndrome. In kindreds with triplication of APP, the clinical disease segregates into two distinct phenotypes: those with classical AD indistinguishable from idiopathic AD and those with vascular dementia and a stroke-like syndrome. The causes of these phenotypic variations remain to be determined.
Aberrant Protein Structure
The formation of aberrant structures by more than 20 different proteins appears to underlie a large disease group, many of which afflict the CNS (Table 1.2). Indeed, deposits of protein aggregates are histological hallmarks of many neurodegenerative diseases and consequently, the proteins involved and the mechanism of their aggregation is under intense scrutiny.
The mechanism by which the attainment of an aberrant protein structure causes disease is still unclear and may involve both the loss of a vital physiological function, and the acquisition of toxic properties. While loss of function can be harmful, toxic gain of function is invariably pathogenic. Toxicity may be direct or indirect. For instance, an aberrant protein structure might bind to a specific receptor directly, causing an inappropriate activation of a cascade that initiates cellular changes, which in turn lead to compromise of cellular function. Alternatively, aberrant structures might acquire properties that allow them to interact with and destabilize cellular membranes or other proteins, thus causing a secondary toxicity. Moreover, accumulation of aberrant proteins may strain the normal mechanisms responsible for controlling protein folding and degradation, resulting in a generalized loss of protein homeostasis and consequent toxicity.
Oligomerization or polymerization of aberrantly folded proteins is concentration dependent. Changes in production, degradation, or clearance of native protein are hypothesized to underlie the assembly of aberrantly folded protein. Once formed, such structures are thought to be the primary event driving pathogenesis (Fig. 1.2). Consequently, many of the therapies under development are designed either to: (1) decrease the quantity of soluble native protein, or (2) remove aberrant protein.

Pathways to aberrant protein structure and aggregation in amyloid related diseases: The process is initiated by denaturation, unfolding, or misfolding (indicated by the transition from blue triangle to red circle). Proteins can form oligomers or amorphous aggregates or small, structured polymers known as protofibrils. Protofibrils mature into amyloid fibrils and then into aggregates of amyloid fibrils. Current data indicate that all aberrant protein structures (i.e., all structures shown other than the native monomer) are toxic
It has been suggested that a threshold of abnormal aggregation must be reached before clinical signs appear, but it is as yet unclear whether deposited protein aggregates or other smaller protein assemblies are the principle mediators of disease. Evidence against deposits of protein aggregates as mediators of disease comes from the finding that many aggregated proteins are found in brains of elderly individuals who die without clinical signs of disease.
Inclusion Bodies
Inclusion bodies are relatively large electron-dense structures that contain membrane-limited protein aggregates. Such deposits often contain ubiquitin-positive material, which is believed to accumulate due to impairment of the ubiquitin-proteasome system (UPS). Under normal conditions, cytoplasmic proteins are tagged for destruction by the enzymatic addition of four or more ubiquitin molecules, but buildup of substrate, decreased efficiency of ubiquitin conjugation, or impaired degradation of ubiquitinated protein can trigger accumulation of partially ubiquitinated protein aggregates. For instance, it is believed that impaired ubiquitination of α-synuclein may explain how mutations in the PRKN gene cause early onset PD. PRKN encodes an E3 ubiquitin ligase (Parkin), which when mutated appears less efficient at ubiquitinating α-synuclein and aggregates of partially ubiquitinated α- accumulate in cytoplasmic structures known as Lewy bodies.
Pathological inclusion bodies are also seen in ALS, FTD, and HD. However, it is not clear if inclusions are purely pathogenic since Marinesco bodies and Hirano bodies are also found in aged asymptomatic individuals. Moreover, several studies have documented the detection of inclusions in functional neurons, implying that these structures may be protective rather than pathogenic.
Altered RNA Metabolism
There is increasing interest in the role of aberrant RNA processing in the pathogenesis of neurodegenerative disease. Mutations in two important genes, FUS and TDP-43, identified in a small percentage of familial ALS and FTD cases, are associated with altered RNA processing. Similarly, loss of function mutations in another RNA regulator, progranulin, has been linked with FTD, which, in turn, may be associated with motor neuron disease in some families. The protein products of TDP-43 and FUS/TLS (FUsed in Sarcoma, Translocated in LipoSarcoma) are both structurally similar to heterogeneous ribonucleoproteins (hnRNP), which are involved in multiple aspects of RNA processing. Mutations in the TAR DNA binding protein, TDP-43, and the protein, FUS/TLS have widespread downstream effects on multiple differentially spliced mRNA species. Consequently, it is anticipated that quite diverse pathogenic pathways are triggered depending on the RNA binding protein and the type of neurons involved. The mutations in the TDP-43 gene are seen in some familial ALS cases, but cytoplasmic inclusions containing ubiquinated and hyperphosphorylated forms of wild-type TDP-43 may be found in cases of sporadic ALS. Inclusions containingTDP-43 are also seen in more than half of all FTD cases, and TDP-43 and FUS/TLS inclusions have been reported in other neurodegenerative conditions including: corticobasal degeneration, sporadic AD and familial AD, Down syndrome, hippocampal sclerosis dementia, familial British dementia, PD, ALS–parkinsonism–dementia complex of Guam and some myopathies.
