
Abstract
Etiology, biology, response to treatment, and outcome greatly differ between adult and childhood cancers. Soft tissue sarcoma encompasses a heterogeneous group of pediatric sarcomas characterized by a high capacity to invade neighboring tissues. Although in the last years the overall survival in childhood cancers has improved to over 70% for the nonmetastatic forms, subgroups of young patients with metastatic and aggressive disease still show a poor outcome. Moreover, survivors often suffer from long-term morbidity due to the effects of therapy. It is widely accepted that soft tissue sarcomas of childhood develop from mesenchymal progenitor cells affected by chromosomal aberrations and mutations in genetic and epigenetic pathways during development. Therefore, pathways driving tissue differentiation are particularly relevant. Among these, the Notch signaling pathway plays one of the major roles. Notch signaling is evolutionarily conserved among species, working as a cell-to-cell communication system strictly defining cell fate, stem cell renewal, and tissue homeostasis during embryo development and in postnatal life. In the present chapter, we describe recent insights on Notch deregulation in the most prominent pediatric soft tissue sarcomas: rhabdomyosarcomas, Ewing sarcomas, and synovial sarcomas. We also summarize the challenges and opportunities in inhibiting Notch signaling for the treatment of this group of tumors.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Notch signaling
- Notch receptors
- Gamma-secretase
- Soft tissue sarcoma
- Rhabdomyosarcoma
- Ewing sarcoma
- Synovial sarcoma
11.1 Introduction
11.1.1 Childhood Versus Adult Cancers
Conversely to adult tumors, whose pathogenesis is related to environment-/age-dependent genetic and epigenetic alterations, pediatric cancers originate from progenitor cells in which developmental pathways governing embryonic life are deregulated. In line with this, tumors of childhood often contain a clonal population of presumably tumor-initiating cells expressing fusion products of genes that guide tissue development.
Increasing knowledge of the landscape of molecular networks involving genetic and epigenetic mechanisms acting in childhood cancers have opened the way to the discovery of novel potential approaches to treat the disease.
Crucial developmental pathways involved in pediatric tumor biology are Sonic Hedgehog (SHH), Wingless (WNT), and Notch signaling. These pathways are fundamental for proper cell differentiation and tissue lineage commitment of progenitor cells and, more importantly, cooperate and cross talk each other (reviewed in [1,2,3,4,5,6]). Considering the crucial role of Notch signaling in developmental processes, it is not surprising that it has been found affected in several diseases ([7,8,9,10,11,12,13,14,15] and reviewed in [16]).
An oncogenic role of Notch signaling has been highlighted for the first time in pediatric acute T-cell leukemia (T-ALL). Indeed, two groups demonstrated that (i) mutations of the Notch1 receptor resulted in the constitutive production of an activated form of Notch1, i.e., the Notch1 intracellular domain, in patients with T-ALL [17], and that (ii) this Notch1 constitutive activation is sufficient for tumorigenesis [18]: an observation confirmed later also in adult cancers [19]. In the last few years, the deregulation of Notch signaling has been shown to be involved in several types of pediatric solid tumors. Recently, we and others have shown Notch signaling abnormalities are pathogenetic events in pediatric soft tissue sarcomas, a heterogeneous group of solid tumors affecting mainly soft tissue and bone of young patients.
As for adult cancers, where several clinical trials with Notch signaling inhibitors are being evaluated, the modulation of the Notch signaling is under preclinical study as an anticancer strategy in this type of pediatric tumors.
11.1.2 Pediatric Soft Tissue Sarcomas
Pediatric soft tissue sarcomas include a group of tumors derived from the mesenchymal compartment that are highly heterogeneous in terms of clinical behavior and genomic alterations [20].
Collectively, they represent about 8–10% of all childhood tumors and about 15% of tumors outside the central nervous system [21]. Multimodal approach with chemotherapy and surgery is the usual treatment of pediatric soft tissue sarcoma, while radiation is rarely used in young children due to its side effects on a growing organism [22]. Advances in treatments have improved the overall survival in all childhood cancers to over 70% today. However, although the prognosis of soft tissue sarcoma has improved considerably, a group of patients still shows a dismal prognosis. Indeed, metastatic forms and subsets of tumors harboring specific oncogenic mutations/chromosomal translocations are often incurable. Additionally, young survivors often suffer from long-term side effects linked to therapy. An additional clinical challenge to eradicate soft tissue sarcomas is due to the high ability of tumor cells to invade the neighboring tissues [22].
Therefore, the scientific community is focusing on finding a therapy that is more specific and less toxic for these young patients. This can be achieved only through the knowledge of the molecular pathogenetic mechanisms responsible for the development and maintenance of these tumors.
The three major groups of pediatric soft tissue sarcomas include rhabdomyosarcoma (RMS), Ewing sarcoma (ES), and synovial sarcoma (SS). Although they have different and peculiar characteristics, experimental evidences clearly indicate that all can develop from mesenchymal progenitor cells affected by chromosomal aberrations and/or gene mutations. It is widely accepted that the dysregulation of the major embryonic developmental molecular pathways plays a fundamental role in the pathogenesis of pediatric soft tissue sarcomas. In agreement, small populations of cells that remain undifferentiated and maintain self-renewal capacity seem to represent the tumor ancestor cells unresponsive to therapy [23, 24].
Therefore, the modulation of developmental pathways regulating stem cell properties, such as the Notch pathway, might be a potential strategy to improve the clinical response of this type of tumors affecting young patients.
In the last several years, we and others have reported preclinical experimental proofs of principle indicating Notch signaling modulation as a potential approach to reduce the tumorigenesis of pediatric soft tissue sarcomas.
11.1.3 Structure of Notch Receptors and Ligands
The Notch pathway is one of the fundamental signaling pathways strictly defining developmental processes regulating cell fate and tissue differentiation and homeostasis in embryo and in the postnatal life. The pathway signals through cell-to-cell interaction between a signal-sending cell (expressing Notch ligands) and a signal-receiving cell (expressing Notch receptors) (Fig. 11.1) [25, 26]. This type of cell communication relies on the particular structure of ligands and receptors.

After proteolytic processing maturation, Notch receptors are expressed on the cell membrane as an extracellular domain (NECD) non-covalently associated with a transmembrane portion and an intracellular domain (NICD). Notch signaling is initiated by a Notch receptor-Delta/Jagged-type (DLL/JAG) ligand interaction between two neighboring cells in trans, which induces two successive proteolytic cleavages. The first one is operated on the S2 site by “a disintegrin and metalloprotease” 10 (ADAM10) or ADAM17, which is followed by an S3 cleavage by a presenilin complex (γ-secretase). The S3 cleavage gives rise to the NICD fragment that translocates into the nucleus, where it binds to a protein complex containing recombination signal-binding protein Jk (RBP-Jk) relieving the repressor complex (CoRep). This event modulates chromatin activity recruiting activators such as MAML1 and converts RBP-Jk from a transcriptional repressor to an activator, leading to the transcription of hairy/enhancer of split (Hes) and Hey family genes, which work as transcriptional repressors. Several stages of the Notch signaling pathway are prone to pharmacological intervention. Decoys, anti-ligand antibodies, anti-receptor-antibodies, γ-secretase inhibitors, and peptide inhibitors are labeled in the red boxes
Notch receptors
While in the fruit fly Drosophila melanogaster only a single Notch gene exists (reviewed in [27]), in mammals four Notch receptors have been identified, i.e., Notch 1–4 [28]. They are encoded by four different gene loci on chromosome (Chr) 9, Chr 1, Chr 19, and Chr 6, respectively, and are about 60% homologous to each other. Each Notch paralog is translated as a single-pass transmembrane protein that is subjected to posttranslational modifications before being expressed on the surface of the cells: a single-chain precursor is cleaved by furin-like proteases in the Golgi compartment (S1 cleavage), resulting in an N-terminal extracellular domain (NECD) and a C-terminal portion encompassing both a Notch transmembrane (NTM) and intracellular domain (NICD). The two fragments are non-covalently reassembled on the Golgi membranes and, then, expressed on the surface of the plasma membrane ([29] and reviewed in [30]).
The NECD is formed by a number of epidermal growth factor (EGF)-like repeats responsible for the binding of ligands [31]. Important under a functional point of view, a specific number of EGF repeats characterize each Notch receptor, Notch1 containing 36 EGF repeats [32], whereas Notch2 presenting 35 EGF repeats [33], Notch3 34 EGF repeats [34], and Notch4, the shorter Notch receptor, only 29 EGF repeats [35]. A negative regulatory region (NRR), composed of three cysteine-rich Lin12/Notch repeats (LNR) [36, 37], followed by a juxtamembrane hydrophobic region, is responsible for the heterodimerization of the NECD and the NTM-NICD portions of the receptor. The LNR regulates the auto-inhibition of the Notch receptor preventing the receptor for being cleaved without binding to the ligand [37, 38].
The intracellular region NICD contains a module, named RAM, which recognizes the recombination signal-binding protein Jk (RBP-Jk) supporting the transcriptional role for the NICD that can interact with the transcriptional coactivator RBP-jK in the CSL complex (RBP-jK/CBF-1/KBF2 in mammals) [39]. The RAM region is followed by seven ankyrin (ANK) repeats important for the interaction with CSL and other transcriptional regulators [40, 41], two nuclear localization signals (NLS) [42], a transactivation domain (TAD) [43], and a C-terminal PEST sequence (rich in proline, glutamic acid, serine, and threonine) [44]. The PEST sequence is highly important since it can be phosphorylated, thus regulating the ubiquitination of the NICD and, consequently, its stability and signaling ability [44]. Notably, the strength of the TAD sequence in transactivating gene transcription is different among the paralogs being strong for Notch1, weak for Notch2, and strong but highly specific for Notch3, while Notch4 does not have a TAD [43, 45]. These differences in the structure and activity explain the diverse and somewhat divergent functions of the Notch receptor family.
Notch ligands
Only two canonical ligands of the Delta-Serrate family are expressed in Drosophila, while mammalian cells express three ligands of the Delta family, Delta-like 1 (DLL1), DLL3, and DLL4 [46,47,48], and two of the Serrate family, JAG1 and JAG2 [49, 50]. All the five mammalian ligands are type I transmembrane proteins containing an N-terminal region and a cysteine-rich domain (DSL for Delta, Serrate, and LAG-2), followed by a number of EGF-like repeats. In particular, the N-terminal region with DSL and the first two EGF-repeats are responsible for the interaction with the EGF-like repeats of Notch receptors ([51, 52] and reviewed in [25]). The structure of the intracellular region of the canonical ligands is not conserved among species and regulates ligand interactions with the cellular cytoskeleton.
Additional noncanonical ligands can interact with and activate Notch receptors, either transmembrane or soluble proteins, such as DLK1, DLK2, DNER, the EGF-like protein7 (EGFL7), or the F3/contactin ([53, 54] and reviewed in [25, 55, 56]). They do not contain a DSL domain but are all characterized by the presence of EGF-like repeats.
Another level of complexity is added by the posttranslational modifications of Notch receptors, operated in the cytoplasmic compartment, which strictly regulate their half-life, selectivity, and activity [25, 57]. Among those are the glycosylation, ubiquitylation, phosphorylation, and acetylation.
Fringe glycosyltransferases, firstly identified in Drosophila, glycosylate specific EGF-like repeats of the Notch heterodimer in the Golgi compartment [58,59,60]: a modification that affects the affinity of the receptor for the ligands, specifically preventing Jagged-dependent activation [61, 62]. Three mammalian fringe enzymes are known, i.e., lunatic fringe (LFNG), manic fringe (MFNG), and radical fringe (RFNG) [63]. It is arguable that dysregulation of these enzymes can lead to imbalance in the expression/activity of Notch components since it can induce the Notch receptors to be cleaved with higher rate than in normal tissue (reviewed in [64]), as demonstrated for breast cancer cells [65].
The lysosomal degradation or, conversely, the recycling to the plasma membrane of the cleaved Notch is regulated by polyubiquitylation, a process governed by several E3 ubiquitin ligases such as Deltex, β-arrestin/Kurtz, Itch, NEDD4 (neural precursor cell expressed developmentally downregulated 4), Cbl (casitas B-lineage lymphoma), and Fbw7/Sel-10 ([66,67,68,69] and reviewed in [70]). The inclusion of Notch in the early endosomes can be regulated by Numb, a cytoplasmic negative regulator of the pathway [71], and it is followed by proteasome-mediated degradation [72]. The phosphorylation of NICD to the ANK and/or PEST domain along with acetylation modulates the stability and the activity of the cleaved receptor [73,74,75,76,77]. Further, NICD can interact in the cytoplasm with several molecules among which Nemo-like kinase NLK, which suppresses Notch signaling [78], or Pin1, which conversely amplifies Notch activation [79,80,81,82].
11.1.4 Notch Signaling Pathway
The Notch signaling is critical in embryos during the differentiation of stem cells when a ligand-expressing cell interacts with a Notch-expressing cell and, then, the former undergoes differentiation while the latter remains in an undifferentiated state [30]. However, the results of this cell-to-cell communication highly depend on the molecular, cellular, and environmental contexts, making a simple mechanism extremely versatile [83,84,85].
