
Abstract
We here summarize the current view of molecular mechanisms involved in the dissemination process of colorectal cancer cells to the liver as deduced from preclinical models. We focus on molecular aspects of the current understanding of the biology of liver metastases formation and survival, both being crucial for identification and validation of possible therapeutic targets and review the latest findings elucidating some features of the liver as a metastatic niche. In more detail, we outline the role of proteases and of major pathways such asc-MET signaling and its modulation by factors such as MACC1 and TIMP1, as well as Notch and TGFβ signaling. The relevance of these signalling pathways during tumor-stroma interactions in this context will be addressed. In addition, the functional role and validation of targets such as PRL3, Trop-2, L1CAM, S100A4, S100P, CD133, LIPC, and APOBEC3G are summarized.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
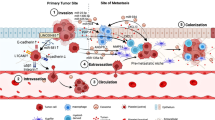
Specific molecular alterations occurring during progression of adenomas to metastatic colorectal cancer (CRC) were elucidated by the pioneering work of Kinzler and Vogelstein [1, 2]. CRC became a role model for molecular events determining the progression of transformed epithelial cells to metastatic carcinoma cells after initiation and subsequent acquisition of multiple tumor-associated mutations due to genomic instability [3]. Depending on the subtype of CRC, specific patterns of chromosomal instability, DNA repair defects or aberrant DNA methylation can be distinguished [3]. These changes culminate in metastasis, which is the cause for the high death toll in patients with CRC [4]. The liver is the major target organ of CRC metastasis as indicated by an investigation covering 4000 tumor patients displaying 11 subtypes of adenocarcinomas [5], however also peritoneal metastasis is not infrequent. While the liver is affected by metastasis in 75 % of these cases, lung metastases were found in only 20 % of these patients [5]. This preference of CRC metastasis can at least partly be explained by the anatomical layout of the vasculature which drains most colon cancer cells through the mesenteric circulation into the liver [6, 7], assuming that the trapping of metastasizing colon cancer cells within the capillary bed of the liver seconds after leaving the colon is a determining mechanism [7, 8]. Indeed, the metastatic dissemination of CRC cells seems to follow, at least in part, a circulation-dependent chronological pattern causing the appearance of liver metastases well before the formation of lung metastases [6]. However, molecular mechanisms may complement or may even overrule the significance of these anatomical aspects according to the well-established ‘seed and soil’ hypothesis [9] that was recently expanded to the concept of pre-metastatic niche formation [10]. The latter addresses the cellular and molecular changes that may occur in the target tissue of metastasis upon the activity of circulating signaling factors as free molecules or packed in exosomes [10] well before a tumor disseminates from the primary site, rendering the target site susceptible to metastatic cells. We here focus on targets that favour dissemination of CRC to the liver (Fig. 1) and discuss approaches for therapeutic intervention. Experimental models are based on syngeneic or xenograft models with a focus on orthotopic liver metastasis models for CRC cells. In a few cases models for experimental metastasis or subcutaneous models are discussed. For target identification and validation CRC cell lines with high tropism of metastasis to the liver in comparison to matching cell lines with low metastatic propensity, have been used. We also describe targets which have been identified by transcriptional profiling of primary CRC versus corresponding liver metastases. We discuss intracellular, plasma-membrane associated, trans-membrane and secreted proteins as targets. They can be derived from tumor cells, but also from stromal cells such as pericytes and fibroblasts.

Schematic display of CRC-related metastasis targets. The circle represents a CRC cell with intracellular, transmembrane and plasma-membrane associated genes. Secreted targets can be derived from tumor cells, stromal cells, or both, depending on the specific target. Abbreviations are explained in the text. ADAM 10 a disintegrin and metalloproteinase 10, APOBEC3G apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G, CD63, CD133 cluster of differentiation 63, 133, CXCR4 CXC chemokine receptor 4, c-MET tyrosine kinase c-MET, HGF hepatocyte growth factor, HIF-1α hypoxia-inducible factor-1α, LIP-C lipase C, L1CAM L1 cell adhesion molecule, MACC1 metastasis associated in colon cancer 1, MMPs matrix metalloproteinases, Notch 1–4 Notch receptors 1–4, PRL3 phosphatase of regenerating liver 3, SDF-1 stromal cell-derived factor-1, S100A4, S100A8, S100P S100 family proteins A4, A8 and 100P, TGFβ transforming growth factor β, TIMP1 tissue inhibitor of metalloproteinase 1, Trop-2 tumor-associated signal transducer 2
Dissemination of CRC to the liver
Metastasis is mediated by an orderly sequence of steps comprising local invasion, intravasation into blood or lymph vessels, survival in the circulation, homing and extravasation, survival of dormancy, and colony formation [11]. As it became evident that anatomical conditions cannot always explain efficacy and pattern of metastasis, it was postulated that the compatibility between the seed (disseminated tumor cells) and the soil (distant sites) are essential for the metastatic process [11]. Metastasis initiation mostly involves remodeling of extracellular matrix (ECM) molecules which compose the basement membrane of epithelial tissues including the colon [12]. Furthermore, metastatic cells have to acquire a migratory phenotype as a pre-requisite for the mobility that underlies initial steps of metastasis [12]. These molecular and morphological changes arise as a consequence of the constant and evolving cross-talk with stromal cells [12], which is critical for the survival of metastatic cells at this time point [12]. In CRC it was consequently found that tumor cells at the invasive front accumulate β-catenin in the nucleus triggering their self-renewal capacity [13–15]. Once in the circulation, CRC cells are able to bind to and cover themselves with platelets leading to better protection from shear stress and immune response [16] and also increasing the likelihood of emboli formation in capillary beds of a possible target organ of metastasis at a distant site [16]. In this process, further pathways are activated which promote specific attachment of these tumor cell/platelet aggregates to endothelial cells [16] rather than just the formation of emboli. In the liver sinusoids, the specific ‘vessels’ in the liver microenvironment, the tumor cells are confronted with different cells that may exhibit some anti-tumoral/anti-metastatic activity which would represent another critical step that successful metastatic tumor cells have to overcome [17–19]. These obstacles include Kupffer cells, specialized macrophages lining the walls of the sinusoids [17] and pit cells, large granular lymphocytes with high cytotoxic activity against tumor cells [18]. In addition, hepatic stellate cells in the peri-sinusoidal space can act as antigen-presenting cells which may elicit an anti-tumoral immune response [19]. After extravasation from the sinusoids, colonization of the liver parenchyma is the final step of metastasis to the liver. In this process it is essential that the tumor cells undergo mesenchymal-epithelial transition (MET), the transition from a spindle-shaped mesenchymal phenotype to a polarized, epithelial phenotype with expression of adhesion molecules allowing successful establishment of a metastatic colony in the new environment [19]. It is still under investigation, how the rather newly coined migrating cancer stem cells (MCSC) surpass these different steps of metastasis and whether their mode of action decisively differs from the current concept [19]. Along these steps of the metastatic cascade, one can categorize the metastasis-associated genes in three groups contributing to metastasis initiation, progression, and virulence, respectively [11, 20, 21]. Those genes with metastasis-initiating function ensure self-renewal, resistance to intermittent hypoxia, migration, and invasion, as well as infiltration of pro-tumorigenic bone marrow-derived cells at the primary site and survival in the circulation. Genes ensuring metastasis progression mediate homing, adhesion and extravasation at the distant site as well as survival of dormancy, re-initiation of proliferation and possibly angiogenesis. The virulence-determining genes can be considered as those determining organ-specific colonization or those emerging genes which are expressed and secreted at the primary site by specific tumors or tumor host-interactions, whose gene products are secreted and act over a distance in future target organs of metastasis in order to create a pre-metastatic niche even before the arrival of tumor cells. The set of responsible genes is likely triggered by the specific type of tumor and the distant sites to be colonized, but probably there are also genes that are generically involved in this process.
