
Key Points
1. Relationship between ADHR, XLH, and ARHP.
2. Clinical assessment of phosphate excretion and TmPi/GFR.
3. FGF-23 and CKD.
4. Acute management of hypophosphatemia.
5. How phosphate needs are met in the growing child.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Key Words
1 Phosphorus Balance
Phosphate is fundamental to cellular metabolism and skeletal mineralization. It is of critical importance when it comes to growth – as a constituent of bone mineral. The body content of phosphorus increases from 0.6% body weight in the newborn to 1% or 600–700 g in the adult reflecting the increasing proportion of mineralized bone and soft tissue per unit of body mass (1). In the growing individual phosphorus balance must be positive to meet the needs of growth. Balance studies indicate that a 1- to 3-month-old infant fed a standard formula retains 32±25 (SD) mg/kg body weight of phosphate (2, 3), while the adult retains zero.
The terms phosphate concentration and phosphorus concentration are often used interchangeably. The plasma phosphate concentration is usually measured in units of mg/dL or mmol/L. The following calculations may be used to convert between these units:
-
1 mmol of phosphate = 31 mg of elemental phosphorus
-
1 mmol/L of phosphate = 3.1 mg/dL (or 31 mg/L) or phosphorus
-
1 mg of phosphorus = 0.032 mmol of phosphate
-
1 mg/dL of phosphorus = 0.32 mmol/L of phosphate
The content of phosphate in plasma, urine, tissue, or foodstuffs is measured and expressed in terms of the amount of elemental phosphorus contained in the specimen or phosphorus concentration. Phosphorus in the form of phosphate ion circulates in blood as an organic form consisting principally of phospholipids and phosphate esters and an inorganic form (Pi). From a clinical standpoint only the inorganic form of phosphate is measured. Ninety percent of plasma Pi is filtered at the glomerulus as phosphate ions, the ratio of HPO4 2– to H2PO4 – depending on pH, or as phosphate complexed with sodium, calcium, or magnesium. The other 10% of the remaining plasma phosphate is as noted protein-bound and not filterable. Correcting the volume occupied by plasma proteins (7%) raises the ultrafiltrate Pi concentration by an additional 7.5%, negating the effect of the reduction in plasma protein-bound Pi. Both in vivo and in vitro measurements show that the ratio of ultrafilterable Pi to total plasma Pi is close to 1 (4).
The serum phosphorus concentration exhibits a circadian rhythm characterized by a rapid decrease in early morning to a nadir shortly before noon, a subsequent increase to a plateau in late afternoon and a small further increase to a peak shortly after midnight (5, 6). Restriction or supplementation of dietary phosphorus induces a substantial decrease or increase, respectively, in serum concentrations of phosphorus during the late morning, afternoon, and evening, but induces less or no change in the morning phosphorus concentration (6). Morning fasting bloods are therefore least affected by dietary changes on the serum phosphorus concentration.
There are substantial differences in serum phosphorus concentrations depending on age. Phosphorus levels are highest in infants, ranging from 4.8 to 7.4 mg/dL in the first 3 months of life and decreasing to 4.5–5.8 mg/dL by 1–2 years of age. In mid-childhood values range from 3.5 to 5.5 mg/dL and decrease to adult values by late adolescence (7–9). In adult males, serum phosphorus levels decrease with age from approximately 3.5 mg/dL at age 20 years to 3.0 mg/dL at age 70 (10). In females values are similar to those of males until after menopause, when they increase slightly from approximately 3.4 mg/dL at age 50 years to 3.7 mg/dL at 70 years of age (10).
1.1 Renal Phosphate Transport
1.1.1 Cellular Aspects
The kidney is the major regulator of Pi homeostasis and can increase or decrease its Pi reabsorptive capacity in response to need. Under normal physiologic conditions, 80–97% of the filtered load of phosphate is reabsorbed by the renal tubule. The rate limiting step in renal Pi reabsorption involves the transport of Pi from the tubular lumen across the apical BBM. Pi then moves across the cell and effluxes at the basolateral membrane. Pi transport across the BBM is saturable, Na-dependent and driven by a Na-gradient (outside > inside) that is maintained by the basolateral membrane-associated Na,K-ATPase. Na/Pi co-transport across the BBM is electrogenic, sensitive to pH, with 10- to 20-fold increases documented when the pH is increased from 6 to 8.5, and the target for physiologic/pathophysiologic regulation. The mechanism of phosphate efflux at the basolateral membrane has not been elucidated. Current data suggest that phosphate can exit the cell down its electrochemical gradient via a low capacity Na/Pi co-transport system that couples flux of one sodium with that of one divalent phosphate ion or a high-capacity phosphate anion exchange mechanism (11).
Approximately 70% of the filtered load of phosphate is reabsorbed by the proximal tubule, with three times as much occurring in the proximal straight tubules as in proximal convoluted tubules (12–14). Due to axial heterogeneity most of the phosphate reabsorption by the proximal tubule occurs within the first 25% of the proximal tubule length. Little or no phosphate transport has been shown to occur in the Loop of Henle. Phosphate reabsorption in the distal nephrons is controversial. Results from micropuncture studies suggest that up to 10% of the filtered phosphate load is reabsorbed by the distal convoluted tubules (15). Other studies have failed to uncover evidence of distal Pi transport (16). Terminal nephrons segments may reabsorb an additional 3–7% of the filtered phosphate load based on the fact that a higher fraction of filtered phosphate remains in the late distal tubules than appears in the final urine (15–17). Although some studies have failed to demonstrate phosphate reabsorption in isolated perfused cortical collecting tubules (18), others have shown a small but significant net efflux of phosphate from this nephron segment (19, 20).
1.1.2 Molecular Characterization
Three classes of Na/Pi co-transporters have been identified in mammalian kidney (21–23). The type I Na/Pi co-transporter (Npt1) is expressed predominantly in the BBM of the proximal tubular cells (24, 25) and mediates fluxes of chloride and organic anions as well as Pi. Dietary phosphorus or PTH does not seem to alter type I Na/Pi co-transporter protein or mRNA expression. Thus, the type I co-transporter is not thought to be a major determinant of proximal tubule phosphate handling.
The type II family of Na/Pi co-transporters exhibit 25% homology with Npt1. The type II co-transporter is largely responsible for renal phosphate reabsorption as indicated by knockout experiments. Targeted inactivation of type II (Npt2) in mice leads to severe phosphate wasting (85% reduction in phosphate reabsorption), hypercalciuria, and skeletal abnormalities. There are three highly homologous isoforms; type IIa (Npt2a) and type IIc (Npt2c) are expressed almost exclusively in the BBM of the renal proximal tubule (26, 27). The type IIb is not expressed in the kidney (28). The type IIb is expressed in the BBM of the small intestine and plays a role in the physiologic regulation of intestinal reabsorption of phosphate (29–31)
1.1.3 Role of Dietary Phosphorus
The dietary intake of phosphorus is one of the most important physiologic regulators of Na/Pi co-transport. An increase in dietary phosphorus is associated with an increased total and fractional urinary excretion of phosphorus. This may occur even in the absence of detectable changes in the serum level and filtered load of phosphorus. PTH plays an important role in this phosphaturic response to a phosphorus load. However, the phosphaturic response may be observed in hypoparathyroid patients as well. In the presence of dietary phosphate restriction or hypophosphatemia, phosphate homeostasis is preserved by short-term intrinsic renal and intestinal adaptations in transport processes, and by more long-term hormonal mechanisms, which regulate the efficiency of phosphate transport in the kidney and intestine. The net effect of these adaptations is virtual abolition of phosphate excretion in the urine, and an increase in the plasma phosphate concentration toward normal. This response is achieved with only a slight increment in the plasma Ca2+ concentration.
The renal tubule can adapt quickly to changes in dietary intake or plasma concentrations of phosphorus. During Pi deprivation the abundance of the apically localized Na/Pi co-transporters, Na/Pi type IIa in the kidney BBM, and Na/Pi type IIb in the intestinal BBM increase with a concomitant increase in Pi reabsorption. These alterations in Na/Pi co-transport take place within hours and are independent of changes in PTH, 1,25 (OH)2D3 or serum calcium levels. This adaptation, in addition, has been shown not to be inhibited by cycloheximide or actinomycin D (inhibitors of protein synthesis), which suggests that new protein synthesis is not required (32).
Adaptation induced after long-term (4 days) phosphorus restriction, on the other hand, has been shown to be inhibited by cycloheximide and actinomycin D, which suggests that new protein synthesis is required for the long-term response. Again there is an adaptive increase in the abundance of the type II Na/Pi co-transporter protein, but also mRNA.
It is important to remember that the renal reabsorption of Pi is very rapidly altered by a number of factors, e.g., paCO2, HCO3, sodium delivery, adrenergic agents, dopamine, etc (see Table 1). These effects may be totally independent of hormones and secreted factors. The well-described hormones involved in the adaptation to high or low Pi diets in both animal models and humans include PTH and 1,25 (OH)2D3.
1.1.4 Role of Parathyroid Hormone
PTH is the major hormonal regulator of renal phosphate reabsorption (see above). Type II Na/Pi transporters represent the major pathway of renal phosphate reabsorption and are the major target with respect to inhibition of proximal tubular reabsorption by PTH. PTH produces a greater inhibition of Pi transport in the proximal straight tubules than proximal convoluted tubules (33).
It also has been demonstrated that the N-amino-terminal fragment PTH sequence 1–34 reproduces all physiologic effects of the PTH sequence 1–84. It has been demonstrated that the amino acid sequences 10–15 and 24–34 of PTH are necessary for binding to the receptor. With regard to the biologic effects of PTH, it has been shown that the first two N-terminal amino acids 1 and 2 are required for activation of the adenylate cyclase–protein kinase A pathway, whereas the amino acid sequences 28–34 are required for the activation of the phospholipase C–protein kinase C pathway. Recent work demonstrated that the addition of PTH 1–34 (which signals through both the protein kinase A and protein kinase C pathways) to either the apical or the basolateral surface of isolated perfused proximal tubules caused internalization of the type II Na/Pi transport protein. On the other hand, PTH 3–34, which signals only through the protein kinase C pathway, has an effect apically but not basolaterally. It can be concluded from these studies that type-II Na/Pi protein transporter activity and internalization are regulated by c-AMP-dependent and independent mechanisms, and that functional PTH receptors are located on both the apical and basolateral membranes of the proximal tubule.
1.1.5 Role of Vitamin D
The main source of vitamin D in humans is endogenous vitamin D3 (also known as cholecalciferol), which is produced by the ultraviolet irradiation of 7-dehydrocholesterol in the skin. Cholecalciferol is metabolized by the liver into 25-hydroxy cholecalci-ferol (25-hydroxyvitamin D3 [25(OH)D3]). After enterohepatic circulation 25(OH)D3 is further metabolized in the kidney to 1,25-dihydroxycholecalciferol (calcitriol [1,25(OH)2D3]), which is the most active known metabolite of vitamin D. It has recently been shown that 25(OH)D3 in complex with its carrier protein, the vitamin D binding protein, is filtered through the glomerulus and reabsorbed in the proximal tubules by the endocytic receptor megalin. Endocytosis is required to preserve 25(OH)D3 and deliver it to the cells as the precursor for generation of 1,25(OH)2D3. PTH seems to act as a trophic hormone in stimulating the production of 1,25(OH)2D3. Thus, with intact parathyroid glands, changes in serum calcium indirectly regulate renal production of 1,25(OH)2D3 by altering the secretion of PTH. In addition there is evidence that calcium acts directly to alter the synthesis of calcitriol. Low serum phosphorus stimulates and high serum phosphorus suppresses the renal formation of 1,25(OH)2D3 independent of PTH. Growth hormone by virtue of increased synthesis of insulin-like growth factor 1 stimulates the activity of 25(OH)D 1α-hydroxylase. Chronic metabolic acidosis also increases serum levels of calcitriol. This latter effect could be mediated by acidosis-induced urinary losses of phosphate leading to cellular phosphate depletion.