It is not yet known if TDP-43 inclusions act as a primary neurotoxin or if they are a cellular by-product of some other toxic event. Similarly, FUS/TLS is a highly active nuclear protein involved in many aspects of RNA metabolism and also plays an important role in mRNA transport along dendrites. Thus, loss of function mutations in FUS/TLS may lead to abnormal synapse function in the motor nerves and abnormal modulation of neuronal plasticity that is essential for proper activity.
Oxidative Stress
In all cells, but particularly highly metabolically active cells such as neurons, there is a constant production and elimination of reactive oxygen species (ROS). At any time, the balance is such that an unusual increase in ROS or loss of antioxidant protection can lead to accumulation of ROS and ensuing cellular damage. High levels of ROS can cause nuclear DNA oxidation and repairing such damage requires substantial expenditure of metabolic energy. If damaged DNA is not adequately repaired, this can lead to cellular dysfunction and apoptosis. Accumulated oxidatively damaged DNA has been observed in AD, PD, ALS, and vascular dementia (VD). Calcium plays an integral role in signaling within the cell and also in maintenance of cellular homeostasis. As part of the role in signaling, ROS activate calcium channels and deactivate calcium pumps. This leads to abnormally high intracellular levels of calcium, which in turn may lead to cell death. Mitochondrial ROS also cause increased uptake of calcium ions with increased membrane permeability, resulting in the release of cytochrome-C, which initiates the apoptotic cascade.
In vivo and in vitro studies have shown that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase derived from microglia play an important role in the generation of ROS. In PD, microglia-specific NADPH oxidases are involved in the production of ROS, which may contribute to the death of dopaminergic neurons. A similar process is seen in ALS, whereby oxygen radicals produced by microglial NADPH oxidase are believed to injure motor neurons. Evidence from the mutant SOD1 mouse model of ALS indicates that genes encoding NADPH oxidase are upregulated in disease and this leads to an increased concentration of ROS in mouse spinal cord tissue.
Mitochondrial Dysfunction
Mitochondria play a crucial role in the production of cellular energy using the respiratory chain. Consequently, accumulated mitochondrial dysfunction is implicated in both normal aging and neurodegeneration. Proposed mechanisms of this effect include failure to meet the energy needs of the cell, calcium misregulation, leading to cell death, over production of ROS, and cytochrome C-induced apoptosis.
Well-documented incidents have shown that ingestion of certain neurotoxins can lead to the sudden onset of clinical syndromes identical to neurodegenerative diseases such as Parkinson disease or Huntington disease. Two such events led to the discovery that some neurotoxins are potent inhibitors of complexes within the mitochondrial respiratory chain. In 1982, seven young drug abusers injected intravenous forms of a synthetic heroin derivative, 1-methyl-4-phenyl-4-propionoxypiperidine (MPPP), and developed the signs and symptoms of PD. This was because of the presence of a contaminant, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). When pure MPTP is injected into animals, it causes specific degenerative of dopaminergic neurons in the substantia nigra pars compacta and produces an irreversible and severe parkinsonism phenotype. After infusion, MPTP crosses the blood–brain barrier and is taken up by glia and serotonergic neurons and converted to MPDP+and then to MPP+. Thereafter, MPP+ is released and specifically taken up by dopaminergic neurons and concentrated in mitochondria where it acts as a potent inhibitor of mitochondrial complex I. Similarly, exposure to 3-nitropropionic acid (3NPA) leads to rapid onset of a Huntington-type syndrome. It is now known that 3NPA is an inhibitor of mitochondrial complex II. Discovery that exogenous neurotoxins can inhibit mitochondrial complexes leading to rapid onset of neurological symptoms suggests that mitochondrial dysfunction plays a central role in these diseases.