Metastasis models
The first crucial step in the development of CRC liver metastasis models was the demonstration of metastasis to the liver of CRC cells implanted into the spleen of nude mice [22] in order to recapitulate the observations in the clinic. Subsequently, highly metastatic CRC cells were selected after implantation of surgical specimen into the subcutis, caecum or spleen and serial passage of cancer cells derived from liver metastases [23]. Tumor cell variants with high metastatic potential reveal higher rates of experimental liver metastases (KM12L4 and KM12SM), while higher rates of spontaneous liver metastasis formation were derived from the parental cell line KM12 [23]. Orthotopic implantation of human carcinomas into nude mice provided reliable models for studies of the biology and therapy of metastasis [24]. A crucial finding was the demonstration that the response of tumors to chemotherapy was modulated by the microenvironment by organ-specific upregulation of multi-drug resistance associated P-glycoprotein [25, 26]. Examination of these models revealed several targets for possible therapeutic intervention. It was shown that expression of epidermal growth factor receptor (EGFR) in CRC cells correlates with their potential to form hepatic metastases [27]. Basic fibroblast growth factor (bFGF), interleukin 8 (IL8), and matrix metalloproteinase 9 (MMP9) [28, 29] have emerged as further targets for treatment of liver metastases derived from CRC. The importance of inhibition of platelet-derived growth factor receptor (PDGFR) signaling pathways in tumor-associated endothelial cells and pericytes for inhibition of CRC liver metastasis was demonstrated [30]. CRC tumors consist of heterogeneous cell populations expressing a combination of factors contributing to tumor cell proliferation and stromal cell activation, such as EGFR, PDGFR, and vascular endothelial growth factor receptor (VEGFR). The rationale of simultaneous targeting of these receptors was adopted and indeed, this was the most efficient strategy of therapy in these models [31]. In addition to these receptor tyrosine kinases, activation of intracellular tyrosine kinases, such as SRC, seems to play an important role in human colon cancer cells with high metastatic potential [32] and the presence of activating SRC mutations in a subset of advanced CRC is indicative of this fact [33].
Liver metastasis and inhibition of MMP’s
As invasive processes in general are a pre-requisite for crucial steps of the metastatic cascade, it was hypothesized that proteases, which degrade components of the basement membrane and interstitial ECM would be an ideal target for intervention with metastasis [34]. Among these proteases, the matrix metalloproteinases (MMP’s) were thought to be specifically attractive as they together can in fact degrade all components of the ECM and the basement membrane including collagen type IV [34]. MMP’s are a class of more than 20 Zn-dependent proteolytic enzymes, which exhibit a very conserved catalytic centre that could easily be explored by chemists to develop highly efficient small molecule broad spectrum inhibitors [34]. MMP inhibitors (MMPI’s) were developed with the expectation to inhibit invasion and spread of tumors. This view could not be held up as broad spectrum MMPI’s failed in clinical trials in cancer patients [35]. Retrospectively, it is agreed that the biology of MMP’s is far more complex than anticipated. It turned out that they exert pro- and anti-tumoral functions via their indirect signaling function due to cleavage of a whole cascade of signaling molecules in the context of the protease web [36–39]. This late insight in mind, it is not surprising anymore that broad spectrum MMPI’s can exhibit unforeseen effects including the promotion of liver metastasis formation in different tumor models [40]. One of the underlying mechanisms of this observation may be based on modulation of the physiology of the liver by MMPI’s in the context of a highly connected protease network. The generation of new microenvironments by inhibition of MMP’s can result in changes of susceptibility to circulating tumor cells. In fact, treatment of mice with a broad-spectrum MMPI led to up-regulation of hepatocyte growth factor (HGF, also known as scatter factor) in the liver [40], which, by induction of the invasive growth programme [50] may increase the virulence of metastasis formation in the liver. Unpredictable perturbation of tissue homeostasis affecting metastasis became also evident when MMP9 was ablated in mice. Upon challenge with lacZ-tagged syngeneic murine colorectal carcinoma cells (CT26-lacZ), an 8 fold increase in experimental liver metastasis was observed as compared to the wild-type control [36, 37]. In fact, an MMP9 ablation-adaptive change of transcripts encoding metastasis-associated functions was observed in MMP9-deficient host tissue. Expression of interleukin 6 (IL6) was up-regulated in the bone marrow and in hematopoietic cells localized in the liver leading to increased IL6 levels in the blood [36, 37]. Such elevated IL6 levels are a negative prognostic marker for survival in CRC patients [41].
While the complexity of interference with MMP activity in the host tissue is still under investigation, specific inhibition of MMP’s in tumor cells revealed effects on their metastatic potential. Liver colonization after knock-down of MMP2 or MMP9 in the tumor cells was investigated in the syngeneic L-CI-5 s lymphoma model [42] which preferentially homes to the liver. MMP2 inhibition resulted in inhibition of metastatic foci growth, whereas MMP9 inhibition suppressed the invasive phase of liver colonisation. Along these lines, MMPI’s with greater selectivity/specificity for MMP9 showed greater efficacy against liver metastasis in the model [43]. However, long-term effects on the host, as seen in the knock out model, rule out the option of long-term treatment of CRC in patients with MMPI’s. While synoptic evaluation of the results regarding unspecific versus specific chemical inhibition of MMP’s, MMP9 knock-out mice and liver colonisation as well as MMP2 and MMP9 knock-down experiments would argue against general conclusions regarding the role of MMP’s in liver metastasis, it is clear that they must be ruled out as primary therapeutic targets. It is true that model-specific features such as, mouse strain, tumor cell type, and mode of tumor cell administration might trigger the specific outcome of liver metastasis experiments. Also one should keep in mind that MMP function is not only mediated by its enzymatic function, but also by protein–protein interactions [39].
Even the intuition that the natural MMPI’s, the tissue inhibitors of metalloproteinases (TIMP1, −2, −3, and −4), should exhibit an anti-metastatic effect, had been challenged in the past few years. Recently, it emerged that TIMP1 can create a pre-metastatic niche specifically in the liver for colon carcinoma cells as well as numerous other tumor cells in the respective syngeneic mouse lines as well as in xenograft models where the systemic TIMP1 levels were experimentally increased to similar levels [44] as were found in cancer patients with bad prognosis [45, 46]. In [46] liver metastases were discussed, but post-operative follow-up was not reported. The experimental observations can now explain the so far unresolved paradox that TIMP1 levels correlate with bad prognosis in CRC patients [44]. Moreover, elevated TIMP1 levels were associated with the extent of liver metastasis and frequency of disease relapse in CRC patients [44–46]. Mice with elevated TIMP1 levels were challenged with syngeneic CT26 colon carcinoma cells and a drastic, liver-specific increase of metastasis was found. Further experiments of this study showed that TIMP1 actually creates a liver-specific pre-metastatic niche, an emerging concept providing an explanation of early metastatic events, where homing and successful colonization of an organ is pre-primed by tumor-secreted factors which alter the cellular and molecular signature of the target organ of metastasis before tumor cells reach the site [47, 48]. In consequence, the TIMP1-induced factors that were specifically up-regulated in the liver such as stromal cell-derived factor-1 (SDF-1), fibronectin, transforming growth factor-β (TGF-β), urokinase-type plasminogen activator (uPA) and S100A8 are potentially new targets for anti-metastatic intervention [44]. In fact, pharmacological interference with the interaction of one of these niche-factors, the cytokine SDF-1, with its receptor CXC chemokine receptor 4 (CXCR4) with the widely used small molecule inhibitor AMD3100 led to abrogation of TIMP1-induced pre-metastatic niche formation in the liver [44].