The effect of vitamin D on the renal handling of phosphorus has been the subject of numerous investigations. One of the difficulties in interpreting the changes in urinary excretion of phosphorus has been related to the calcemic actions of vitamin D, which by suppressing PTH secretion, indirectly alter the renal handling of phosphorus. 1,25(OH)2D3 may have a phosphaturic or phosphate sparing effect based on the state of phosphate balance. When hypophosphatemic due either to vitamin D deficiency or phosphate deprivation or when baseline phosphate excretion is high (e.g., volume expansion, administration of PTH or calcitonin) 1,25(OH)2D3 is antiphosphaturic. On the other hand, 1,25(OH)2D3 is phosphaturic with hyperphosphatemia or in phosphate replete states. The duration of 1,25(OH)2D3 therapy also affects phosphate transport. Chronic administration of 1,25(OH)2D3 is associated with a decrease in renal phosphate reabsorption. This response is thought to be a consequence of an increase in intestinal phosphate and positive phosphate balance, which, in turn, induces an adaptive decrease in phosphate reabsorption by the proximal tubule. Acute 1,25(OH)2D3 administration is associated with a change in the lipid composition of the membrane and increases renal phosphate reabsorption – but also depends on the experimental conditions of the study such as prior administration of vitamin D, PTH, and phosphorus balance of the organism.
Type IIa co-transport protein has been thought to be the target of 1,25(OH)2D3 regulation. In vitamin D deficient, but not parathyroidectomized rats, 1,25(OH)2D3 increased renal type II-mediated co-transport, mRNA, and protein levels (34).
1.1.6 Role of Growth Hormone
Growth hormone (GH) has been shown to be a factor that increases the renal reabsorption of phosphate. When GH is elevated or administered on a chronic basis to adult humans or adult animals there is a reduction in the urinary Pi excretion and elevation in plasma Pi levels. The removal of GH in the adult rat (through hypophysectomy) causes a significant decline in the maximum capacity to reabsorb Pi (TmPi) by the whole kidney and results in increased phosphaturia. However, this effect cannot be attributed solely to GH because all pituitary hormones were removed. In addition, Hammerman et al. (35) reported that the effects of pharmacologic doses of GH results in a selective stimulation of proximal tubular BBM Na/Pi transport systems.
Mulroney et al. (36) using a peptidic antagonist to GH-releasing factor to suppress the pulsatile release of GH from the anterior pituitary over a 2-day period showed a doubling of the urinary excretion of Pi and attenuation of somatic body growth. These effects were attributed to a decrease in the TmPi. On the other hand, short-term use of the GH-releasing factor antagonist had no effect of lowering the TmPi and increasing urinary Pi excretion. Woda et al. (37) have demonstrated that using the GH-releasing factor antagonist for 48 h is associated with a 30% reduction in Vmax of Pi transport in proximal tubule BBM prepared from weaning rats, implicating GH in proximal tubule Pi uptake. Furthermore, these authors have shown that the mechanism for the enhanced Na/Pi co-transport activity in the juvenile rat appears to be through the action of GH on the expression of proximal tubular BBM type IIa Na/Pi co-transporter protein.
1.1.7 Other Factors
A number of other hormones also target the type II co-transporter in the regulation of renal Pi handing. Amongst these are thyroid hormone, dexamethasone, epidermal growth factor, insulin, etc. Please see Table 1 for additional factors affecting the renal transport of Pi.
1.1.8 The Role of Phosphatonins in Phosphate Homeostasis
In the last several years a new class of regulators have surfaced called phosphatonins that control systemic phosphate homeostasis linking bone metabolism and mainly the renal handling of phosphate. A number of different molecules have been identified such as FGF-23 (fibroblast growth factor 23) mainly expressed in regions of active bone formation and remodeling (particularly in osteocytes and lining cells), FGF-7 (fibro-blast growth factor 7), MEPE (matrix extracellular phosphoglycoprotein) expressed in odontoblasts and osteocytes embedded in mineralized matrix, and more specifically its carboxy-terminal MEPE/acidic serine–aspartate-rich MEPE associated motif (ASARM peptide), and sFRP4 (secreted frizzled related protein-4). These phosphatonins, and especially FGF-23, decrease serum phosphate via two simultaneous actions in the proximal convoluted tubules: they decrease the expression of the Na/Pi co-transporters, thus increasing phosphate excretion and they reduce the production of 1,25(OH)2D3, thus decreasing the ability of the intestine to absorb phosphate (Fig. 1).

Hormonal regulation of serum phosphate by parathyroid hormone, vitamin D3, and FGF-23. Serum phosphate levels are influenced by dietary intake and intestinal absorption, the rate of renal excretion of reabsorption, respectively, and skeletal deposition and release from bone. Changes in serum phosphate concentration alter the calcium × phosphate product and a fall in free calcium triggers release of parathyroid hormone (PTH). PTH inhibits renal phosphate reabsorption and at the same time stimulates 1α-hydroxylase expression and activation of 1,25(OH)2D3 from its inactive precursor 25(OH)D3. The active vitamin D3 stimulates in turn renal and intestinal phosphate absorption as well as deposition of phosphate in bone. An increase in serum phosphate concentrations also directly increases FGF-23 synthesis and release from bone independent from the PTH axis. FGF-23 lowers phosphate concentrations by inhibiting renal and intestinal phosphate transport as well as preventing activation of 1,25(OH)2D3, which in turn controls levels of bone synthesis of FGF-23 (from Wagner (134) (Fig. 1)).
Phosphatonins were first identified in patients with tumor-induced osteomalacia (TIO). Patients with TIO typically exhibit low serum Pi concentrations, normal or slightly low serum calcium concentrations, normal PTH concentrations, inappropriately low 1,25(OH)2D3 concentrations, renal Pi wasting, and rickets and osteomalacia. It was demonstrated that conditioned medium from a tumor associated with TIO produced a factor or factors that inhibited Na+–Pi-dependent transport in cultured OK cells (38). Several laboratories subsequently showed that FGF-23, sFRP-4, FGF-7, and MEPE are present in these tumors and contribute to the phosphaturia associated with this syndrome (39–47).
FGF23 has been studied most extensively and clearly has been established as a potent regulator of systemic phosphate transport and balance at the level of bone, intestine, and kidney (Figs. 2 and 3). FGF-23 inhibits Na+–Pi uptake in cultured renal epithelial cells and also inhibits Pi reabsorption when infused into rodents in vivo (39, 43, 48, 49). Similar findings are reported for sFRP-4, MEPE (42), and FGF-7 (43).

Structure and function of FGF-23. FGF-23 is a protein with 251 amino acids. There is a signal peptide with 24 amino acids in the N-terminal portion of the FGF-23 protein. A part of FGF-23 is cleaved between Arg179 and Ser180 by furin recognizing Arg176-X-X-Arg179 motif. FGF-23 has an FGF homology region in the N-terminal portion of this processing site. FGF-23 reduces serum phosphate by suppressing proximal tubular phosphate reabsorption and intestinal phosphate absorption (from Fukumoto (135) (Fig. 2)).

Regulation and function of FGF-23. FGF-23 is released from bone upon elevated serum phosphate levels. To be biologically active, FGF-23 requires O-glycosylation by GALNT3 (UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3). FGF-23 lowers serum phosphate levels by decreasing the expression of renal (Na/Pi-IIa and Na/Pi-IIc) and intestinal (Na/Pi-IIb) phosphate transporters thereby reducing phosphate uptake from diet and increasing renal excretion of phosphate. Furthermore, FGF-23 inhibits 1α-hydroxylase expression in kidney reducing the final step in vitamin D3 activation and thereby prevents a compensatory increase in intestinal and renal phosphate transport. High vitamin D3 levels itself may also directly stimulate FGF-23 synthesis providing for a regulatory circuit and feedback mechanism. FGF-23 is cleaved and inactivated by subtilisin-like proprotein convertases requiring a recognition motif consisting of Arg176-X-X-Arg179 at position 176. PHEX is not directly involved in FGF-23 degradation but may indirectly contribute to cleavage (from Wagner, CA (134) (Fig. 2)).
In addition to inhibiting Pi reabsorption in the kidney FGF-23 alters vitamin D metabolism such that serum 1,25(OH)2D3 concentrations are reduced or fail to increase despite the presence of hypophosphatemia (43, 49). The reduction in serum concentrations of 1,25(OH)2D3 reduces Pi absorption in the intestine and possibly in the kidney as well, suggesting an inhibitory effect of these proteins on 25-hydroxyvitamin D 1α-hydroxylase activity (43, 49). As might be expected, in FGF-23 knockout mice, 25-hydroxyvitamin D 1α-hydroxylase expression is increased and elevated serum levels of 1,25(OH)2D3 cause significant hypercalcemia and hypophosphatemia. MEPE, on the other hand, increases circulating 1,25(OH)2D3 concentrations.
The intake of dietary phosphorus and serum Pi concentrations might be expected to play a role in the regulation of phosphatonin concentrations. In humans, however, short-term alterations in dietary intake do not appear to influence concentrations of FGF-23. In rodents, on the other hand, following changes in dietary Pi, the data suggest that FGF-23, PTH, and 1,25(OH)2D3 are all involved in the adaptation to dietary Pi (50).
1,25(OH)2D3 in turn has been shown to regulate FGF-23 in rats. Saito et al. (51) showed that serum FGF-23 levels increased following the administration of 1,25(OH)2D3 to intact rats in a dose-dependent manner. There, in addition, was a direct correlation between the serum phosphorus concentrations and serum FGF-23 concentrations. In thyroparathyroidectomized rats, 1,25(OH)2D3 also increased serum FGF-23 concentrations. In the thyroparathyroidectomized rats, serum FGF-23 levels were at the low end of normal despite elevated serum phosphorus concentrations. In contrast in hypoparathyroid humans, serum FGF-23 levels are elevated.
FGF-23 interacts with FGF receptors that belong to type 1 transmembrane phosphotyrosine kinase receptors to elicit its biological response (52). Recent studies indicate that FGF-23 requires Klotho, as a co-factor for receptor activation (52, 53). The Klotho/Klotho gene encodes a single-pass membrane protein, which has homologies to β-glucosidases (54–57). Two transcripts formed through alternative RNA splicing are transcribed from the gene and encode a membrane or secreted klotho protein (54). Klotho is expressed in several tissues including the kidney, reproductive tissues, and brain (56). The role of Klotho as an FGF-23 receptor is supported by the fact that Klotho-deficient mice have a phenotype similar to FGF-23 null mice (55).
In summary, FGF-23, sFRP-4, MEPE, and FGF-7 have been shown to inhibit Pi reabsorption. FGF-23 and sFRP-4 also modulate the synthesis of 1,25(OH)2D3. FGF-23 synthesis, in turn, is regulated by 1,25(OH)2D3 as 1,25(OH)2D3 action is required for maintaining normal FGF-23 production in osteoblasts. It also seems that FGF-23 acts on the parathyroid gland to inhibit both PTH biosynthesis and secretion. By inhibiting the circulating level of PTH, FGF-23 thus appears to counteract its inhibition of tubular phosphate reabsorption and enhance its suppression of 1,25(OH)2D3 biosynthesis. Data in rodents show that FGF-23 and sFRP-4 concentrations may be regulated by the intake of dietary Pi. The data in human subjects are much less clear with respect to the effects of dietary Pi on the concentrations of these peptides. Thus, FGF-23 is in the position to keep serum phosphate levels low and to act as a bone phosphate sensor controlling intestinal and renal phosphate handling.
1.2 Renal Phosphate Transport – Changes as a Function of Age
The kidneys of infants and children reabsorb a high fraction of filtered Pi appropriate to the needs of the growing child. Spitzer and Barac-Nieto (58) recently reviewed the body of literature pertaining to the role of the newborn kidney in Pi balance. Early experiments in dogs (59) and rats (60) had suggested that this high reabsorptive capacity is intrinsic to the kidneys. That this is indeed the case was demonstrated by Johnson and Spitzer (61) in isolated kidneys containing phosphate concentrations varying between 3 and 15 mg/dL. The slopes of the regression lines describing the relationship between the filtered load of Pi and the amount reabsorbed per unit of kidney weight (Fig. 4) illustrate that at any filtered load of Pi the kidney of the newborn reabsorbed almost four times as much Pi than that of the adult. Micropuncture experiments by Kaskel et al. (62) confirmed the avid nature of Pi reabsorption by the newborn. Studies at comparable locations along the proximal tubule revealed that the fraction of filtered Pi reabsorbed was significantly higher in immature than in mature guinea pigs (Fig. 5). Eighty-five percent of the age-related difference in the renal reabsorption of Pi could be explained by the higher rates of Pi reabsorption in the proximal segments of the nephron and the remaining 15% by differences in reabsorption at more distal nephron sites. Woda et al. (63) also examined the renal tubular sites of increased phosphate transport as well as Na/Pi expression in the juvenile rat. Renal micropuncture experiments were performed in acutely thyroparathyroidectomized adult (>14 weeks old) and juvenile (4 weeks old) male rats fed a normal phosphate or low phosphate diet. Phosphate reabsorption was greater in the proximal convoluted and straight tubules of the juvenile compared to the adult fed a normal phosphate diet.