Excitotoxicity
Glutamate is an important excitatory neurotransmitter in the CNS and its misregulation has been implicated in the development of neurodegenerative diseases. Glutamate has essential roles in synaptic transmission and plasticity, which are important in learning and memory as well and sensory and motor functions. Transmission of glutamate is mediated through three major receptors – N-methyl-d-aspartate receptors (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptors, and kainite receptors. The glutamate excitotoxicity hypothesis postulates that excessive synaptic glutamate causes over-activation of the postsynaptic NMDA and AMPA receptors resulting in neuronal death. High glutamate levels, which continuously activate postsynaptic receptors, may lead to increased intracellular calcium and catabolic enzyme activity. Downstream effects can include depolarization of mitochondrial membrane, activation of the caspase system, and production of reactive oxidation species, all of which culminate in cell death (Fig. 1.3). Excessive synaptic glutamate may be potentiated because of a fault in the cellular glutamate reuptake system. Excitatory amino-acid transporter 2 (EAAT2) is a glutamate transporter involved in cerebral glutamate transport. It has been postulated that some patients with ALS have decreased expression of this protein. Similar studies carried out in patients with AD have also shown a reduction in EAAT 2 expression. It has been shown that GLUR2, an AMPA glutamate receptor subtype responsible for calcium permeability into the postsynaptic cell, is not expressed in motor neurons affected by ALS because of a defect in the editing process for messenger RNA encoding the GLUR2 receptor. Absence of a functional GLUR2 subunit allows calcium influx into the postsynaptic cell and results in cellular damage.

Proposed mechanisms of glutamate-induced excitotoxicity
Parkin, which is the gene product of PARK2, has regulator effects on excitatory glutaminergic synapese. Abnormalities in parkin production can lead to enhanced synaptic activity and may even trigger an increase in the number of glutamate receptors. Excessive glutaminergic activity may be responsible for nigral excitotoxicity.
Given this evidence, it would seem beneficial to downregulate glutamate activity in patients affected by neurodegenerative disease. Riluzole has a direct and indirect blocking effect on glutamate receptor activation and is proven to slow the progression of ALS. Unfortunately, no other anti-glutamate agent has been successful in disease treatment.
Neuroinflammation and Microglial Activation
An epidemiological study carried out in 1980s was the first to postulate an association between inflammation and neurodegeneration. The study demonstrated that the incidence of AD was lower in patients with rheumatoid arthritis (RA) who had been on long-term anti-inflammatory treatment compared to those who had not. Since then, detailed descriptions of systemic and CNS specific proinflammatory cascades have fueled the hypothesis that neuroinflammation plays an active role in the process of neurodegeneration. Microglia are CNS-specific macrophages, derived from myeloid precursor cells, which enter the CNS during embryogenesis. The primary function of this subset of immune cells is to protect the brain from extrinsic pathogens and processes. Recently, a number of experiments have shown that activated microglia can cause irreversible damage to tissues of the CNS. Microglia in the deactivated or resting state are in surveillance mode. They constantly sample the surrounding milieu to detect signals associated with injured brain tissue. A number of factors have been identified that upregulate microglia to the activated state.
During the process of activation, microglia are highly plastic and differ in morphology and phenotype depending on the nature of the insult-causing activation. Microglia may remain in the activated state for prolonged periods. However, it is unlikely that microglial activation is the primary cause of any neurodegenerative process. It is more likely that an initial challenge induces an inflammatory cascade, which in turn initiates maladaptive processes and positive feedback loops that cause further pathological inflammation.
Postmortem examination of brain tissue from PD patients has demonstrated activated microglia in the substantia nigra pars compacta. In AD, neuroinflammation is considered a downstream effect of abnormal protein production. Aβ causes upregulation and activation of microglia leading to an inflammatory cascade. This cascade sets out to respond to abnormal protein accumulation but causes damage as a by-product of activation.
An analogy on the macroscopic scale can be drawn to acute brain injury such as a stroke, where reparative attempts by the brain tissue can cause edema, which in turn leads to an increase in intracranial pressure and even death. In the same way, processes that set out to reverse neurodegeneration may actually contribute to secondary damage. Understanding the balance between protective and destructive capabilities of microglia has led to trials seeking to slow progression of neurodegenerative diseases by dampening down neuroinflammatory processes.
Disrupted Axonal Transport
In neurons, proteins and lipids are manufactured in the cell body and are transported along axonal projections, which can extend over a meter in length, to synaptic terminals. Conversely, neurotrophic factors transported from synaptic terminals help to regulate cellular function. This process is called fast axonal transport and is an essential part of cellular homeostasis.