Tyrosine kinase c-MET signaling in tumor cells or liver tissue and generation of liver metastases
c-MET is a pan-epithelial transmembrane tyrosine kinase which can be activated by its ligand HGF [49]. It is overexpressed and activated in many types of carcinomas including colon carcinomas [49–52]. c-MET can be activated by autocrine and paracrine mechanisms [49, 50]. Overexpression of c-MET in tumor cells can lead to ligand-independent activation of the c-MET signaling pathway, resulting in hyperactivation of phosphatidylinositol-3-kinase (PI3 K) and mitogen-activated protein kinase (MAPK) signaling [49]. In tumor cells, this elicits the ‘invasive growth programme’ including induction of proliferation, migration, survival, angiogenesis and morphogenesis [50]. Another level of liver metastasis-promoting c-MET signaling is its modulation of the target tissue of metastasis. This alternative parameter impacting on metastasis formation in the liver is based on the finding that ectopic TIMP1 expression in the liver [53] or elevation of systemic levels of TIMP1 in the host organism [44] formed a potent metastatic niche [53] or even a pre-metastatic niche [44], respectively, leading to increased liver metastasis. In the case of ectopic expression of TIMP1 in liver tissue, cell surface-associated c-MET accumulates by the activity of TIMP1 to inhibit the c-MET sheddase, a disintegrin and metalloproteinase 10 (ADAM-10) [53–55]. The resulting increased c-MET signaling in the liver tissue led to expression of several pro-metastatic factors in this target tissue of metastasis [53] including the protease uPA [56]. Experiments with uPA-ablated mice indicated the crucial role of uPA for the recruitment of neutrophilic cells with elevated TIMP1 levels to the liver and also in increased availability of HGF [56]. HGF promotes a scattered and very invasive pattern of metastases after i.v. inoculation of syngeneic lymphoma cell line LCI.5 s or human fibrosarcoma cells HT 1080 in the liver [56]. In vitro, knock-down of tumor cell-derived TIMP1 prevented HGF-induced phosphorylation of c-MET, ADAM 10 knock-down increased c-MET phosphorylation in metastatic foci and scattering into the surrounding liver parenchyma. TIMP1 expression and c-MET signaling were co-localized within the metastatic colonies and expressed by tumor cells [55].
Similar molecular events take place on the surface of tumor cells themselves rendering them more aggressive. TIMP1 is able to induce hypoxia-inducible factor 1-α (HIF-1α) signaling in tumor cells [57, 58]. Also on tumor cells, TIMP1 induces inhibition of ADAM 10, thereby preventing the shedding of c-MET. This results in cell-associated c-MET accumulation and signaling in host as well as in tumor cells. Making use of i.v. inoculated L-CI.5 s lymphoma cells which home to the liver, it was demonstrated that tumor cell-derived TIMP1 is necessary for maintenance of metastasis-promoting c-MET signaling via inhibition of ADAM 10 [55].
The mode of action of TIMP1 leading to stimulation of liver metastasisis likely more complex, as TIMP1 can exert metalloproteinase inhibition-independent functions. TIMP1 as a cell-binding protein recently was shown to interact in vitro with cluster of differentiation 63 (CD63) by activating the PI3 K signalling pathway dependent on Akt8 virus oncogene cellular homolog (Akt) activation [62] or with CD63 and integrin β1, independent of Akt activation [59, 60]. In fact, we recently found that TIMP1 induces HIF-1α and miR-210 in tumor cells in vitro via CD63, resulting in increased tumorigenicity of the cells [62]. HIF-1α as a mediator of the hypoxic state promotes pro-invasive mechanisms by upregulation of miR-210 which inhibits respiration by inhibition of the subunit D of the succinate dehydrogenase complex (SDHD), regulates the switch to anaerobic glycolysis and induces cell-cycle arrest by inhibition of transcription factor E2F3 in vitro [61–63].
The tumor-promoting function of TIMP1 is still neglected in this field of research [64]. However, the experimental evidence for TIMP1 as a driver of liver metastasis in the past few years explains its strong and independent association with shorter survival time in CRC patients [45, 46]. On the other hand, low TIMP1 was found to be significantly associated with objective response and survival in CRC patients receiving a combination of irinotecan, 5 fluoro-uracil and folinic acid [65].
The relevance of c-MET and its impact on CRC becomes even more evident as it was identified as a transcriptional target of metastasis associated in colon cancer (MACC1) [66]. The src homology 3 (SH3) domain and proline-rich region containing protein MACC1 promotes proliferation, invasion, and HGF-induced scattering of colon cancer cells in vitro and metastasis in mouse models. Mutants of MACC1 lacking the SH3 domain of the proline-rich motif were functionally inactive. MACC1 is a predictor of CRC metastasis. 5 year survival was 80 % for MACC1 low-expressors, but 15 % for individuals who showed high MACC1 expression in their primary tumors [66]. MACC1 mRNA levels were identified as a predictor of recurrence after resection of CRC liver metastasis [67]. Circulating MACC1 transcripts in CRC patients are predictors of metastasis and prognosis [68]. Altogether, c-MET and MACC1 are metastatic pacemakers in CRC patients [69, 70] and therefore important targets for prognosis and therapy of CRC patients. In this context, it was shown that micro RNA-143 (miR-143) targets MACC1, resulting in inhibition of invasion and migration of colon cancer cells in vitro [71].
Phosphatase of regenerating liver (PRL3)
Dysregulation of protein tyrosine phosphatases resulting in aberrant tyrosine phosphorylation has been implicated in development of cancer and its progression [72, 73]. The family of phosphatases of regenerating liver (PRL) is composed of three members. They are farnesylated proteins, which are associated with the inner leaflet of the plasma membrane [72, 73]. Overexpression of PRL3 promotes tumor cell migration, invasion, and metastasis based on Akt activation and subsequent upregulation of MMP’s and EGFR activation as shown in vitro and in vivo [74, 75]. Activation of Akt is based on downregulation of phosphatase and tensin homolog (PTEN) and activation of PI3 K signaling resulting in promotion of epithelial-mesenchymal transition (EMT) in vitro [76]. In a study with 18 patient samples, global gene expression profiling has implicated PRL3 in metastases of CRC to the liver as it is the only gene whose expression was restricted to samples from metastases while no expression in normal colon mucosa and non-metastatic carcinomas [77] was found. PRL3 expression is specific for CRC metastases and was not detected in metastases of other types of cancer [77]. Interestingly, PRL3 can be found in the vasculature of tumors regardless of the tumor type [78], indicating a role in tumor angiogenesis. In fact, the multitasking phosphatase PRL3 is able to promote endothelial recruitment and formation of new blood vessels [79]. It was shown that PRL3 is activated by loss of TGFβ signaling, which often occurs during progression of CRC, by releasing inhibition of its transcription [80].
Taken together, PRL3 seems to be a promising target for treatment of liver metastases derived from CRC, since PRL3 is only expressed during development of the heart, blood vessels and erythrocyte precursors, but not in the corresponding mature tissues [81] so that side effects are unlikely. Proof-of-concept experiments have shown that intracellularly delivered antibodies directed against PRL3 inhibit lung metastasis of human colorectal HT-116 and A2780 ovarian cancer cells in xenograft models and B16 melanoma cells with high PRL3 expression after tail vein injection into syngeneic C57Bl6 mice [81, 82]. In these settings the PRL3 expression status was highly predictive. However, in the context of CRC-derived liver metastasis, PRL3 has not been functionally validated in a respective in vivo system. Functional validation of PRL3 as a target would justify the discovery and development of small molecule PRL3 inhibitors.