Regression lines and 95% confidence limits of the relationship between the reabsorption of phosphate (Pi) and the filtered load of Pi by the isolated perfused kidneys of newborn (y = 1.25x + 0.09) and mature (y = 0.34x + 3.1) guinea pigs (used with permission from Johnson and Spitzer (61)).

Neiberger and Barac-Nieto (64) further studied the mechanism underlying the high renal reabsorption of Pi during growth looking at the Na+–Pi co-transport system in luminal brush-border membrane vesicles (BBMV) obtained from the kidneys of newborn and adult animals (Fig. 6). At both ages, most transport of Pi into vesicles was found to be Na+ gradient dependent. The uptake was concentrative, i.e., the intravesical Pi concentration exceeded the equilibrium concentration as long as an inwardly directed Na+ gradient subsisted across the brush-border membrane. As the gradient dissipated, the vesicular Pi content diminished to reach equilibrium. The initial rate of Na+-dependent Pi uptake was linear with time and was much higher from vesicles obtained from newborn than from adult animals. Kinetic analysis of the initial transport rates revealed that the Vmax of the Na+–Pi was substantially higher in BBMV from newborns (650 pmol/s per mg protein) than those of adults (144 pmol/s per mg protein). The Km, a measure of the apparent affinity of the co-transporter for Pi did not differ with age. The capacity for Na+–Pi co-transport across the luminal brush-border membrane of the renal proximal tubular cells was reported to be four-fold higher in the newborn than the adult. Dietary modifications in which the diet was supplemented with Pi for 3 days decreased by about 50% the Vmax for Na+–Pi co-transport in renal BBMV from adult animals but only about 25% in the newborn. On the other hand, low dietary Pi resulted in a doubling of the Na+–Pi co-transport capacity in renal BBMV of the adult (from 144 to 318 pmol/mg per s), but no significant change in the Vmax of the Na+–Pi co-transport system of the newborn. Based on these studies Spitzer and Barac-Nieto (58) conclude that the newborn Na+–Pi co-transport system is characterized by a high transport capacity and low adaptability to changes in dietary Pi. Woda et al. (63) similarly showed greater Pi uptake in BBMV from both superficial and outer juxtamedullary cortices of juvenile rats. Western blot analysis revealed a 2- and 1.8-fold higher amount of Na/Pi-2 protein in the superficial and outer juxtamedullary cortices, respectively, in juvenile rats. Immunofluorescence microscopy also indicated that Na/Pi-2 expression was present in the proximal tubule BBM to a greater extent in juvenile rats. These features of the co-transport system may explain, in part, the hyperphosphatemia observed in newborns fed a diet of cow’s milk, which is rich in Pi (75).

Time course of Pi (0.1 mM) uptake in brush-border membrane vesicles (BBMV) of 3- to 14-day-old and >57-day-old guinea pigs; Circles, uptake in the presence of 100 mM Na+ gradient. Triangles, uptake in the presence of 100 mM inwardly directed KCl gradient (used with permission from Neiberger et al. (64)).
The high capacity of the developing kidney for Pi reabsorption appears to persist independent of extracellular factors known to modulate renal Pi transport in vivo. As Bojour and Fleish (65) present evidence that renal phosphate reabsorption may be affected by total body stores of Pi, Barac-Nieto et al. (66) set out to determine if the high Pi demand associated with growth may, similar to a reduction in Pi supply, result in a low level of intracellular Pi, and that this low level is responsible for initiating and maintaining a high renal Pi co-transport capacity. Nuclear Magnetic Resonance (NMR) spectroscopy and chemical methods were used to determine the effect of age and Pi intake on intracellular Pi in the perfused kidney. The findings from these studies indicate that changes in intracellular Pi related to age or diet are not a consequence of changes in abundance or maximum mobility of Na+–Pi co-transporters and are consistent with earlier observations. In animals fed a normal phosphate diet intracellular Pi was twice as high (1.85±0.23 vs. 0.90±0.02 mM) while the fractional reabsorption of Pi was lower (0.70 vs. 0.90) in >4-week-old than in <1-week-old animals. Diet induced changes in intracellular Pi were associated with changes in Vmax of similar magnitude in mature and immature animals, but in opposing directions. However, as seen in Fig. 7, at any given intracellular Pi concentration, Vmax was substantially higher in microvilli prepared from kidneys of newborn than of older animals.

Relationship between intracellular Pi ([Pi]) in kidney and Vmax in renal microvilli of newborn guinea pigs fed standard (open triangle) or high (solid triangle) Pi diets and mature guinea pigs fed standard (open circles) or low (solid circle) Pi diets (used with permission from Barac-Nieto et al. (66)).
Woda et al. (63) also showed that dietary phosphate restriction in juvenile rats resulted in a significant increase in Na/Pi expression in the proximal tubule BBM as well as the expression of intracellular NaPi-2 protein. Dietary phosphate restriction in the juvenile rat upregulated BBM Na/Pi-2 expression, which was associated with the further increase in proximal tubular Pi reabsorption. As a result of this study and others (67, 68) a specific type IIa-related Na/Pi co-transporter protein was postulated to account for the high Pi transport rate in weanling animals. Evidence for this was obtained by antisense experiments and transport expression in Xenopus oocytes. When mRNA isolated from kidney cortices of rapidly growing rats was treated with type IIa transporter antisense oligonucleotides or was depleted of type IIa-specific mRNA by a subtraction hybridization procedure, Na+-dependent Pi uptake was still detected in injected oocytes (67). The type IIa transporter-depleted mRNA contained an mRNA species that showed some sequence homology to the type IIa transporter encoding message. This is compatible with the fact that young type IIa knock-out mice lacking type IIa mRNA and protein retain the capacity to reabsorb phosphate at a rate that cannot be explained by the presence of type I and III Na/Pi transporter (69). Segawa et al. (67) isolated a cDNA from the human and rat kidney that encodes a growth-related Na+-dependent Pi co-transporter, type IIc. The transport activity was dependent on extracellular pH. In electrophysiological studies, type IIc Na/Pi was electroneutral, while type IIa was highly electrogenic (see above). In Northern blotting analysis, the type IIc protein was shown to be localized at the apical membrane of the proximal tubular cells in superficial and corticomedullary nephrons of the weanling rat kidney. Hybrid depletion experiments showed that type IIc could function as a Na/Pi co-transporter in weanling animals, with its role reduced in adults. Segawa’s studies (67, 68) suggest that the type IIc is a growth-related renal Na/Pi co-transporter, which has a high affinity for Pi and is electroneutral.
Parathyroid hormone in the adult decreases the excretion of Pi by inhibiting BBM Na/Pi co-transport. The response to PTH is different during early post-natal life. Linarelli (70) showed that infusion of PTH into newborn babies resulted in minimal depression of the tubular reabsorption of Pi. In the isolated kidney of the newborn guinea pig addition of PTH to the perfusion fluid caused a marked increase in tubular calcium (Ca2+) reabsorption and in urinary excretion of c-AMP, but had little effect on the excretion of Pi.
Growth hormone equally plays an important role in renal reabsorption of Pi during development. Woda et al. (71) studied the regulation of renal Na/Pi-2 expression and tubular phosphate reabsorption in the juvenile rat and have demonstrated that GH is responsible independently of PTH, for the enhanced Pi uptake in both the proximal convoluted tubule and straight tubule of juvenile rats on a normal Pi diet. In addition, the authors found that GH contributes significantly to the blunted phosphaturic response to PTH in the juvenile rat. The proposed mechanism for the enhanced Na/Pi co-transport activity appears to be through the action of GH on the expression of proximal tubular BBM type IIa Na/Pi transporter protein. Overall phosphate handling by the immature kidney is regulated so that phosphate retention is promoted to meet the phosphorus needs of the growing organism.
2 Clinical Assessment Of Renal Phosphorus Excretion
Under normal physiologic conditions 80–97% of the filtered load of phosphate is reabsorbed by the renal tubules (72). For those solutes not undergoing tubular secretion, the difference, 100 – FEPi, where FEPi represents the fractional excretion of phosphate, is the percentage of phosphate that is reabsorbed or the fractional tubular reabsorption of Pi (TRPi). If renal function is normal and dietary intake average, calculation of the TRPi gives a rough guide as to whether or not tubular Pi reabsorption is normal or not. The TRPi may be calculated on random urine samples without the need for a timed urine collection. Calculation of the TRPi requires drawing a blood sample at the time of the urine collection. The TRPi is calculated as follows: TRPi (%) = (1 – Upi × Pcr/Ucr × Ppi) × 100, where Upi and Ppi represent the plasma and urinary concentrations of Pi and Pcr and Ucr represent the plasma and urinary concentrations of creatinine, respectively.
The TRPi is markedly influenced by changes in GFR as well as dietary phosphate intake, and a more reliable means of assessing phosphate reabsorption is to measure the TmPi/GFR. Clearance studies in humans and experimental animals show that as the filtered load of phosphate is progressively increased, phosphate reabsorption rises until a maximum tubular reabsorptive rate for phosphate (Pi), or TMPi, is reached, after which phosphate excretion increases in proportion to its filtered load (73). The TmPi/GFR or the maximum tubular reabsorption of phosphate per unit volume of GFR therefore is thought to be the most reliable measure of overall tubular reabsorptive capacity. Ideally, the TmPi should be calculated by performing a phosphate titration study. However, the TmPi may be calculated using a more practical method. Walton and Bijvoet (74) have shown that TmPi/GFR can be derived from the TRPi and plasma Pi and have produced a nomogram to simplify the calculation. The validity of the Walton–Bijvoet nomogram was questioned in children who are known to have higher concentrations of PPi and lower GFRs (7, 8, 75, 76). Brodehl et al. (77) have shown that the Walton–Bijvoet nomogram, while derived from studies in normal adults, gives good agreement with directly measured TmPi/GFR in infants and children at high rates of filtered Pi load. Brodehl et al. (14) also showed that as predicted on theoretical grounds TmPi/GFR could be calculated from the formula TmPi/GFR = PPi – (UPi × PCr/UCr). For clinical evaluation this equation may be used regardless of the phosphate load. Stark et al. (78) showed that there were no differences between morning fasting and non-fasting serum phosphate values. Furthermore, timed urinary collections are not necessary.
3 Hypophosphatemic Disorders
The causes of hypophosphatemia may be classified into one of three groups: increased urinary phosphate excretion, decreased GI absorption of phosphate, and shifts of phosphorus from the extracellular compartment (Table 2). Mild hypophosphatemia does not usually result in symptoms. Serum phosphate levels generally have to be below 1 mg/dL for patients to become symptomatic. A number of the clinical manifestations of severe hypophosphatemia may be seen in Table 3. The focus of this chapter is on a number of the disorders that result in increased urinary phosphate excretion and treatment of these conditions. Treatment: For the other causes listed in Table 2 the clinical circumstances will generally suggest whether severe underlying phosphate deficiency is present, as serum levels may not always be a good reflection of body stores. In some patients who may be hypophosphatemic from antacid or diuretic use correction of the underlying cause may be all that is necessary. In mild to moderate hypophosphatemia (∼2 mg/dL) oral repletion with skim milk (0.9 mg phosphorus per mL) or Neutra-phos, Neutra-phos K, or Fleet phosphorus soda preparations may be useful. Intravenous phosphorus repletion is reserved for severe (∼1 mg/dL) hypophosphatemia. The most frequently recommended regime is to administer 2.5 mg/kg body weight (0.08 mmol/kg body weight) of elemental phosphorus over a 6-h period for severe asymptomatic hypophosphatemia and 5 mg/kg body weight (0.16 mmol/kg body weight) of elemental phosphorus over a 6-h period for severe symptomatic hypophosphatemia. Parenteral administration should be discontinued when the serum phosphorus concentration is greater than 2 mg/dL.