When a canonical ligand on a cell binds to the specific EGF-like repeats of a Notch receptor on a neighboring cell (in trans), the resulting mechanical stretch favors the cleavage (at site S2) of the heterodimeric portion just outside the plasma membrane by the a disintegrin and metalloprotease 10 (ADAM10) or 17 (ADAM17) [86]. A requirement for this process is the ubiquitination and subsequent endocytosis of the ligand (reviewed by [87]). Then, the remaining membrane-tethered intermediate, named NEXT (Notch extracellular truncation), is subsequently cleaved in an intracellular region (at sites S3 and S4) by a γ-secretase complex formed by four subunits [88,89,90,91,92,93]. This last cleavage results in an intracellular activated form, NICD, which translocates to the nucleus, binds the CSL complex (RBP-Jk/CBF-1/KBF2 in mammals) and activates the transcription of canonical Notch target genes [94]. To do so, the CSL/Notch complex recruits several transcriptional coactivators such as Mastermind-like 1 (MAML1) and the acetyltransferases CBP/p300 or PCAF/GCN5 ([41, 95,96,97] and reviewed in [98]). The canonical target genes belong to the basic helix-loop-helix (bHLH) families of hairy/enhancer of split (Hes) and Hey (subfamily of Hes, related with YRPW motif) repressors [25]. The result is the transcriptional repression of multiple differentiation genes. Interestingly, conversely to the classical view based on the recruitment of NICD by RBP-jK already bound to the DNA in a repressor state [83, 99], more recently the group of Tajbakhsh demonstrates that in mammalian myoblasts (i) NICD recruits free RBP-jK to the chromatin on specific enhancers, while (ii) the amount of RBP-jK constitutively bound to the DNA is unaffected by Notch activation [100]. This finding further highlights the importance of the cellular and molecular context for the regulation and effects of Notch signaling pathway. In addition to the Hes and Hey genes, Notch signaling can activate in a context-/tissue-dependent manner the transcription of, among others, Deltex or members of NF-kB family, the cyclin-dependent kinase inhibitor p21Cip1, cyclin D1 or MYC [101,102,103,104,105,106]. Notch signaling can be also activated in a noncanonical way that can be (i) independent from CSL, (ii) independent from the S3 cleavage, or (iii) in the absence of Notch cleavage and NICD formation (reviewed in [55, 107, 108]). Finally, ligand-receptor interactions on the same cell can be also in cis and results in inhibition of the signaling [109,110,111,112]. Importantly, the structural molecular features of Notch components that allow several types of modifications concurring to the diverse mechanisms of signalization represent a platform for therapeutical interventions with modulators of the pathway (Fig. 11.1). Notably, being Notch signaling tissue- and context-specific and paralogs similar but not identical, the signal triggered by different Notch receptors in different tissues is somewhat specific and can be even opposite (reviewed in [25]).
11.2 Notch Signaling Deregulation in Pediatric Soft Tissue Sarcomas
11.2.1 Notch Signaling in Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) is the most common soft tissue tumor of childhood of myogenic origins accounting for about 8% of all pediatric tumors [113]. Despite the expression of the master regulators of skeletal muscle differentiation such as MYOD and myogenin, also used for diagnostic purposes to exclude other small round blue cell tumors, RMS cells do not differentiate in multifiber structures and proliferate indefinitely ([114, 115] reviewed in [116]). To date, we and others have shown that the modulation of differentiation represents a potential approach to restore the cell cycle checkpoints inhibiting tumor cell proliferation [117, 118]. However, as shown in genetically modified mice models (GEMM) of spontaneous RMS, this sarcoma could originate from a heterogeneous group of mesenchymal-derived cells, even if mesenchymal precursors with different degrees of skeletal muscle commitment have been implicated as the major tumor-prone subset [119,120,121,122,123,124]. Two major histological subtypes are included in pediatric RMS: the embryonal (ERMS) and the alveolar (ARMS) variants. ERMS represents about 70–75% of all cases of pediatric RMS and primarily affects young children arising in the head and neck and retroperitoneum and showing, when nonmetastatic, a good prognosis with an overall survival of about 80% [125, 126]. ERMS is characterized by somatic gene mutations in the RAS gene family, TP53, FGFR4, PIK3CA, CTNNB1, FBXW7, and BCOR, associated with genomic instability including loss of imprinting and loss of heterozygosity of specific chromosomal regions, among which the Chr. 11p.15 region, and gain of regions of chromosomes 2, 7, 8, 11, 12, 13, and 20 [127,128,129]. Moreover, ERMS pathogenesis has been related to mutation/dysfunction of components of one of the major developmental pathways, i.e., Hedgehog [130,131,132,133]. Interestingly, the MYOD gene has been shown to be mutated in a group of older adolescent with an aggressive form of ERMS [134]. The p.Leu122Arg substitution leads to a MYOD protein capable to activate gene transcription in a “MYC-like” manner, once more highlighting the strong involvement of malfunction of myogenic factors in RMS. Collectively, these findings emphasize the heterogeneous molecular features of the ERMS variant. An about 20% of ARMS behave clinically and show molecular alterations similar to the ERMS subtype [127, 135], whereas the majority of ARMS is characterized by clonal cell populations with specific chromosomal translocations, defining a subset of RMS clinically and molecularly different from fusion-negative RMS [127, 135].
The most frequent chromosomal translocations in ARMS are t(2;13) (q35;q14) or t(1;13) (q36;q14), which result in the expression of the two oncogenic proteins PAX3-FOXO1 and PAX7-FOXO1, respectively [136, 137]. Both are transcription factors formed by the DNA-binding domain of PAX3/7 and the transactivation domain of FOXO1. The result is a constitutive activation of a PAX3/7 transcriptional gene profile. In addition, PAX3-FOXO1 acquires transcriptional ability that is absent in PAX3 alone (reviewed in [138]). Fusion-positive ARMS affects mainly older children and adolescents arising in legs and trunk. The expression of the fusion proteins is a negative prognostic factor per se independent from histology, identifying a subset of patients at high risk frequently with metastatic disease at diagnosis. Fusion-positive ARMS but also metastatic fusion-negative RMS represent a challenge for clinicians since they are often unresponsive to treatments with a high chance to recur. The demonstration of the expression of the fusion products is entering the clinical practice to help in the risk stratification of patients, and, more recently, the Shipley group demonstrated that those patients characterized by a PAX3-FOXO1 protein expression are at ultrahigh risk showing a 5-year overall survival (OS) less than 15% [139]. Taken together, these clinical data indicate that to halt the disease, it is imperative to hamper PAX3-FOXO1 activity. Despite improvements in the therapeutic strategies, the outcome of high-risk patients remains poor. Therefore, the need of a deeper knowledge of the mechanisms underlying the development and progression/recurrence of RMS is urgent. However, transcription factors such as PAX3-FOXO1 are difficult to target. Therefore, targeting PAX3-FOXO1 downstream molecules could be an acceptable approach to block its signaling. The developmental networks appear to be good targets due to their involvement in the differentiation of mesenchymal cells in addition to the PAX3 program. In particular, Notch signaling plays one of the major roles among the crucial regulators of skeletal muscle differentiation, maintenance, and homeostasis, both in embryo and in the postnatal life [140].
To date, several recent experimental findings by our group and other laboratories demonstrate that Notch signaling pathway is deregulated in RMS (Table 11.1). The first evidence of an implication of a Notch component in RMS stems from the work of Sang et al. [151] showing that the Notch target gene HES1, encoding for a transcriptional repressor, was able to halt the muscle-like differentiation when expressed in fibroblasts engineered with a plasmid encoding MYOD. This effect was reversed by treatment with a γ-secretase inhibitor (GSI), which blocks the cleavage of Notch receptors, or by silencing a corepressor working with HES1, i.e., TLE1/groucho. HES1 transcripts were then shown to be overexpressed in RMS tumors and cell lines compared to normal skeletal muscle tissue. Then, the authors elegantly demonstrated that inhibition of the HES1 function using either a mutant HES1, defective in the DNA binding, or a dominant-negative HES1 form, lacking the domain that mediates the interaction between HES1 and its corepressors, halted cell proliferation and facilitated muscle-like differentiation of fusion-positive ARMS cell lines [151]. Similar results, associated to a diminution of the levels of HES1, were obtained inhibiting Notch signaling with a GSI, establishing that the effects seen were, at least in part, dependent from the Notch signaling activation.
Subsequently, the group of Gallego published a report showing a general deregulation in transcripts of the Notch pathway in 37 primary RMS samples, irrespective of the fusion status [152]. The authors showed significant upregulation of Notch2 and HEY1 compared to normal muscles. No overt difference in the levels of Notch4 and Notch1 transcripts in RMS compared to control tissues was seen, while HES1 transcripts resulted modestly overexpressed in ERMS. However, the expression of the HES1 protein by immunohistochemistry was more elevated in RMS either fusion-negative or fusion-positive compared to muscle tissues. Interestingly, HES1 expression levels well correlated with the invasive capabilities of RMS cells with the lowest expression in low-invasive ERMS cell lines, and highest expression in PAX3-FOXO1 cells, which are the most invasive subtype [152]. The importance of Notch signaling in the invasive features of RMS cells was then confirmed either (i) inhibiting the γ-secretase-dependent cleavage of Notch receptors with several GSIs or (ii) transfecting RMS cells with a dominant-negative form of MAML1 (dnMAML1), which forms inactive RBP-jK/NICD/MAML1 complexes on DNA [158]. In both cases, HES1 transcript and protein levels were negatively affected by each of the two approaches, supporting the view of a Notch-dependent direct or indirect mechanism for HES1 overexpression. In a more recent work, Belyea et al. [153], interrogating previously published gene expression datasets [135], showed a marked upregulation of HEY1 transcripts in ERMS compared not only to muscle tissues but also to ARMS samples. The results were confirmed in ERMS cell lines with respect to fusion-positive ARMS cells. The authors investigated the protein levels of HEY1 along with those of nuclear Notch1 in primary samples by immunohistochemistry and found that both were remarkably higher in ERMS compared to ARMS or to normal muscle tissue. HEY1 or Notch1 genetic depletion through shRNAs led to impaired ERMS cell proliferation in vitro and enhanced expression of the differentiation gene myogenin, particularly when cells were cultured in differentiation medium (low serum). However, despite the upregulation of myogenin and the phenotypic changes from round- to spindle-shaped cells, only a few myofiber-like structures were formed in these experimental conditions. Since Notch1 downregulation induced HEY1 decrease, suggesting that HEY1 was directly or indirectly targeted by Notch1 signaling in ERMS cells, the Notch1-HEY1 axis seems to be a regulator of cell cycle rather than of terminal differentiation in the ERMS context [153]. These effects were phenocopied by two GSIs and, more importantly, rescued in GSI-treated cells by vector-induced N1ICD forced expression, supporting the hypothesis of a Notch1-specific effect. Moreover, these approaches worked also in in vivo models of ERMS xenografts, which showed reduced tumor growth for those formed by cells depleted of Notch1 or in animals treated with a GSI [153]. This last treatment resulted in the reduction of Notch1 levels in tumor samples, confirming the involvement of the Notch paralog signaling in the development of tumor masses [153]. Recently, the RBP-jK transcription factor has been shown to indicate a trend for a bad prognosis in RMS patients [135], and its modulation in ERMS cells clarified that Notch signaling aberrant functions in ERMS relies partly on a canonical signaling [155]. In fact, RBP-jK knockdown in ERMS cells downregulated HES1 expression and reduced colony formation in soft agar, while its overexpression behaved in the opposite manner [155]. ERMS cells depleted of RBP-jK formed smaller tumors in vivo and showed downregulation of pro-proliferative markers associated with upregulation of the cyclin-dependent inhibitor p21Cip1 [155].
The metastatic behavior is recognized as extremely important for the response to therapy and outcome of RMS patients, and metastasis formation has been related to Notch activation in cancer [159,160,161]. Therefore, starting from the findings of a correlation of HES1 or HEY1 levels with cell invasion in vitro in RMS cell lines, the Rome group further clarified the role of Notch1 and HES1 in the invasiveness of RMS cells [162]. Pharmacological treatment with a GSI of one fusion-negative- and one PAX7-FOXO1- and one PAX3-FOXO1-positive cell line led to a marked decrease of cell adhesion on two different substrates and negatively modulated N-cadherin and α9-integrin transcriptional expression, together with those of the Notch target gene HES1, resulting in the lowering of protein levels [162]. These findings were in agreement with the observation that Notch1 and Notch3 upregulate N-cadherin in melanoma cells [163, 164]. In patients with RMS, a positive correlation between N-cadherin and α9-integrin with HES1 was seen. In line with the hypothesis of an involvement of Notch signaling in this phenomenon, RMS cells transfected with a plasmid expressing a dominant-negative form of MAML1 [152] showed a response similar to that of cells treated with a GSI. Conversely, RMS cells in which an exogenous DLL1 was forcedly overexpressed, thus leading to Notch signaling over-activation, enhanced the expression of all the three genes. These effects appeared quite specific since the level of the usual partner of α9-integrin, i.e., β1-integrin, was unaffected. Interestingly from a translational point of view, the authors showed that cell adhesion on fibronectin and the invasive capabilities of the cells in vitro were markedly reduced using an anti-N-cadherin-blocking antibody, whereas anti-α9-integrin-blocking antibody was able to impair only the tumor cell adhesive properties. Chromatin-immunoprecipitation assays demonstrated a possible direct regulation of Notch1 on the two gene promoters. However, HES1 seemed also to bind those promoters, but its role in regulating these genes should be clarified in future studies. This pro-invasive role of Notch signaling in RMS seems to be counteracted by the restoration of the expression of miR-203, a microRNA often downregulated epigenetically by promoter hypermethylation in RMS primary samples and cell lines and re-expressed after treatment with the DNA methyltransferase 1 inhibitor 5-AZA [157]. When miR-203 was re-expressed in vitro in one ERMS and one PAX3-FOXO1 ARMS cell line, it inhibited cell proliferation inducing the myogenic conversion of the tumor cells, decreasing the levels of the transcription factor p63, an inducer of JAG1 and of HES1. Similar results were obtained silencing p63. These findings suggest that the promyogenic role of miR-203 relies, at least in part, on its ability to down-modulate p63. Moreover, miR-203 forced expression blocked both cell migration and invasion. Tumor growth in vivo was also hampered in RMS cells overexpressing miR-203 or in ERMS-xenografted mice treated with 5-AZA. It could be interesting to evaluate whether the re-expression of miR-203 could have similar effects in vivo also in PAX3-FOXO1 ARMS cells, which are less prone to differentiate.