L1 cell adhesion molecule (L1CAM)
L1CAM, a member of the L1 family of adhesion molecules, is composed of an extracellular domain consisting of six Ig-like domains and five fibronectin III domains, a transmembrane domain and a cytoplasmic domain [83]. L1CAM expression is involved in axon guidance and neural crest migration and is associated with the invasive front of colon carcinomas [84].
It was shown that MMP2 and MMP9 mediate liver metastasis of the murine lymphoma cell line L-CI.5 s [85]. Increased migration, invasion, and proliferation in vitro as well as increased experimental and spontaneous metastasis to the liver and transcriptional activation of MMP2 and MMP9 could be linked to expression of L1CAM [85, 86], because knock-down of L1CAM abolished these phenomena. Analogous observations were made with the ovarian carcinoma cell line SKOV3ip. The MMP2- and MMP9-specific inhibitor SB-3CT reduced migration and invasion in vitro as well as experimental metastasis to the lungs [87].
Two splice variants have been identified for L1CAM: a full-length form (FL-L1CAM) and an evolutionary conserved splice variant lacking exons 2 and 27 [88]. Exon 2 is involved in homophilic and heterophilic binding to neural ligands contactin and TAX-1 and the cytoplasmic sequence encoded by exon 27 is necessary for clathrin-dependent endocytosis in vitro [89, 90]. Only FL-L1CAM was able to promote experimental metastasis of HT1080 fibrosarcoma, SKOV3ip, and L-CI.5 s to the lungs and to the liver, respectively [91]. Metastasis formation correlated with increased invasive potential, elevated MMP2 and MMP9 expression in vitro, and enhanced proteolytic activity of these MMPs in vivo. These findings might indicate that FL-L1CAM activates distinct signaling pathways upon internalisation due to its endosomal location. It was shown in vitro that L1CAM is able to interact with membrane molecules via its extracellular domain or with other proteins via its cytoplasmic domain [92, 93]. The therapeutic potential of L1CAM antibodies for treatment of cancer has been evaluated [94–96]. They were shown to inhibit growth and dissemination of ovarian carcinoma cells in the peritoneal cavity. L1CAM is a mediator of resistance to chemotherapy and therefore, treatment of pancreatic and ovarian xenografts by combined treatment of L1CAM antibodies and cytotoxic drugs showed increased therapeutic response [97]. From the clinical point of view, a strong correlation between expression of L1CAM in CRC patients and decreased survival has been demonstrated by independent groups [98, 99], confirming the role of L1CAM in liver metastasis. These findings are supported by preclinical data which underline the role of L1CAM in motility and β-catenin/Wnt signaling [100–102].
Tumor-associated calcium signal transducer 2 (Trop-2)
Trop-2 is a transmembrane pan-epithelial antigen related to epithelial cellular adhesion molecule (EpCAM,Trop-1) which is overexpressed in many types of cancer. It has been identified as an oncogene and its upregulation was shown to stimulate cancer growth in vitro and in vivo [103, 104]. Trop-2 can activate the MAPK pathway and support a protumoral signaling network [105, 106]. A possible context of Trop-2 as a metastasis-associated target was indicated by the observation that high levels of Trop-2 correlate with a higher frequency of liver metastasis in 16 CRC patients [107]. Independent from these observations, it was shown that elevated expression of Trop-2 was associated with survival, disease recurrence, and liver metastasis of patients with CRC [108]. Trop-2 was validated as a driver of CRC-related liver metastasis in an experimental in vivo system [109]. After transfection of the CRC cell line KM12SM with Trop-2, the cells gained a dramatically increased metastatic potential as they formed liver metastases after orthotopic injection. Surprisingly, deletion of the whole cytoplasmic domain of Trop-2 increased the metastatic propensity, whereas deletion of the pleckstrin-homology binding domain motif diminished metastasis to the liver. Further validation experiments are required in order to substantiate this issue. Due to the broad expression of Trop-2 in epithelial tissues, therapeutic intervention with antibodies or antibody-related moieties such as antibody–drug conjugates or antibody-fusion proteins with immunotoxins may not be a feasible approach due to possible toxicity issues.
S100 family proteins S100A4 and S100A8
The S100 family consists of 24 members exhibiting a broad range of intracellular and extracellular functions such as regulation of calcium homeostasis and cytoskeletal components, phosphorylation of proteins as well as induction of gene expression when they act as transcription factors [110, 111]. Some S100 proteins are secreted or released and regulate cell functions in an autocrine and paracrine manner via activation of cell surface receptors, such as the receptor of advanced glycation end products (RAGE), G-protein coupled receptors, toll-like receptor 4 (TLR4), and heparin sulphate proteoglycan [100, 111]. S100A4 can interact with epidermal growth factor (EGF) and bFGF, thus enhancing the activity of the appropriate receptors as shown by in vitro experiments [110, 111].
Expression of S100A4 is significantly associated with progression and liver metastasis of CRC patients [112, 113]. An important contribution in this context is the finding that S100A4 is able to bind to tropomyosin and non-muscle myosin II in a Ca-dependent manner, promoting disassembly of myosin filaments in vitro [110]. Systemic RNA knock-down of S100A4 in human CRC cells such as HCT 116, SW 620 and DLD-1 reduces liver metastasis formation in xenograft mouse models [114]. Moreover, S100A4 has been identified in vitro and in vivo as a target of β- catenin/TCF signaling which is often deregulated in CRC [115]. Interestingly, another member of the S100 family, S100A8, is specifically involved in TIMP1-induced pre-metastatic niche formation in liver [44]. It was among those factors that correlated with the increased susceptibility of the liver to tumor cells, including colon carcinoma cells [44]. This suggests that S100 proteins can account for the promotion of liver-specific metastases in patients that have a TIMP1-associated bad prognosis of CRC.
Profiling of highly metastatic versus low-metastatic colon cancer cell lines reveals further genes promoting liver metastasis of CRC cells
These genes were identified by functional selection in an orthotopic mouse model of CRC [116]. The experimental approach was based on non-metastatic CRC cell lines SW480 and SW620 with only lymph node metastasis, their inoculation into the colon of nude mice, isolation of liver lesions (L-1 and L-2) and reinoculation for confirmation of the metastatic phenotype. Transcriptomic microarray analysis of the cell lines [116] and knockdown experiments revealed that four genes are essential mediators of liver metastasis: S100P, CD133, lipase C (LIP-C), and apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G (APOBEC3G). These genes are significantly increased in metastatic colonies as compared to non-metastatic primary tumors. Some impact of these four genes on metastasis is already known:
S100P has been previously identified as a prognostic marker in CRC patients [117]. S100P regulates Ca-signaling transduction to mediate cytoskeletal interactions and is induced by the prostaglandin E2 (PGE2)/EP4) receptor signaling pathway in colon cancer cells [118]. Ectopic expression of S100P in SW 480 CRC promotes invasion and metastasis and decreases sensitivity to 5 FU in vitro [119].
LIP-C is a hydrolase involved in lipoprotein uptake and lipoprotein homeostasis [120]. It has been shown that monoacylglycerol lipase regulates the fatty acid network and promotes cancer pathogenesis [121]. This finding indicates that cancer cells can co-opt a lipolytic enzyme to translate their lipogenic state into pro-tumorigenic signals in vitro and in vivo [121, 122].
CD133 is a stem cell marker in CRC [123]. Expression of CD133 or CD133 and CXCR4 on cancer cells correlates with the presence of lymph node metastasis in CRC patients [124]. However, the role of CD133 in colon cancer metastasis is still controversial, since both CD133+ and CD133− cancer stem cells can initiate tumor formation and the impact on the formation of metastatic colonies is not clear [124].
