
Abstract
Lung cancer is the deadliest malignancy in the world. The Notch signaling pathway plays an important role in both normal lung development and the pathobiology of lung cancer. By understanding the function of the Notch pathway in normal development, we can begin to appreciate the intricate role that it plays in lung cancer. The complexity of Notch signaling includes multiple Notch receptors and ligands, posttranslational modifications affecting Notch receptor function, and significant cross talk with other signaling pathways. Dysregulation of the Notch signaling pathway occurs in every type of lung cancer, but the specific role of the Notch pathway in the different subtypes of lung cancer is still unclear. There is evidence that Notch can act in a pro-tumorigenic manner under some circumstances and in an anti-tumorigenic manner under others. Notch can facilitate tumor growth and proliferation, apoptosis, cell differentiation, survival, immune response, angiogenesis, cancer stem cell biology, and chemoresistance. Understanding how Notch naturally usurps these mechanisms to promote or suppress tumors can provide new insights regarding therapeutic intervention while minimizing toxicity.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Introduction to the Notch Signaling Pathway in Lung Cancer
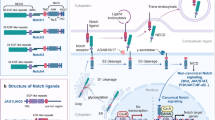
Notch signaling plays a prominent role in early lung development promoting cell fate determination, cell differentiation and the coordination of alveolar development. In humans, there are four Notch receptors (NOTCH1, NOTCH2, NOTCH3, and NOTCH4) and five ligands (DLL1, DLL3, DLL4, JAGGED1, and JAGGED2). Notch receptors and ligands are membrane bound and act in both a juxtacrine and autocrine manner. Notch receptors are first synthesized as precursor polypeptides that are cleaved in the Golgi apparatus by a furin-like convertase (S1 cleavage). The resulting extracellular domain (ECD) and intracellular domain (ICD) are maintained by a non-covalent bond between the N- and C- terminal halves and present at the cell surface. The second proteolytic cleavage site, S2, is buried within the negative regulatory region (NRR). Notch ligands DLL1, DLL4, JAGGED1, and JAGGED2 transactivate the Notch receptor and induce a conformational change that exposes the NRR and triggers the second cleavage (S2) by ADAM10(Kuz)/17(TACE) protease. Cleavage by the γ-secretase complex at a third site (S3) releases the ICD, which translocates to the nucleus and regulates gene expression by cooperating with the DNA binding protein CSL (CBF-1/SU(H)/Lag-1) and co-activator MamL1-3 (Fig. 10.1).

The Notch signaling cascade does not rely on an enzymatic amplification step. Instead precise stoichiometry of receptor-ligand complexes is required for Notch activation [3]. This likely allows for the precise regulation of Notch signaling and partially explains the sensitivity of Notch signals to small perturbations.
Aberrant Notch signaling has been reported in 20% of all cancers [4]. Likewise, 25% of small cell lung cancer (SCLC) [5] tumors and 33% of non-small cell lung cancer (NSCLC) [6] tumors have altered Notch signaling, and is correlated with poor overall survival [7, 8]. It has become increasingly clear that the outcomes of Notch signaling alterations are context dependent and can have opposing roles in different subtypes of lung cancer. The divergent impact of Notch receptor or ligand expression at the RNA or protein level may be related to its context-dependent role as oncogene or tumor suppressor [9,10,11,12,13,14,15]. Given the complexity of Notch signaling, modulated by the expression of multiple combinations of Notch receptors and ligands and their state of posttranslational modification, numerous target genes and crosstalk with other signaling cascades, it is crucial to understand Notch biology to predict the outcome of Notch therapies [12]. Pan-inhibition of Notch receptors or their ligands may not be optimal, and therapies that target individual receptors or ligands may be necessary. Successful development of targeted and combination therapies will require a better understanding of the role of each Notch receptor and ligand in each tumor and how targeting them affects different aspects of cell behavior.
10.2 Modulation of Notch Signaling by Posttranslational Modification
Regulation of the Notch receptor and Notch-ICD occurs throughout maturation with signaling and turnover affected by a number of posttranslational modifications that include glycosylation, ubiquitination, and phosphorylation events [3, 6, 16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. How these modifications affect Notch activity, signaling, and turnover is not yet fully understood.
With the exception of the loss of NUMB, a negative regulator of Notch that promotes ubiquitylation and degradation of NOTCH1, posttranslational modifications of Notch have not been extensively studied in the context of lung cancer [6]. One study identified manic fringe as a tumor suppressor in lung cancer [30]. Because JAGGED1 is often upregulated in lung cancer and manic fringe was found to be downregulated, the authors hypothesized that manic fringe expression in lung cancer would suppress Notch-Jagged activation. They found that re-expressing manic fringe downregulated NOTCH3 signaling through increased protein turnover. More studies are needed to better understand the role of modifications in the context of lung cancer. Mechanisms such as posttranslational modifications can alter Notch signaling activity without affecting Notch expression itself and thus represent potential targets for therapeutic modulation.
10.3 Notch Signaling in Normal Lung Development and Homeostasis
Notch plays an integral role in the development of the lung, a stratified structure composed of a number of specialized cells each with specific functions (Fig. 10.2).

The tracheobronchial tree
Notch pathway genes are expressed during tracheobronchial bud formation and regulate proximal and distal cell fates. Within the budding epithelium, NOTCH1, JAGGED1, and JAGGED2 expressions are localized to distal areas of the bud, whereas DLL1 expression occurs proximally [31, 32]. This pattern suggests that Notch signaling mediates cell fate determination along the proximodistal axis. In mouse embryos, pan-Notch inhibition using a γ-secretase inhibitor has been shown to disrupt the proximodistal axis of the budding lung epithelium by causing an expansion of distal progenitors and loss of proximal structure formation [31].
Notch signaling regulates the development of undifferentiated precursor populations into specialized cell types. In basal cells, NOTCH1-mediated lateral inhibition appears to regulate the adoption of a club (secretory), ciliated, or pulmonary neuroendocrine cell (PNEC) fate [33]. Morimoto et al. found that deletion of the Notch effector protein RBPJ (CSL) redirects cells from a club fate to a ciliated fate. They also found that NOTCH2 determines club cell fate independently of NOTCH1 and NOTCH3 [33, 34]. Moreover, using an injury model, the authors found that CC10-positive club cells arise from a population of CC10-negative cells that activate the Notch signaling pathway and develop into club cells [33].
Similarly, Notch signaling controls the binary cell fate decision of neuroendocrine versus non-neuroendocrine cells [34, 35]. Morimoto et al. found that NOTCH1, NOTCH2, and NOTCH3 contribute to development of PNECs and observed a mutually exclusive relationship between expression of HES1, which is expressed in non-neuroendocrine cells, and ASH1, which is expressed in neuroendocrine cells. PNECs express DLL1, which activates Notch receptors on adjacent cells to produce HES1. HES1 functions as a transcriptional repressor of ASH1, which is required for neuroendocrine cell differentiation. Thus pulmonary neuroendocrine cells that express the Notch ligand DLL1 suppress adjacent cells from developing into pulmonary neuroendocrine cells themselves [35, 36]. Morimoto et al. propose that this traditional model may be incomplete and suggest that DLL1 expression by pulmonary neuroendocrine cells does not merely inhibit adjacent cells from developing a neuroendocrine cell phenotype but rather drives the development of a specialized group of cells surrounding the neuroendocrine cells. They call these specialized cells stage-specific embryonic antigen-1 (SSEA-1)-positive, peri-neuroepithelial body, Notch-active, CC10-negative cells (SPNCs) [34]. Deletion of JAGGED1 in non-neuroendocrine cells has also been reported to increase the number of neuroendocrine cells [36]. Zhang et al. hypothesize that JAGGED1 may be able to activate Notch receptors in neighboring SPNCs and prevent their adoption of a neuroendocrine cell fate [36]. Recent studies by Lafkas et al. demonstrate that under normal conditions, JAGGED1 prevents differentiated secretory cells from adopting a ciliated fate; on the other hand, inhibition of JAGGED1 promotes the conversion of secretory cells to a ciliated fate [37]. It appears that DLL3 may act as a negative regulator of Notch and DLL1 by redirecting them to internal degradation pathways [38, 39]. DLL3 is a direct downstream target of ASCL1, a basic helix-loop-helix (bHLH) transcription factor involved in neuronal cell differentiation [40, 41]. Saunders et al. suggest that DLL3 is associated with a neuroendocrine cell phenotype and contributes to neuroendocrine tumorigenesis [42]. They found that targeting DLL3 with an antibody-drug conjugate (ADC) known as SC16LD6.5 (Rova-T) suppressed tumor growth in SCLC patient-derived xenograft (PDX)’s [42].
Pulmonary goblet cell fate is also regulated by Notch. In murine airway tracheal explant studies, addition of the Notch agonist Dll4 increased the percentage of Muc5ac-positive expressing goblet cells [43]. Presumably overexpression of Notch1-ICD increased goblet cell numbers in the proximal airways driving a goblet cell fate over a ciliated one [43]. Conversely when a diazepine inhibitor of γ-secretase (DBZ) was added to mouse tracheal explants, the fraction of ciliated cells increased, and the number of mucus-secreting cells decreased [43].