Previously, our findings unveiled a role for Notch3 in RMS [154]. Genetic downregulation of Notch3 by silencing in fusion-negative and fusion-positive RMS cell lines overexpressing nuclear Notch1–3-activated forms compared to myoblasts resulted in a blockade of cell cycle in the G1 phase and formation of myofiber-like structures even when the cells were cultured in medium containing serum. In agreement with this phenotype, p21 was upregulated together with members of the differentiation machinery such as myogenin, MHC, and troponin. Moreover, p38MAPK, AKT, and mTOR were activated as during myogenesis. In parallel, HES1 levels were decreased suggesting that Notch3 can have a direct or indirect effect on its expression. Concordantly, HES1 depletion mimicked, as already reported by Sang et al. [151], as well as reinforced the effects of Notch3 silencing, while, conversely, its forced overexpression partially overcame them. Moreover, silencing Notch3 even in a fraction of cells inhibited tumor growth in vivo. Interestingly, (i) the depletion of Notch1, which was also hyperactivated in RMS cell lines, reduced the proliferation of the cells and, only in fusion-negative cells, favored the formation of some myotube-like structures, but was ineffective in fusion-positive cells; and (ii) the knockdown of Notch2, whose levels were higher in myoblasts, reduced the expression of myogenin and led to HES1 levels upregulation.
Consistent with a role of Notch3 in RMS, tumor cells forcedly expressing an exogenous N3ICD form proliferated faster in vitro and formed more colonies in soft agar irrespective of their fusion status [156]. Notably, the antiproliferative effects of a GSI were counteracted by N3ICD overexpression. We also confirmed that N3ICD influences tumor growth in vivo showing that PAX3-FOXO1/N3ICD xenografted cells produced bigger masses with a higher expression of Ki67 and HES1 [156]. Of note, we also showed that HES1 and Notch3 protein levels correlated with those of Ki67 in samples from RMS patients [156].
Since a very low number of mutations of Notch paralogs have been found in RMS primary samples [128, 129, 165], it is arguable that the hyperactivation of Notch receptors in tumor cells could be due to other reasons such as to the binding to the Notch ligands. As a matter of fact, downregulating DLL1 and JAG1, whose transcripts were found expressed in RMS cell lines [153] and primary specimens [135, 155], led to the inhibition of cell proliferation of ERMS and PAX3-FOXO1 ARMS cells associated with the lowering of N3ICD and HES1 levels [154, 156]. Summarizing all these results, it appears clear that a general dysregulation of the Notch signaling characterizes the RMS setting opening the way to potential targeted therapy for this sarcoma.
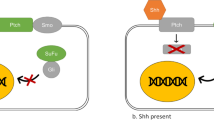
One of the characteristics of Notch signaling is the capacity to cross talk with several key pathways that regulate stem cell fate and are involved in cancer pathogenesis and maintenance. The Hedgehog pathway is one of the major regulators of the myogenesis in vertebrates, by maintaining the expression of the myogenic regulatory factors (MRFs) such as MYF5 and modulating survival and proliferation of developing myoblasts [166]. In particular, it supports the proliferation of myogenic precursors repressing terminal differentiation and apoptosis [167,168,169]. A dysregulation of Hedgehog seems to be one of the drivers of ERMS development, as highlighted by studies in humans and GEMM [170,171,172,173]. Recently, alterations of Hedgehog signaling have been recently shown to be interconnected to that of Notch in the pathogenesis of ERMS [132]. In this work the authors demonstrated that in mice heterozygous for the negative regulator of Hedgehog signaling ptch1, which spontaneously develop ERMS, the cells of origin of the tumor are derived from those expressing the Notch ligand Dll1 and concomitantly negative for Myf5, myogenin, and Pax3 expression [132]. This type of cells is prone to undergo myogenic differentiation but is not yet stably committed. These results, on one hand, imply that Hedgehog and Notch cross talk to define the fate of some cells during myogenesis, and on another hand highlight the importance of the molecular degree of differentiation and commitment for subsets of cells to behave as tumor-initiating RMS cells, as already demonstrated by the group of Keller [122, 174]. Importantly under a translational point of view, Hedgehog signaling activation is able to induce HES1 expression in both mesodermal and neural cells independently from Notch, suggesting combinatorial inhibition of the two pathways [175].
Several points on the impact of Notch signaling deregulation in RMS remain to be investigated among which the expression of protein levels of Notch ligands in RMS patients, its role in the invasiveness and metastasis in in vivo models, and its effects in GEMM of RMS. However, it appears evident that this signaling pathway could be activated in both ARMS and ERMS thus representing a potential target for therapy in both RMS variants.
11.2.2 Notch Signaling in Ewing Sarcoma
Ewing sarcoma (ES) is the second most common bone and soft tissue sarcoma of childhood. It arises most commonly in adolescents showing a median age of 15 years, even if cases of ES in neonates and infants have been reported [176, 177]. The most frequently affected sites are the lower extremities and pelvis for bone and the trunk and extremities for soft tissue disease. It is an aggressive malignancy, metastatic at diagnosis in about 25% of young patients [176]. Improvements in therapy have enhanced the survival rates for localized forms, but the outcome and disease-free survival of patients with metastatic disease remain poor [178,179,180]. ES often shows gains of chromosomes 8, 12, 20, and 1q, losses of 1p36 and 16q, and homozygous deletion of CDKN2A , but the mutation rate is low and mostly involves STAG2 or TP53 (5–20% of cases), making finding actionable therapeutic targets difficult ([181,182,183] and reviewed in [180]). In about 90% of cases, ES is characterized by typical chromosomal translocations t(11;22)(q24;q12) resulting in the fusion of the amino-terminal-encoding portion of EWSR1 to the carboxyl-terminal DNA-binding domain of the FLI1 gene of the ETS family genes, generating the EWS-FLI1 fusion product with transcriptional regulatory functions ([184, 185] and reviewed in [186]). The translocation can involve several different portions of the genes, due to different breakpoints in each of the sequences, but without effects on the prognosis [187, 188]. Variants of fusion products involving or not EWS have also been observed in a number of cases (reviewed in [186]). When expressed in a “permissive” cell of origin context, i.e., mesenchymal- and neural crest-derived progenitors, EWS-FLI1 shows transforming capacity [185, 189,190,191,192]. EWS-FLI1 is a transcription factor with higher potency compared to FLI1 that binds to ETS consensus sequences across the genome [185, 193] and whose mechanism of action has been recently unraveled. It binds several types of chromatin regions, from promoters to intra- and intergenic regions, repressing but also inducing a high number of genes [194,195,196,197] with a function that can be context-dependent [198]. When exogenously expressed in murine fibroblasts, EWS-FLI1 induced the transcription of the Notch signaling enzymatic component Mfng [145], a result in agreement with transcript MNFG upregulation found in ES patients (Table 11.1) [146], even if in human ES cells, the transcriptional effect on MNFG is weaker [199]. Recently, the group of Kovar unveiled the mechanism through which EWS-FLI1 was able to overcome cell cycle arrest in a context of wild-type TP53 [147]. The authors demonstrated that, by repressing the expression of the Notch ligand JAG1, EWS-FLI1 reduced the activation of Notch3 necessary for the induction of the Notch target gene HEY1 that, in turn, stabilized and activated p53 [147]. Indeed, in TP53 wild-type ES cell lines, (i) EWS-FLI1 silencing promoted p53 and p21Cip1 expression followed by cell cycle arrest; (ii) this effect was associated with the induction of JAG1 and HEY1, often barely expressed in ES primary samples; (iii) Notch2 and Notch3 were expressed in both ES cell lines and primary samples, and Notch3 resulted activated only in TP53 wild-type cells by JAG1; and (iv) in EWS-FLI1-depleted cells, JAG1 or HEY1 silencing, treatment with a GSI, or expression of the negative regulator of Notch, NUMB, prevented p53 and p21Cip1 induction, while forced expression of either exogenous JAG1, HEY1, or N3ICD reversed the effects. Therefore, in ES cells with wild-type TP53, Notch signaling seems to act as an onco-suppressor stabilizing p53 with an unknown mechanism involving HEY1. Interestingly, when Notch signaling was inhibited in the presence of EWS-FLI1, no HEY1 expression was observed, suggesting that the pathway could be inactive under these cell conditions [147]. This was consistent with the observation of a lack of nuclear expression of NICD and HES1 in ES tumors, despite the mRNA upregulation of the latter [148]. Moreover, the transcriptional overexpression of HES1 was independent from Notch activation and also from EWS-FLI1 expression.
ES pathogenesis implies an aberrant chromatin remodeling due to the influence of the fusion proteins on epigenetic machinery (reviewed in [186]). Accordingly, pharmacological inhibition of the lysine demethylase LSD1 (or KDM1A), upregulated in a large cohort of sarcomas including ES, led to p53 expression in ES cell lines through the methylation of Lys 4 on histone H3 (H3K4) followed by cell cycle arrest [200]. In other cell systems, LSD1 is able, as a component of a corepressor complex with the deacetylase SIRT1, to inhibit Notch signaling by recruiting the RBP-jK complexes and repressing the expression of Notch target genes, including HEY1 [201,202,203]. Starting from this observation, the same group sheds light on the mechanism of p53 induction after EWS-FLI1 depletion showing that ectopically expressed HEY1 prevented the expression of the deacetylase SIRT1, which in turn was responsible for the posttranslational modification that leads to p53 destabilization and deactivation [149]. This effect was obtained also by ectopic expression of NICD, demonstrating that it is Notch-dependent, and also demonstrated in other cell contexts in which Notch signaling can act similarly, such as B-cell tumors and primary human keratinocytes lacking HEY1 expression. Consistently, genetic and pharmacologic inhibition of SIRT1 was sufficient to increase p53 acetylation and target genes activation, in ES cells in the presence of EWS-FLI1, resulting in tumor cell death, while its overexpression reverted the phenotype [149]. An antitumorigenic effect was also seen in vivo after pharmacological treatment of xenografted zebrafish models. Finally, the screening of about 400 ES human tumor samples by immunohistochemistry showed that SIRT1 expression could be correlated to disseminated disease due to the highest levels of staining in metastatic patients. Thus, on one hand, this work unveils a novel epigenetic Notch-dependent mechanism to regulate cell cycle and on the other hand points to SIRT1 as a pharmacologically targetable factor in ES. Although EWS-FLI1 is necessary for tumorigenesis, it requires a “permissive” cellular background for transformation. Among the involved adjuvant molecules is CD99 [204], a cell surface protein involved in cell migration, proliferation, and differentiation [205, 206]. As a matter of fact, EWS-FLI1 is able to upregulate CD99 that, in turn, facilitates the oncogenic function of the fusion protein [204, 207, 208]. However, although CD99 contributes to the oncogenic phenotype defined by the fusion gene, EWS-FLI1 is able to induce a neuroblastic phenotype while CD99 counteracts this effect [204]. Since ES cells are unable to completely differentiate, a recent work demonstrates that a network CD99-miR-34a-Notch-NF-kB underpins the mechanism underlying the anti-differentiative phenotype and suggests novel avenues for intervention [150].
The work showed that CD99, by inducing the expression of the Notch ligand DLL1, resulted in Notch1 and Notch3 activation paralleled by a concomitant activation of NF-kB, all effects prevented by CD99 depletion or GSI treatment. In turn, the CD99-dependent activity of NF-kB, or NF-kBp65 forced overexpression in a CD99 knockdown context, affected the neural phenotype due to the presence of EWS-FLI1, whereas, conversely, its silencing enhanced the proneural differentiation [150]. Elegantly, the authors then demonstrate that all the molecular and phenotypic effects of CD99 depletion, including Notch components regulation, can be phenocopied by a microRNA previously involved in ES and able to regulate Notch signaling, i.e., miR-34a [209,210,211,212], which was induced by CD99 knockdown. Thus, the presence of CD99 prevented miR-34a expression thus allowing Notch and NF-kB activation [150]. Interestingly, Notch and NF-kB pathways cross talk in several systems mainly in a noncanonical way (reviewed in [213]), which is in agreement with the inactivation of canonical Notch signaling found in ES, despite the expression of Notch receptors [147]. Strikingly, the effects of CD99 expression spread to neighboring cells through exosomes bearing CD99 from ES cells, and, consequently, when CD99 was depleted, exosomes lacking CD99 and containing high levels of the induced miR-34a carried a proneural signal to the target cells. These important results are in agreement with a previous report showing that Notch signaling inhibition induced neuroectodermal differentiation of tumor xenografts in ES with low impact on tumor cell proliferation [146]. Taken together, the reported findings further complicate the scenario of an role of Notch signaling in ES, showing an antiproliferative but also anti-differentiative role for this pathway. The predominance of a canonical versus noncanonical Notch signaling activation depends on the molecular context of the cells and deserves further investigations.