3.1 Hereditary Hypophosphatemic Rickets with Hypercalciuria
Hereditary hypophosphatemic rickets with hypercalciuria (HHRH) is an autosomal recessive disorder characterized by hypophosphatemia secondary to renal Pi wasting, an appropriate increase in the serum concentration of 1,25(OH)2D3 with associated intestinal calcium hyperabsorption and hypercalciuria, and rickets and osteomalacia (Table 4). The most probable mechanism for the hypercalciuria in this disorder is increased intestinal calcium absorption. The increased renal phosphate clearance (TmP/GFR) is usually 2–4 standard deviations below the age-related normal range.
Initial clinical studies suggested that HHRH was a primary renal Pi wasting disorder since all abnormalities, with the exception of decreased renal Pi reabsorption, are completely corrected by dietary Pi supplementation. The Npt2a gene and a fragment of the Npt2a promoter gene, though, were not found to have mutations in affected individuals. The disease locus was mapped to human chromosome region 9q34, which contains Slc34A3, the gene encoding the type IIc Na/Pi co-transporter. The mutation is predicted to truncate the type IIc protein in the first transmembrane domain and to result in complete loss of function in individuals homozygous for the deletion. Compound heterozygotes are similarly affected supporting the conclusion that this disease is caused by Slc34A3 mutations affecting both alleles. The phosphaturic factor FGF-23 is at normal or low-normal serum levels in patients with HHRH, further supporting a primary renal defect as the cause of the disease.
It is not entirely clear why loss of function of the less abundant type IIc Na/Pi co-transporter causes rickets and osteomalacia in humans where as mutations in the more abundant type IIa Na/Pi co-transporter elicits a mild skeletal phenotype that lacks the typical features of rickets and osteomalacia in mice (79). Possibly the type IIc co-transporter may be a more important regulator of Pi homeostasis in humans than mice. Differences in developmental expression patterns of type IIa and type IIc Na/Pi co-transporters in relation to skeletal maturation, which is critically dependent on Pi supply may also be at issue. Extrarenal expression of type IIa and type IIc Na/Pi co-transporters in addition may have an impact on skeletal phenotype.
Treatment: Patients with HHRH frequently develop renal stones as a result of increased urinary excretion of both calcium and phosphate. Long-term phosphate supplementation as the sole therapy leads, with the exception of the persistently decreased TmP/GFR, to reversal of the clinical and biochemical abnormalities. Elemental phosphorus (as Neutra-Phos or Neutra-Phos K) is administered several times daily. The addition of 1,25(OH)2D3 runs the risk of development of nephrocalcinosis and nephrolithiasis. The goal of therapy is to improve mineralization of osteoid, and to decrease circulating levels of 1,25(OH)2D3, thereby reducing the intestinal absorption of calcium.
3.2 Tumor-Induced Osteomalacia (TIO)
TIO is a rare paraneoplastic syndrome with symptoms including chronic muscle and bone pain, weakness, and fatigue in association with a high risk of fragility fractures due to osteomalacia. The pathogenesis of TIO results from the production and secretion of the phosphaturic tumor factors (phosphatonins), which as noted above specifically inhibit Na/Pi co-transport. FGF-23, sFRP-4, FGF-7, and MEPE all have been identified in relation to this condition. FGF-23, the first to be identified in TIO by Shimada et al. (80), is normally produced in bone and is markedly elevated in the tumors and serum of patients with TIO. As a consequence of the elevated FGF-23 the renal expression of Npt2a and Npt2c is decreased and there is reduced renal reabsorption of Pi (with resultant decrease in serum Pi; Table 4). FGF-23 downregulates the conversion of 25(OH)D3 to 1,25(OH)2D3 (with ensuing rickets and osteomalacia) (80). Cure of the disease phenotype and decreases in FGF-23 serum concentrations after tumor removal in patients with TIO support the role of FGF-23 in the pathogenesis of this condition.
Treatment: Location of the tumor in TIO is often difficult and may require extensive and repeated surveys with conventional imaging techniques. Magnetic resonance skeletal survey may be improved by using magnetic resonance gradient recall echo imaging. Alternatively, sst-based molecular imaging scintigraphies have been developed, based on the possibility that these tumors are somatostatin receptor positive (sst1–sst5). Finally, the anatomical localization of the tumor may be more precise when combining PET and CT using a using a radiopharmaceutical compound coupling octreotide, a DOTA chelator, and 68gallium (81). Unless the location of the tumor has been found and its size enables surgical removal treatment usually consists of chronic oral treatment with phosphate and calcitriol. There are preliminary reports that Cinacalcet may also be of benefit to individuals with this condition (82).
3.3 Autosomal Dominant Hypophosphatemic Rickets (ADHR), X-Linked Hypophosphatemic Rickets (XLH), and Autosomal Recessive Hypophosphatemic Rickets (ARHP)
ADHR, XLH, and ARHP are characterized by hypophosphatemia, decreased renal Pi reabsorption and rickets, and osteomalacia. These disorders are inherited, and are easily distinguished from HHRH by the absence of appropriate increases in the serum concentration of 1,25(OH)2D3 in the setting of hypophosphatemia, and the absence of associated intestinal calcium reabsorption and hypercalciuria (Table 4).
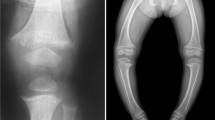
ADHR is characterized by hypophosphatemia due to isolated renal phosphate wasting. Children with ADHR present with skeletal defects including severe bowing of long bones and widening of metaphyseal regions of the bones that is most prominent at costochondral joints. Interestingly, study of a large family cohort with ADHR has revealed evidence of incomplete penetrance, variable onset (pre-pubertal vs. post-pubertal, and spontaneous recovery of renal phosphate reabsorption) (83–85).
ADHR is caused by heterozygous mutations in the gene encoding FGF-23 (86, 87). Mutations identified in ADHR are missense mutations and in each case, the mutation alters an arginine residue at either position 176 or 179. The mutations, which involve a proprotein convertase (furin) cleavage site, prevent the proteolytic processing of FGF-23 to its inactive N- and C-terminal peptides. Mutant FGF-23 proteins exhibit increased stability, and are more active than wild-type FGF-23 in vivo (88), and are likely present at elevated concentrations in ADHR patients (89). In these patients FGF-23 acts by not only suppressing the reabsorption of phosphate in the proximal tubule, but also the biosynthesis of 1,25(OH)2D3. The latter is consistent with the inappropriately low or normal levels of 1,25(OH)2D3 observed in patients with ADHR.
XLH (vitamin D resistant rickets) is the most common inherited phosphate wasting disorder with a prevalence of 1/20,000. The defective gene is on the X-chromosome, but female carriers are affected so that it is an X-linked dominant disorder. It frequently becomes manifest during late infancy when the child begins walking. The patient develops skeletal deformities that primarily include bowing of the long bones and widening of the metaphyseal region. The latter is most common at costochondral junctions (rachitic rosary). These deformities are associated with diminished growth velocity, often resulting in short stature. Later in life patients show osteomalacia, enthesopathy (calcified ligaments and teno-osseous junctions), degenerative joint disease, and continued dental disease in particular tooth decay and dental abscesses.
Early parabiosis and kidney cross-transplantation experiments showed that there was a circulating hypophosphatemic factor present in the serum of Hyp mice (the mouse homolog of human XLH) (90–92). In Hyp mice, the defect in renal phosphate reabsorption is a consequence of a decrease in BBM abundance of Npt2a and Npt2c co-transporter proteins (93). In addition, Pi regulation of the renal enzymes involved in the synthesis and catabolism of 1,25(OH)2D3 is abnormal in the mouse mutants. These findings are consistent with the action of phosphatonins.
Genetic linkage analysis has revealed inactivating mutations in PHEX, a gene located on Xp22. PHEX (PHosphate regulating gene with homology to Endopeptidases on the X chromosome) protein is expressed in various tissues, including the kidney, but is most abundant in mature osteoblasts and odontoblasts. There is significant sequence homology between PHEX and members of the M13 family of zinc metallopeptidases, which are integral membrane glycoproteins that show proteolytic activity immediately outside the cell. Since it does not appear to circulate it has been suggested that PHEX mediates inactivation by proteolytic cleavage of FGF-23. Serum FGF-23 concentrations are, in fact, elevated in about two-thirds of patients with XLH (94, 95) and in all Hyp mice. The Hyp phenotype is dependent on FGF-23 (96). The phenotype of FGF-23-null mice is indistinguishable from the phenotype of animals that are null for both PHEX and FGF-23 further supporting the fact that FGF-23 is required for the development of hypophosphatemia and is downstream from PHEX (97, 98). Hyp mice, which have been injected with inactivating antibodies to FGF-23, normalized their blood phosphorus concentrations and, furthermore, healed their rachitic changes, also supporting the conclusion that PHEX is directly or indirectly involved in the metabolism of FGF-23 (99). PHEX-dependent cleavage of FGF-23, however, has not been yet demonstrated in vivo and there is limited data in vitro supporting this possibility (48, 100). Alternatively, there is data to suggest that accelerated FGF-23 synthesis rather than decreased FGF-23 degradation may characterize this disorder (97).
ARHP: Study of the clinical and biochemical abnormalities of affected individuals with ARHP indicates a great deal of similarity to ADHR and XLH. Clinical features include rickets, skeletal deformities, dental defects, and affected individuals develop osteosclerotic bone lesions and enthesopathies later in life. Hypophosphatemia resulting from renal phosphate wasting is accompanied by normal or low 1,25(OH)2D3 levels and high alkaline phosphatase levels. PTH is normal and urinary calcium excretion is normal. FGF-23 levels appear to be either elevated or normal in ARHP, which is inappropriate to the low serum phosphorus levels. The families of these patients showed no mutations in FGF-23 (causing its resistance to degradation) or the PHEX gene that is linked to the degradative pathway of FGF-23. Subsequently, mutations in the dentin matrix protein 1 (DMP-1) were identified (101, 102). DMP-1 is widely expressed but particularly abundant in bone where it is synthesized by osteoblasts. It is involved in the regulation of transcription in undifferentiated osteoblasts. DMP-1 belongs to the SIBLING protein family, which includes osteopontin, matrix extracellular phosphoglycoprotein (MEPE), bone sialoprotein II, and dentin sialoprotein, and whose genes are clustered on chromosome 4q21. DMP-1 undergoes phosphorylation during the early phase of osteoblasts maturation and is subsequently exported into the extracellular matrix where it regulates the nucleation of hydroxyapatite.
Of the several DMP1 mutations identified, one mutation alters the translation initiation codon (MIV), two mutations are located in different intron–exon boundaries, and three are frameshift mutations within exon 6. These mutations all appear to be inactivating.
Studies using a DMP-1 knock-out mouse model showed highly elevated levels of FGF-23 in bone and serum. Moreover, DMP-1 deficient mice showed abnormal maturation of osteoblasts to osteocytes and an altered structure of bone, dentine, and cartilage, as well as hypophosphatemia and osteomalacia (102). Thus DMP1 may play a dual role in phosphate homeostasis. It may act as a negative regulator of FGF-23. In addition, as DMP-1 has a well established importance in osteoblast function, loss of DMP-1 function in osteoblasts, and extracellular matrix may also contribute to the bone abnormalities typical of ARHP.
Treatment Strategies for XLH, ADHR, ARHP: Treatment for the above hypophosphatemic conditions depends on the underlying genetic defect. Those disorders caused by genetic mutations associated with low or inappropriately normal 1,25(OH)2D3 levels (as a consequence of elevated FGF-23 that suppresses the renal 25-hydroxyvitamin D 1α-hydroxylase activity) are generally treated with oral phosphate and oral 1,25(OH)2D3. This includes XLH, ADHR, and ARHP. Elemental phosphorus (as Neutra-phos or Neutra-phos K) is administered several times daily. Since the oral phosphate leads to the development of secondary hyperparathyroidism, 1,25(OH)2D3 (available as calcitriol in capsular form or liquid) is also given. Therapy with 1,25(OH)2D3 is adjusted to avoid the development of hypercalcemia and hypercalciuria, yet to maximize the suppression of PTH synthesis and secretion. The therapeutic goal is to maintain serum calcium and PTH levels within the normal range, to improve alkaline phosphatase activity, and to prevent the development of increased calcium excretion. Phosphate therapy in some cases is limited by the development of diarrhea and abdominal discomfort. A renal ultrasound should be performed before treatment and subsequently at 1–2 year intervals. Radiographs of the knees and over the hand/wrist should be performed before treatment and subsequently at yearly intervals.