In the distal lung, Notch signaling regulates alveolar development that is necessary for alveoli formation [44]. Constitutive activation of Notch1 in the distal lung epithelium stops alveolar development [43]. Distal cysts form and cells within these structures stop expressing alveolar markers [43]. Similarly, constitutive expression of Notch3-ICD in the distal lung epithelium arrests alveolar epithelium differentiation, stalls maturation of Type II pneumocytes, and prevents the formation of Type I pneumocytes in the lungs of transgenic mice [45]. This study suggests that constitutive expression of the Notch3 receptor is essential for proper microvasculature development in the alveoli of the embryonic lung.
While Notch signaling plays a critical role in cell fate determination, maintenance of adult airways, and tissue architecture, it has also been investigated for its role in stem cell maintenance [46,47,48]. Throughout the lung, specific stem/progenitor cells have been identified that are capable of self-renewal and regeneration into specialized cell types [46]. The role of Notch in lung stem cells was recently reviewed by Carraro et al. [46]. In cancer, the lung epithelium undergoes pathological remodeling with large changes to the proportion of cell types [49,50,51], recapitulating what happens during development. Notch contributes to the dedifferentiated state of tumor cells [52]. A deeper understanding of the mechanisms regulating lung maintenance and repair by stem cells is needed for the development of new therapies.
10.4 Notch Signaling in Lung Tumorigenesis: Preclinical and Clinical Relevance
The contribution of Notch signaling to lung tumorigenesis is poorly understood. Notch’s oncogenic role in lung cancer was supported by the discovery of a chromosome 15:19 translocation in a case of poorly differentiated lung cancer in 2000 [53]. The position of this translocation upstream of the NOTCH3 locus on chromosome 19 was associated with massive overexpression of NOTCH3 [53]. While translocation-mediated oncogene activation is common in leukemia and increasingly recognized in other solid tumor types, this was the first reported case of a translocation in a cancer of epithelial origin and the first to implicate NOTCH3 as an oncogene in lung cancer.
Notch has been implicated as both an oncogene and a suppressor in lung cancer. These contrasting roles may be a result of the complexity of the pathway, interactions with other signaling pathways, lack of specific inhibitors, and the fact that Notch signaling is context-dependent. For example, NOTCH1 can play opposing roles in different subtypes of lung cancer (Table 10.1). A review of Notch mutation rates and copy number alterations is provided in Table 10.2. The tumor microenvironment can also influence Notch’s role in cancer, as Notch exerts opposing effects in the same tissue type under hypoxic versus normoxic conditions [67].
10.4.1 Role of Notch in Small Cell Lung Cancer (SCLC)
SCLC comprises 15% of all lung cancers and typically arises in heavy smokers [68]. SCLC is an aggressive neuroendocrine carcinoma that is homogeneously poorly differentiated, has a very high mitotic rate, arises in the central airways, and infiltrates the bronchial airways. SCLC is distinguished by a rapid growth rate and early spread to regional lymph nodes and distant sites. While chemotherapy is often temporarily effective, recurrence is nearly universal, with death occurring within weeks or months [69].
Notch signaling has a tumor-suppressive role in PNECs [70] including the neuroendocrine cells in SCLC and other neuroendocrine tumors [5, 54]. For example, overexpression of active NOTCH1 or NOTCH2 caused growth arrest of SCLC cells [54]. There are two known mechanisms of Notch-mediated tumor suppression in SCLC [71]. The first mechanism occurs through the transcriptional regulatory cascade whereby Notch signaling causes transactivation of HES1, a transcriptional repressor of hASH1, which leads to repression of neural determination and differentiation genes. The second mechanism involves a novel pathway of NOTCH1 signaling that enhances hASH1 ubiquitination and targets it for degradation through a proteasome-dependent pathway.
Since SCLC is rarely treated surgically, it has been difficult to acquire a large number of high-quality surgical resections that are needed for large genomic studies. In 2012 two independent studies performed comprehensive genomic characterization of SCLC tumors [72, 73]. In an analysis of 36 primary human SCLC samples, Rudin et al. found mutations clustering in the Notch (NOTCH1, NOTCH2, and NOTCH3) family genes [72]. Scientists at the University of Cologne in Germany sequenced the genome of 110 resected SCLCs [5]. Using unsupervised hierarchical clustering of tumor transcriptomes, they observed that the majority (53/69) of tumors had high expression of neuroendocrine markers and low Notch pathway activity as indicated by high levels of DLK1, a noncanonical inhibitor of Notch signaling, and ASCL1 whose expression is inhibited by active Notch signaling [5]. Damaging mutations were enriched in the extracellular domains of Notch receptors suggesting a tumor-suppressive role of Notch in SCLC. In concordance with the earlier study by Rudin et al., the University of Cologne’s study determined that Notch family genes were affected by predicted functional genomic alterations in 25% of tumors [5]. A review of mutation rates in SCLC from the University of Cologne study is provided in Table 10.2 [5]. It is possible that alterations in other pathway genes could make the frequency of functional Notch inactivation even higher. Notch receptor/ligand mutations were mutually exclusive of mutations in other frequently altered pro-tumorigenic genes such as CREBBP, EP300, TP73, RBL1, and RBL2. In the University of Cologne dataset, NOTCH1 and JAGGED1 (p = 0.02) as well as DLL1 and DLL4 (p = 0.04) had a significant association toward co-occurrence [63]. Mutations in Notch were not significantly associated with the total number of mutations, overall survival, or other clinical parameters.
Lim et al. identified that activation of Notch in SCLC models leads some cells to undergo a neuroendocrine to non-neuroendocrine shift [55]. These non-neuroendocrine cells are slow growing, chemoresistant and stimulate neuroendocrine tumor cell growth [55]. This lineage switch requires the expression of the Notch-targeted transcription factor Rest (NRSF), an inhibitor of neuroendocrine fate [55].
Activation of Notch1 or Notch2 signaling in murine SCLC models is associated with increases in Hes1 expression, suppression of neuroendocrine differentiation, and significantly reduced tumor formation [5]. Consistent with earlier studies, expression of NOTCH1-ICD inhibited tumor growth, and expression of NOTCH2-ICD prolonged overall survival. In a SCLC cell line, inhibition of NOTCH3 promoted tumor growth supporting a tumor-suppressive role [59]. Taken together this data supports a tumor-suppressive role for Notch in SCLC that parallels its role as a regulator of lineage specification in PNECs during lung development. The finding of frequent DLL3 overexpression in SCLC (a suppressive Notch ligand) supports this hypothesis [42].
10.4.2 Role of Notch in Non-small Cell Lung Cancer (NSCLC)
10.4.2.1 Altered Expression, Mutations, and SNPs
Many studies have characterized mutations in Notch genes that are involved with the pathogenesis of NSCLC [6, 74,75,76]. A 2015 article by Guo et al. reviewed the role of Notch in lung cancer [77]. In a cohort of 49 NSCLC cancers, Westhoff et al. found a subset of patients had NOTCH1 gain-of-function mutations [6]. The authors reported that 30% of NSCLC tumors lose expression of NUMB, a negative regulator of Notch, whose loss leads to increased NOTCH1 expression and activity [6]. A review of mutation rates in NSCLC from The Cancer Genome Atlas (TCGA) is provided in Table 10.2 [65, 66].
Multiple studies have examined the value of Notch signaling as a prognostic indicator in patients with NSCLC [8, 78,79,80]. Studies show that NSCLCs have higher NOTCH1 expression compared to normal lung tissue and that the expression of NOTCH1 is positively correlated with disease progression, metastasis, and poorer overall survival [78]. Mariscal et al. showed that high NOTCH1 expression in circulating tumor cells is a negative prognostic factor for progression-free survival, suggesting its potential utility in liquid biopsy [81]. Another recent study found that patients with lung adenocarcinoma have higher NOTCH2 expression, which is positively correlated with recurrence. This study identified high NOTCH1 and NOTCH3 expression as negative prognostic indicators in adenocarcinoma [82].
A meta-analysis by Yuan et al. examining 3663 patients across 19 studies found that high expression of NOTCH1 was associated with higher tumor, lymph node, and metastasis (TNM) stage and higher risk of lymph node metastasis [78]. NOTCH1 and NOTCH3 overexpression was linked to poor overall survival (NOTCH1, HR, 1.29; 95% CI, 1.06–1.57, p = 0.468, and I2 = 0.0%; NOTCH3, HR, 1.57; 95%CI, 1.04–2.36, p = 0.445, and I2 = 0.0%). The study also identified that DLL4 expression and HES1 expression were associated with poor overall survival in NSCLC. There was no association found between DLL1 and DLL3 expression and overall survival.