11.2.3 Notch Signaling in Synovial Sarcoma
Synovial sarcoma (SS) is a soft tissue sarcoma developing most commonly in the lower limbs of adolescents and young adults and showing a high metastatic potential ([214]; and reviewed in [215, 216]). It accounts for about 10–20% of all soft tissue sarcomas in young patients [217]. SS includes three histological subtypes: monophasic (only spindle cells), biphasic (both spindle and epithelial cells), and poorly differentiated. In addition to the soft tissue adjacent to the joints (i.e., synovial), it can develop in extra-synovial tissues. Localized disease can be treated by surgical intervention followed by adjuvant radiotherapy, but it often shows early and even late recurrences with 50% 10-year disease-free survival [218]. Molecularly, SS is characterized by the chromosomal translocation t(X,18; p11,q11) involving SS18 (previously SYT) on chromosome 18q11 and either SSX1, SSX2, or very rarely SSX4 on chromosome Xp11. The results are fusion proteins formed by almost all the SS18 sequence with the C-terminal portion of the SSX paralogs. That SS18-SSX proteins are the oncogenic drivers of the malignancy was demonstrated by the observations that their expression in vitro is sufficient to transform the cells, while their silencing reverts the malignant phenotype [219, 220]. SS is considered to be derived from mesenchymal stem cells in which the fusion proteins behave as oncogenes [221, 222]. In agreement with the importance of a specific cell of origin for tumorigenesis, the SS1-SSX oncoproduct induces spontaneous SS in transgenic mice in vivo with 100% penetrance when expressed in mesenchymal-derived progenitors expressing Myf5 [223]. However, conversely to myogenic sarcomas, no expression of myogenic markers has been unveiled in SS murine models or in SS patients. The evidence of the presence of the fusion both in primary and metastatic lesions and the apoptotic effects linked to its depletion concur to suggest a master role for SS18-SSX in the development of SS [220, 224]. Although SS18 is a transcriptional activator and SSX functions as a repressor and both bind several partners, SS18-SSX does not contain a DNA-binding domain making difficult the identification of direct target genes [219]. However, it acts as a transcriptional regulator controlling gene expression by chromatin remodeling (reviewed in [225]). Indeed, both SS18 and SS18-SSX associate with the SWI/SNF chromatin remodeling complex, which in normal cells/tissues facilitates gene transcriptional programs creating nucleosome-depleted regions at core promoters and regulatory regions [226,227,228,229]. The inclusion of SS18-SSX fusion products in the SWI/SNF complex dysregulates the function of the complex [229]. This is due to the repressor intrinsic properties of the SSX portion that can interact with gene repressor complexes, thus behaving in an opposite manner compared to SS18 itself [230]. SS shows no additional chromosomal imbalance in young patients; however it is characterized by a high expression of components of molecular pathways strictly involved in early embryogenesis. Among these are WNT, Hedgehog, BMP, and Notch pathways. Studies aimed at unveiling binding partners for the SS18-SSX factor demonstrated an interaction of the SSX portion with the corepressor TLE1 (Table 11.1) [144]. TLE genes encode for TLE1–4 proteins that are corepressors and, in particular TLE1, components of the Notch signaling the regulate stemness of embryonic progenitors during development. As a matter of fact, TLE1 is recruited by the Notch target HES1 on promoters to prevent gene expression [231]. SS18-SSX/TLE1 complex was found linked to ATF2, a transcriptional activator and DNA-binding protein, and was able to turn the ATF2 activator program in a repressor program [144]. The ultimate result is the repression of apoptotic/cell cycle blocker genes EGR1, p21Cip1, and ATF3 and the promotion of tumor cell survival, which was impaired by SS18-SSX silencing. The intrinsic mechanism of this effect on ATF2 was related to the interaction of SS18-SSX with the polycomb repressor complexes PRC2 and PRC1 [232], whose repressor activity was further enhanced by the presence of TLE1 in the complex. A deregulated transcript expression of TLE1 has been found by expression profiling experiments in primary SS [141] and the nuclear expression of the protein confirmed by immunohistochemistry [142, 233]. To date, the evidence of an overexpression of TLE1 has currently entered the clinical use to discriminate among other soft tissue sarcomas [143]. In addition to TLE1, also other Notch-related factors have been shown to be upregulated in SS, such as Notch1 and JAG1 [141], although no evidence for functional roles for these proteins in SS pathogenesis has been described so far. However, results from a randomized Phase I/II clinical trial using the GSI RO4229097 in association with the Hedgehog inhibitor vismodegib for adult and adolescent patients with advanced and metastatic sarcomas, among which SS (Table 11.2), will give some information about the potentiality of Notch signaling inhibition in SS.
11.3 Approaches to Inhibit Notch Signaling
Considering the structure, regulation, and function of Notch components, several steps of the signaling pathway can be targeted for inhibition.
11.3.1 γ-Secretase Inhibitors (GSI)
The most widely used approach to hamper Notch signaling is based on the inhibition of γ-secretase activity resulting in Notch cleavage blockade. GSI showed antitumorigenic activities in various cancer cells in preclinical models, and some of them are currently in clinical trials for oncologic diseases, mostly for adult patients. However, over the last years, several Phase I/II studies have been started involving also pediatric and adolescent oncologic patients (Table 11.2).
MK-0752 is a clinical GSI that was evaluated in several Phase I clinical trials for treatment in pediatric and adult malignancies (Table 11.2) [234,235,236]. Another GSI, RO4929097 [237], was evaluated in several NCI-sponsored Phase I/II clinical trials for treatment of solid tumors and T-ALL (Table 11.2). RO4929097 has been used in combinatorial adjuvant regimens with other anticancer drugs, and it is recently in Phase I/II associated with vismodegib, an inhibitor of Hedgehog signaling, for treatment of advanced and metastatic sarcomas for adults and pediatric patients (Table 11.2).
The Phase II clinical trial with the GSI PF-03084014 for pediatric patients is ongoing for desmoid tumors and aggressive fibromatosis and is progressing to a Phase II for T-ALL and solid tumors (Table 11.2) [238]. In preclinical models, GSIs have shown also anti-angiogenic effects that could contribute to their efficacy in vivo. However, (i) GSI are unable to discriminate among Notch receptors and (ii) γ-secretase have a plethora of targets, and, thus, these chemicals can have off-target effects in vivo [239]. Among these, the most evident is the goblet cell metaplasia of the small intestine due to Notch2 inhibition in the intestinal epithelial stem cells compartment. Even if this effect can be partly prevented by coadministration of glucocorticoids, often the treatment with GSI) requires a lowering in the doses and intermittent administration. Moreover, the evidence of Notch target inhibition in tumor tissue, to decide the dose escalation, is often difficult since the modulated clinical targets not always are the Notch targets found in preclinical studies but can depend on the tissue-context of the patient.
11.3.2 Antibodies Against Notch Signaling Components
Although all Notch paralogs have similar mechanisms of signalization, paralog-specific and even opposite downstream effects have been reported [154, 240,241,242,243,244,245,246]. Therefore, specific monoclonal antibodies against individual receptors or ligands have been developed so far. Although no Notch monoclonal antibody has been evaluated in pediatric tumors, some of them are being evaluated in clinical trials for adult tumors. The binding of the Notch component by the antibody results in the blockade of interaction between the receptor and the ligand and hampers the activation of the signaling. Among the antibodies against DLL4, the ligands responsible for the sprouting of endothelial cells and formation of new vessels that have been evaluated in clinical trials for adult malignancies are MEDI0639 (NCT01577745, recruiting Phase I), OMP-21M18 (NCT01189929; NCT01952249; NCT01189942; NCT01189968 Phase I and Ib), and REGN421 (Phase I completed, showing good tolerability and two partial responses [247]). The specific antibody OMP-52M51 against Notch1 is in clinical trial Phase I, NCT01778439; NCT01703572) and the antibody OMP-59R5 against Notch2/3 in Phase I/II trials (NCT01647828; NCT01859741).
11.3.3 Blocking Peptides
Preclinical studies demonstrated that it is possible to interfere with the transcriptional machinery of Notch signaling with inhibitory peptides. This is the case of a dnMAML1 used to block the RBP-jK-dependent transcription due to Notch activation. Stapled peptides competing with MAML are able to prevent gene transcription in murine models of T-ALL [248, 249]. One characteristic of this stapled peptide is the ability to bind also to preassembled Notch1–CSL complexes to inhibit the binding of the endogenous MAML1 [249]. These peptides have relatively small size and are highly cell-permeable. However, if they target only the transcriptional activity of Notch signaling, they can be ineffective in cancers in which the Notch pathway works in a noncanonical way. Nonetheless, dnMAML peptides could act also sequestering NICD in the cytoplasm, thus hampering also noncanonical roles of the cleaved protein.
11.3.4 Decoys
Soluble forms of the extracellular domains of Notch receptors and their ligands have been studied as decoys to inhibit the signaling. Decoys function by binding to endogenous ligands or receptors preventing the endogenous counterpart to be bound, and, since it lacks intracellular domains, the signaling of the pathway is completely abrogated [250,251,252]. Interestingly, endogenous soluble Notch ligands can be produced by metalloproteases, but their physiologic role still needs to be clarified [253, 254].
11.4 Conclusions
In conclusion, we summarized the role of Notch signaling in pediatric soft tissue sarcomas, giving an overview of the potentiality in targeting the pathway. Notch signaling plays a major role in the determination and homeostasis of tissues of mesenchymal origin in the embryo and postnatal life. Here we highlighted a role of Notch signaling deregulation in pediatric soft tissue sarcomas in the preclinical setting, reporting evidence that Notch modulation regulates cell proliferation, differentiation, and motility/invasion of tumor cells. To date, the majority of approaches against Notch signaling activation rely on the use of GSI even if promising monoclonal antibodies and cell-permeable small molecules are being developed for adult cancers. It is arguable that the pharmacokinetics properties and the biodistribution of decoys and antibodies are the limiting factors for their therapeutic application. Interestingly, for those patients with tumors in which Notch pathway works as a tumor suppressor, such as in EWS, agents stimulating its activity or downstream effects should be considered. In summary, potentially a Notch-based therapy might represent one of the future personalized strategies for young patients with soft tissue sarcomas.
Abbreviations
- DLL1, 3, 4:
-
Delta-like 1, 3, 4
- ES:
-
Ewing sarcoma
- GEMM:
-
Genetically engineered mice models
- GSI:
-
Gamma secretase inhibitors
- MAML1:
-
Mastermind-like 1
- NEC:
-
Notch extracellular domain
- NEXT:
-
Notch extracellular truncation
- NICD:
-
Notch intracellular domain
- NTM:
-
Notch transmembrane domain
- RMS:
-
Rhabdomyosarcoma
- SS:
-
Synovial sarcoma
References
Visweswaran, M., Pohl, S., Arfuso, F., Newsholme, P., Dilley, R., Pervaiz, S., et al. (2015). Multi-lineage differentiation of mesenchymal stem cells – To Wnt, or not Wnt. The International Journal of Biochemistry & Cell Biology, 68, 139–147.
Happe, C. L., & Engler, A. J. (2016). Mechanical forces reshape differentiation cues that guide cardiomyogenesis. Circulation Research, 118(2), 296–310.
Briscoe, J., & Small, S. (2015). Morphogen rules: Design principles of gradient-mediated embryo patterning. Development, 142(23), 3996–4009.
Luo, S. X., & Huang, E. J. (2015). Dopaminergic neurons and brain reward pathways: From neurogenesis to circuit assembly. The American Journal of Pathology, 186(3), 478–488.
Li, X. Y., Zhai, W. J., & Teng, C. B. (2015). Notch signaling in pancreatic development. International Journal of Molecular Sciences, 17(1), 48.
Luxan, G., D’Amato, G., MacGrogan, D., & de la Pompa, J. L. (2016). Endocardial Notch signaling in cardiac development and disease. Circulation Research, 118(1), e1–e18.
Krantz, I. D., Colliton, R. P., Genin, A., Rand, E. B., Li, L., Piccoli, D. A., et al. (1998). Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. American Journal of Human Genetics, 62(6), 1361–1369.
McDaniell, R., Warthen, D. M., Sanchez-Lara, P. A., Pai, A., Krantz, I. D., Piccoli, D. A., et al. (2006). NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. American Journal of Human Genetics, 79(1), 169–173.
Federico, A., Bianchi, S., & Dotti, M. T. (2005). The spectrum of mutations for CADASIL diagnosis. Neurological Sciences, 26(2), 117–124.