The fourth gene identified in the SW480 and SW620-based model, APOBEC3G, belongs to the family of cellular cytidine deaminases which deaminate cytidine to uridine in the single-strand DNA substrate [125]. Resulting G to A hypermutations at hot spots in the viral RNA destroy the coding and replicative capacity of the virus [125]. In respect to CRC liver metastasis it was shown that APOBEC3G induces expression of MMP2 by depression of miR29 mediated protein translation inhibition [126].The enzymatic activity of APOBEC3G can be suppressed by recruitment into high-molecular-weight ribonucleoprotein (RNP) complexes [127].
Notch pathway
The importance of Notch signaling in the context of cancer pathogenesis is well-documented [128], but the role of this pathway in liver metastasis of CRC remains to be studied in more detail. There is evidence that inhibition of Notch signaling interferes with CRC liver metastasis [129]. The amino-terminal enhancer of split (Aes) gene product, a member of the family of transcriptional co-repressors, was found to be decreased in liver metastasis compared to primary colon tumors in mice and humans. Aes inhibits Notch signaling in vitro by converting active recombining binding protein suppressor of hairless (rbpj) active transcription complexes into repression complexes on insoluble nuclear matrix. Notch signaling in the context of liver metastasis was shown to stimulate transendothelial migration [129].
TGFβ signaling
The TGFβ signaling pathway has emerged as an other possible target to interfere with CRC liver metastasis [130, 131]. TGFβ signaling is mediated by contraction of sma and mad 2, 3 and 4 (Smad 2, 3, and 4), which are translocated to the nucleus to regulate transcription. Loss of Smad 4 function, as shown with Smad4-null MC 38 and SW 620 colon cancer cells, results in a functional shift of TGFβ from a tumor suppressor to a tumor promoter [132]. TGFβ-receptor tyrosine kinase inhibitor LY2109761 was shown to inhibit liver metastasis of these cell lines by reducing expression of MMP2, uPA, and cyclooxygenase 2 as well as the reduction of E-cadherin expression [130]. Expression of Smad 4 in these cell lines reduced metastasis significantly [132].
Additional experiments support the view that the balance between Smad 4/Smad 7 and the TGFβ pathway may be critical for the metastatic process [133]. In a spleen injection model of liver metastasis, it was shown that abrogation of Smad signaling by ectopic expression of Smad 7 induces liver metastasis. Cells expressing Smad 7 migrated to the liver [133]. Increased junctional staining of claudin 1, claudin 4 and E-cadherin was found in liver metastasis.
The role of cancer-associated fibroblasts (CAF’s) in the context of liver metastasis of CRC has been investigated [134]. Although tumor cells are often non-responsive to TGFβ due to mutational inactivation of the components of the TGFβ pathway [135], CAF’s can respond to TGFβ and secrete tumor-promoting cytokines such as IL11, which can bind to its receptor gp130 on tumor cells and can activate STAT3 signaling in tumor cells, promoting their survival as liver metastases [134].
Concluding remarks
Overall, it should be kept in mind that the mechanisms for invasion and metastasis to the liver can vary between different individual CRC diseases and therefore the involved pathways have to be resolved case-by-case. Also, one should realize that animal models are useful for discovery of new targets and pathways, but are often rather poor models for treatment modalities of human disease.
We have analysed RNA expression of MACC1, MET, PRL3, S100P and TIMP1 in CRC in comparison to matching normal colon mucosa in publicly available RNA Seq data from the Cancer Genome Atlas (TCGA) (Fig. 2). All of them are dramatically overexpressed in tumor tissue. However, we did not find a correlation between progression of primary CRC’s as mesasured by clinical stage annotation and an increase in steady-state RNA levels of these genes (Fig. 3).The elucidation of the status of these genes in liver metastases, preferentially matching with the primary tumor is a pending issue.

Expression of selected genes in colon cancer in comparison to normal colon mucosa. Data were derived from the Cancer Genome Atlas. This cohort represents 421 Colon adenocarcinoma primary tumor samples as well as 40 matched normal samples. Expression was measured by whole transcriptome sequencing and values provided represent normal read counts (log2) as derived from the Broad FIREHOSE portal. ‘Solid tissue normal’ represents matched adjacent normal tissue of cancer patients. Data are shown for MACC1, MET, PRL3, S100P, and TIMP1. For all those genes expression levels between tumor and matched normal tissue are significantly different (t test, p value < 0.0001)

RNA expression of selected genes in different stages of colorectal cancer progression. Data were derived from whole transcriptome RNA sequencing and values shown represent normalized read counts (log2) as provided by the Broad FIREHOSE portal. This cohort represents 421 colon adenocarcinoma primary tumor samples (72 stage i, 34 stage ii, 124 stage iia, 9 stage iib, 22 stage iii, 13 stage iiia, 52 stage iiib, 36 stage iiic, 43 stage iv, 16 stage iva). Expression is shown in correlation to pathological stage annotation. Selected genes: MACC1, MET, PRL3, S100P and TIMP1
Prevention of liver metastasis of CRC after resection of the primary tumor based on new molecular targets might be a future option. It is of high clinical relevance that it is not yet possible to identify prospectively those patients (20–30 %) belonging to the high risk group with locally restricted stage II colon cancer but will ultimately experience disease recurrence [136]. In the subset of metastatic cases who undergo liver-metastasis resections, half were cured and the other half relapsed with metastases elsewhere in the liver and/or in the lungs. Thus the latter half of the patients could have been cured if early metastasis could be blocked post-operatively by metastasis-prevention, e.g. by employing adjuvant therapies to surgery [137]. Also, identification of a validated gene expression signature that is predictive for the metastatic potential would be helpful for the design of clinical trials [136]. Another clinical objective is the conversion of non-resectable to resectable disease based on new targets expressed in liver metastasis. A significantly higher resection rate (25 vs. 7.4 %) was observed in a randomized controlled clinical trial of cetuximab plus chemotherapy in patients with KRAS wild-type non-resectable colorectal liver-limited metastasis [138]. In addition to direct targeting of liver metastasis, identification of new pathways conferring dependence of liver metastasis on stromal interactions for their maintainance and propagation is an exciting issue for further studies. It is presently unclear, whether the targets described in this review can be translated into clinically relevant targets.