The studies reviewed by Yuan [78], Westhoff [6], and Andersen [80] stand out for their size and significance. Westhoff et al. identified NOTCH1 expression as a poor prognostic marker, and the Andersen et al. study identified NOTCH1 and HIF1α co-expression as a poor prognostic marker. Similarly, a 2007 study by Jiang et al. showed that JAGGED1 expression was correlated with lymph node metastasis [79]. Jiang et al. also found that high NOTCH1 expression in adenocarcinoma samples was associated with poorer overall survival and that high co-expression of NOTCH1 and VEGF-A was associated with poorer overall survival in all types of NSCLC. In squamous cell carcinoma, low DLL4 expression was an indicator of poor prognosis [7, 8].
According to dbSNP, the single-nucleotide polymorphism (SNP) rs2229968(V1671I) occurs in African American ancestry populations with a frequency of approximately 3.4% but not in populations of European ancestry [83]. A study by Bollig-Fischer et al. observed that in 472 patients (137 African American ancestry, 335 European ancestry) with NSCLC, the frequency of NOTCH1 V1671I was increased in the African American (9%) versus European ancestry (0%) population (p < 0.0001) [84]. These results from Bollig-Fisher associate this SNP with a higher risk of cancer. Another study by Lee and colleagues suggest that the DTX1 rs1732786A>G promoter region polymorphism may affect DTX1 expression and is associated with better overall survival and disease-free survival [85]. Results from Quan et al. suggest that the NOTCH1 SNP rs3124599 may be associated with a predisposition to SCLC in northeast Chinese non-smoking women but had no prognostic effect [86].
10.4.2.2 Notch Signaling in Adenocarcinoma
Several studies have shown that NOTCH1 directly contributes to lung adenocarcinoma carcinogenesis and is critical for invasion, metastasis, and malignant transformation [9,10,11, 56]. Allen et al. demonstrated that continuous expression of activated Notch1-ICD in the alveolar epithelium of transgenic mice induced lung adenoma formation [56]. After seven days of induction of Notch1-ICD expression, mice began to develop alveolar hyperplasia, which progressed to adenoma after eight months. When crossed with mice overexpressing Myc in the alveolar epithelium, adenocarcinoma developed. Further studies by Baumgart et al. demonstrated that the loss of Notch1 substantially reduced tumor formation in mouse lung adenocarcinoma models driven by KrasG12D mutations [9]. In agreement with these studies, Licciulli et al. demonstrated Notch1 function is required for tumor initiation through suppression of p53-mediated apoptosis [10]. Following knockdown of the individual NOTCH1-3 receptors in vitro, Licciulli et al. found a dramatic decrease in cell numbers only after NOTCH1 knockdown [10]. In KrasG12D Notch1flox/flox mice, six weeks after tumor initiation, KrasG12D mice with the conditional Notch1 knocked out had two lung lesions versus 13 lesions in the KrasG12D control animals. Additionally, substantially lower tumor-to-lung ratios were observed in mice without Notch1 function. These combined findings demonstrate the role of NOTCH1 in tumor initiation and promotion of lung adenocarcinoma.
In contrast NOTCH2 has been demonstrated to mediate differentiation and function as a tumor suppressor in lung adenocarcinoma. Conditional ablation of Notch2 in vivo led to upregulation of β-catenin and development of a higher number of tumors in a shorter period of time [9]. Furthermore, Notch2 has been shown to regulate E-cadherin levels, cell migration, and invasiveness.
Evidence for an oncogenic role of NOTCH3 is provided by experiments that demonstrated in vitro and in vivo suppression of NOTCH3 results in loss of the malignant phenotype [60]. NOTCH3 is elevated in 30–40% of primary lung tumors and frequently co-expressed with EGFR [61, 87]. In cells co-expressing NOTCH3 and EGFR, NOTCH3 suppression sensitizes cells to EGFR inhibitors. Studies by Haruki et al. showed that expression of dominant-negative (DN) NOTCH3 receptor, with a nonfunctional intracellular domain, antagonized NOTCH3 signaling, slowed growth, and induced apoptosis [61]. While all four Notch receptors are present in tumor propagating cells, studies by Zheng et al. showed that only NOTCH3 played a functionally non-redundant role in tumor cell propagation in Kras-driven NSCLC [88]. A study by Arasada et al. showed that NOTCH3 is tyrosine phosphorylated in an EGFR-dependent manner, the functional consequences of which still need to be determined [89]. The authors also demonstrated that erlotinib-mediated EGFR inhibition increased the cancer stemlike cell population and was dependent on activation of NOTCH3 [89]. Knocking down NOTCH3, but not NOTCH1, was shown to eliminate the erlotinib-induced ALDH+ stemlike population, which also suggests a non-redundant role for NOTCH3 in this process [89].
Likewise, NOTCH4 expression has been linked to cancer stem cells in adenocarcinoma models [90]. The frequency of NOTCH4 alterations in white non-Hispanics in adenocarcinoma is approximately 5.5% but approximately 20% in the Hispanic/Latino cohort. Moreover, 7/12 (58.4%) of amino acid substitutions occurred in the NRR of Notch [62]. Expression of one of these NRR domain mutations (P1663Q) by Gordian et al. in the lung adenocarcinoma, A549 cell line model, suggests NOTCH4 may have an oncogenic role in lung adenocarcinoma [62].
10.4.2.3 Notch Signaling in Squamous Cell Carcinoma
To date, the role for Notch receptors in lung squamous cell carcinoma has focused on NOTCH1 signaling but very little on other Notch receptors. Downregulation of NOTCH1 is often associated with dysfunctional (or aberrant) squamous cell differentiation and the development of squamous cell carcinoma [91]. However, early studies demonstrated that Notch signaling drove cell cycle arrest and differentiation in keratinocytes and that loss of NOTCH1 in epidermal keratinocytes promoted tumorigenesis [92, 93]. Subsequent studies by Nicolas et al. demonstrated that conditional ablation of Notch1 in the mouse epidermis resulted in epidermal hyperplasia, skin carcinoma, and basal and squamous carcinomas, thus implying a tumor-suppressive role for NOTCH1 [14]. While the tumor-suppressive role for NOTCH1 has been primarily studied in skin cancer, Li et al. reported that an increase in NOTCH1 signaling in lung squamous cell carcinoma was associated with squamous lung cell differentiation and corresponded with a lengthened survival, low grade, and low stage [94]. Interestingly, studies have shown inhibition of Notch1 in a Kras-driven mouse model of lung cancer strongly decreased adenocarcinoma formation but promoted squamous hyperplasia in the alveoli [11].
The TCGA dataset for lung squamous cell carcinoma identified alterations in Notch receptors in 39% (69/178) of cases [74,75,76]. Additionally a comparative genomic analysis by Kim et al. of 104 squamous cell carcinoma tumors from East Asia with 178 tumors from mostly white patients from the United States suggests that the frequency of Notch mutations in squamous cell carcinoma may vary by ethnic group [57]. Although the frequency of mutations in NOTCH1 (7% and 9%) was similar between the two cohorts, NOTCH2 mutations occurred in 4% of East Asian versus 13% of tumors from the Unites States and found that 10% East Asian tumors versus 7% of tumors from the United States had mutations in NOTCH3 [57, 66]. Although these results were not statistically significant, a larger study in lung squamous cell carcinoma may be able to identify the frequency of Notch alterations among ethnic groups. Furthermore Kim et al. found that eight of the 17 samples with NOTCH1 mutations had truncating mutations suggesting loss of function [57]. Moreover, NOTCH1 mutations have been reported in cutaneous squamous cell carcinoma and head and neck squamous cell carcinoma [74,75,76]. Experiments by Brooks et al. show that ER-ß is a direct positive regulator of NOTCH1 expression in lung keratinocyte-derived squamous cell carcinoma cells [58]. The authors demonstrate that in vitro and in vivo overexpression of ER-ß induces NOTCH1 expression and suppresses proliferation in lung squamous cell carcinoma [58]. This finding is consistent with clinical epidemiological studies that have shown in postmenopausal women that estrogen exposure is associated with reduced risk of NSCLC and that nuclear ER-ß expression is a positive prognostic marker for male NSCLC patients [95,96,97].
10.4.3 Conflicting Roles of Notch in Cancer Subtypes
Despite ample experimental evidence for an oncogenic role for NOTCH1 in NSCLC, conflicting data exist [58, 77, 98, 99]. A study by Zheng et al. demonstrated Notch signaling inhibited growth of A549 lung adenocarcinoma cells suggesting a tumor suppressive rather than oncogenic role for Notch in lung adenocarcinoma [100]. Other studies support an oncogenic role of NOTCH1 in squamous lung cancer [101]. Wael et al. used siRNAs to knockdown NOTCH1 in the adenocarcinoma (A549) and lung squamous cell carcinoma (H2170) cell line and reported that knockdown of NOTCH1 had a tumor-suppressive function in the lung adenocarcinoma (A549) cells and no effect on biological functions in the H2170 squamous cell carcinoma line [99]. One possible explanation for these apparently conflicting data is that the Notch output is highly context and cell of origin dependent. The precise underlying mechanisms of this difference remain to be unraveled.