Dysfunction in the axonal transport system was first studied in large motor neurons. A decreased level of kinesin-mediated anterograde transport and retrograde dynein-mediated transport was observed in patients with ALS. Since then, research has shown that SOD1 aggregates interact with the retrograde transport system in a way that may lead to axonal dysfunction (Fig. 1.4). A decrease in retrograde transport could lead to toxicity at synaptic terminals as well as a loss of positive feedback from factors that stimulate neuronal survival. Mitochondrial distribution could be affected, resulting in a mismatch in energy provision. The accumulation of damaged mitochondria at synaptic terminals could cause increased terminal ROS production.

Schematic model of how mutant SOD1 aggregates lead to altered axonal transport in ALS
Conclusion
While arguments can be made regarding pathophysiological processes that make neurons vulnerable to degeneration, we still do not know fully why some people are physiologically more susceptible to specific neurodegenerative diseases, and why different neurodegenerative diseases cluster within some kindreds. It is clear that the common neurodegenerative conditions share similar processes. Increased understanding of the pathophysiologic processes for neurodegenerative diseases is likely to lead to disease-modifying therapeutic interventions that may be beneficial over a range of different clinical phenotypes.
Further Reading
Al-Chalabi A, Enayat ZE, Bakker MC et al. Association of apolipoprotein E epsilon 4 allele with bulbar-onset motor neuron disease. Lancet 1996;347:159–160
Beetend K, Sleegers K, Van Broeckhoven C. Current status on Alzheimer disease molecular genetics: from past, to present to future. Human Molecular Genetics 2010 (Advanced access April 2010)
Boillee S, Cleveland DW. Revisiting oxidative damage in ALS: microglia, Nox and mutant SOD1. The Journal of Clinical Investigation 2008;118:474–478
Dickson DW. Linking selective vulnerability to cell death mechanisms in Parkinson’s disease. The American Journal of Pathology ; 2007;170(1):16–19
Fallis BA, Hardiman O.. Aggregation of neurodegenerative disease in ALS kindreds. Amyotrophic Lateral Sclerosis 2009;10(2):95–98
Gilbert RM, Fahn S, Mitsumoto H, Rowland LP. Parkinsonism and motor neuron diseases: Twenty-seven patients with diverse overlap syndromes. Mov Disord. 2010 Jul 28. [Epub ahead of print]
Heidler-Gary J, Hillis AE. Distinctions between the dementia in amyotrophic lateral sclerosis with frontotemporal dementia and the dementia of Alzheimer’s disease. Amyotrophic Lateral Sclerosis 2007;8(5):276–282
Kawahara Yukio, Ito Kyoto, Sun Hui, Aizawa Hitoshi, Kanazawa Ichiro, Kwak Shin. RNA editing and death of motor neurons. Nature 2004;Vol 427
Langman’s Medical Embryology, 8th edition (2000) Thomas W Sadler. Lippincott Williams & Wilkins
Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. European Journal of Physiology 2010;460:525–542
Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nature reviews—Neuroscience 2006;7:278–294
Morfini GA, Burns M, Binder LI et al. The Journal of CLinical Neuroscience 2009;29(41):127776–12786
Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nature Reviews—Neurology 2010;6:193–201
Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: What is it and where are we? The Journal of Clinical Investigation 2003;111:3–10
Robbins Basic Pathology, 7th edition (2003) Vinay Kumar, Ramzi S. Cotran, Stanley J. Robbin. Elsevier Science
Ross CA, Porter MA. Protein aggregation and neurodegenerative disease. Nature Medicine 2004;S10–S17
Schon EA, Manfredi G. Neuronal degeneration and mitochondrial dysfunction. The Journal of Clinical Investigation 2003;111(3):303–312
Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Annals of Neurology 2010;67:291–300
Tovar-y-Roma LB, Santa-Cruz LD, Tapia R. Experimental models for the study of neurodegeneration in amyotrophic lateral sclerosis. Molecular Neurodegeneration 2009;4:31
Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21–22. American Journal of Human Genetics 1994;55(6):1159–1165
Xinkum W, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Frontiers in Aging Neuroscience 2010;2(12)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Verlag London Limited
About this chapter
Cite this chapter
Byrne, S.C., Rowland, L.P., Vonsattel, J.P.G., Welzel, A.T., Walsh, D.M., Hardiman, O. (2011). Common Themes in the Pathogenesis of Neurodegeneration. In: Hardiman, O., Doherty, C. (eds) Neurodegenerative Disorders. Springer, London. https://doi.org/10.1007/978-1-84996-011-3_1
Download citation
DOI: https://doi.org/10.1007/978-1-84996-011-3_1
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-84996-010-6
Online ISBN: 978-1-84996-011-3
eBook Packages: MedicineMedicine (R0)