References
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87:159–170
Velculescu VE, Vogelstein B, Kinzler KW (2000) Analysing uncharted transcriptomes with SAGE. Trends Genet 16:423–425
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361:2449–2460
Hölzl D, Eckel R, Engel J (2009) Colorectal cancer metastasis. Frequency, prognosis and consequences. Chirurg 80:331–340
Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenz R, Abbruzzese JL (2006) Metastatic patterns in adenocarcinoma. Cancer 106:1624–1633
Wan L, Pantel K, Kang Y (2013) Tumor metastasis: moving new biological insights into the clinic. Nat Med 19:1450–1564
Deneve E, Rietdorf S, Ramos J et al (2013) Capture of viable circulating tumor cells in the liver of colorectal cancer patients. Clin Chem 59:1384–1392
MacDonald IC, Groom AC, Chambers AF (2002) Cancer spread and micrometastasis development: quantitative approaches for in vivo models. BioEssays 24:885–893
Padget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 1:571–573
Sceneay J, Smith MJ, Möller H (2013) The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 32:449–464
Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9:274–284
Zeisberg M, Neilson EG (2009) Biomarkers for epithelial-mesenchymal transitions. J Clin Invest 119:1429–1437
Brabletz T, Jung A, Hermann K, Günther K, Hohenberger W, Kirchner T (1998) Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res Pract 194:701–704
Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T (2001) Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98:10356–10361
Kirchner T, Brabletz T (2000) Patterning and nuclear beta-catenin expression in the colonic adenoma-carcinoma sequence. Analogies with human gastrulation. Am J Pathol 157:1113–1121
Placke T, Kopp HG, Salih HR (1996) The wolf in sheeps clothing: platelet derived “pseudo-self” impairs cancer cell “missing self” recognition by NK cells. Oncoimmunology 1:557–559
Bayon LG, Izquierdo MA, Sirovich I, van Rooijen N, Beelen RH, Meijer S (1996) Role of Kupffer cells in arresting circulating tumor cells and controlling metastatic growth in the liver. Hepatology 23:1224–1231
Luo DZ, Vermijlen D, Ahisali B, Triantis V, Plakoutsi G, Braet F, Vanderkerken K, Wisse E (2000) On the biology of pit cells, the liver-specific NK cells. World J Gastroenterol 6:1–11
Wellner UF, Keck T, Brabletz T (2010) Liver metastases: pathogenesis and oncogenesis. Chirurg 81:551–556
Chiang AC, Massague J (2008) Molecular basis of metastasis. N Engl J Med 359:2814–2823
Paschos KA, Majeed AW, Bird NC (2014) Natural history of hepatic metastases from colorectal-pathological pathways with clinical significance. World J Gastroenterol 20:3719–3737
Giavazzi R, Jessup JM, Campbell DE, Walker SM, Fidler IJ (1986) Experimental nude mouse model of human colorectal cancer liver metastases. J Natl Cancer Inst 77:1303–1308
Morikawa K, Walker SM, Jessup JM, Fidler IJ (1988) In vivo selection of highly metastatic cells from surgical specimens of different human primary colon carcinomas in nude mice. Cancer Res 48:1943–1948
Fidler IJ (1991) Orthotopic implantation of human colon carcinomas into nude mice provides a valuable model for the biology and therapy of metastasis. Cancer Metastasis Rev 10:229–243
Morikawa K, Walker SM, Nakajima M, Pathak S, Jessup JM, Fidler IJ (1988) Influence of organ environment on the growth, selection and metastasis of human colon carcinoma cells in nude mice. Cancer Res 48:6863–6871
Fidler IJ, Wilmanns C, Staroselsky A, Radinsky R, Dong Z, Fan D (1994) Modulation of tumor cell response to chemotherapy by the organ environment. Cancer Metastasis Rev 13:209–243
Radinski R, Risin S, Fan D, Dong Z, Bielenberg D, Bucana CD, Fidler IJ (1995) Level and function of epidermal growth factor receptor predict the metastatic potential of human colon carcinoma cells. Clin Cancer Res 1:19–31
Kitadai Y, Bucana CD, Ellis LM, Anzai H, Tahara E, Fidler IJ (1995) In situ mRNA hybridization technique for analysis of metastasis-related genes in human colon carcinoma cells. Am J Pathol 147:1238–1247
Takahashi Y, Llis LM, Wilson MR, Bucana CD, Kitadai Y (1996) Progressive upregulation of metastasis-related genes in human colon cancer cells implanted into the cecum of nude mice. Oncol Res 8:163–169
Kitadai Y, Sasaki T, Kuwai T, Nakamura T, Bucana CD, Fidler IJ (2006) Targeting the expression of platelet-derived growth factor receptor by reactive stroma inhibits growth and metastasis of human colon carcinoma. Am J Pathol 169:2054–2065
Kuwai T, Nakamura T, Sasaki T, Kitadai Y, Kim JS, Langley RR, Fan D, Wang X, Do KA, Kim SJ, Fidler IJ (2008) Targeting EGFR, VEGFR, and PDGFR on colon cancer cells is required for therapy. Clin Exp Metastasis 25:477–489
Mao W, Irby R, Coppola D, Fu L, Wloch M, Turner J, Yu H, Garcia R, Jove R, Yeatman TJ (1997) Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 15:3083–3090
Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TH (1999) Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet 21:187–190
Brinckerhoff CE, Matrisian LM (2002) Matrix metalloproteinases: a tail of a frog that became a prince. Nat Rev Mol Cell Biol 390:91–97
Overall CM, Lopez-Otin C (2007) Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat Rev Cancer 2:657–672
Krüger A (2009) Functional genetic mouse models: promising tools for investigation of the proteolytic internet. Biol Chem 390:91–97
Krüger A, Kates RE, Edwards DR (2009) Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. Biochim Biophys Acta 1803:95–102
Han J, Gao B, Jin X, Li Z, Sun Y, Song B (2012) Small interfering RNA down-regulation of β-catenin inhibits invasion of colon cancer cells in vitro. Med Sci Monit 18:BR273–BR280
Damodharan U, Ganesan R, Radhakrishnan UC (2011) Expression of MMP2 and MMP9 gelatinases in human colon cancer cells. Appl Biochem Biotechnol 165:1245–1252
Krüger A, Soeltl R, Sopov I, Kopitz C, Artl M, Magdolen V, Harbeck N, Gänsbacher B, Schmitt M (2001) Hydroxamate-type matrix metalloprotease inhibitor batimastat promotes liver metastasis. Cancer Res 61:1272–1275
Clinchi B, Fransson A, Druvefors B, Hellsten A, Hakansson A, Gustavson B, Sjödahl R, Hakanson L (2007) Preoperative interleukin-6 production by mononuclear blood cells predicts survival after radical surgery for colorectal carcinoma. Cancer 109:1742–1749
Gerg M, Kopitz C, Schaten S et al (2008) Distinct functionality of tumor cell-derived gelatinases during formation of liver metastases. Mol Cancer Res 6:341–351
Arlt M, Kopitz C, Pennington C et al (2002) Increase in gelatinase-specificity of matrix metalloprotease inhibitors correlates with antimetastatic efficacy in a T-cell lymphoma model. Cancer Res 62:5543–5550
Seubert B, Grünwald B, Kobuch J et al (2014) TIMP1 creates a pre-metastatic niche in the liver through SDF/CXCR4-dependent neutrophil recruitment in mice. Hepatology 61:238–248
Moller Sorensen N, Veigaard Sorensen I, Ornbierg Würtz S et al (2008) Biology and potential clinical implications of tissue inhibitor of metalloproteinases-1 in colorectal cancer treatment. Scand J Gastroenterol 43:774–786
Holten-Andersen MN, Stephens RW, Nielsen HJ et al (2000) High preoperative plasma tissue inhibitor of metalloproteinases-1 levels are associated with short survival of patients with colorectal cancer. Clin Cancer Res 6:4292–4299