Hallmarks of cancer such as the tumor microenvironment and interaction with the immune system may be involved in mechanisms that favor an oncogenic versus tumor-suppressive role for Notch in different cellular contexts.
10.4.4 Notch and the Tumor Microenvironment
The tumor microenvironment, including oxygen levels, angiogenesis, paracrine signaling, and immune cells, may contribute to the apparent discrepancies in experimental findings associated with the role of Notch in NSCLC. Maintenance of normal oxygen concentrations is important for normal lung physiology, and tissue hypoxia is common in many tumors including NSCLC [102,103,104].
Regulation of mitochondrial metabolism may also depend on the interaction between tumor cells and the microenvironment. Hypoxia has an important role in lung cancer progression and been shown to decrease therapeutic efficacy of some forms of radiotherapy and chemotherapy [102, 105,106,107]. Notch signaling in lung tumor cell lines is dramatically elevated under hypoxic conditions [108], and Notch signaling is necessary to maintain tumor cells in an undifferentiated state and allow them to survive in these hypoxic microenvironments [109]. Under normoxic conditions, NOTCH1 expression promotes apoptosis, but in hypoxic conditions, NOTCH1 signaling stimulates cell survival by inhibiting PTEN and activating the IGF-1R pathway [110]. Lung adenocarcinoma studies have shown that inhibition of the mitochondrial electron transport chain induced cell cycle arrest and triggered apoptosis [111]. In contrast, a recent comparison of the metabolic phenotype of lung adenocarcinoma and squamous cell carcinoma identified elevated expression of the GLUT1 glucose transporter selectively in lung squamous cell carcinoma [112]. While squamous cells were sensitive to glucose deprivation, adenocarcinoma cells exhibited glucose intolerance [112]. Different phenotypes such as hypoxic versus normoxic have different metabolic requirements. Researchers speculate that differences in signaling pathways mediated in part by Notch may drive divergent metabolic phenotypes [113]. Studies such as these underscore the complexity of the pathway and the importance of the controlling the tumor microenvironment for studies focused on Notch signaling therapeutics.
Studies in mammary epithelial cells indicate that the phenotypic response to Notch is determined by the degree of pathway activation [114]. It is likely that the amount of Notch signaling in lung cancer similarly affects the balance between growth-stimulating and growth-suppressing effects [114]. In squamous cell carcinoma keratinocytes, high levels of Notch1 cause growth arrest but at low levels cause transformation [115]. It is likely that mechanisms have evolved for cells with excessive Notch expression to undergo programmed cell death [108]. A study by Chen et al. suggested that under hypoxic conditions, which potentiate the strength of Notch signaling, total Notch1 protein levels increase, but active Notch1 levels remain relatively unchanged. The low levels of active Notch1-ICD may be a reflection of rapid activation-degradation to prevent pathway hyperactivation and maintain Notch signaling homeostasis [108]. These studies suggest that this pathway, like most other crucial pathways, is self-regulating/negatively regulating and underscores the fragile nature and context dependency of Notch signaling.
10.5 Notch and the Immune Response to Lung Cancer
The immune system plays a critical role in the suppression of tumors, and immune evasion is a hallmark of malignancy [116,117,118]. Notch signaling has been found to play a critical role in normal immune system activation and T cell differentiation (Fig. 10.3). Proliferating helper T cells develop into two major subtypes known as TH1 and TH2 cells. TH1 helpers are host immunity effectors against bacterial and protozoa, while TH2 helpers are host immunity effectors against extracellular parasites. A new lineage of T cells has been designated as TH18 cells that produce proinflammatory cytokines and are thought to play an essential role in host defense against extracellular bacteria and fungi and be involved in autoimmune disease.

Schema of Notch’s role directing T cell fate. (Figure illustrated by Mikhail Dikov, Translational Therapeutics, The Ohio State University)
Notch has been implicated as a general coactivator of T cells [119] and a pathway that favors polarization of activated macrophages toward the M1 state [120, 121], both of which could augment the host immune response against cancer in the local microenvironment. This signaling is often ligand-specific, as specific ligands can elicit the development of different immune cells. DLL4 expressed by antigen-presenting cells specifically directs the differentiation and activation of CD8+ T cells [122, 123]. On the other hand, expression of JAGGED1 by antigen-presenting cells induces a T regulatory phenotype and subsequent immune suppression [124]. Expression of DLL1 or DLL4 leads to induction of T cell development and suppression of B cell development [125, 126]. Moreover, DLL4 is the essential and non-redundant ligand for NOTCH1 in T cell development [127]. Reduction in DLL1 and DLL4 in the hematopoietic microenvironment allows tumors to escape T cell immunity by elevating circulating VEGF [128]. This tumor-mediated suppression of T cell activity can be reversed by activation of Notch signaling using multivalent Dll1 in mouse xenograft models [129]. This activation is specific to the DLL1 ligand. For example, since DLL3 does not activate Notch signaling, it is not presumed to have an effect on T cell maturation [130]. JAGGED1 and JAGGED2 expression is thought to induce a Th2 fate [131]. NOTCH1 activation also directs T cells to a Th17 fate, although the specific ligand interaction mediating this fate is not known [132].
Notch1 and Notch2 are the receptors that mediate this signaling axis. It has been shown that Notch1 and Notch2 are necessary for the proliferation of activated CD8+ T cells and tumor-infiltrating T cells [133]. T cell differentiation is primarily mediated through Notch1. Eliminating Notch1 signaling results in impaired T cell development and increased B cell development [134]. Conversely, inducing constitutive Notch1 signaling promotes T cell development and reduces B cell development [135]. Furthermore, myeloid-derived suppressor cells, another immune-suppressing cell, block Notch1 and Notch2 expression in T cells, and this suppression can be circumvented by exogenously expressing Notch1-ICD in transgenic mouse models [133].
DLL1 plays a critical role in immunotherapy. DLL1 stimulation increases the number of infiltrating T cells and decreases the number of immune-suppressing regulatory T cells [129]. Combination of multivalent DLL1 and bortezomib, which sensitizes tumors to death signals, has been shown to restore the ability of the immune system to attack lung cancer cells [129]. Treatment with DLL1 has also been shown to improve progression-free survival in mice when combined with erlotinib by inducing T cell immunity [129, 136]. Importantly, DLL1 therapy does not have a proliferative or clonogenic effect on lung cancer cells, which suggests that Notch pathways can be targeted in the immune compartment without promoting tumor growth or aggressiveness.
In contrast to these studies, it has been shown that continued activation of the Notch signaling pathway in CD8+ T cells results in an increase in PD-1 expression and suppression of T cell activation [137]. Notch1-ICD specifically occupies the Pdcd1 promoter and can thus downregulate T cell activation [137]. Moreover, combination dosing of PD-1 and anti-DLL4 in a syngeneic CT26 model resulted in enhanced long-term memory, reduced suppressive functions of MDSC and Tregs, increased CD8 T cells, IL2 and IFN-γ levels [138].
Because of the complexity of these roles and the difficulty of measuring Notch activation and downstream effects in tumors, there remains uncertainty about the contribution of Notch signaling in stromal cells to cancer biology. Nonetheless, some intriguing preclinical data have emerged that suggest that modulation of host responses with selective Notch pathway inhibitors holds therapeutic promise. For example, short-term treatment with Dll4- or Notch1-blocking antibodies in the immediate posttransplant setting abrogates graft-versus-host disease without any measurable deleterious effect on graft-versus-leukemia activity [139], possibly because of a role for Dll4 expressed on lymph node stromal cells in the priming of Notch-expressing T cells [140]. Further investigation of Notch effects on host immunity in various disease settings clearly appears to be merited.
10.6 Notch as a Therapeutic Target
Many features unique to the Notch pathway must be considered when developing cancer therapies targeting Notch. The first key feature is that the Notch signaling cascade does not rely on an enzymatic amplification step by a phospholipase, nucleotide cyclase, or protein kinase [115, 141]. Instead, the Notch signaling cascade is triggered by receptor-ligand interaction and regulated by a series of proteolytic cleavages, protein stability, and cellular compartment changes. The “strength” of the Notch signal is proportional to the nuclear accumulation of cleaved intracellular “active” Notch [115]. As a consequence, Notch signaling is dose-dependent and can be regulated by factors that control the expression of ligands, expression of receptors, export of ligands and receptors to the membrane, receptor-ligand interaction, proteolytic cleavage, and endocytosis. Thus, complete shutdown of the pathway may be neither necessary nor ideal to achieve therapeutic effect [115]. A second key feature is that the intracellular half-life of cleaved intracellular “active” Notch is very short, resulting in a pulse of gene regulation and implying that intermittent inhibition may be sufficient to disrupt Notch signaling [52, 141]. The third key feature is that Notch signaling in the lung is context-dependent. While Notch signaling has an oncogenic role in adenocarcinoma, it is a tumor suppressor in SCLC and squamous cell carcinoma.