3.4 Other Genetic Disorders of Hypophosphatemia
Phosphaturia, hypophosphatemia, and rickets/osteomalacia have been reported in fibrous dysplasia (FD). The disorder is characterized by fibrous skeletal lesions and associated localized mineralization defects. FD can result in pain, fracture, and deformity in affected areas. FD is a classic feature of McCune Albright Syndrome (MAS; triad of precocious puberty, café-au-lait lesions and polyostotic bone dysplasia). FD and MAS are caused by activating mutations of GNAS1, the gene encoding the alpha-subunit of the stimulatory G protein. While somatic activating mutations in GNAS1 gene are responsible for this disease it is unclear whether or not increased cyclic AMP level causes enhanced expression of FGF-23. Recent studies confirm that FD by itself may also be associated with increased FGF-23 levels, in turn, inversely correlated with serum phosphorus, and 1,25(OH)2D3 levels. Treatment: Treatment with bisphosphonates has been shown to reduce serum FGF-23 levels, which result in a reduction of renal phosphate wasting. The mechanisms underlying the reduction of FGF-23 by bisphosphonates are unclear.
Borzani et al. (103) a number of years ago reported a patient with a condition called Hypophosphatemic Bone Disease (HBD). This disorder of phosphate metabolism previously has been described by Scriver et al. (104, 105) in a kindred with autosomal dominant inheritance. Frymoyer and Hodgkin (106) describe a similar kindred with X-linked disease. Although the condition is in some ways analogous to XLH, there are important differences between the two diseases. For example, there is selective impairment in the tubular reabsorption of phosphate in HBD but the defect is less severe and it is clearly different from that described in XLH. Clinical manifestations of HBD appear in infancy, but the dwarfism and the bone changes are less severe than in XLH at comparable concentrations of serum phosphorus in the two diseases. While in both conditions there is osteomalacia of endostal trabecular bone, only in XLH is florid rickets present, affecting the epiphyses and compromising linear growth. The phosphaturic response to PTH infusion is abnormal in qualitative aspects, but it is present in HBD, and this differs from that described in XLH. Treatment: The treatment with oral phosphates and 1,25(OH)2D3 in patients with HBD is accompanied by increases in serum phosphorus, with improved tubular reabsorption of phosphate and bone healing; this combination of responses is not present in XLH.
Linear nevus sebaceous syndrome (LNSS)/epidermal nevus syndrome (ENS) or Schimmelpenning–Feuerstein–Mims syndrome is another rare condition with hypophosphatemia that may lead to the development of rickets. Two recent case reports have described elevated FGF-23 levels in two patients. While the skin lesions appeared to be the source of the FGF-23 in one patient (107), the bone lesions were thought to secrete FGF-23 in the other patient (108).
3.5 Other Causes of Hypophosphatemic Rickets
As discussed above ADHR, ARHR, XLH, TIO, and hypophosphatemic rickets osteomalacia associated with MAS/FD are characterized by hypophosphatemia and low 1,25(OH)2D3 levels. FGF-23 levels are basically high in patients with these hypophosphatemic diseases. However, the mechanisms of excess actions of FGF-23 in these disorders are variable. Adolescent hypophosphatemic osteomalacia is similar to XLH except for the fact that with this condition patients develop the hypophosphatemic rickets at an advanced age, compared with the classic X-linked disturbance. Increased levels of biologically active FGF-23 has been observed in this group of patients (109). This is a diagnosis of exclusion and careful follow-up of the patients is required as TIO is a much more common condition and the responsible tumor is often small and difficult to locate on initial careful clinical examination. The mechanism for the elevated FGF-23 level in this condition is unclear. Treatment: Drug therapy is based on phosphate supplements together with a large dose of 1,25(OH)2D3.
3.5.1 Vitamin D-Dependent Rickets, Type I
25-Hydroxy vitamin D3-1α hydroxylase deficiency, also known as vitamin D-dependent rickets, type 1 (VDDR-1) is inherited as an autosomal recessive disorder. It is characterized by the early onset of rickets with hypocalcemia and is caused by mutations of the 25-hydroxy vitamin D3-1α hydroxylase gene. The human gene encoding the 1α-hydroxylase is located on chromosome 12q14, and comprises nine exons, and eight introns. The enzyme is specifically expressed in the proximal tubule. It results from inactivating mutations. Vitamin D is metabolized by sequential hydroxylations in the liver (25-hydroxylation) and the kidney (1α-hydroxylation). Hydroxylation of 25-hydroxyvitamin D3 is mediated by 25-hydroxy vitamin D3-1α hydroxylase in the kidney (Fig. 8). Patients usually appear normal at birth and develop muscle weakness, tetany, convulsions, and rickets starting at 2 months of age. Serum phosphorus and calcium levels are low, and PTH levels are high with low to undetectable levels of 1,25(OH)2D3. Patients with this disorder have elevated levels of 25(OH)D3 as opposed to children with deprivational rickets where 25(OH)D3 levels are reduced or absent. Treatment: Treatment with physiologic doses of 1,25(OH)2D3 results in healing of the rickets with restoration of the plasma phosphate, calcium, and PTH levels.

Schematic representation of the molecular genetic basis of three inherited forms of rickets. Vitamin D-dependent rickets type 1 (VDDR-1) is secondary to mutations in the 1α-hydroxylase gene. This gene is responsible for the 1α-hydroxylation of 25-hydroxyvitamin D3 (25(OH)D3) that occurs in the proximal renal tubule. This 1α-hydroxylation is catalyzed by 25-hydroxyvitamin D3-1α-hydroxylase (1α-hydroxylase), a mitochondrial cytochrome P450 enzyme that is subject to complex regulation by parathyroid hormone, calcium, phosphorus, and 1,25-hydroxyvitamin D3 (1,25(OH)2D3) itself. Vitamin D-dependent rickets type 2 or hereditary 1,25(OH)2D3-resistant rickets is thought to be due in many cases to a mutation in the gene for the vitamin D receptor. In contrast X-linked hypophosphatemic (XLH) rickets results from loss-of-function mutations in the PHEX gene (from Bonnardeaux and Bichet (136) (Fig. 40.5)).
3.5.2 Hereditary 1,25(OH) 2D3-Resistant Rickets (Vitamin D-Dependent Rickets, Type II)
This rare autosomal recessive disorder is similar to selective deficiency of 1,25(OH)2D3. It usually presents with rachitic changes not responsive to vitamin D treatment (with either 1,25(OH)2D3 or 1(OH)D3) with elevated circulating levels of 1,25(OH)2D3, thus differentiating it from vitamin D-dependent rickets, type 1 (Fig. 8). Alopecia of the scalp or the body is seen in approximately 50% of families with this condition. In a subset of affected families the disease has been found to be due to mutations in the vitamin D-receptor gene. In one family a nonsense mutation coding for a premature stop codon in exon 7 of the gene encoding the vitamin D receptor was identified, resulting in the absence of the ligand-binding domain (110). In other families the genetic abnormality has been a point mutation within the steroid-binding domain of the vitamin D-receptor gene (111). In another patient with type II vitamin D-resistant rickets with normal receptor function, failure of 1,25(OH)2D3 to stimulate the enzyme 25(OH)2D3-24-hydroxylase was demonstrated (112–114). The latter may represent a step in the physiological action of 1,25(OH)2D3 that is lacking in some patients with type II vitamin D-dependent rickets.
Treatment: In contrast to patients with vitamin D-dependent rickets, type I, in type II serum 1,25(OH)2D3 is elevated and the patients either respond to pharmacologic doses of 1,25(OH)2D3 or do not respond at all. Prolonged periods of therapy are usually required. Parenteral therapy with 1,25(OH)2D3 with the administration of oral or parenteral calcium is often necessary. The response to therapy likely is dependent on the exact defect.
3.6 Osteoglophonic Dysplasia (OGD)
OGD is an autosomal dominant disorder characterized by skeletal abnormalities including craniosynostosis, prominent supraorbital ridges and mild facial hypoplasia, rhizomelic dwarfism, and non-ossifying bone lesions. Affected individuals have hypophosphatemia due to renal phosphate wasting associated with inappropriately normal 1,25(OH)2D3 levels (115, 116). White et al. (117) recently identified several heterozygous missense mutations in fibroblast growth factor receptor 1 (FGFR1) that are located within or close to the receptor’s membrane spanning domain. These mutations all affect amino acid residues that are highly conserved across species and seem to lead to constitutive receptor activation (117, 118). It is thought that the skeletal lesions develop because the constitutive activation of the FGFR1 leads to an up-regulation of FGF-23 secretion in the metaphyseal growth plate. The elevated FGF-23 results in the renal phosphate wasting seen in this condition. Patients with a worse radiographic picture seem to have the most profound hypophosphatemia. Treatment: Prosthetic dental replacement has been difficult because of distorted jaw relationship and large alveolar ridges (119). Craniofacial reconstruction may be compromised by obstruction of the nasal airways, difficulty in intubation, and postoperative respiratory problems.
3.7 Post-renal Transplant Hypophosphatemia
Persistent hypophosphatemia has been noted in some patients with chronic kidney disease following renal transplantation, despite relatively modest increases in PTH levels. In these patients FGF-23 levels have been noted to be elevated, and it is possible that FGF-23 plays a role in the hypophosphatemia seen in this situation. Treatment: Generally elemental Phosphorus (as Neutra-phos or Neutra-phos K) is administered several times daily. Phosphorus supplementation may aggravate mild hyperparathyroidism, if present; if hyperparathyroidism is absent, phosphate supplementation is the recommended therapy.
4 Hyperphosphatemic Disorders
The causes of hyperphosphatemia may be classified into three groups: decreased urinary phosphate excretion, redistribution of phosphate, and exogenous administration of phosphate (Table 5). Clinical manifestations of hyperphosphatemia are seen in Table 6. Pseudohyperphosphatemia may be seen in states of paraproteinemia. Hyperlipidemia, hyperbilirubinemia, and sample dilution are much more rare causes of pseudohyperphosphatemia. Treatment: Dietary phosphate restriction and oral phosphate binders are generally used for treatment of chronic hyperphosphatemia. Chronic hyperphosphatemia is most commonly seen in chronic kidney disease (see below). Chronic hyperphosphatemia may also be seen in association with tumoral calcinosis/hyperostosis hyperphosphatemia syndrome (see below) and is treated similarly. Acute hyperphosphatemia in association with hypocalcemia requires immediate attention. Severe hyperphosphatemia as seen in patients with acutely reduced renal function, particularly in those with tumor lysis syndrome, may require hemodialysis or a continuous form of renal replacement therapy. Volume expansion also may increase urinary phosphate excretion as can the administration of diuretics such as acetazolamide. Redistribution of phosphorus from the intracellular to the extracellular space can sometimes be rapidly corrected by the administration of glucose and insulin.
4.1 Familial Tumoral Calcinosis (FTC)
Another condition for which the basis of disease has become clearer is tumoral calcinosis. Patients with this condition manifest hyperphosphatemia, mild hypercalcemia, reduced renal phosphate excretion, and elevated 1,25(OH)2D3 concentrations (120, 121). The physical chemical product of calcium × phosphorus is greater than 70 and soft tissue calcifications are present. Extraskeletal calcifications including periarticular, vascular, and other soft tissue calcium deposits are present in patients with this syndrome. Affected individuals report recurrent painful subcutaneous masses often resulting in ulceration leading to sinus tracts and infection. Masses as much as 1 kg in weight are reported. Three different types of mutations have been reported with this condition. The first type occurs in the gene encoding UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3 (GalNAc transferase 3; GALNT3; mapped to 2q24-q31). GalNAc transferase 3 is a Golgi-associated biosynthetic enzyme, which initiates mucin-type O-glycosylation of proteins. O-glycosylation of FGF-23 by GalNAc transferase 3 is essential for the secretion of intact FGF-23 because O-glycosylation at a subtilisin-like proprotein convertase recognition sequence motif prevents cleavage of FGF-23 (120). Some patients with this syndrome have low concentrations of FGF-23, but high concentrations of FGF-23 fragments. It has been thought that the fragments lack biological activity, but in vivo studies have shown that carboxyl-terminal fragments maintain their biological activity (122). This has caused uncertainty as to precisely how GalNAc transferase 3 mutations cause the syndrome.
A second gene for FTC, encoding FGF-23, has also been found. A missense mutation in the FGF-23 gene abrogates FGF-23 function by absent or extremely reduced secretion of intact FGF-23 (123, 124). A third group of mutations resulting in FTC occurs in the gene for Klotho, which encodes the co-receptor for FGF-23 (125). This results in a diminished ability of FGF-23 to signal via its cognate FGF receptors.