Sparrow, D. B., Chapman, G., Wouters, M. A., Whittock, N. V., Ellard, S., Fatkin, D., et al. (2006). Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. American Journal of Human Genetics, 78(1), 28–37.
Lee, S. J., Kim, K. H., Pak, S. C., Kang, Y. N., Yoon, G. S., & Park, K. K. (2015). Notch signaling affects biliary fibrosis via transcriptional regulation of RBP-jkappa in an animal model of chronic liver disease. International Journal of Clinical and Experimental Pathology, 8(10), 12688–12697.
Bansal, R., van Baarlen, J., Storm, G., & Prakash, J. (2015). The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Scientific Reports, 5, 18272.
Vieira, N. M., Elvers, I., Alexander, M. S., Moreira, Y. B., Eran, A., Gomes, J. P., et al. (2015). Jagged 1 rescues the duchenne muscular dystrophy phenotype. Cell, 163(5), 1204–1213.
Lafkas, D., Shelton, A., Chiu, C., de Leon Boenig, G., Chen, Y., Stawicki, S. S., et al. (2015). Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature, 528(7580), 127–131.
Hu, B., Wu, Z., Bai, D., Liu, T., Ullenbruch, M. R., & Phan, S. H. (2015). Mesenchymal deficiency of Notch1 attenuates bleomycin-induced pulmonary fibrosis. The American Journal of Pathology, 185(11), 3066–3075.
Louvi, A., & Artavanis-Tsakonas, S. (2012). Notch and disease: A growing field. Seminars in Cell & Developmental Biology, 23(4), 473–480.
Ellisen, L. W., Bird, J., West, D. C., Soreng, A. L., Reynolds, T. C., Smith, S. D., et al. (1991). TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell, 66(4), 649–661.
Pear, W. S., Aster, J. C., Scott, M. L., Hasserjian, R. P., Soffer, B., Sklar, J., et al. (1996). Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. The Journal of Experimental Medicine, 183(5), 2283–2291.
Weng, A. P., Ferrando, A. A., Lee, W., JPt, M., Silverman, L. B., Sanchez-Irizarry, C., et al. (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science, 306(5694), 269–271.
Bovee, J. V., & Hogendoorn, P. C. (2010). Molecular pathology of sarcomas: Concepts and clinical implications. Virchows Archiv, 456(2), 193–199.
Grunewald, T. G., & Fulda, S. (2016). Editorial: Biology-driven targeted therapy of pediatric soft-tissue and bone tumors: Current opportunities and future challenges. Frontiers in Oncology, 6, 39.
Thacker, M. M. (2013). Malignant soft tissue tumors in children. The Orthopedic Clinics of North America, 44(4), 657–667.
Walter, D., Satheesha, S., Albrecht, P., Bornhauser, B. C., D’Alessandro, V., Oesch, S. M., et al. (2011). CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS One, 6(5), e19506.
De Vito, C., Riggi, N., Cornaz, S., Suva, M. L., Baumer, K., Provero, P., et al. (2012). A TARBP2-dependent miRNA expression profile underlies cancer stem cell properties and provides candidate therapeutic reagents in Ewing sarcoma. Cancer Cell, 21(6), 807–821.
Espinoza, I., & Miele, L. (2013). Notch inhibitors for cancer treatment. Pharmacology & Therapeutics, 139(2), 95–110.
Teodorczyk, M., & Schmidt, M. H. (2015). Notching on cancer’s door: Notch signaling in brain tumors. Frontiers in Oncology, 4, 341.
Dominguez, M. (2014). Oncogenic programmes and Notch activity: An ‘organized crime’? Seminars in Cell & Developmental Biology, 28, 78–85.
Lardelli, M., Williams, R., & Lendahl, U. (1995). Notch-related genes in animal development. The International Journal of Developmental Biology, 39(5), 769–780.
Blaumueller, C. M., Qi, H., Zagouras, P., & Artavanis-Tsakonas, S. (1997). Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell, 90(2), 281–291.
Hori, K., Sen, A., & Artavanis-Tsakonas, S. (2013). Notch signaling at a glance. Journal of Cell Science, 126(Pt 10), 2135–2140.
Rebay, I., Fleming, R. J., Fehon, R. G., Cherbas, L., Cherbas, P., & Artavanis-Tsakonas, S. (1991). Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell, 67(4), 687–699.
del Amo, F. F., Gendron-Maguire, M., Swiatek, P. J., Jenkins, N. A., NG, C., & Gridley, T. (1993). Cloning, analysis, and chromosomal localization of Notch-1, a mouse homolog of Drosophila Notch. Genomics, 15(2), 259–264.
Weinmaster, G., Roberts, V. J., & Lemke, G. (1992). Notch2: A second mammalian Notch gene. Development, 116(4), 931–941.
Lardelli, M., Dahlstrand, J., & Lendahl, U. (1994). The novel Notch homologue mouse Notch 3 lacks specific epidermal growth factor-repeats and is expressed in proliferating neuroepithelium. Mechanisms of Development, 46(2), 123–136.
Uyttendaele, H., Marazzi, G., Wu, G., Yan, Q., Sassoon, D., & Kitajewski, J. (1996). Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development, 122(7), 2251–2259.
Aster, J. C., Simms, W. B., Zavala-Ruiz, Z., Patriub, V., North, C. L., & Blacklow, S. C. (1999). The folding and structural integrity of the first LIN-12 module of human Notch1 are calcium-dependent. Biochemistry, 38(15), 4736–4742.
Gordon, W. R., Vardar-Ulu, D., Histen, G., Sanchez-Irizarry, C., Aster, J. C., & Blacklow, S. C. (2007). Structural basis for autoinhibition of Notch. Nature Structural & Molecular Biology, 14(4), 295–300.
Sanchez-Irizarry, C., Carpenter, A. C., Weng, A. P., Pear, W. S., Aster, J. C., & Blacklow, S. C. (2004). Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Molecular and Cellular Biology, 24(21), 9265–9273.
Tamura, K., Taniguchi, Y., Minoguchi, S., Sakai, T., Tun, T., Furukawa, T., et al. (1995). Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Current Biology, 5(12), 1416–1423.
Lubman, O. Y., Korolev, S. V., & Kopan, R. (2004). Anchoring notch genetics and biochemistry; structural analysis of the ankyrin domain sheds light on existing data. Molecular Cell, 13(5), 619–626.
Nam, Y., Sliz, P., Song, L., Aster, J. C., & Blacklow, S. C. (2006). Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell, 124(5), 973–983.
Lieber, T., Kidd, S., Alcamo, E., Corbin, V., & Young, M. W. (1993). Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes & Development, 7(10), 1949–1965.
Kurooka, H., Kuroda, K., & Honjo, T. (1998). Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Research, 26(23), 5448–5455.
Rechsteiner, M. (1988). Regulation of enzyme levels by proteolysis: The role of pest regions. Advances in Enzyme Regulation, 27, 135–151.
Ong, C. T., Cheng, H. T., Chang, L. W., Ohtsuka, T., Kageyama, R., Stormo, G. D., et al. (2006). Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. The Journal of Biological Chemistry, 281(8), 5106–5119.
Bettenhausen, B., Hrabe de Angelis, M., Simon, D., Guenet, J. L., & Gossler, A. (1995). Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development, 121(8), 2407–2418.
Dunwoodie, S. L., Henrique, D., Harrison, S. M., & Beddington, R. S. (1997). Mouse Dll3: A novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development, 124(16), 3065–3076.
Shutter, J. R., Scully, S., Fan, W., Richards, W. G., Kitajewski, J., Deblandre, G. A., et al. (2000). Dll4, a novel Notch ligand expressed in arterial endothelium. Genes & Development, 14(11), 1313–1318.
Lindsell, C. E., Shawber, C. J., Boulter, J., & Weinmaster, G. (1995). Jagged: A mammalian ligand that activates Notch1. Cell, 80(6), 909–917.
Shawber, C., Boulter, J., Lindsell, C. E., & Weinmaster, G. (1996). Jagged2: A serrate-like gene expressed during rat embryogenesis. Developmental Biology, 180(1), 370–376.
Parks, A. L., Stout, J. R., Shepard, S. B., Klueg, K. M., Dos Santos, A. A., Parody, T. R., et al. (2006). Structure-function analysis of delta trafficking, receptor binding and signaling in Drosophila. Genetics, 174(4), 1947–1961.
Shimizu, K., Chiba, S., Kumano, K., Hosoya, N., Takahashi, T., Kanda, Y., et al. (1999). Mouse jagged1 physically interacts with notch2 and other notch receptors. Assessment by quantitative methods. The Journal of Biological Chemistry, 274(46), 32961–32969.
Schmidt, M. H., Bicker, F., Nikolic, I., Meister, J., Babuke, T., Picuric, S., et al. (2009). Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nature Cell Biology, 11(7), 873–880.
Hu, Q. D., Ang, B. T., Karsak, M., Hu, W. P., Cui, X. Y., Duka, T., et al. (2003). F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell, 115(2), 163–175.
Ayaz, F., & Osborne, B. A. (2014). Non-canonical notch signaling in cancer and immunity. Frontiers in Oncology, 4, 345.
D’Souza, B., Meloty-Kapella, L., & Weinmaster, G. (2010). Canonical and non-canonical Notch ligands. Current Topics in Developmental Biology, 92, 73–129.
Guruharsha, K. G., Kankel, M. W., & Artavanis-Tsakonas, S. (2012). The Notch signalling system: Recent insights into the complexity of a conserved pathway. Nature Reviews. Genetics, 13(9), 654–666.
Bruckner, K., Perez, L., Clausen, H., & Cohen, S. (2000). Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature, 406(6794), 411–415.
Moloney, D. J., Panin, V. M., Johnston, S. H., Chen, J., Shao, L., Wilson, R., et al. (2000). Fringe is a glycosyltransferase that modifies Notch. Nature, 406(6794), 369–375.
Moloney, D. J., Shair, L. H., Lu, F. M., Xia, J., Locke, R., Matta, K. L., et al. (2000). Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. The Journal of Biological Chemistry, 275(13), 9604–9611.
Okajima, T., Xu, A., & Irvine, K. D. (2003). Modulation of notch-ligand binding by protein O-fucosyltransferase 1 and fringe. The Journal of Biological Chemistry, 278(43), 42340–42345.
Panin, V. M., Papayannopoulos, V., Wilson, R., & Irvine, K. D. (1997). Fringe modulates Notch-ligand interactions. Nature, 387(6636), 908–912.
Cohen, B., Bashirullah, A., Dagnino, L., Campbell, C., Fisher, W. W., Leow, C. C., et al. (1997). Fringe boundaries coincide with Notch-dependent patterning centres in mammals and alter Notch-dependent development in Drosophila. Nature Genetics, 16(3), 283–288.
Pakkiriswami, S., Couto, A., Nagarajan, U., & Georgiou, M. (2016). Glycosylated Notch and cancer. Frontiers in Oncology, 6, 37.
Xu, K., Usary, J., Kousis, P. C., Prat, A., Wang, D. Y., Adams, J. R., et al. (2012). Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell, 21(5), 626–641.
Mukherjee, A., Veraksa, A., Bauer, A., Rosse, C., Camonis, J., & Artavanis-Tsakonas, S. (2005). Regulation of Notch signalling by non-visual beta-arrestin. Nature Cell Biology, 7(12), 1191–1201.
Qiu, L., Joazeiro, C., Fang, N., Wang, H. Y., Elly, C., Altman, Y., et al. (2000). Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. The Journal of Biological Chemistry, 275(46), 35734–35737.
Sakata, T., Sakaguchi, H., Tsuda, L., Higashitani, A., Aigaki, T., Matsuno, K., et al. (2004). Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Current Biology, 14(24), 2228–2236.
Jehn, B. M., Dittert, I., Beyer, S., von der Mark, K., & Bielke, W. (2002). c-Cbl binding and ubiquitin-dependent lysosomal degradation of membrane-associated Notch1. The Journal of Biological Chemistry, 277(10), 8033–8040.
Conner, S. D. (2016). Regulation of Notch signaling through intracellular transport. International Review of Cell and Molecular Biology, 323, 107–127.
Santolini, E., Puri, C., Salcini, A. E., Gagliani, M. C., Pelicci, P. G., Tacchetti, C., et al. (2000). Numb is an endocytic protein. The Journal of Cell Biology, 151(6), 1345–1352.
McGill, M. A., & McGlade, C. J. (2003). Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. The Journal of Biological Chemistry, 278(25), 23196–23203.
Foltz, D. R., Santiago, M. C., Berechid, B. E., & Nye, J. S. (2002). Glycogen synthase kinase-3beta modulates notch signaling and stability. Current Biology, 12(12), 1006–1011.
Ingles-Esteve, J., Espinosa, L., Milner, L. A., Caelles, C., & Bigas, A. (2001). Phosphorylation of Ser2078 modulates the Notch2 function in 32D cell differentiation. The Journal of Biological Chemistry, 276(48), 44873–44880.
Fryer, C. J., White, J. B., & Jones, K. A. (2004). Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Molecular Cell, 16(4), 509–520.
Popko-Scibor, A. E., Lindberg, M. J., Hansson, M. L., Holmlund, T., & Wallberg, A. E. (2011). Ubiquitination of Notch1 is regulated by MAML1-mediated p300 acetylation of Notch1. Biochemical and Biophysical Research Communications, 416(3–4), 300–306.