Additionally, the outcome of aberrant Notch activity is dependent on the spatial and temporal context of Notch activation. During the initial stages of tumor initiation, Notch signaling can prevent tumor formation, while in later stages of tumor development, Notch activation is required for maintenance of the tumor [142]. Furthermore, Notch activity can exert opposing effects in the same tissue under different microenvironmental conditions such as hypoxia.
The most established method of therapeutically targeting Notch signaling has been through inhibition of γ-secretase in tumors with Notch gain-of-function mutations. However, this approach needs to be carefully considered for use in cancers such as SCLC and squamous cell carcinoma, where NOTCH1 has been identified as a tumor suppressor. When designing therapies for the Notch pathway, it is important to consider the broad implications and multiple effects that Notch may have in different cell types. Clearly, having well-defined patient-stratification biomarkers is required for the development of effective Notch inhibitors.
A multitude of therapeutic approaches have been explored that include γ-secretase inhibitors (GSIs) , antibodies against Notch ligands, therapies targeting the Notch receptor negative regulatory region (NRR), and antibodies against Notch receptors. Table 10.3 summarizes drugs in development, and Table 10.4 summarizes clinical trials targeting the Notch signaling pathway.
10.6.1 γ-Secretase Inhibitors (GSIs)
To date, the most widely studied inhibitors of the Notch pathway are γ-secretase inhibitors. γ-secretase inhibitors were originally developed to treat Alzheimer’s disease. γ-secretase inhibitors target all four Notch receptors, ligands including DLL1 and JAGGED2, as well as many other proteins involved in Notch signal transduction, transcriptional regulation, and differentiation [149,150,151,152]. A number of small molecules that inhibit the γ-secretase complex with variable specificity and selectivity have been developed. These γ-secretase inhibitors inhibit Notch signaling by preventing presenilin-1 substrate binding and S3 proteolytic cleavage of Notch receptors. Early studies with γ-secretase inhibitors suggested that these agents might be useful as Notch-targeted therapies.
In vitro, γ-secretase inhibitors induce apoptosis in human lung cancer lines. For example, under hypoxia, which potentiates the strength of Notch signaling in adenocarcinoma, Chen et al. demonstrated that treatment with the γ-secretase inhibitor MRK-003 caused a potent apoptotic response as early as 48 h after treatment [108]. Reintroduction of active NOTCH1 led to a twofold reduction in cell death despite MRK-003 treatment [108], supporting an essential role for NOTCH1 in survival of adenocarcinoma cells. Similarly, Westhoff et al. demonstrated that primary cell cultures with NOTCH1 gain-of-function mutations were selectively killed by treatment with γ-secretase inhibitor’s DAPT and MRK-003 [6]. Additionally, Konoshi et al. investigated the in vitro and in vivo properties of MRK-003 in NSCLC and showed that MRK-003 inhibited NOTCH3 signaling, reduced tumor cell proliferation, and induced apoptosis [60]. Loss of NOTCH3 rendered the γ-secretase inhibitor ineffective, suggesting that the antitumor effect was NOTCH3 dependent [60]. Downregulation of pMAPK following MRK-003 treatment suggested that NOTCH3 may regulate apoptosis by modulating pERK and the pro-survival BCL-2 proteins [153]. A study by Kaur and colleagues demonstrated that SCLC cell lines are not responsive to γ-secretase inhibitors alone or in combination with etoposide or carboplatin [154].
In vivo studies have demonstrated that γ-secretase inhibitors slow the growth of subcutaneous lung cancer xenografts. In an adenocarcinoma xenograft model, Paris and colleagues demonstrated that treatment with the γ-secretase inhibitor DAPT resulted in dose-dependent inhibition of proliferation and differentiation [143]. Using a transgenic lung GEMM, Maraver et al. also demonstrated the efficacy of γ-secretase inhibitors [144]. Likewise, the γ-secretase inhibitor, LSN-4111575, exhibited antitumor efficacy in a KRASG12V-driven NSCLC mouse model [144]. Treatment with the γ-secretase inhibitor correlated with decreased expression of HES1, a Notch target gene and a negative regulator of DUSP1, a phosphatase that acts on the MAPKs [144]. The researchers demonstrated that increased expression of DUSP1 led to a decrease in pERK without changes in phosphorylated MEK [144]. In human lung tumor samples, high HES1 and low DUSP1 are associated with a poor outcome. Ambrogio et al. found that in KRAS-driven lung adenocarcinoma, inhibition of DDR1 in combination with Notch (using either LY-411575 or demcizumab) showed a survival benefit in patient-derived xenografts [155]. The authors hypothesized that this synergy was due to Notch and DDR1s combined role maintaining MAPK activity in KRAS-driven lung adenocarcinoma. Interestingly, the γ-secretase inhibitor JLK-6, a mechanism-based inhibitor of serine proteases, does not affect the Notch pathway and was shown to have a dose-dependent antitumor effect in lung adenocarcinoma xenografts [143]. Additionally JLK-6 inhibited the growth and vascularization of the human lung adenocarcinoma xenografts [143].
γ-secretase inhibitors have been evaluated in Phase I clinical trials in lung cancer. A Phase I clinical trial (NCT01193881) involving 16 patients with Stage IV or recurrent NSCLC was carried out to evaluate Roche’s γ-secretase inhibitor R04929097 [156, 157] in combination with erlotinib. The study showed improved median progression-free survival (PFS) for patients with a prior history of progression on erlotinib alone (64 versus 42 days) and four patients had stable disease at six weeks. While the combination was considered safe and feasible [156], adverse events included hypophosphatemia, rash, neuropathic pain and nausea. The manufacturer discontinued the development of R04929097 following completion of the Phase I cohort. The Bristol-Myers Squibb pan-Notch/γ-secretase inhibitor, BMS-906024, is currently in Phase I clinical trial (NCT01653470) alone and in combination with paclitaxel, FOLFIRI, or carboplatin plus paclitaxel to determine safe and tolerable dose in patients with solid tumors including lung cancer.
γ-secretase inhibitors have many advantages including ease of administration, oral bioavailability and low cost. Two main challenges facing the development of γ-secretase inhibitors include (a) substrate specificity since γ-secretase cleaves more than 60 substrates in addition to Notch [158] and (b) toxicity since pan-Notch inhibition has been linked with severe GI toxicity. These problems could be circumvented by developing substrate-specific γ-secretase inhibitors. Recent efforts have targeted Nicastrin, one of the subunits of γ-secretase that appears to be a main discriminator in γ-secretase substrate selectivity. Nicastrin is the target of the novel monoclonal antibody A5226A, developed by Hayashi et al. at the University of Tokyo. Hayashi et al. have demonstrated A5226A reduced cell viability of A549 cells [145]. Intriguingly, A5226A treatment further reduced the viability of DAPT-treated A549 cells [145].
10.6.2 Notch Antibodies
A number of biologics have been approved for the treatment of lung cancer. Moreover, a number of antibodies targeting Notch ligands, Notch receptors, and the NRR of Notch receptors are in clinical development. Some of these drugs have shown promising results in early clinical trials.
10.6.2.1 Ligand-Targeted Antibodies
Inhibitory antibodies directed against Notch ligands DLL3, DLL4, JAGGED1, and JAGGED2 have been developed. Preclinical studies by Genentech using an anti-DLL4 antibody, YW152F, demonstrated targeting DLL4/NOTCH1 signaling could have a profound impact by suppressing tumor angiogenesis and growth [146, 159]. Unfortunately, these studies also identified a number of significant safety concerns associated with this approach. In vitro studies with the anti-DLL4 antibody showed dysregulation of endothelium-specific genes as well as genes critical for proliferation and cell cycle regulation, while in vivo mice developed histopathological changes in the liver, sinusoidal dilation, and centrilobular hepatocyte atrophy. These preclinical findings suggest an essential role of DLL4 for maintaining the structural and functional integrity of the liver sinusoidal epithelium and hepatocyte homeostasis [159]. Genentech has also generated synthetic therapeutic antibodies targeting JAGGED1 and JAGGED2 [37]. JAGGED1 is overexpressed in many cancer types and has been implicated as a target for antitumor therapy [160]. Future studies will need to assess the clinical implications of using these antibodies in the context of lung cancer.