Treatment: Treatment of FTC has not been very successful. Apart from surgery, no modality has been shown to be efficient in managing the calcium deposition of this condition. Low phosphate diet, phosphate-binding antacids, and radiation therapy have been tried. A recent report has suggested that the phosphate-binding agent sevelamer in combination with the carbonic anhydrase inhibitor acetazolamide may be of benefit (126).
4.2 Hyperostosis Hyperphosphatemia Syndrome (HHS)
HHS is a rare metabolic disorder characterized by hyperphosphatemia, inappropriately normal or elevated 1,25(OH)2D3 and cortical hyperostosis. Pain in the long bones is associated with erythema and warmth of the overlying skin. Typical radiographic features of affected bones include cortical hyperostosis, diaphysitis, and periosteal apposition. Prior to gene identification HHS and FTC were thought to share a common pathologic mechanism based on the fact that cortical hyperostosis and ectopic calcifications co-existed in some patients (127–129). HHS is caused by mutations in the GalNAc transferase 3 (GALNT3), which encodes UDP-N-acetyl-α-d-galactosamine: polypeptide N-acetylgalactosaminyltransferase 3. These inactivating mutations and the low FGF-23 levels found in HHS are the same as that seen in FTC, providing evidence that HHS and FTC are two different phenotypic manifestations of the same disorder. The different phenotypic manifestations in these disorders are thought to result from GALNT3 mutations expressed in different environments or genetic backgrounds.
4.3 Chronic Kidney Disease
The ability of the kidneys to control Pi becomes impaired at glomerular filtration rates of approximately 50–60 mL/min. As the glomerular filtration continues to fall a number of changes occur that affect phosphorus balance, the most important being a decrease in calcitriol level due to deficient 1α-hydroxylation with consequent lower intestinal calcium absorption, hypocalcemia, and stimulation of PTH production. There is also a decrease in the filtered amount of phosphorus with resultant hyperphosphatemia, hypocalcemia, and again stimulation of PTH production. The maintenance of normal levels of Pi when the GRF is between 50 and 30 mL/min has been thought to occur at the expense of continued increase in PTH secretion. FGF-23 production, though, is increased as well. Moreover, the increase in FGF-23 levels correlate with the decline in glomerular filtration rate (130–132). The elevated PTH levels will enhance urinary clearance of phosphorus by lowering proximal tubular reabsorption, thus returning plasma levels to normal, but at the expense of the development of secondary hyperparathyroidism (the classical trade-off hypothesis) and also the higher FGF-23 level, which in itself inhibits the 1α-hydroxylation of 25(OH)D3, resulting in further lowering of calcitriol levels and more stimulation of PTH production. Whether the elevated serum FGF-23 levels found in chronic kidney disease are sufficient to correct the hyperphosphatemia of early and advanced chronic kidney disease is not clear. The normal regulatory mechanisms are unable to compensate for phosphorus retention once the glomerular filtration rate falls below approximately 50–30 mL/min. At this point there is a subtle rise in serum phosphorus. Frank hyperphosphatemia becomes manifest once the patient’s chronic kidney disease reaches the need for dialysis where the lack of significant kidney function combined with the inefficiency of the dialysis therapy in facilitating phosphorus clearance results in positive phosphate balance unless the amount of absorbed phosphorus is diminished through diet as well as the use of phosphate binders. The secondary hyperparathyroidism causes the development of osteitis fibrosa cystica, which presents radiographically as subperiosteal bone resorption. These lesions most commonly are seen in the middle phalanges of the hands, distal ends of the clavicles, and proximal ends of the tibia. The role of FGF-23 in osteitis fibrosa cystica has not been established. As renal failure advances hyperphosphatemia assumes a major role in the aggravation of secondary hyperparathyroidism. The serum levels of 1,25(OH)2D3 decrease and the intestinal absorption of calcium is low. In many of these patients with advanced renal failure, the hyperplastic parathyroid glands begin not to respond to physiologic regulation and become refractory to treatment. The parathyroid glands may become “autonomous,” which may require surgical removal of excessive parathyroid tissue.
Treatment: Strategies to lower plasma phosphorus in chronic kidney disease include dietary phosphate (dietary protein) restriction and the use of medications (phosphate binders) that inhibit intestinal absorption of phosphorus. These agents form poorly soluble complexes with phosphorus in the intestinal lumen. They are most effective when administered concomitantly with meals. Medications that inhibit the absorption of phosphorus include calcium, magnesium, iron and lanthanum salts, and sevelamer hydrochloride. Long-term use of aluminum-based binders have been associated with dementia, refractory anemia, and osteomalacia. If they are used, the duration of therapy should be limited to 2–3 months. The concomitant use of citrate compounds should be avoided as citrate enhances the intestinal absorption of aluminum. Soft tissue and vascular calcification has been found to be associated with a higher serum calcium level and higher calcium intake. The concurrent administration of vitamin D sterols further increases this risk. This has limited the use of calcium-containing binders to 1500–2000 mg/day from both dietary and medication sources (133).
4.4 Case Scenario 1
A 14-month-old male infant presents to his primary care physician’s office with failure to thrive. The mother was noted to have short stature. On physical exam of the infant bowing of the lower extremities was noted. Length was 68 cm (<5th percentile for age). Weight was 9.0 kg (12th percentile for age). Initially, a chemistry panel was obtained which was significant for a serum potassium of 4.0 mmol/L, total CO2 of 24 mmol/L, serum calcium of 9.5 mg/dL, serum phosphorus of 2.6 mg/dL, and alkaline phosphatase of 806 IU/L. Because of the low serum phosphorus and elevated alkaline phosphatase his PCP was prompted to send additional laboratory studies including a PTH, spot urine for phosphate, spot urine for calcium, 25-hydroxyvitamin D, and 1,25-dihydroxyvitamin D. The PTH level was 80 pg/mL (normal range 12–88 pg/mL), 1,25-dihydroxyvitamin D 60 pg/mL (normal range 27–71 pg/mL), and 25-hydroxyvitamin D 26 ng/mL (normal range 13–67 ng/mL). The total reabsorption of phosphate was 45.5% (normal ~80–97%) and the urine calcium creatinine ratio 0.5 (normal range for age <0.6). An x-ray of the long bones confirmed rachitic changes
Autosomal recessive hypophosphatemic rickets (ARHR), X-linked hypophosphatemic rickets (XLH), autosomal dominant hypophosphatemic rickets (ADHR), hereditary hypophosphatemic rickets with hypercalciuria (HHRH), and tumor-induced osteomalacia (TIO) share many of the biochemical features noted in Table 4. In this patient the normal PTH level and absence of hypercalciuria excludes a diagnosis of HHRH. The family history excludes ARHR as this condition is autosomal recessive. The laboratory findings, though, do not distinguish between XLH, ADHR, and TIO. FGF-23 is the circulating phosphaturic factor in XLH, ADHR, and TIO. In XLH the circulating level of FGF-23 is thought to be determined by the rate of its proteolytic cleavage by PHEX protease, while in ADHR there appears to be a gain in function mutation in FGF-23 and in TIO an overproduction of FGF-23. As TIO is an acquired form of hypophosphatemic rickets caused by a variety of benign mesenchymal tumors that secrete FGF-23, this diagnosis seems quite unlikely in this scenario. FGF-23 levels do not help in distinguishing between XLH and ADHR even if they were available. Further family history will help distinguish between the two. As it stands there is insufficient history above, as XLH is an X-linked dominant condition. Sequence analysis for the entire coding region for both of these conditions, though, is available since mutations in these conditions have been identified and will differentiate between the two when the family history is not sufficient to do so.
The treatment for both involves a combination of oral phosphorus and 1,25(OH)2D3. The daily need for phosphorus supplementation is 1–3 g of elemental phosphorus divided into four to five doses. Frequent dosing helps maintain the serum phosphorus level throughout the day, but also decreases the incidence of diarrhea. Calcitriol is administered at a dose of 30–70 ng/kg/day divided into two doses. Complications of treatment occur when there is not an adequate balance between phosphorus supplementation and calcitriol. Phosphorus excess, by decreasing enteral calcium absorption, may lead to secondary hyperparathyroidism and a worsening of the bone disease. Excess calcitriol leads to hypercalciuria and nephrocalcinosis and may even result in hypercalcemia. Laboratory monitoring as well as periodic renal ultrasounds to check for nephrocalcinosis of treatment is essential. Normalization of alkaline phosphatase is a more useful way to monitor therapeutic response than serum phosphorus. For children with significant short stature, growth hormone is an effective treatment option.
4.5 Case Scenario 2
A 17-7/12-year-old young lady presents with blurry vision. She was taken to an ophthalmologist and retina specialist who found her to have cotton wool exudates and ischemia of the retina. Her physical examination otherwise was unremarkable with the exception of her blood pressure, which was 177/121 mmHg. She was admitted to the hospital for management of the blood pressure and further evaluation. Blood pressure was brought under control with Procardia and Atenolol.
Her laboratory evaluation revealed the following: serum Na 139 mmol/L, K 3.8 mmol/L, chloride 109 mmol/L, CO2 19 mmol/L, glucose 102 mg/dL, BUN 61 mg/dL, creatinine 4.9 mg/dL, total protein 6.2 g/dL, albumin 3.7 g/dL, calcium 8.4 mg/dL, and phosphorus 5.7 mg/dL. Spot urine: protein 53, creatinine 27. UAs were 30–100 mg/dL for protein. Based on her serum creatinine the estimated creatinine clearance was 18 mL/min/1.73 m2. By renal US the kidneys were small (<5th percentile for age). A PTH was subsequently obtained that was 275 pg/mL (normal range 12–88 pg/mL).
The fact that the kidneys are small for age indicates that this patient has chronic kidney disease. Her kidney disease appears to be far advanced in view of the low creatinine clearance. As would be anticipated normal renal regulatory mechanisms are unable to compensate, and she has started to develop phosphorus retention. Phosphorus retention and hyperphosphatemia develop in virtually all patients with advanced chronic kidney disease. In this aged patient current recommendations are that the phosphorus be kept between 3.5 and 5.5 mg/dL. Hyperphosphatemia leads to secondary hypocalcemia as seen here by causing calcium precipitation, by decreasing the production of 1,25(OH)2D3, and by decreasing intestinal calcium absorption. The hypocalcemia has resulted in the development of secondary hyperparathyroidism. The calcium phosphorus product in this patient is 48, fortunately, making the risk for ectopic calcifications low.
Treatment of the hyperphosphatemia in this case involves a reduction in dietary protein and introduction of phosphate binders. Calcium carbonate and calcium acetate are both used widely for this purpose. Sevelamer is a synthetic polymer that binds phosphorus within the lumen of the gastrointestinal tract and reduces its absorption as well. Small doses of calcitriol are useful for enhancing intestinal calcium absorption with correction of the hypocalcemia. Calcitriol therapy lowers plasma PTH levels in patients who already have biochemical evidence of secondary hyperparathyroidism. Calcium concentrations need to be measured regularly to avoid the complication of hypercalcemia.
References
Forbes GB (1987) Body Composition. Springer, Berlin, Heidelberg, New York, pp. 170–182.
Hohenhauer L, Rosenberg TS, Oh W (1970) Calcium and phosphorus homeostasis on the first day of life. Biol Neonate, 15:49–56.
Nordin BCE (1976) Calcium, phosphate and magnesium metabolism. Churchill Livingstone, New York, p. 78.
Harris CA, Baer PG, Chirito E, Dirks JH (1974) Composition of mammalian glomerular filtrate. Am J Physiol, 227:972–976.
Markowitz M, Rotkin L, Rosen JF (1981) Circadian rhythms of blood minerals in humans. Science, 213:672–674.
Portale AA, Hallorhan BP, Morris RC Jr. (1987) Dietary intake of phosphorus modulates the circadian rhythm in serum concentrations of phosphorus: implications for the renal production of 1,25-dihydroxyvitamin D. 80:1147–1154.
Brodehl J, Gellissen K, Weber HP (1982) Postnatal development of tubular phosphate reabsorption. Clin Nephrol, 17:163–171.
Arnaud SB, Goldsmith RS, Stickler GB, et al. (1973) Serum parathyroid hormone and blood minerals; interrelationships in normal children. Pediatr Res, 7:485–493.