Palermo, R., Checquolo, S., Giovenco, A., Grazioli, P., Kumar, V., Campese, A. F., et al. (2012). Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene, 31(33), 3807–3817.
Ishitani, T., Hirao, T., Suzuki, M., Isoda, M., Ishitani, S., Harigaya, K., et al. (2010). Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nature Cell Biology, 12(3), 278–285.
Rustighi, A., Tiberi, L., Soldano, A., Napoli, M., Nuciforo, P., Rosato, A., et al. (2009). The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nature Cell Biology, 11(2), 133–142.
Rustighi, A., Zannini, A., Tiberi, L., Sommaggio, R., Piazza, S., Sorrentino, G., et al. (2014). Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Molecular Medicine, 6(1), 99–119.
Baik, S. H., Fane, M., Park, J. H., Cheng, Y. L., Yang-Wei Fann, D., Yun, U. J., et al. (2015). Pin1 promotes neuronal death in stroke by stabilizing Notch intracellular domain. Annals of Neurology, 77(3), 504–516.
Franciosa, G., Diluvio, G., Del Gaudio, F., Giuli, M. V., Palermo, R., Grazioli, P., et al. (2016). Prolyl-isomerase Pin1 controls Notch3 protein expression and regulates T-ALL progression. Oncogene, 35(36), 4741–4751.
Cho, S., Lu, M., He, X., Ee, P. L., Bhat, U., Schneider, E., et al. (2011). Notch1 regulates the expression of the multidrug resistance gene ABCC1/MRP1 in cultured cancer cells. Proceedings of the National Academy of Sciences of the United States of America, 108(51), 20778–20783.
Wang, Z., Li, Y., Ahmad, A., Banerjee, S., Azmi, A. S., Kong, D., et al. (2011). Down-regulation of Notch-1 is associated with Akt and FoxM1 in inducing cell growth inhibition and apoptosis in prostate cancer cells. Journal of Cellular Biochemistry, 112(1), 78–88.
Zhao, B., Zou, J., Wang, H., Johannsen, E., Peng, C. W., Quackenbush, J., et al. (2011). Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proceedings of the National Academy of Sciences of the United States of America, 108(36), 14902–14907.
Brou, C., Logeat, F., Gupta, N., Bessia, C., LeBail, O., Doedens, J. R., et al. (2000). A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Molecular Cell, 5(2), 207–216.
Musse, A. A., Meloty-Kapella, L., & Weinmaster, G. (2012). Notch ligand endocytosis: Mechanistic basis of signaling activity. Seminars in Cell & Developmental Biology, 23(4), 429–436.
Saxena, M. T., Schroeter, E. H., Mumm, J. S., & Kopan, R. (2001). Murine notch homologs (N1-4) undergo presenilin-dependent proteolysis. The Journal of Biological Chemistry, 276(43), 40268–40273.
Schroeter, E. H., Kisslinger, J. A., & Kopan, R. (1998). Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature, 393(6683), 382–386.
Chen, F., Hasegawa, H., Schmitt-Ulms, G., Kawarai, T., Bohm, C., Katayama, T., et al. (2006). TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature, 440(7088), 1208–1212.
Zhang, Y. W., Luo, W. J., Wang, H., Lin, P., Vetrivel, K. S., Liao, F., et al. (2005). Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. The Journal of Biological Chemistry, 280(17), 17020–17026.
Lee, S. F., Shah, S., Yu, C., Wigley, W. C., Li, H., Lim, M., et al. (2004). A conserved GXXXG motif in APH-1 is critical for assembly and activity of the gamma-secretase complex. The Journal of Biological Chemistry, 279(6), 4144–4152.
Prokop, S., Shirotani, K., Edbauer, D., Haass, C., & Steiner, H. (2004). Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. The Journal of Biological Chemistry, 279(22), 23255–23261.
Fortini, M. E., & Artavanis-Tsakonas, S. (1994). The suppressor of hairless protein participates in notch receptor signaling. Cell, 79(2), 273–282.
Wu, L., Aster, J. C., Blacklow, S. C., Lake, R., Artavanis-Tsakonas, S., & Griffin, J. D. (2000). MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nature Genetics, 26(4), 484–489.
Wallberg, A. E., Pedersen, K., Lendahl, U., & Roeder, R. G. (2002). p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Molecular and Cellular Biology, 22(22), 7812–7819.
Kurooka, H., & Honjo, T. (2000). Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. The Journal of Biological Chemistry, 275(22), 17211–17220.
Kitagawa, M. (2016). Notch signalling in the nucleus: Roles of Mastermind-like (MAML) transcriptional coactivators. Journal of Biochemistry, 159(3), 287–294.
Wang, Z., Ahmad, A., Li, Y., Azmi, A. S., Miele, L., & Sarkar, F. H. (2011). Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Research, 31(4), 1105–1113.
Castel, D., Mourikis, P., Bartels, S. J., Brinkman, A. B., Tajbakhsh, S., & Stunnenberg, H. G. (2013). Dynamic binding of RBPJ is determined by Notch signaling status. Genes & Development, 27(9), 1059–1071.
Choi, J. W., Pampeno, C., Vukmanovic, S., & Meruelo, D. (2002). Characterization of the transcriptional expression of Notch-1 signaling pathway members, Deltex and HES-1, in developing mouse thymocytes. Developmental and Comparative Immunology, 26(6), 575–588.
Oswald, F., Liptay, S., Adler, G., & Schmid, R. M. (1998). NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Molecular and Cellular Biology, 18(4), 2077–2088.
Cheng, P., Zlobin, A., Volgina, V., Gottipati, S., Osborne, B., Simel, E. J., et al. (2001). Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. Journal of Immunology, 167(8), 4458–4467.
Rangarajan, A., Talora, C., Okuyama, R., Nicolas, M., Mammucari, C., Oh, H., et al. (2001). Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. The EMBO Journal, 20(13), 3427–3436.
Ronchini, C., & Capobianco, A. J. (2001). Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): Implication for cell cycle disruption in transformation by Notch(ic). Molecular and Cellular Biology, 21(17), 5925–5934.
Weng, A. P., Millholland, J. M., Yashiro-Ohtani, Y., Arcangeli, M. L., Lau, A., Wai, C., et al. (2006). c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes & Development, 20(15), 2096–2109.
Andersen, P., Uosaki, H., Shenje, L. T., & Kwon, C. (2012). Non-canonical Notch signaling: Emerging role and mechanism. Trends in Cell Biology, 22(5), 257–265.
Sanalkumar, R., Dhanesh, S. B., & James, J. (2010). Non-canonical activation of Notch signaling/target genes in vertebrates. Cellular and Molecular Life Sciences, 67(17), 2957–2968.
de Celis, J. F., & Bray, S. (1997). Feed-back mechanisms affecting Notch activation at the dorsoventral boundary in the Drosophila wing. Development, 124(17), 3241–3251.
Li, Y., & Baker, N. E. (2004). The roles of cis-inactivation by Notch ligands and of neuralized during eye and bristle patterning in Drosophila. BMC Developmental Biology, 4, 5.
Miller, A. C., Lyons, E. L., & Herman, T. G. (2009). cis-Inhibition of Notch by endogenous Delta biases the outcome of lateral inhibition. Current Biology, 19(16), 1378–1383.
Becam, I., Fiuza, U. M., Arias, A. M., & Milan, M. (2010). A role of receptor Notch in ligand cis-inhibition in Drosophila. Current Biology, 20(6), 554–560.
Loeb, D. M., Thornton, K., & Shokek, O. (2008). Pediatric soft tissue sarcomas. The Surgical Clinics of North America, 88(3), 615–627. vii.
Tapscott, S. J., Thayer, M. J., & Weintraub, H. (1993). Deficiency in rhabdomyosarcomas of a factor required for MyoD activity and myogenesis. Science, 259(5100), 1450–1453.
De Giovanni, C., Landuzzi, L., Nicoletti, G., Lollini, P. L., & Nanni, P. (2009). Molecular and cellular biology of rhabdomyosarcoma. Future Oncology, 5(9), 1449–1475.
Parham, D. M., & Barr, F. G. (2013). Classification of rhabdomyosarcoma and its molecular basis. Advances in Anatomic Pathology, 20(6), 387–397.
Taulli, R., Bersani, F., Foglizzo, V., Linari, A., Vigna, E., Ladanyi, M., et al. (2009). The muscle-specific microRNA miR-206 blocks human rhabdomyosarcoma growth in xenotransplanted mice by promoting myogenic differentiation. The Journal of Clinical Investigation, 119(8), 2366–2378.
Ciarapica, R., Carcarino, E., Adesso, L., De Salvo, M., Bracaglia, G., Leoncini, P. P., et al. (2014). Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer, 14, 139.
Kikuchi, K., Taniguchi, E., Chen, H. I., Svalina, M. N., Abraham, J., Huang, E. T., et al. (2013). Rb1 loss modifies but does not initiate alveolar rhabdomyosarcoma. Skeletal Muscle, 3(1), 27.
Chen, E. Y., Dobrinski, K. P., Brown, K. H., Clagg, R., Edelman, E., Ignatius, M. S., et al. (2013). Cross-species array comparative genomic hybridization identifies novel oncogenic events in zebrafish and human embryonal rhabdomyosarcoma. PLoS Genetics, 9(8), e1003727.
Hettmer, S., Liu, J., Miller, C. M., Lindsay, M. C., Sparks, C. A., Guertin, D. A., et al. (2011). Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proceedings of the National Academy of Sciences of the United States of America, 108(50), 20002–20007.
Abraham, J., Nunez-Alvarez, Y., Hettmer, S., Carrio, E., Chen, H. I., Nishijo, K., et al. (2014). Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes & Development, 28(14), 1578–1591.
Nitzki, F., Zibat, A., Frommhold, A., Schneider, A., Schulz-Schaeffer, W., Braun, T., et al. (2011). Uncommitted precursor cells might contribute to increased incidence of embryonal rhabdomyosarcoma in heterozygous Patched1-mutant mice. Oncogene, 30(43), 4428–4436.
Hatley, M. E., Tang, W., Garcia, M. R., Finkelstein, D., Millay, D. P., Liu, N., et al. (2012). A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell, 22(4), 536–546.
Meza, J. L., Anderson, J., Pappo, A. S., Meyer, W. H., & Children’s Oncology G. (2006). Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: The Children’s Oncology Group. Journal of Clinical Oncology, 24(24), 3844–3851.
Arndt, C. A., Stoner, J. A., Hawkins, D. S., Rodeberg, D. A., Hayes-Jordan, A. A., Paidas, C. N., et al. (2009). Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. Journal of Clinical Oncology, 27(31), 5182–5188.
Williamson, D., Missiaglia, E., de Reynies, A., Pierron, G., Thuille, B., Palenzuela, G., et al. (2010). Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. Journal of Clinical Oncology, 28(13), 2151–2158.
Chen, X., Stewart, E., Shelat, A. A., Qu, C., Bahrami, A., Hatley, M., et al. (2013). Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell, 24(6), 710–724.
Shern, J. F., Chen, L., Chmielecki, J., Wei, J. S., Patidar, R., Rosenberg, M., et al. (2014). Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discovery, 4(2), 216–231.
Hahn, H., Wojnowski, L., Zimmer, A. M., Hall, J., Miller, G., & Zimmer, A. (1998). Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nature Medicine, 4(5), 619–622.
Zibat, A., Missiaglia, E., Rosenberger, A., Pritchard-Jones, K., Shipley, J., Hahn, H., et al. (2010). Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene, 29(48), 6323–6330.
Nitzki, F., Cuvelier, N., Drager, J., Schneider, A., Braun, T., & Hahn, H. (2016). Hedgehog/Patched-associated rhabdomyosarcoma formation from delta1-expressing mesodermal cells. Oncogene, 35(22), 2923–2931.
Pressey, J. G., Anderson, J. R., Crossman, D. K., Lynch, J. C., & Barr, F. G. (2011). Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: A report from the Children’s Oncology Group. Pediatric Blood & Cancer, 57(6), 930–938.
Kohsaka, S., Shukla, N., Ameur, N., Ito, T., Ng, C. K., Wang, L., et al. (2014). A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K-AKT pathway mutations. Nature Genetics, 46(6), 595–600.
Davicioni, E., Anderson, J. R., Buckley, J. D., Meyer, W. H., & Triche, T. J. (2010). Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: A report from the children’s oncology group. Journal of Clinical Oncology, 28(7), 1240–1246.
Galili, N., Davis, R. J., Fredericks, W. J., Mukhopadhyay, S., Rauscher, F. J., 3rd, Emanuel, B. S., et al. (1993). Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nature Genetics, 5(3), 230–235.
Davis, R. J., D’Cruz, C. M., Lovell, M. A., Biegel, J. A., & Barr, F. G. (1994). Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Research, 54(11), 2869–2872.
Marshall, A. D., Picchione, F., Geltink, R. I., & Grosveld, G. C. (2013). PAX3-FOXO1 induces up-regulation of Noxa sensitizing alveolar rhabdomyosarcoma cells to apoptosis. Neoplasia, 15(7), 738–748.
Missiaglia, E., Williamson, D., Chisholm, J., Wirapati, P., Pierron, G., Petel, F., et al. (2012). PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. Journal of Clinical Oncology, 30(14), 1670–1677.
Mourikis, P., & Tajbakhsh, S. (2014). Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Developmental Biology, 14, 2.