More recently, clinical trials of Regeneron’s anti-DLL4 antibody enoticumab (REGN421) and OncoMed Pharmaceutical’s antibody demcizumab (OMP-21M18) have shown dose-limiting adverse toxicity (nausea, abdominal pain, hypertension, fatigue and headache) [161] in clinical trials despite promising antitumor activity in the preclinical setting. No clinical benefit was seen in the Phase I clinical trial of enoticumab (NCT00871559). However, partial response was observed in a NSCLC bronchioloalveolar carcinoma patient with a β-catenin mutation [162]. Because tumor angiogenesis involves both VEGF and Notch, future studies may want to target both the VEGF and Notch pathway to increase efficacy or specifically investigate use of the DLL4 inhibitor in specific subtypes of cancers such as lung cancer patients with dysregulated Notch/β-catenin signaling. Anti-DLL4 treatment (OMP-21M18 targeting human and 21R30 targeting mouse) combination with chemotherapy inhibited tumor growth and appeared to decrease the frequency of tumor-initiating cells in a series of NSCLC PDX models [163]. Phase Ib clinical trials (NCT01189968) of demcizumab in combination with carboplatin and pemetrexed (Alimta) in patients with non-squamous NSCLC demonstrated promising results (RECIST response rate of 50%) and benefit for patient survival (overall clinical benefit rate of 88%). These results are being confirmed in an ongoing Phase II trial known as DENALI (NCT02259582) of demcizumab with carboplatin and pemetrexed in first-line non-squamous NSCLC patients [164].
Antibody-drug conjugates (ADCs) are therapies that combine the precision of an antibody linked with the cytotoxic power of a payload. DLL3 is a promising target for the treatment of SCLC, since DLL3 is expressed by both SCLC and large cell neuroendocrine carcinoma (LCNEC) tumors and tumor-initiating cells but is not expressed by normal cells [42]. AbbVie Stemcentrx is evaluating SC15LD6.5 that consists of an anti-DLL3 monoclonal antibody conjugated to a potent DNA-damaging pyrrolobenzodiazepine (PBD) dimer toxin. SC15LD6.5 dosed at one mg/kg, intravenous (iv), once a week for a total of four weeks (qwX4) demonstrated durable complete regression for up to 144 days of observation in preclinical mouse xenograft models [42]. Mechanism-of-action studies suggest that the rapid tumor debulking is a result of DLL3 expression on most tumor cells and suggest that the durability in response to SC16LD6.5 is due to eradication of DLL3-expressing tumor-initiating cells (TICs) . Moreover, SC16LD6.5 is efficacious in relapsed and refractory SCLC PDX models and is thus a potentially promising option for patients in the setting of second and third-line treatment. The safety and efficacy of SC16LD6.5 have been evaluated in an ongoing Phase 1 clinical trial (NCT01901653) in recurrent or refractory high-grade pulmonary and/or neuroendocrine cancer patients [165]. Rova-T demonstrated an acceptable safety profile in this trial and showed a confirmed objective response in 10/26 patients with elevated tumor DLL3 expression. Phase 1/2/3 trials with Rova-T are active (NCT03026166, NCT02819999, NCT03033511).
10.6.2.2 Targeted Antibodies Against the Negative Regulatory Region (NRR) of the Notch Receptor
The NRR domain plays a critical role in preventing activation of the receptors in the absence of ligand. The three cysteine-rich Lin12 Notch repeats (LNR) and heterodimerization (HD) domain of the NRR interact to keep the S2 cleavage site buried and prevent cleavage. During canonical Notch signaling, ligand binding to the receptor at the extracellular domain triggers a conformational change in the NRR exposing the S2 site for cleavage by ADAM proteases [166]. Li et al. demonstrated the potential of antibodies to target and stabilize the NRR domain in an “inactive” conformation where the S2 cleavage site remains buried within the NRR [167]. The inhibitory antibodies they identified by functional screening reverted the phenotypes of 293T cells induced by NOTCH3 signaling [167]. Scientists at Genentech have also developed antibodies to the NRR domain of NOTCH1 (NRR1) and NOTCH2 (NRR2). Antibodies targeted to the NRR1 demonstrated antitumor efficacy and decreased tumor angiogenesis in a Calu-6 lung adenocarcinoma mouse xenograft model [148]. Using ligand-competitive assays, researchers at Merck demonstrated that NRR-specific antibodies blocked JAGGED2-stimulated NOTCH1 activity, presumably by stabilizing the NRR domain in an auto-inhibited conformation [168]. While the NRR antibodies maximally inhibited NOTCH1 signaling, they did not significantly inhibit ligand-stimulated NOTCH2 or NOTCH3 [168]. Moreover, the NRR antibodies developed by Merck had variable efficacy in colorectal carcinoma and T-ALL cell lines, suggesting binding of the NRR is complex and that epitope masking by glycosylation or other posttranslational modifications of NOTCH1 may be cell type specific [168].
10.6.2.3 Receptor-Targeted Antibodies
The extracellular domains (ECD) of the four Notch receptors are composed of EGF-like repeats, which are of variable length. For example, NOTCH1 and NOTCH2 each contain 36 EGF-like repeats, whereas NOTCH3 contains 34, and NOTCH4 contains 29 EGF-like repeats in the extracellular domain. Antibodies that target the ligand-binding domain and compete with the endogenous ligand represent a promising novel therapeutic approach.
Tarextumab (OMP-59R5) which targets the ligand-binding domain of NOTCH2 and NOTCH3 has recently completed Phase IIb trials (NCT01859741) in SCLC [169]. It demonstrated dose-dependent antitumor efficacy and corresponding biomarker-driven activity in a Phase I trial. However the randomized 145 Phase 2 PINNACLE clinical trial in combination with chemotherapy (etoposide plus cisplatin or carboplatin) in previously untreated SCLC patients with extensive disease showed no benefit over placebo [170]. The median progression-free survival for tarextumab plus chemotherapy was 5.6 months versus 5.5 months for the placebo plus chemotherapy group. Median overall survival was 9.4 months in the treated versus 10.3 months (HR = 1.01) in the placebo group. The overall response rates were 68.5% in the treated and 70.8% in the placebo group. The Notch biomarkers that were evaluated in the study (HES1, HES6, HEY1, HEY2, and NOTCH3) failed to identify a subset of patients with treatment effect on progression-free or overall survival [170]. Furthermore, increased diarrhea and thrombocytopenia were observed in the placebo arm.
There is some early evidence that Notch overexpression may predict response to tarextumab, a NOTCH2/3 inhibitor, in SCLC. This comes from a Phase Ib dose escalation trial of tarextumab in SCLC [171]. In this study, patients were treated with etoposide, platinum, and tarextumab. NOTCH3 expression was determined by RT-PCR. Extensive disease patients with high NOTCH3 expression fared better with tarextumab than patients with low NOTCH3 expression. Although this trend was striking, it was not significant due to small patient numbers. Further analysis of NOTCH3 as a predictive marker of tarextumab response will take place when the Phase II trial of the drug is completed.
Another human monoclonal antibody brontictuzumab (OMP-52M51) targets the ligand-binding domain of NOTCH1 and demonstrated antitumor efficacy in early studies [172]. One potential concern was that chronic reduction of Notch1 signaling in mice promoted widespread vascular tumor formation in preclinical studies [173]. Additionally enrollment for Phase 1B clinical trials in combination with trifluridine/tipiracil in third-line colorectal patients was abruptly ended and was not tolerated in that patient population [170]. A clinical trial for brontictuzumab in relapsed or refractory solid tumors (including SCLC) with activated NOTCH1 has been completed (OncoMed Pharmaceuticals, NCT01778439). Preliminary findings demonstrated general tolerability at 1.5 mg/kg Q3W. The main toxicity was off-target diarrhea, but patients also exhibited fatigue, nausea and vomiting. In patients with high NOTCH1, there were a few patients with stable disease 42.9% (6/14), and one (7.1%; 1/14) patient had partial response [174]. Patients with low NOTCH1 did not respond and had stable 9.1% (1/11) or progressive disease 90.9% (10/11) [174]. Oncomed has halted clinical trials with brontictuzumab.
The Notch receptor is also an ideal target for antibody-drug conjugates. Pfizer is currently evaluating the safety and efficacy of their non-inhibitory anti-NOTCH3 antibody-drug conjugate PF-06650808 in a Phase I clinical trial (NCT02129205) for patients with solid tumors that have a history of metastatic triple-negative breast cancer [175].
Monoclonal antibodies provide a number of advantages including improved half-life, immune-mediated efficacy through antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, as well as specificity allowing members of the Notch signaling pathway to be targeted with high affinity. While antibodies are generally well tolerated, it is critical to identify the right combination of tumor and drug to derive maximal therapeutic benefit. Additional challenges with antibody-targeted therapies are their complex dose-response curves in vivo and long half-lives. While small molecules are typically excreted in hours, most antibodies remain in circulation for days. This may pose a problem, as the consequences of sustained inhibition of the Notch signaling pathway are not fully understood. For example, studies with anti-Dll4 antibodies in rat models have shown that prolonged blockade of the ligand resulted in severe disruption of normal tissue homeostasis, activation of endothelial cells and led to development of vascular/endothelial-based tumors resembling hemangioblastoma in the heart and lung [147]. These studies raise significant concern that chronic blockade of Notch signaling pathways may disrupt normal organ homeostasis and potentially produce disease in multiple organs. In that case, single-chain antibodies with short half-lives may be preferable.