Greenberg BG, Winters RW, Graham JB (1960) The normal range of serum inorganic phosphorus and its utility as discriminant in the diagnosis of congenital hypophosphatemia. J Clin Endocrinol, 20:364–379.
Keating FR Jr, Jones JD, Elveback LR, et al. (1969) The relation of age and sex to distribution of values in healthy adults of serum calcium, inorganic phosphorus, magnesium, alkaline phosphatase, total proteins, albumin and blood urea. J Lab Clin Med, 73:825–834.
Mount DB, Yu ASL (2008) Transport of inorganic solutes: sodium, chloride, potassium, magnesium, calcium and phosphate. In: Brenner BM Levine SA (eds.) Brenner and Rector’s The Kidney. Philadelphia. Saunders Elsevier, pp. 156–213.
Straum BB, Hamburger RJ, Goldberg M (1972) Tracer microinjection study of renal tubular phosphate reabsorption in the rat. J Clin Invest, 51:2271–2276.
Strickler JC, Thompson DD, Klose RM, Giebisch G (1964) Micropuncture study of inorganic phosphate excretion in the rat. J Clin Invest, 43:1596–1607.
Dennis VW, Woodhall PB, Robinson RR (1976) Characteristics of phosphate transport in isolated proximal tubule. Am J Physiol, 231:979–985.
Pastoriza-Munoz E, Colindres RE, Lassiter WE, Lechene C (1978) Effect of parathyroid hormone on phosphate reabsorption in rat distal convolution. Am J Physiol, 235:F321–F330.
Lang F, Greger R, Marchand GR, Knox FG (1977) Stationary microperfusion study of phosphate reabsorption in proximal and distal nephrons segments. Pflugers Arch, 368:45–48.
De Rouffignac C, Morel F, Moss N, Roinel N (1973) Micropuncture study of water and electrolyte movements along the loop of Henle in psammomys with special reference to magnesium, calcium and phosphorus. Pflugers Arch, 344:309–326.
Dennis VW, Bello-Reuss E, Robinson RR (1977) Response of phosphate transport to parathyroid hormone in segments of rabbit nephrons. Am J Physiol, 233:F29–F38.
Shareghi GA, Agus ZS (1982) Phosphate transport in the light segment of the rabbit cortical collecting duct. Am J Physiol, 242:F379–F384.
Peraino RA, Suki WN (1980) Phosphate transport by isolated rabbit cortical collecting tubule. Am J Physiol, 238:F358–F362.
Murer H, Hernando N, Biber J (2000) Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev, 80: 1373–1409.
Murer H, Forster I, Biber J (2004) The sodium-phosphate cotransporters family SLC34. Pflugers Arch, 447:763–767.
Collins JF, Bai I, Ghishan FK (2004) The SLC20 family of proteins: dual functions as sodium phosphate cotransporters and viral receptors. Pfleugers Arch, 447:647–652.
Yabuuchi H, Tamai I, Morita K, Kouda T, Miyamoto K, Takeda E, Tsuji A (1998) Hepatic sinusoidal membrane transport of anionic drugs mediated by anion transporter Npt1. J Pharmacol Exp, 286:1391–1396.
Jutabha P, Kanai Y, Hosoyamada M, Chairoungdua A, Kim DK, Iribe Y, Babu E, Kim JY, Anzai N, Chatsudthipong V, Endou H (2003) Identification of a novel voltage-driven organic anion transporter present at apical membrane of renal proximal tubule. J Biol Chem, 278:27930–27938.
Custer M, Lotscher M, Biber J, Murer H, Kaissling B (1994) Expression of Na–Pi cotransport in rat kidney: localization by RT-PCR and immunochemistry. Am J Physiol, 266:F767–F774.
Segawa H, Kaneko I, Takahashi A, Kuwahata M, Ito M, OhkidoI, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi cotransporters. J Biol Chem, 277:19665–19672.
Hilfiker H, Hattenhauer O, Traebert M, Forster I, Murer H, Biber J (1998) Characterization of a murine type II sodium-phosphate cotransporters expressed in mammalian intestine. Proc Natl Acad Sci USA, 95:14564–14569.
Hattenhauer O, Traebert M, Murer H, Biber J (1999) Regulation of small intestinal Na–P(i) type IIb cotransporters by dietary phosphate intake. Am J Physiol Gastrointest Liver Physiol, 277:G756–G762.
Xu H, Bau I, Collins JF, Ghishan FK (2002) Age dependent regulation of rat intestinal type IIb sodium phosphate cotransporters by 1,25(OH)2 vitamin D3. Am J Physiol Cell Physiol, 282:C487–C493.
Katai K, Miyamoto K, Kishida S, Segawa H, Nii T, Tanaka H, Tanai Y, Arai H, Tatsumi S, Morita K, Taketani Y, Takeda E (1999) Regulation of intestinal Na+-dependent phosphate cotransporters by a low-phosphate diet and 1,25-dihydroxyvitamin D3. Biochem J, 343 (Pt3): 705–712.
Levine BS, Ho LD, Pasiecznik K, et al. (1986) Renal adaptation to phosphorus deprivation; characteristics of early events. J Bone Minerl Res, 1: 33–40.
Haas JA, Berndt T, Knox FG (1978) Nephron heterogeneity of phosphate reabsorption. Am J Physiol, 234:F287–F290.
Taketani Y, Segawa H, Chikamori M, et al. (1998) Regulation of type II renal Na+-dependent inorganic phosphate transporters by 1,25-dihydroxyvitamin D3- identification of a vitamin D- responsive element in the human NAPI-3 gene. J Biol Chem, 273: 14575–14581.
Hammerman MR, Karl IE, Hruska KA (1980) Regulation of canine renal vesicle Pi transport by growth hormone and parathyroid hormone. Biochem Biophys Acta, 603: 322–355.
Mulroney SE, Lumpkin MD, Haramati A (1989) Antagonist to GH-releasing factor inhibits growth and renal Pi reabsorption in immature rats. Am J Physiol, 257:F29–F34.
Woda CB, Halaihel N, Wilson PV, Haramati A, Levi M, Mulroney SE (2004) Regulation of renal NaPi-2 expression and tubular phosphate reabsorption by growth hormone in the juvenile rat. Am J Physiol Renal Physiol, 287:F117–F123.
Cai Q, Hodgson SF, Kao PC, Lennon VA, Klee GG, et al. (1994) Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med, 330:1645–1649.
Berndt TJ, Schiavi S, Kumar R (2005) “Phosphatonins” and the regulation of phosphorus homeostasis. Am J Physiol Renal Physiol, 289:F1170–F1182.
ADHR Consortium (2000) Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF23. Nat Genet, 26:345–348.
Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, et al. (2000) MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics, 67:54–68.
Rowe PS, Kumagai Y, Gutierrez G, Garrett IR, Blacher R et al. (2004) MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone, 34:303–319.
Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, et al. (2003) Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest, 112:785–794.
De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, et al. (2002) Tumors associated with oncogenic osteomalacia express gene important in bone and mineral metabolism. J Bone Miner Res, 17:1102–1110.
Carpenter TO, Ellis BK, Insogna KI, Philbrick WM, Sterpka J, Shimkets R (2005) FGF7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab, 90: 1012–1020.
Schiavi SC, Kumar R (2004) The phosphatonin pathway; new insights in phosphate homeostasis. Kidney Int, 65: 1–14.
Schiavi SC, Moe OW (2002) Phosphatonins: a new class of phosphate regulating proteins. Curr Opin Nephrol Hypertens, 11: 423–430.
Bowe AE, Finnegan R, de Beur SM, Cho J, Levine MA, Kumar R (2001) FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun, 284: 977–981.
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2001) Cloning and characterization of FGF-23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA, 98:6500–6505.
Perwad F, Azam N, Zhang MY, Yamashita T, Tenenhouse HS, Portale AA (2005) Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology, 146:5358–5364.
Saito H, Maeda A, Ohtomo S, Hirata M, Kusano K, et al. (2005) Circulating FGF-23 is regulated by 1α,25-dihydoxyvitamin D3 and phosphorus in vivo. J Biol Chem, 280:2543–2549.
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum M, Schiavi S, Hu MC, Moe OW, Kuro-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem, 281:6120–6123.
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature, 444:770–774.
Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima, Y (1998) Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett, 424:6–10.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Obyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI (1997) Mutation of mouse klotho gene leads to syndrome resembling ageing. Nature, 390:45–51.
Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, Nakamura K, Iida A, Anazawa H, Koh N, Iwana A, Imura A, Fujimori T, Kur-o M, Hanai N, Takeshige K, Nabeshima Y, (2000) Establishment of anti-klotho monoclonal antibodies and detection of Klotho protein in kidneys. Bichem Biophy Res Commun, 267:597–602.
Torres PU, Prie D, Molina-Bletry V, Beck L, Silve C, Friedlander G (2007) Klotho: an antiaging protein involved in mineral and vitamin D metabolism. Kidney Int, 71:730–737.
Spitzer A, Barac-Nieto M (2001) Ontogeny of renal phosphate transport and the process of growth. Pediatr Nephrol, 16:763–771.
Russo JC, Nash MA (1980) Renal response to alterations in dietary phosphate in the young beagle. Biol Neonate, 38:1–10.
Caverzasio J, Bonjour J-P, Fleisch J (1982) Tubular handling of Pi in young, growing, and adult rates. Am J Physiol, 242:F705–F710.
Johnson V, Spitzer A (1986) Renal reabsorption of Pi during development: whole kidney events. Am J Physiol, 251:F251–F256.
Kaskel FJ, Kumar AM, Feld LG, Spitzer A (1988) Renal reabsorption of phosphate during development: tubular events. Pediatr Nephrol, 2:129–134.
Woda C, Mulroney SE, Halaihel N, Sun L, Wilson PV, Levi M, Haramati A (2001) Renal tubular sites of increased phosphate transport and NaPi-2 expression in the juvenile rat. Am J Physiol Regulatory Integrative Comp Physiol, 280:R1524–R1533.
Neiberger RE, Barac-Nieto M Spitzer A (1989) Renal reabsorption of phosphate during development: transport kinetics in BBMV. Am J Physiol, 257:F268–F274.
Bonjour JP, Fleish H (1980) Tubular adaptation to the supply and requirement of phosphate. In: Massry SG, Fleisch H (eds.) Renal Handling of Phosphate. Plenum Press, New York, pp. 243–264.
Barac-Nieto M, Dowd TL, Gupta RK, Spitzer A (1991) Changes in NMR visible kidney cell phosphate with age and diet: relationship to phosphate transport. Am J Physiol, 261:F153–F162.
Segawa H, Kaneko I, Takahasgi A, Kuwahata M, Ito M, Ohkido I, Tatsumi S, Miyamoto K (2002) Growth-related renal type II Na/Pi Cotransporter. J Biol Chem, 277:19665–19672.
Ohkido I, Segawa H, Yanagida R, Nakamura M, Miyamoto K (2003) Cloning, gene structure and dietary regulation of the type-IIc Na Pi cotransporters in the mouse kidney. Pflugers Archiv, 446:106–115.
Silverstein DM, Barac-Nieto M, Murer H, Spitzer A (1997) A putative growth-related renal Na+–Pi cotransporters. Am J Physiol Regul Integr Comp Physiol, 273:R928–R933.
Linarelli LG (1972) Nephron urinary cyclic AMP levels and developmental renal responsiveness to parathyroid hormone. Pediatrics, 50:14–32.
Woda C, Mulroney SE, Levi M, Halaihel N, Haramati A (1999) Nephron sites of phosphate (Pi) reabsorption in the juvenile rat (Abstract). J Am Soc Nephrol, 10:614A–615A.
Moe SM, Sprague SM (2008) Mineral bone disorders in chronic kidney disease. In: Brenner BM Levine SA (eds.) Brenner and Rector’s The Kidney. Saunders, Elsevier, Philadelphia, pp. 1784–1813.
Bijvoet OLM (1969) Relation of plasma phosphate concentration to renal tubular reabsorption of phosphate. Clin Sci, 37:23–36.
Walton RJ, Bijvoet OLM (1975) Nomogram for the derivation of renal tubular threshold phosphate concentration. Lancet, 2:309–310.
McCrory WW, Forman CW, McNamara H, Barnett HL (1952) Renal excretion of inorganic phosphate in newborn infants. J Clin Invest, 31:357–366.
Arant BS (1978) Developmental patterns of renal functional maturation compared in the human neonate. J Pediatr, 92:705–712.