Francis, P., Namlos, H. M., Muller, C., Eden, P., Fernebro, J., Berner, J. M., et al. (2007). Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: Hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics, 8, 73.
Terry, J., Saito, T., Subramanian, S., Ruttan, C., Antonescu, C. R., Goldblum, J. R., et al. (2007). TLE1 as a diagnostic immunohistochemical marker for synovial sarcoma emerging from gene expression profiling studies. The American Journal of Surgical Pathology, 31(2), 240–246.
Jagdis, A., Rubin, B. P., Tubbs, R. R., Pacheco, M., & Nielsen, T. O. (2009). Prospective evaluation of TLE1 as a diagnostic immunohistochemical marker in synovial sarcoma. The American Journal of Surgical Pathology, 33(12), 1743–1751.
Su, L., Sampaio, A. V., Jones, K. B., Pacheco, M., Goytain, A., Lin, S., et al. (2012). Deconstruction of the SS18-SSX fusion oncoprotein complex: Insights into disease etiology and therapeutics. Cancer Cell, 21(3), 333–347.
May, W. A., Arvand, A., Thompson, A. D., Braun, B. S., Wright, M., & Denny, C. T. (1997). EWS/FLI1-induced manic fringe renders NIH 3T3 cells tumorigenic. Nature Genetics, 17(4), 495–497.
Baliko, F., Bright, T., Poon, R., Cohen, B., Egan, S. E., & BA, A. (2007). Inhibition of notch signaling induces neural differentiation in Ewing sarcoma. The American Journal of Pathology, 170(5), 1686–1694.
Ban, J., Bennani-Baiti, I. M., Kauer, M., Schaefer, K. L., Poremba, C., Jug, G., et al. (2008). EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Research, 68(17), 7100–7109.
Bennani-Baiti, I. M., Aryee, D. N., Ban, J., Machado, I., Kauer, M., Muhlbacher, K., et al. (2011). Notch signalling is off and is uncoupled from HES1 expression in Ewing’s sarcoma. The Journal of Pathology, 225(3), 353–363.
Ban, J., Aryee, D. N., Fourtouna, A., van der Ent, W., Kauer, M., Niedan, S., et al. (2014). Suppression of deacetylase SIRT1 mediates tumor-suppressive NOTCH response and offers a novel treatment option in metastatic Ewing sarcoma. Cancer Research, 74(22), 6578–6588.
Ventura, S., Aryee, D. N., Felicetti, F., De Feo, A., Mancarella, C., Manara, M. C., et al. (2016). CD99 regulates neural differentiation of Ewing sarcoma cells through miR-34a-Notch-mediated control of NF-kappaB signaling. Oncogene, 35(30), 3944–3954.
Sang, L., Coller, H. A., & Roberts, J. M. (2008). Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science, 321(5892), 1095–1100.
Roma, J., Masia, A., Reventos, J., Sanchez de Toledo, J., & Gallego, S. (2011). Notch pathway inhibition significantly reduces rhabdomyosarcoma invasiveness and mobility in vitro. Clinical Cancer Research, 17(3), 505–513.
Belyea, B. C., Naini, S., Bentley, R. C., & Linardic, C. M. (2011). Inhibition of the Notch-Hey1 axis blocks embryonal rhabdomyosarcoma tumorigenesis. Clinical Cancer Research, 17(23), 7324–7336.
Raimondi, L., Ciarapica, R., De Salvo, M., Verginelli, F., Gueguen, M., Martini, C., et al. (2012). Inhibition of Notch3 signalling induces rhabdomyosarcoma cell differentiation promoting p38 phosphorylation and p21(Cip1) expression and hampers tumour cell growth in vitro and in vivo. Cell Death and Differentiation, 19(5), 871–881.
Nagao, H., Setoguchi, T., Kitamoto, S., Ishidou, Y., Nagano, S., Yokouchi, M., et al. (2012). RBPJ is a novel target for rhabdomyosarcoma therapy. PLoS One, 7(7), e39268.
De Salvo, M., Raimondi, L., Vella, S., Adesso, L., Ciarapica, R., Verginelli, F., et al. (2014). Hyper-activation of Notch3 amplifies the proliferative potential of rhabdomyosarcoma cells. PLoS One, 9(5), e96238.
Diao, Y., Guo, X., Jiang, L., Wang, G., Zhang, C., Wan, J., et al. (2014). miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. The Journal of Biological Chemistry, 289(1), 529–539.
Kitagawa, M., Oyama, T., Kawashima, T., Yedvobnick, B., Kumar, A., Matsuno, K., et al. (2001). A human protein with sequence similarity to Drosophila mastermind coordinates the nuclear form of notch and a CSL protein to build a transcriptional activator complex on target promoters. Molecular and Cellular Biology, 21(13), 4337–4346.
Hu, Y. Y., Zheng, M. H., Zhang, R., Liang, Y. M., & Han, H. (2012). Notch signaling pathway and cancer metastasis. Advances in Experimental Medicine and Biology, 727, 186–198.
Liu, Z. H., Dai, X. M., & Du, B. (2015). Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biology & Therapy, 16(3), 353–359.
Weidle, U. H., Birzele, F., & Kruger, A. (2015). Molecular targets and pathways involved in liver metastasis of colorectal cancer. Clinical & Experimental Metastasis, 32(6), 623–635.
Masia, A., Almazan-Moga, A., Velasco, P., Reventos, J., Toran, N., Sanchez de Toledo, J., et al. (2012). Notch-mediated induction of N-cadherin and alpha9-integrin confers higher invasive phenotype on rhabdomyosarcoma cells. British Journal of Cancer, 107(8), 1374–1383.
Liu, Z. J., Xiao, M., Balint, K., Smalley, K. S., Brafford, P., Qiu, R., et al. (2006). Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Research, 66(8), 4182–4190.
Wang, T., Holt, C. M., Xu, C., Ridley, C., POJ, R., Baron, M., et al. (2007). Notch3 activation modulates cell growth behaviour and cross-talk to Wnt/TCF signalling pathway. Cellular Signalling, 19(12), 2458–2467.
Shukla, N., Ameur, N., Yilmaz, I., Nafa, K., Lau, C. Y., Marchetti, A., et al. (2012). Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clinical Cancer Research, 18(3), 748–757.
Gustafsson, M. K., Pan, H., Pinney, D. F., Liu, Y., Lewandowski, A., Epstein, D. J., et al. (2002). Myf5 is a direct target of long-range Shh signaling and Gli regulation for muscle specification. Genes & Development, 16(1), 114–126.
Straface, G., Aprahamian, T., Flex, A., Gaetani, E., Biscetti, F., Smith, R. C., et al. (2009). Sonic hedgehog regulates angiogenesis and myogenesis during post-natal skeletal muscle regeneration. Journal of Cellular and Molecular Medicine, 13(8B), 2424–2435.
Koleva, M., Kappler, R., Vogler, M., Herwig, A., Fulda, S., & Hahn, H. (2005). Pleiotropic effects of sonic hedgehog on muscle satellite cells. Cellular and Molecular Life Sciences, 62(16), 1863–1870.
Bren-Mattison, Y., & Olwin, B. B. (2002). Sonic hedgehog inhibits the terminal differentiation of limb myoblasts committed to the slow muscle lineage. Developmental Biology, 242(2), 130–148.
Hahn, H., Nitzki, F., Schorban, T., Hemmerlein, B., Threadgill, D., & Rosemann, M. (2004). Genetic mapping of a Ptch1-associated rhabdomyosarcoma susceptibility locus on mouse chromosome 2. Genomics, 84(5), 853–858.
Calzada-Wack, J., Schnitzbauer, U., Walch, A., Wurster, K. H., Kappler, R., Nathrath, M., et al. (2002). Analysis of the PTCH coding region in human rhabdomyosarcoma. Human Mutation, 20(3), 233–234.
Bridge, J. A., Liu, J., Weibolt, V., Baker, K. S., Perry, D., Kruger, R., et al. (2000). Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: An intergroup rhabdomyosarcoma study. Genes, Chromosomes & Cancer, 27(4), 337–344.
Tostar, U., Malm, C. J., Meis-Kindblom, J. M., Kindblom, L. G., Toftgard, R., & Unden, A. B. (2006). Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. The Journal of Pathology, 208(1), 17–25.
Rubin, B. P., Nishijo, K., Chen, H. I., Yi, X., Schuetze, D. P., Pal, R., et al. (2011). Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell, 19(2), 177–191.
Ingram, W. J., McCue, K. I., Tran, T. H., Hallahan, A. R., & Wainwright, B. J. (2008). Sonic Hedgehog regulates Hes1 through a novel mechanism that is independent of canonical Notch pathway signalling. Oncogene, 27(10), 1489–1500.
Esiashvili, N., Goodman, M., & Marcus, R. B., Jr. (2008). Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. Journal of Pediatric Hematology/Oncology, 30(6), 425–430.
van den Berg, H., Dirksen, U., Ranft, A., & Jurgens, H. (2008). Ewing tumors in infants. Pediatric Blood & Cancer, 50(4), 761–764.
Grier, H. E., Krailo, M. D., Tarbell, N. J., Link, M. P., Fryer, C. J., Pritchard, D. J., et al. (2003). Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. The New England Journal of Medicine, 348(8), 694–701.
Womer, R. B., West, D. C., Krailo, M. D., Dickman, P. S., Pawel, B. R., Grier, H. E., et al. (2012). Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children’s Oncology Group. Journal of Clinical Oncology, 30(33), 4148–4154.
Gaspar, N., Hawkins, D. S., Dirksen, U., Lewis, I. J., Ferrari, S., Le Deley, M. C., et al. (2015). Ewing sarcoma: Current management and future approaches through collaboration. Journal of Clinical Oncology, 33(27), 3036–3046.
Roberts, P., Burchill, S. A., Brownhill, S., Cullinane, C. J., Johnston, C., Griffiths, M. J., et al. (2008). Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing’s sarcoma family of tumors: A study by the United Kingdom Cancer Cytogenetics and the Children’s Cancer and Leukaemia Group. Genes, Chromosomes & Cancer, 47(3), 207–220.
Postel-Vinay, S., Veron, A. S., Tirode, F., Pierron, G., Reynaud, S., Kovar, H., et al. (2012). Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nature Genetics, 44(3), 323–327.
Tuna, M., Ju, Z., CI, A., & Mills, G. B. (2012). Soft tissue sarcoma subtypes exhibit distinct patterns of acquired uniparental disomy. BMC Medical Genomics, 5, 60.
Delattre, O., Zucman, J., Plougastel, B., Desmaze, C., Melot, T., Peter, M., et al. (1992). Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature, 359(6391), 162–165.
May, W. A., Gishizky, M. L., Lessnick, S. L., Lunsford, L. B., Lewis, B. C., Delattre, O., et al. (1993). Ewing sarcoma 11;22 translocation produces a chimeric transcription factor that requires the DNA-binding domain encoded by FLI1 for transformation. Proceedings of the National Academy of Sciences of the United States of America, 90(12), 5752–5756.
Lawlor, E. R., & Sorensen, P. H. (2015). Twenty years on: What do we really know about Ewing sarcoma and what is the path forward? Critical Reviews in Oncogenesis, 20(3–4), 155–171.
van Doorninck, J. A., Ji, L., Schaub, B., Shimada, H., Wing, M. R., Krailo, M. D., et al. (2010). Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: A report from the Children’s Oncology Group. Journal of Clinical Oncology, 28(12), 1989–1994.
Le Deley, M. C., Delattre, O., Schaefer, K. L., Burchill, S. A., Koehler, G., Hogendoorn, P. C., et al. (2010). Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: Prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. Journal of Clinical Oncology, 28(12), 1982–1988.
Riggi, N., Cironi, L., Provero, P., Suva, M. L., Kaloulis, K., Garcia-Echeverria, C., et al. (2005). Development of Ewing’s sarcoma from primary bone marrow-derived mesenchymal progenitor cells. Cancer Research, 65(24), 11459–11468.
Riggi, N., Suva, M. L., & Stamenkovic, I. (2009). Ewing’s sarcoma origin: From duel to duality. Expert Review of Anticancer Therapy, 9(8), 1025–1030.
Riggi, N., Suva, M. L., De Vito, C., Provero, P., Stehle, J. C., Baumer, K., et al. (2010). EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes & Development, 24(9), 916–932.
von Levetzow, C., Jiang, X., Gwye, Y., von Levetzow, G., Hung, L., Cooper, A., et al. (2011). Modeling initiation of Ewing sarcoma in human neural crest cells. PLoS One, 6(4), e19305.
May, W. A., Lessnick, S. L., Braun, B. S., Klemsz, M., Lewis, B. C., Lunsford, L. B., et al. (1993). The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Molecular and Cellular Biology, 13(12), 7393–7398.
Bilke, S., Schwentner, R., Yang, F., Kauer, M., Jug, G., Walker, R. L., et al. (2013). Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Research, 23(11), 1797–1809.
Patel, M., Simon, J. M., Iglesia, M. D., Wu, S. B., McFadden, A. W., Lieb, J. D., et al. (2012). Tumor-specific retargeting of an oncogenic transcription factor chimera results in dysregulation of chromatin and transcription. Genome Research, 22(2), 259–270.
Owen, L. A., Kowalewski, A. A., & Lessnick, S. L. (2008). EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS One, 3(4), e1965.