10.6.3 Radiotherapy
Although radiation has been reported to induce Notch activation, very little is known about the relationship between radiation and the Notch pathway. Studies have proposed a synergistic effect of Notch-targeted therapy following radiation therapy of lung cancer in vitro and in vivo. Mizugaki et al. combined γ-secretase inhibitor and radiation in escalating doses in three Notch-expressing NSCLC cell lines (H460, A549, and H1395) [176]. γ-secretase inhibitor treatment following radiation suppressed growth most effectively in vitro and in vivo. The combination induced apoptosis via MAPK and Bcl2 family proteins. They found that blocking activation of Notch by using γ-secretase inhibitors after radiation treatment prevents Notch-induced radiation resistance. Ikezawa et al. determined that the induction of NOTCH3 following radiotherapy is caused by HIF-1α and found that co-treatment with HIF inhibitor YC-1 improved radiosensitivity of tumors in conjunction with a γ-secretase inhibitor [177].
10.6.4 Summary of Therapeutic Approaches
Each of the therapeutic approaches described above has potential advantages and disadvantages. While γ-secretase inhibitors have demonstrated preclinical activity, their clinical utility for lung cancer remains to be demonstrated. Targeted and selective biologics including antibody-drug conjugates (ADCs) are likely to be the treatment of choice with fewer side effects. Alternatively, bispecific antibodies may offer a useful strategy for Notch-targeted drug development in the future, for example, combining a Notch antibody with a T cell antibody to direct the immune response to Notch-expressing tumor cells. It will be critical to have a strong companion biomarker package in place to select patients and optimize clinical benefit. A better understanding of the transcriptional, translational, and posttranslational regulation of the Notch signaling pathway is needed to understand the implications that these specific therapies may have for lung cancer patients. The cellular context of the cancer will be very important, as roles of Notch in adenocarcinoma will be very different than in squamous cell carcinoma. The ability to identify subgroups of cancer patients that will benefit from Notch-targeted therapies will be essential if they are to be of clinical utility.
10.7 Targeting Notch in Cancer Stem Cells and Rational Combinations of Therapies with Notch Inhibitors
As discussed in Section 10.3, the Notch signaling pathway has been shown to have a role in the survival and maintenance of stem cell populations in the lung [46,47,48, 185]. Cancer stemlike cells are undifferentiated cancer cells that share properties of normal stem cells such as self-renewal, asymmetric cell division, clonogenicity, and resistance to chemotherapies. Cancer stemlike cells are thought to exist in small numbers as a distinct population of cells within tumors. Their quiescent nature allows them to escape standard therapies and even after periods of apparent complete remission cause recurrence or metastasis. Targeted therapeutics to eradicate cancer stemlike cells should theoretically result in more durable responses and improve survival outcomes.
Cancer stemlike cells can be isolated on the basis of the expression of cancer stemlike cell markers, but the accuracy and relevance of these markers remains controversial [186, 187]. Research has identified CD44 [188], CD133 [189,190,191] and aldehyde dehydrogenase (ALDH) [192,193,194] as potential markers of lung cancer stemlike cells [195]. Sullivan et al. compared the expression of three putative lung cancer stemlike cell markers (ALDH1A1, ALDH3A1 and CD133) and found that the markers identified distinct tumor subpopulations [194]. Elevated expression of NOTCH1, NOTCH2, NOTCH3, HES1, and HEY1 in ALDH+ cells versus ALDH− cells, indicated Notch pathway activation in the ALDH+ subpopulation [194]. NOTCH3 had the strongest correlation with ALDH+ expression in NSCLC and suppression of NOTCH3 expression reduced the ALDH positive population and reduced tumor cell proliferation [194]. Other studies have supported a role of ALDH as a marker for stemlike lung cancer cells. Studies by Li et al. isolated ALDH+ lung cancer cells and demonstrated the population was capable of colony formation, proliferation, growth, migration and resulted in tumors in mice that represented characteristics of the parental lung cancer cells [193]. Futhermore, γ-secretase inhibitor therapy reduced the ADLH+ population, supporting a role of Notch signaling in maintenance of the cancer stemlike cell population [194]. In addition to NSCLC, Notch drives stemlike properties in esophageal adenocarcinoma [196]. Another research group has identified that MAP17-mediated sequestration of Numb promotes Notch signaling and cancer stemlike cell phenotypes [197].
Experimental studies have found that pretreatment of H460 and H661 lung cancer cell lines with low-dose cisplatin resulted in enrichment of CD133+cells and appeared to be mediated through Notch signaling [198]. Pretreatment with the γ-secretase inhibitor, DAPT, or a NOTCH1-targeted shRNA reduced enrichment of CD133+cells and increased sensitivity to doxorubicin and paclitaxel. Additionally, re-expression of NOTCH1-ICD was shown to reverse the action of DAPT on drug sensitivity. Immunohistochemistry of relapsed NSCLC lung tumors that had been treated with cisplatin showed a significant increase in CD133+ expression in three out of six patients. In vivo studies demonstrated cisplatin treatment increased NOTCH1 cleavage and suggested that cisplatin-induced enrichment of CD133+ cells was mediated through activation of Notch signaling.
It has also been shown that treating lung cancer cells with erlotinib enriches ALDH+ stemlike cells through activation of Notch3 [89]. Similarly, a study by Rosell et al. identified that gefitinib treatment in EGFR-mutant cell lines induced YAP1-Notch signaling in a compensatory manner [199]. These studies may shed light on the perplexing observation that the addition of EGFR TKIs to curative-intent therapies (chemoradiation [200] or surgery [201]) actually results in an increased risk of death. Clinical benefit in these situations may depend on the elimination of cancer stemlike cells as well as the primary tumor. Together, these studiessuggest that dual targeting of the stem cells using Notch therapies together with targeted TKI’s or standard chemotherapies may result in more durable responses in lung cancer patients.
10.8 Conclusion
Notch plays essential role in early lung development, maintenance of adult airways, and cancer. Genome sequencing studies have identified Notch mutations in a small percentage of lung cancers. Recent literature has highlighted the significant role that Notch signaling plays in lung tumorigenesis even in the absence of mutational evidence. Despite the low number of mutations, dysregulation of the Notch signaling pathway is associated with 25% of SCLC and 33% of NSCLCs. This makes Notch an attractive target for therapy since it plays critical roles in the regulation of tumor growth and proliferation, apoptosis, cell differentiation, survival, immune response, angiogenesis, cancer stem cell biology, and chemoresistance. Unfortunately, efforts to target Notch therapeutically still face a number of hurdles. Successful development of targeted therapies that can be used in combination with other approaches will require a deeper understanding of the mechanisms that enable different cell-type specific responses of Notch in lung cancer.
References
Radtke, F., Schweisguth, F., & Pear, W. (2005). The Notch ‘gospel’. EMBO Reports, 6(12), 1120–1125.
Gazave, E., et al. (2009). Origin and evolution of the Notch signalling pathway: An overview from eukaryotic genomes. BMC Evolutionary Biology, 9(1), 249.
Aster, J. C., Pear, W. S., & Blacklow, S. C. (2017). The varied roles of notch in cancer. Annual Review of Pathology: Mechanisms of Disease, 12, 245–275.
Leiserson, M. D., et al. (2015). Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nature Genetics, 47(2), 106–114.
George, J., et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature, 524(7563), 47–53.
Westhoff, B., et al. (2009). Alterations of the Notch pathway in lung cancer. Proceedings of the National Academy of Sciences, 106(52), 22293–22298.
Capaccione, K. M., & Pine, S. R. (2013). The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis, 34(7), 1420–1430. p. bgt127.
Donnem, T., et al. (2010). Prognostic impact of Notch ligands and receptors in nonsmall cell lung cancer. Cancer, 116(24), 5676–5685.
Baumgart, A., et al. (2015). Opposing role of Notch1 and Notch2 in a Kras G12D-driven murine non-small cell lung cancer model. Oncogene, 34(5), 578.
Licciulli, S., et al. (2013). Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Research, 73(19), 5974–5984.
Xu, X., et al. (2014). The cell of origin and subtype of K-Ras-induced lung tumors are modified by Notch and Sox2. Genes & Development, 28(17), 1929–1939.
Duan, L., et al. (2006). Growth suppression induced by Notch1 activation involves Wnt—β-catenin down-regulation in human tongue carcinoma cells. Biology of the Cell, 98(8), 479–490.
Radtke, F., Fasnacht, N., & MacDonald, H. R. (2010). Notch signaling in the immune system. Immunity, 32(1), 14–27.
Nicolas, M., et al. (2003). Notch1 functions as a tumor suppressor in mouse skin. Nature Genetics, 33(3), 416–421.
Leong, K. G., & Karsan, A. (2006). Recent insights into the role of Notch signaling in tumorigenesis. Blood, 107(6), 2223–2233.
Espinoza, I., & Miele, L. (2013). Notch inhibitors for cancer treatment. Pharmacology & Therapeutics, 139(2), 95–110.
Stanley, P., & Okajima, T. (2010). Chapter four-roles of glycosylation in Notch signaling. Current Topics in Developmental Biology, 92, 131–164.