Brodehl J, Krause A, Hoyer PF (1988) Assessment of maximal tubular phosphate reabsorption: comparison of direst measurement with the nomogram of Bijvoet. Pediatr Nephrol, 2: 183–189.
Stark H, Eisenstein B, Tieder M, Rachmel A, Alpert, G. (1986) Direct measurement of TP/GFR: a simple and reliable parameter of renal phosphate handling. Nephron, 44:125–128.
Gupta A, Tenenhouse HS, Hoag HM, Wang D, Khadeer MA, Namba N, Feng X, Hruska KA (2001) Identification of the type II Na+–Pi cotransporters (Npt2) in the osteoclast and the skeletal phenotype of Npt2-/-. Bone, 29:467–476.
Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, et al. (2001) Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA, 98:6500–6505.
Hesse E, Moessinger E, Rosenthal H, et al. (2007) Oncogenic osteomalacia: exact tumor localization by co-registration of position emission and computed tomography. J Bone Miner Res, 22:158–162.
Geller JL, Khosravi A, Kelly MH, Riminucci M, Adams JS, Collins MT (2007) Cinacalcet in the management of tumor-induced osteomalacia. J Bone Miner Res, 22:931–937.
Econs M, McEnery P (1997) Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate wasting disorder. J Clin Endocrinol Metab, 82:674–681.
Econs M, McEnery P, Lennon F, Speer M (1997) Autosomal dominant hypophosphatemic rickets is linked to chromosome 12p13. J Clin Invest, 100: 2653–2657.
Imel EA, Hui SL, Econs MJ (2007) FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Mineral Res, 22:520–526.
ADHR Consortium T, White KE, Evans WE, O’Riordan JLH, Speer MC, Econs MJ, et al. (2000) Autosomal dominant hypophosphatemic rickets is associated with mutations in FGF23. Nat Genet, 26:345–348.
Kruse K, Woelfel D, Strom T (2001) Loss of renal phosphate wasting in a child with autosomal dominant hypophosphatemic rickets caused by a FGF23 mutation. Horm Res, 55:305–308.
White KE, Carn G, Lorenz-Depieteux B, Benet-Pages A, Strom TM, Econs MJ (2001) Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int, 60:2079–2086.
Imel EA, Econs MJ (2005) Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol, 16:2565–2575.
Meyer RA Jr, Meyer MH, Gray RW (1989) Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res, 4:493–500.
Meyer RA Jr, Tenenhouse HS, Meyer MH, Klugerman AH (1989) The renal phosphate transport defect in normal mice parabiosed to X-linked hypophosphatemic mice persists after parathyroidectomy. J Bone Miner Res, 4:523–532.
Nesbitt T, Coffman TM, Griffiths R, Drezner MK (1992) Cross-transplantation of kidneys in normal and Hyp mice. Evidence that the Hyp mouse phenotype is unrelated to an intrinsic renal defect. J Clin Invest, 89:1453–1459.
Tenenhouse HS, Martel J, Gauthier C, Segawa H, Miyamoto K (2003) Differential effects of Npt2a gene ablation and the X-linked Hyp mouse mutation on renal expression of type IIc Na/Pi cotransporters. Am J Physiol, 285:F1271–F1278.
Jonsson K, Zahradnik R, Larsson T, White K, Sugimoto T, Imanishi Y, et al. (2003) Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med, 348:1656–1662.
Weber T, Liu S, Indridason O, Quarles L (2003) Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res, 18:1227–1234.
Liu S, Brown T, Zhou J, Xiao Z, Awad H, Guilak F, et al. (2005) Role of matrix extracellular phosphoglycoprotein in the pathogenesis of X-linked hypophosphatemia. J Am Soc Nephrol, 16:91645–91653.
Sitara D, Razzaque MS, Hesse M, Yoganathan S, Taguchi T, Erben RG, et al. (2004) Homozygous ablation of fibroblast growth factor-23 results in hypophosphatemia and impaired skeletogenesis and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol, 23:421–432.
Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD, (2006) Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab, 291:E38–E49.
Aono Y, Shimada T, Yamazaki Y, Hino R, Takeuchi M, Fujita T, et al. (2003) The neutralization of FGF-23 ameliorates hypophosphatemia and rickets in Hyp mice. Meeting of the American Society for Bone and Mineral research. Minneapolis, Minnesota, p. 1056.
Benet-Pages A, Lorenz-Depiereux B, Zischka H, White K Econs M, Strom T (2004) FGF23 is processed by proprotein convertases but not by PHEX. Bone, 35:455–462.
Lorenz-Depiereux B, Bastepe M, Benet-Pages A, Amyere M, Wagenstaller J, Muller-Barth U, Badenhoop K, Kaiser SM, Rittmaster RS, Shlossberg AH, Oliveras JL, Loris C, Ramos FJ, Glorieux F, Vikkula M, Juppner H, Strom TM (2006) DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet, 38:1248–1250.
Feng JQ, Ward LM, Liu S, Lu Y Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE (2006) Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet, 38:1310–1315.
Borzani M, Mauri I, Roggero P, Auriti I Facchini R (1983) Non-rachitic hypophosphatemic osteopathy. Pediatr Med Chir, 5:99–102.
Scriver CR, MacDonald W, Reade T, et al. (1977) Hypophosphatemic nonrachitic bone disease; an entity distinct from X-linked hypophosphatemia in the renal defect, bone involvement, and inheritance. Am J Med Genet, 1:101–117.
Scriver CR, Reade T, Halal F, et al. (1981) Autosomal hypophosphatemic bone disease responds to 1,25(OH)2D3. Arch Dis Child, 56:203–207.
Frymoyer JW, Hodgkin W (1977) Adult-onset vitamin D-resistant hypophosphatemic osteomalacia. J Bone Joint Surg, 59: 101–106.
Hoffman W, Juppner H, Deyung B, O’dorisio M, Given K (2005) Elevated fibroblast growth factor-23 in hypophosphatemic linear nevus sebaceous syndrome. Am J Genet, 134: 233–236.
Heike C, Cunningham M, Steiner R, Wenkert D, Hornung R, Gruss J, et al. (2005) Skeletal changes in epidermal nevus syndrome: does focal bone disease harbor clues concerning pathogenesis? Am J Med Genet, 139:67–77.
Hoshino C, Satoh, N, Sugawara S, Kurivama C, Kikuchi A, Ohta M (2008) Sporadic adult-onset hypophosphatemic osteomalacia caused by excessive action of fibroblast growth factor 23. Intern Med, 47:453–457.
Cockerill FJ, Hawa NS, Yousaf N, et al. (1997) Mutations in the vitamin D receptor gene in three kindreds associated with hereditary vitamin D resistant rickets. J Clin Endocrinol Metab, 82:3156–3160.
Malloy, PJ, Hochberg Z, Tiosano D, et al. (1990) The molecular basis of hereditary 1,25(OH)2 vitamin D3 resistant rickets in seven families. J Clin Invest, 86:2071–2079.
Griffin E, Zerwekh JE (1983) Impaired stimulation of 25(OH) vitamin D-24-hydroxylase in fibroblasts from a patient with vitamin D-dependent rickets type II: form of receptor positive resistance of 1,25-dihydroxyvitamin D3. J Clin Invest, 72:1190–1199.
Haussler MR, Haussler CA, Jurutka PWL (1997) The vitamin D hormone and its nuclear receptor, molecular action and disease states. J Endocrinol, 154 Suppl:557–573.
Silver J, Popovtzer MM (1984) Hypercalcemia with elevated dihydroxycholecalciferol levels and hypercalciuria. Arch Intern Med, 144:162–163.
Beighton P, Cremin BJ, Kozlowski K (1980) Osteoglophonic dwarfism. Pediatr Radiol, 10:46–50.
Beighton P (1989) Osteoglophonic dysplasia. J med Genet, 26:572–576.
White K, Cabral J, Evans W, Ichikawa S, Davis S, Ornitz D, et al. (2003) A missense mutation in FGFR1 causes a novel syndrome: craniofacial dysplasia with hypophosphatemia (CFDH). J Bone Miner Res, 18 Suppl 2:S4.
Farrow E, Davis S, Mooney S, Beighton P, Mascarenhas L, Gutierrez Y, et al. (2006) Extended mutational analysis of FGFR1 in osteoglophonic dysplasia. Am J Med Genet, 140:537–539.
Roberts TS, Stephen L Beighton P (2006) Osteoglophonic dysplasia; dental and orthodontic implications. Orthod Craniofac Res, 9:153–156.
Topaz O, Shurman DL, Bergman R, Indelman M, Ratajczak P, Mizrachi M, Khamaysi Z, Behar D, Petronius D, Friedman V, Zelikovic I, Raimer S, Metzker A, Richard G, Sprecher E (2004) Mutations in GALNT3, encoding a protein involved in o-linked glycosylation, cause of familial tumoral calcinosis. Nat Genet, 36:579–581.
Ichikawa S, Lyles K, Econs M (2005) A novel GALNT3 mutations in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab, 90:2420–2423.
Berndt TJ, Craig TA, McCormick DJ, Lanske B, Sitara D, Razzaque MS, Pragnell M, Bowe AE, O’Brien SP, Schiavi SC, Kumar R (2007) Biological activity of FGF-23 fragments. Pflugers Arch, 454:615–623.
Araya K, Fukomoto S, Backenroth R, Takeuchi Y, Nakayama K, Ito N, Yoshii N, Yamazaki Y, Yamashita T, Silver J, Igarashi T, Fujita T (2005) A novel mutation in fibroblast growth factor (FGF)23 gene as a cause of tumoral calcinosis. J Endocrinol Metab, 90:5523–5527.
Larsson T, Davis SI, Garringer HJ, Mooney SD, Dramann MS, Cullen MJ, White KE (2005) Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differently processed. Endocrinology, 146:3883–3891.
Ichikawa S, Imel EA, Kreiter ML, Yu X, Mackenzie DS, Sorenson AH, Goetz R, Mohammadi M, White KE, Econs MJ (2007) A homozygous missence mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest, 117:2684–2691.
Garringer HJ, Fisher C, Larsson TE, et al. (2006) The role of mutant UDP-N-acetyl-alpha-d-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab, 91:4037–4042.
Narchi H (1997) Hyperostosis with hyperphosphatemia: evidence of familial occurrence and association of tumoral calcinosis. Pediatrics, 99:745–748.
Clarke E, Swischuk LE, Hayden CK Jr (1984) Tumoral calcinosis, diaphysitis and hyperphosphatemia. Radiology, 151:643–646.
Wilson MP, Lindsley CB, Warady BA, Johnson JA (1989) Hyperphosphatemia associated with cortical hyperostosis and tumoral calcinosis. J Pediatr, 114:1010–1013.
Pande S, Ritter CS, Rothstein M, Wiesen K, Vassiliadis J, Kumar R, Schiavi SC, Slatapolskt E, Brown AJ (2006) FGF-23 and sFRP-4 in chronic kidney disease and post-renal transplantation. Nephron Physiol, 104:23–32.
Imanishi Y, Inaba M, Nakatsuka, Nagasue K, Okuno S, Yoshihara A, Miura M, Miyauchi A, Kobayashi K, Miki T, Shoji T, Ishimura E, Nishizawa Y (2004) FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int, 65:1943–1946.
Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int, 64: 2272–2279.
Block GA, Port FK (2000) Re-evaluation of risks associated with hyperphosphatemia and hyperparathyroidism in dialysis patients: recommendations for a change in management. Am J Kidney Dis, 35:1226–1237.
Wagner CA (2007) Novel insights into the regulation of systemic phosphate homeostasis and renal phosphate excretion. J Nephrol, 20:130–134.
Fukumoto S (2008) Physiological regulation and disorders of phosphate metabolism-pivotal role of fibroblast growth factor 23. Inter Med, 47:337–343.
Bonnardeaux A, Bichet DG (2008) Inherited disorders of the renal tubule. In: Brenner BM LevineSA (eds.) Brenner and Rector’s The Kidney. Saunders Elsevier. Philadelphia, pp. 1390–1427.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Humana Press, a part of Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Johnson, V.L. (2010). Disorders of Phosphorus Homeostasis. In: Feld, L., Kaskel, F. (eds) Fluid and Electrolytes in Pediatrics. Nutrition and Health. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60327-225-4_6
Download citation
DOI: https://doi.org/10.1007/978-1-60327-225-4_6
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-60327-224-7
Online ISBN: 978-1-60327-225-4
eBook Packages: MedicineMedicine (R0)