Stoll, G., Surdez, D., Tirode, F., Laud, K., Barillot, E., Zinovyev, A., et al. (2013). Systems biology of Ewing sarcoma: A network model of EWS-FLI1 effect on proliferation and apoptosis. Nucleic Acids Research, 41(19), 8853–8871.
Zwerner, J. P., Guimbellot, J., & May, W. A. (2003). EWS/FLI function varies in different cellular backgrounds. Experimental Cell Research, 290(2), 414–419.
Smith, R., Owen, L. A., Trem, D. J., Wong, J. S., Whangbo, J. S., Golub, T. R., et al. (2006). Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell, 9(5), 405–416.
Bennani-Baiti, I. M., Machado, I., Llombart-Bosch, A., & Kovar, H. (2012). Lysine-specific demethylase 1 (LSD1/KDM1A/AOF2/BHC110) is expressed and is an epigenetic drug target in chondrosarcoma, Ewing’s sarcoma, osteosarcoma, and rhabdomyosarcoma. Human Pathology, 43(8), 1300–1307.
Mulligan, P., Yang, F., Di Stefano, L., Ji, J. Y., Ouyang, J., Nishikawa, J. L., et al. (2011). A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Molecular Cell, 42(5), 689–699.
Wang, J., Scully, K., Zhu, X., Cai, L., Zhang, J., Prefontaine, G. G., et al. (2007). Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature, 446(7138), 882–887.
Di Stefano, L., Walker, J. A., Burgio, G., Corona, D. F., Mulligan, P., Naar, A. M., et al. (2011). Functional antagonism between histone H3K4 demethylases in vivo. Genes & Development, 25(1), 17–28.
Rocchi, A., Manara, M. C., Sciandra, M., Zambelli, D., Nardi, F., Nicoletti, G., et al. (2010). CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. The Journal of Clinical Investigation, 120(3), 668–680.
Schenkel, A. R., Mamdouh, Z., Chen, X., Liebman, R. M., & Muller, W. A. (2002). CD99 plays a major role in the migration of monocytes through endothelial junctions. Nature Immunology, 3(2), 143–150.
Alberti, I., Bernard, G., Rouquette-Jazdanian, A. K., Pelassy, C., Pourtein, M., Aussel, C., et al. (2002). CD99 isoforms expression dictates T cell functional outcomes. The FASEB Journal, 16(14), 1946–1948.
Miyagawa, Y., Okita, H., Nakaijima, H., Horiuchi, Y., Sato, B., Taguchi, T., et al. (2008). Inducible expression of chimeric EWS/ETS proteins confers Ewing’s family tumor-like phenotypes to human mesenchymal progenitor cells. Molecular and Cellular Biology, 28(7), 2125–2137.
Hu-Lieskovan, S., Zhang, J., Wu, L., Shimada, H., Schofield, D. E., & Triche, T. J. (2005). EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Research, 65(11), 4633–4644.
Nakatani, F., Ferracin, M., Manara, M. C., Ventura, S., Del Monaco, V., Ferrari, S., et al. (2012). miR-34a predicts survival of Ewing’s sarcoma patients and directly influences cell chemo-sensitivity and malignancy. The Journal of Pathology, 226(5), 796–805.
Marino, M. T., Grilli, A., Baricordi, C., Manara, M. C., Ventura, S., Pinca, R. S., et al. (2014). Prognostic significance of miR-34a in Ewing sarcoma is associated with cyclin D1 and ki-67 expression. Annals of Oncology, 25(10), 2080–2086.
Li, Y., Guessous, F., Zhang, Y., Dipierro, C., Kefas, B., Johnson, E., et al. (2009). MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Research, 69(19), 7569–7576.
Bu, P., Chen, K. Y., Chen, J. H., Wang, L., Walters, J., Shin, Y. J., et al. (2013). A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell, 12(5), 602–615.
Osipo, C., Golde, T. E., Osborne, B. A., & Miele, L. A. (2008). Off the beaten pathway: The complex cross talk between Notch and NF-kappaB. Laboratory Investigation, 88(1), 11–17.
Ducimetiere, F., Lurkin, A., Ranchere-Vince, D., Decouvelaere, A. V., Péoc'h, M., Istier, L., et al. (2011). Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One, 6(8), e20294.
Palmerini, E., Paioli, A., & Ferrari, S. (2014). Emerging therapeutic targets for synovial sarcoma. Expert Review of Anticancer Therapy, 14(7), 791–806.
Nielsen, T. O., Poulin, N. M., & Ladanyi, M. (2015). Synovial sarcoma: Recent discoveries as a roadmap to new avenues for therapy. Cancer Discovery, 5(2), 124–134.
Herzog, C. E. (2005). Overview of sarcomas in the adolescent and young adult population. Journal of Pediatric Hematology/Oncology, 27(4), 215–218.
Sultan, I., Rodriguez-Galindo, C., Saab, R., Yasir, S., Casanova, M., & Ferrari, A. (2009). Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: An analysis of 1268 patients. Cancer, 115(15), 3537–3547.
Nagai, M., Tanaka, S., Tsuda, M., Endo, S., Kato, H., Sonobe, H., et al. (2001). Analysis of transforming activity of human synovial sarcoma-associated chimeric protein SYT-SSX1 bound to chromatin remodeling factor hBRM/hSNF2 alpha. Proceedings of the National Academy of Sciences of the United States of America, 98(7), 3843–3848.
Carmody Soni, E. E., Schlottman, S., Erkizan, H. V., Uren, A., & Toretsky, J. A. (2014). Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clinical Orthopaedics and Related Research, 472(3), 874–882.
Naka, N., Takenaka, S., Araki, N., Miwa, T., Hashimoto, N., Yoshioka, K., et al. (2010). Synovial sarcoma is a stem cell malignancy. Stem Cells, 28(7), 1119–1131.
Cironi, L., Provero, P., Riggi, N., Janiszewska, M., Suva, D., Suva, M. L., et al. (2009). Epigenetic features of human mesenchymal stem cells determine their permissiveness for induction of relevant transcriptional changes by SYT-SSX1. PLoS One, 4(11), e7904.
Haldar, M., Randall, R. L., & Capecchi, M. R. (2008). Synovial sarcoma: From genetics to genetic-based animal modeling. Clinical Orthopaedics and Related Research, 466(9), 2156–2167.
Panagopoulos, I., Mertens, F., Isaksson, M., Limon, J., Gustafson, P., Skytting, B., et al. (2001). Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes, Chromosomes & Cancer, 31(4), 362–372.
Zollner, S. K., Rossig, C., & Toretsky, J. A. (2015). Synovial sarcoma is a gateway to the role of chromatin remodeling in cancer. Cancer Metastasis Reviews, 34(3), 417–428.
Thaete, C., Brett, D., Monaghan, P., Whitehouse, S., Rennie, G., Rayner, E., et al. (1999). Functional domains of the SYT and SYT-SSX synovial sarcoma translocation proteins and co-localization with the SNF protein BRM in the nucleus. Human Molecular Genetics, 8(4), 585–591.
Kato, H., Tjernberg, A., Zhang, W., Krutchinsky, A. N., An, W., Takeuchi, T., et al. (2002). SYT associates with human SNF/SWI complexes and the C-terminal region of its fusion partner SSX1 targets histones. The Journal of Biological Chemistry, 277(7), 5498–5505.
Middeljans, E., Wan, X., Jansen, P. W., Sharma, V., Stunnenberg, H. G., & Logie, C. (2012). SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS One, 7(3), e33834.
Kadoch, C., & Crabtree, G. R. (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell, 153(1), 71–85.
Soulez, M., Saurin, A. J., Freemont, P. S., & Knight, J. C. (1999). SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene, 18(17), 2739–2746.
Grbavec, D., & Stifani, S. (1996). Molecular interaction between TLE1 and the carboxyl-terminal domain of HES-1 containing the WRPW motif. Biochemical and Biophysical Research Communications, 223(3), 701–705.
Su, L., Cheng, H., Sampaio, A. V., Nielsen, T. O., & Underhill, T. M. (2010). EGR1 reactivation by histone deacetylase inhibitors promotes synovial sarcoma cell death through the PTEN tumor suppressor. Oncogene, 29(30), 4352–4361.
Valente, A. L., Tull, J., & Zhang, S. (2013). Specificity of TLE1 expression in unclassified high-grade sarcomas for the diagnosis of synovial sarcoma. Applied Immunohistochemistry & Molecular Morphology, 21(5), 408–413.
Macy, M. E., Sawczyn, K. K., Garrington, T. P., Graham, D. K., & Gore, L. (2008). Pediatric developmental therapies: Interesting new drugs now in early-stage clinical trials. Current Oncology Reports, 10(6), 477–490.
Zweidler-McKay, P. A. (2008). Notch signaling in pediatric malignancies. Current Oncology Reports, 10(6), 459–468.
Fouladi, M., Stewart, C. F., Olson, J., Wagner, L. M., Onar-Thomas, A., Kocak, M., et al. (2011). Phase I trial of MK-0752 in children with refractory CNS malignancies: A pediatric brain tumor consortium study. Journal of Clinical Oncology, 29(26), 3529–3534.
Luistro, L., He, W., Smith, M., Packman, K., Vilenchik, M., Carvajal, D., et al. (2009). Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Research, 69(19), 7672–7680.
Messersmith, W. A., Shapiro, G. I., Cleary, J. M., Jimeno, A., Dasari, A., Huang, B., et al. (2015). A Phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. Clinical Cancer Research, 21(1), 60–67.
De Strooper, B., Iwatsubo, T., & Wolfe, M. S. (2012). Presenilins and gamma-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 2(1), a006304.
Bigas, A., Martin, D. I., & Milner, L. A. (1998). Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Molecular and Cellular Biology, 18(4), 2324–2333.
Fan, X., Mikolaenko, I., Elhassan, I., Ni, X., Wang, Y., Ball, D., et al. (2004). Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Research, 64(21), 7787–7793.
Shimizu, K., Chiba, S., Saito, T., Kumano, K., Hamada, Y., & Hirai, H. (2002). Functional diversity among Notch1, Notch2, and Notch3 receptors. Biochemical and Biophysical Research Communications, 291(4), 775–779.
Nefedova, Y., Cheng, P., Alsina, M., Dalton, W. S., & Gabrilovich, D. I. (2004). Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood, 103(9), 3503–3510.
Graziani, I., Eliasz, S., De Marco, M. A., Chen, Y., Pass, H. I., De May, R. M., et al. (2008). Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Research, 68(23), 9678–9685.
Sun, Y., Lowther, W., Kato, K., Bianco, C., Kenney, N., Strizzi, L., et al. (2005). Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene, 24(34), 5365–5374.
Verginelli, F., Adesso, L., Limon, I., Alisi, A., Gueguen, M., Panera, N., et al. (2015). Activation of an endothelial Notch1-Jagged1 circuit induces VCAM1 expression, an effect amplified by interleukin-1beta. Oncotarget, 6(41), 43216–43229.
Chiorean, E. G., LoRusso, P., Strother, R. M., Diamond, J. R., Younger, A., Messersmith, W. A., et al. (2015). A phase I first-in-human study of enoticumab (REGN421), a fully human Delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clinical Cancer Research, 21(12), 2695–2703.
Weng, A. P., Nam, Y., Wolfe, M. S., Pear, W. S., Griffin, J. D., Blacklow, S. C., et al. (2003). Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Molecular and Cellular Biology, 23(2), 655–664.
Moellering, R. E., Cornejo, M., Davis, T. N., Del Bianco, C., Aster, J. C., Blacklow, S. C., et al. (2009). Direct inhibition of the NOTCH transcription factor complex. Nature, 462(7270), 182–188.
Funahashi, Y., Hernandez, S. L., Das, I., Ahn, A., Huang, J., Vorontchikhina, M., et al. (2008). A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Research, 68(12), 4727–4735.
Varnum-Finney, B., Wu, L., Yu, M., Brashem-Stein, C., Staats, S., Flowers, D., et al. (2000). Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. Journal of Cell Science, 113(Pt 23), 4313–4318.
Small, D., Kovalenko, D., Kacer, D., Liaw, L., Landriscina, M., Di Serio, C., et al. (2001). Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src-dependent chord-like phenotype. The Journal of Biological Chemistry, 276(34), 32022–32030.
Six, E., Ndiaye, D., Laabi, Y., Brou, C., Gupta-Rossi, N., Israel, A., et al. (2003). The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and gamma-secretase. Proceedings of the National Academy of Sciences of the United States of America, 100(13), 7638–7643.
LaVoie, M. J., Fraering, P. C., Ostaszewski, B. L., Ye, W., Kimberly, W. T., Wolfe, M. S., et al. (2003). Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. The Journal of Biological Chemistry, 278(39), 37213–37222.
Acknowledgments
This work was financially supported by the Italian Association for Cancer Research (AIRC IG 15312).
Conflict of Interest/Disclosures
No conflict of interest
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
Cossetti, C., Gualtieri, A., Pomella, S., Carcarino, E., Rota, R. (2018). Notch Signaling in Pediatric Soft Tissue Sarcoma. In: Miele, L., Artavanis-Tsakonas, S. (eds) Targeting Notch in Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-8859-4_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-8859-4_11
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-8857-0
Online ISBN: 978-1-4939-8859-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)