Psaila B, Lyden D (2009) The metastatic niche: adapting the foreign soil. Nat Rev Cancer 9:285–293
Peinado H, Lavotshkin S, Lyden D (2011) The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 21:139–146
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G (2012) Targeting MET in cancer: rationale and progress. Nat Rev Cancer 12:89–103
Liu Y, Yu X-F, Zou J, Luo Z-H (2015) Prognostic value of c-MET in colorectal cancer: a meta-analysis. World J Gastroenterol 21:3706–3710
Liu Y, Li Q, Zhu L (2012) Expression of hepatocyte growth factor and c-MET in colon cancer: correlation with clinicopathological features and overall survival. Tumori 98:105–112
Luraghi P, Schelter F, Krüger A, Boccaccio C (2012) The MET oncogene as a therapeutic target in cancer invasive growth. Front Pharmacol 3:164
Kopitz C, Gerg M, Bandapalli OR et al (2007) Tissue inhibitor of metalloproteinases-1 promotes liver metastasis by induction of hepatocyte growth factor signaling. Cancer Res 67:8615–8623
Murphy G (2011) Tissue inhibitors of metalloproteases. Genome Biol 12:233
Schelter F, Grandl M, Seubert B et al (2011) Tumor cell-derived Timp-1 is necessary for maintaining metastasis-promoting Met signaling via inhibition of Adam-10. Clin Exp Metastasis 28:793–802
Schrötzlmair F, Kopitz C, Halbgewachs B (2010) Tissue inhibitor of metalloproteinases-1-induced scattered liver metastasis is mediated by host-derived urokinase type plasminogen activator. J Cell Mol Med 14:2760–2770
Schelter F, Halbgewachs B, Bäumler P et al (2010) Tissue inhibitor of metalloproteinases-1 induced scattered liver metastasis is mediated by hypoxia-inducible factor-1α. Clin Exp Metastasis 28:91–99
Schelter F, Gerg M, Halbgewachs B et al (2010) Identification of survival-independent metastasis enhancing role of hypoxia-inducible factor-1 alpha with a hypoxia-tolerant tumor cell line. J Biol Chem 285:26182–26189
Jung KK, Liu XW, Chirco R, Fridman R, Kim HR (2006) Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J 25:3934–3942
Toricelli M, Melo FH, Peres GB, Siulva DC, Jasiulionis MG (2013) Timp1 interacts with beta-1 integrin and CD63 along melanoma genesis and confers anoikis resistance by activating PI3 K signaling pathway independently of Akt phosphorylation. Mol Cancer 12:51
Puissegur MP, Mazure NM, Bertero P et al (2011) miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ 18:465–478
Cui A, Seubert B, Stahl E et al (2014) Tissue inhibitor of metalloproteinases-1 induces a protumorigenic increase of miR-210 in lung adenocarcinoma cells and their exosomes. Oncogene in press
Cui H, Grosso S, Schelter F, Mari B, Krueger A (2012) On the prometastatic stress response in cancer therapies: evidence for a positive cooperation between TIMP-1, HIF-1α and miR-210. Front Pharmacol 3:134
Mason SD, Joyce JA (2011) Proteolytic networks in cancer. Trends Cell Biol 21:228–237
Sorensen NM, Byström P, Christensen IJ et al (2007) TIMP-1 is significantly associated with objective response and survival in metastatic colorectal cancer patients receiving a combination of irinotecan, 5-fluoruracil, and folinic acid. Clin Cancer Res 13:4117–4122
Stein U, Walther W, Arlt F et al (2009) MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat Med 15:59–67
Isella C, Mellano A, Galimi F et al (2013) MACC1 mRNA levels predict cancer recurrence after resection of colorectal cancer liver metastases. Ann Surg 257:1089–1095
Stein U, Burock S, Herrmann P et al (2012) Circulating MACC1 transcripts in colorectal cancer patient plasma predict metastasis and prognosis. PLoS ONE 7:e49249
Stein U, Dahlmann M, Walther W (2010) MACC1 – more then metastasis? Facts and predictions about a novel gene. J Mol Med 88:11–18
Arlt F, Stein U (2009) Colon cancer metastasis: MACC1 and Met as metastatic pacemakers. Int J Biochem Cell Biol 41:2356–2359
Zhang Y, Wang Z, Chen M et al (2012) MicroRNA-143 targets MACC1 to inhibit cell invasion and migration in colorectal cancer. Mol Cancer 11:23
Ostman A, Hellberg C, Böhmer FD (2006) Protein-tyrosine phosphatases and cancer. Nat Rev Cancer 6:307–320
Bessette DC, Qui D, Pallen C (2008) PRL PTPs: mediators and markers of cancer prognosis. Cancer Metastasis Rev 27:231–252
Lee SK, Han YM, Yun J et al (2012) Phosphatase of regenerating liver-3 promotes migration and invasion by upregulating matrix metalloproteinases-7 in human colorectal cancer cells. Int J Cancer 131:E190–E203
Al-Aidaroos AQ, Yuen HD, Guo K (2013) Metastasis-associated PRL-3 induces EGFR activation and addiction in cancer cells. J Clin Invest 123:3459–3471
Al-Aidaroos AQ, Zeng Q (2010) PRL3 phosphatase and cancer metastasis. J Cell Biochem 111:1087–1098
Saha S, Bardelli A, Buckhaults P et al (2001) A phosphatase associated with metastasis of colorectal cancer. Science 294:1343–1346
Bardelli A, Saha S, Sager JA et al (2003) PRL-3 expression in metastatic cancers. Clin Cancer Res 9:5607–5615
Zeng Q, Dong JM, Guo K et al (2003) PRL-3 and PRL-1 promote cell migration, invasion and metastasis. Cancer Res 63:2716–2722
Jiang Y, Liu XQ, Rajput A et al (2011) Phosphatase PRL-3 is a direct regulatory target of TGFβ in colon cancer metastasis. Cancer Res 71:234–244
Guo K, Tang JP, Tan CP, Wang H, Zeng Q (2008) Monoclonal antibodies target intracellular PRL phosphatases to inhibit cancer metastases in mice. Cancer Biol Ther 7:750–757
Guo K, Tang JP, Al-Aidaroos AQ et al (2012) Engineering the first chimeric antibody in targeting intracellular PRL-3 oncoprotein for cancer therapy in mice. Oncotarget 3:158–171
Weidle UH, Eggle D, Klostermann S (2009) L1-CAM as a target for treatment of cancer with monoclonal antibodies. Anticancer Res 29:4919–4931
Gavert S, Conacci-Sorell M, Gast D et al (2005) L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon tumors. J Cell Biol 168:633–642
Gerg M, Kopitz C, Schaten S et al (2008) Distinct functionality of tumor-cell derived gelatinases during formation of liver metastases. Mol Cancer Res 6:341–351
Weinspach D, Seubert B, Schaten S et al (2014) Role of L1 cell adhesion molecule (L1CAM) in the metastatic cascade: promotion of dissemination, colonization and metastatic growth. Clin Exp Metastasis 31:87–100
Lim IT, Brown S, Mobashery S (2004) A convenient synthesis of a selective gelatinase inhibitor as an antimetastatic agent. J Org Chem 69:3572–3573
Coutelle O, Nyakatura G, Tsudien S et al (1998) The neural cell adhesion molecule L1: genomic organisation and differential splicing is conserved between man and the pufferfish fugu. Gene 208:7–15
De Angelis E, Brummendorf T, Cheng L, Lemmon V, Kenwrick S (2001) Alternative use of a mini exon of the L1 gene affects L1 binding to neural ligands. J Biol Chem 276:32738–32742
Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V (1998) The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via its clathrin-mediated pathway. J Neurosc 18:5311–5321
Hauser S, Bickel L, Weinspach D et al (2011) Full-length L1CAM and not its ∆2∆27 splice variant promotes metastasis through induction of gelatinase expression. PLoS One 6:e18989
Gast D, Riedle S, Kiefel H et al (2008) The RGD integrin binding site in human L1-CAM is important for nuclear signaling. Exp Cell Res 314:2411–2418
Gavert N, Ben-Shmuel A, Lemmon V, Brabletz T, Ben-Zeév A (2010) Nuclear factor-kappa B signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J Cell Sci 123:2135–2143
Gast D, Riedle S, Issa Y et al (2008) The cytoplasmic part of L1-CAM controls growth and gene expression in human tumors that is reversed by therapeutic antibodies. Oncogene 27:1281–1289
Arlt M, Novak-Hofer I, Gast D et al (2006) Efficient inhibition of intra-peritoneal tumor growth and dissemination of human ovarian carcinoma cells in nude mice by anti-L1-cell adhesion molecule monoclonal antibody treatment. Cancer Res 66:936–943
Wolterink S, Moldenhauer G, Fogel M et al (2010) Therapeutic antibodies to human L1CAM: functional characterization and application in a mouse model for ovarian carcinoma. Cancer Res 70:2504–2515
Schäfer H, Dieckmann C, Kornijenko O et al (2012) Combined treatment of L1CAM antibodies and cytotstatic drugs improve the therapeutic response to pancreatic and ovarian carcinoma. Cancer Lett 319:66–82
Kaifi JT, Reichelt U, Quaas A et al (2007) L1 is associated with micrometastatic spread and poor outcome in colorectal cancer. Mod Pathol 20:1183–1190
Boo YJ, Park JM, Kim J et al (2007) L1 expression as a marker for poor diagnosis, tumor progression, and short survival in patients with colorectal cancer. Ann Surg Oncol 14:1703–1711
Gavert N, Sheffer M, Raveh S et al (2007) Expression of L1-CAM and ADAM 10 in human colon cancer cells induces metastasis. Cancer Res 67:7703–7712
Gavert N, Conacci-Sorrell M, Gast D et al (2005) L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol 168:633–642
Gavert N, Ben-Shmuel A, Raveh S, Ben-Zeév A (2008) L1-CAM in cancerous tissues. Expert Opin Biol Ther 8:1749–1757
Trerotola M, Cantanelli P, Guerra E et al (2013) Upregulation of Trop-2 quantitatively stimulates human cancer growth. Oncogene 32:222–233
Wang J, Day R, Dong Y, Weintraub SJ, Michel L (2008) Identification of Trop-2 as an oncogene and an attractive target in colon cancers. Mol Cancer Ther 7:280–285
Cubas R, Zhang S, Li M, Chen C, Yao Q (2010) Trop2 expression contributes to tumor pathogenesis by activating the ERK MAPK pathway. Mol Cancer 9:253
Guerra E, Trerotola M, Aloisi AL et al (2013) The Trop-2 signaling network in cancer growth. Oncogene 32:1594–1600
Ohmachi T, Taneka Miori K, Inoue H, Yanaga K, Mori M (2006) Clinical significance of TROP2 expression in colorectal cancer. Clin Cancer Res 12:3057–3063
Fang YH, Lu ZH, Wang GQ et al (2007) Elevated expression of MMP7, TROP2, and Survivin are associated with survival, disease recurrence, and liver metastasis of colon cancer. Int J Colorectal Dis 24:875–884
Alberti S, Trerotola G, Vaca R et al (2007) TROP2 is a major determinant of growth and metastatic spreading of human cancer. J Clin Oncol 25(18S):10510
Donato R, Cannon BR, Sorci G et al (2013) Functions of S100 proteins. Curr Mol Med 1:24–57
Sack U, Walther W, Scuderio D et al (2011) S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol Biol Cell 22:3344–3354
Cho YG, Kim CJ, Nam SW (2005) Overexpression of S100A4 is closely associated with progression of colorectal cancer. World J Gastroenterol 11:4852–4856
Boye K, Nesland JM, Sandstad B, Maelandsmo GM, Flatmark K (2007) Nuclear S100A4 is a novel prognostic marker in colorectal cancer. Eur J Cancer 46:2919–2925
Dahlmann M, Sack U, Herrmann P et al (2012) Systemic shRNA mediated knock down of S100A4 in colorectal cancer xenografted mice reduces metastasis formation. Oncotarget 3:783–797
Stein U, Arlt F, Walther W et al (2006) The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology 131:1486–1500
Ding Q, Chang CJ, Xie X et al (2011) APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J Clin Invest 121:4526–4536
Wang Q, Zhang YN, Lin GL et al (2012) S100P, a potential novel prognostic marker in colorectal cancer. Oncol Rep 28:303–310
Chandramouli A, Mercado-Pimentel ME, Hutchinson A et al (2010) The induction of S100P expression by the prostaglandin E2 (PGE2/EP4 receptor signaling pathway in colon cancer cells. Cancer Biol Ther 28:303–310
Dong L, Wang F, Yin X et al (2014) Overexpression of S100P promotes colorectal cancer metastasis and decreases chemosensitivity to 5-FU in vitro. Mol Cell Biochem 389:257–264
Wong H, Schotz MC (2002) The lipase gene family. J Lipid Res 43:993–999
Nomura DK, Long JZ, Niessen S et al (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140:49–61
Notarnicola M, Messa C, Caruso MG (2012) A significant role of lipogenic enzymes in colorectal cancer. Anticancer Res 32:2585–2590
Silinsky J, Grimes C, Driscoll T et al (2013) CD 133+ and CXCR4+ colon cancer cells as a marker for lymph node metastasis. J Surg Res 185:113–118
Shmelkov SV, Butler JM, Hooper AT et al (2008) CD133 expression is not restricted to stem cells, and both CD133+ and CD133− colon cancer cells initiate tumors. J Clin Invest 118:2111–2120
Jaszczur M, Bertram JG, Pham P, Scharff MD, Goodman MF (2013) AID and Apobec3G haphazard deamination and mutational diversity. Cell Mol Life Sci 70:3089–3108
Huang J, Liang Z, Yang B, Tian H, Ma J, Zhang H (2007) Derepression of microRNA-mediated protein translation inhibition by apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3G (APOBEC3G) and its family members. J Biol Chem 282:33632–33640
Gallois-Montbrun S, Kramer B, Swanson CM et al (2007) Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol 81:2165–2178
Ranganathan P, Weaver KL, Capobianco AJ (2011) Notch signalling in solid tumors: a little bit of everything but not all the time. Nat Rev Cancer 11:338–351
Sonoshita M, Aoki M, Fuwa H et al (2011) Suppression of colon cancer metastasis by Aes through inhibition of Notch signaling. Cancer Cell 19:125–137
Katz LH, Li Y, Chen J-S et al (2013) Targeting TGFβ signaling in cancer. Exp Opin Ther Targets 17:743–760
Pickup M, Novitsky S, Moses HL (2013) The role of TGFβ in the tumor microenvironment. Nat Rev Cancer 13:788–799
Zhang B, Halder SK, Kashikar ND et al (2010) Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 138:969–980
Halder SK, Rachakonda G, Deane NG, Datta PK (2008) Smad7 induces hepatic metastasis in colorectal cancer. Br J Cancer 99:957–965
Calon A, Espinet E, Palomo-Ponce S et al (2012) Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 22:571–584
Markowitz SD, Bertagnolli MM (2009) Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med 361:2449–2460
Shibayama M, Maak M, Nitsche U et al (2011) Prediction of metastasis and recurrence in colorectal cancer based on gene expression analysis: ready for the clinic? Cancers 3:2858–2869
Taketo MM (2011) Reflections on the spread of metastasis to cancer prevention. Cancer Prev Res 4:324–328
Ye L-C, Liu T-S, Ren L et al (2013) Randomized controlled clinical trial of cetuximab plus chemotherapy for patients with KRAS unresectable colorectal liver-limited metastasis. J Clin Oncol 31:1931–1938
Acknowledgments
This work was supported by Grants to A.K. from Deutsche Forschungsgemeinschaft (KR2047 1-2, KR2047 1-3, KR2047 3-1, and KR2047 4-2) and the European Union Research Framework Programme 7 (HEALTH-2007-201279/Microenvimet, and NMP-2010-263307/SaveMe).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Weidle, U.H., Birzele, F. & Krüger, A. Molecular targets and pathways involved in liver metastasis of colorectal cancer. Clin Exp Metastasis 32, 623–635 (2015). https://doi.org/10.1007/s10585-015-9732-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10585-015-9732-3