Shi, S., & Stanley, P. (2003). Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proceedings of the National Academy of Sciences of the United States of America, 100(9), 5234–5239.
Fernandez-Valdivia, R., et al. (2011). Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development, 138(10), 1925–1934.
Takeuchi, H., & Haltiwanger, R. S. (2014). Significance of glycosylation in Notch signaling. Biochemical and Biophysical Research Communications, 453(2), 235–242.
Andersson, E. R., & Lendahl, U. (2014). Therapeutic modulation of Notch signalling [mdash] are we there yet? Nature Reviews. Drug Discovery, 13(5), 357–378.
Le Bras, S., Loyer, N., & Le Borgne, R. (2011). The multiple facets of ubiquitination in the regulation of notch signaling pathway. Traffic, 12(2), 149–161.
O’Neil, J., et al. (2007). FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ-secretase inhibitors. The Journal of Experimental Medicine, 204(8), 1813–1824.
Matsuno, K., et al. (2002). Involvement of a proline-rich motif and RING-H2 finger of Deltex in the regulation of Notch signaling. Development, 129(4), 1049–1059.
Matsuno, K., et al. (1995). Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development, 121(8), 2633–2644.
Espinosa, L., et al. (2003). Phosphorylation by glycogen synthase kinase-3β down-regulates Notch activity, a link for Notch and Wnt pathways. Journal of Biological Chemistry, 278(34), 32227–32235.
McGill, M. A., & McGlade, C. J. (2003). Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. Journal of Biological Chemistry, 278(25), 23196–23203.
Housden, B. E., et al. (2013). Transcriptional dynamics elicited by a short pulse of notch activation involves feed-forward regulation by E (spl)/Hes genes. PLoS Genetics, 9(1), e1003162.
Lamar, E., et al. (2001). Nrarp is a novel intracellular component of the Notch signaling pathway. Genes & Development, 15(15), 1885–1899.
Yi, F., Amarasinghe, B., & Dang, T. P. (2013). Manic fringe inhibits tumor growth by suppressing Notch3 degradation in lung cancer. American Journal of Cancer Research, 3(5), 490–499.
Tsao, P.-N., et al. (2008). γ-secretase activation of Notch signaling regulates the balance of proximal and distal fates in progenitor cells of the developing lung. Journal of Biological Chemistry, 283(43), 29532–29544.
Kong, Y., et al. (2004). Functional diversity of notch family genes in fetal lung development. American Journal of Physiology-Lung Cellular and Molecular Physiology, 286(5), L1075–L1083.
Morimoto, M., et al. (2010). Canonical Notch signaling in the developing lung is required for determination of arterial smooth muscle cells and selection of Clara versus ciliated cell fate. Journal of Cell Science, 123(2), 213–224.
Morimoto, M., et al. (2012). Different assemblies of Notch receptors coordinate the distribution of the major bronchial Clara, ciliated and neuroendocrine cells. Development (Cambridge, England), 139(23), 4365–4373.
Ito, T., et al. (2000). Basic helix-loop-helix transcription factors regulate the neuroendocrine differentiation of fetal mouse pulmonary epithelium. Development, 127(18), 3913–3921.
Zhang, S., et al. (2013). Jagged1 is the major regulator of notch-dependent cell fate in proximal airways. Developmental Dynamics, 242(6), 678–686.
Lafkas, D., et al. (2015). Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature, 528(7580), 127–131.
Chapman, G., et al. (2011). Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Human Molecular Genetics, 20(5), 905–916.
Serth, K., et al. (2015). O-fucosylation of DLL3 is required for its function during somitogenesis. PLoS One, 10(4), e0123776.
Henke, R. M., et al. (2009). Ascl1 and Neurog2 form novel complexes and regulate Delta-like3 (Dll3) expression in the neural tube. Developmental Biology, 328(2), 529–540.
Augustyn, A., et al. (2014). ASCL1 is a lineage oncogene providing therapeutic targets for high-grade neuroendocrine lung cancers. Proceedings of the National Academy of Sciences, 111(41), 14788–14793.
Saunders, L. R., et al. (2015). A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Science Translational Medicine, 7(302), 302ra136.
Guseh, J. S., et al. (2009). Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development, 136(10), 1751–1759.
Xu, K., et al. (2010). Lunatic Fringe-mediated Notch signaling is required for lung alveogenesis. American Journal of Physiology-Lung Cellular and Molecular Physiology, 298(1), L45–L56.
Dang, T. P., et al. (2003). Constitutive activation of Notch3 inhibits terminal epithelial differentiation in lungs of transgenic mice. Oncogene, 22(13), 1988–1997.
Carraro, G., & Stripp, B. R. (2015). A new Notch for lung stem cells. Cell Stem Cell, 16(2), 107–109.
Pardo-Saganta, A., et al. (2015). Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell, 16(2), 184–197.
Vaughan, A. E., et al. (2015). Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature, 517(7536), 621–625.
Randell, S. H. (2006). Airway epithelial stem cells and the pathophysiology of chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society, 3(8), 718–725.
Rock, J. R., Randell, S. H., & Hogan, B. L. (2010). Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Disease Models & Mechanisms, 3(9–10), 545–556.
Shi, W., Chen, F., & Cardoso, W. V. (2009). Mechanisms of lung development: Contribution to adult lung disease and relevance to chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society, 6(7), 558–563.
Custodio, A., & Barriuso, J. (2014). What is the meaning of notch pathway and how can we selectively do the targeting? In Stem cells in Cancer: Should we believe or not? (pp. 23–65). Dordrecht: Springer.
Dang, T. P., et al. (2000). Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. Journal of the National Cancer Institute, 92(16), 1355–1357.
Sriuranpong, V., et al. (2001). Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Research, 61(7), 3200–3205.
Lim, J. S., et al. (2017). Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature, 545(7654), 360.
Allen, T. D., et al. (2011). Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma. Cancer Research, 71(18), 6010–6018.
Kim, Y., et al. (2014). Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. Journal of Clinical Oncology, 32(2), 121–128.
Brooks, Y. S., et al. (2014). Multifactorial ERβ and NOTCH1 control of squamous differentiation and cancer. The Journal of Clinical Investigation, 124(5), 2260.
Hassan, W. A., et al. (2016). Evaluation of role of Notch3 signaling pathway in human lung cancer cells. Journal of Cancer Research and Clinical Oncology, 142(5), 981–993.
Konishi, J., et al. (2007). γ-Secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Research, 67(17), 8051–8057.
Haruki, N., et al. (2005). Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Research, 65(9), 3555–3561.
Gordian, E., et al. (2017). Novel oncogenic function of Notch4 in Hispanic lung cancer. In AACR. Proceedings: AACR annual meeting 2017. Washington, DC.: http://cancerres.aacrjournals.org/content/77/13_Supplement/4456.short.
Cerami, E., et al. (2012). The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery, 2, 401–404. https://doi.org/10.1158/2159-8290.CD-12-0095. PubMed: 22588877.
Gao, J., et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling, 6(269), pl1.
Ding, L., et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature, 455(7216), 1069–1075.
Network, C. G. A. R. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489(7417), 519–525.
Barse, L., & Bocchetta, M. (2015). Non-small-cell lung carcinoma: Role of the Notch signaling pathway. Lung Cancer (Auckl), 6, 43–53.
Society, A. C. Lung cancer (Non-Small Cell): What is non-small cell lung cancer. 2016 03/04/2015 12 January 2016]. Available from: http://www.cancer.org/cancer/lungcancer-non-smallcell/detailedguide/non-small-cell-lung-cancer-what-is-non-small-cell-lung-cancer.
van Meerbeeck, J. P., Fennell, D. A., & De Ruysscher, D. K. (2011). Small-cell lung cancer. The Lancet, 378(9804), 1741–1755.
Kunnimalaiyaan, M., & Chen, H. (2007). Tumor suppressor role of Notch-1 signaling in neuroendocrine tumors. The Oncologist, 12(5), 535–542.
Sriuranpong, V., et al. (2002). Notch signaling induces rapid degradation of achaete-scute homolog 1. Molecular and Cellular Biology, 22(9), 3129–3139.
Rudin, C. M., et al. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature Genetics, 44(10), 1111–1116.
Peifer, M., et al. (2012). Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature Genetics, 44(10), 1104–1110.
Wang, N. J., et al. (2011). Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proceedings of the National Academy of Sciences, 108(43), 17761–17766.
Agrawal, N., et al. (2011). Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science, 333(6046), 1154–1157.
Stransky, N., et al. (2011). The mutational landscape of head and neck squamous cell carcinoma. Science, 333(6046), 1157–1160.
Guo, L., et al. (2015). Roles of NOTCH1 as a therapeutic target and a biomarker for lung cancer: Controversies and perspectives. Disease Markers, 2015, 520590.
Yuan, X., et al. (2015). Meta-analysis reveals the correlation of Notch signaling with non-small cell lung cancer progression and prognosis. Scientific Reports, 5, 10338.
Jiang, X., et al. (2007). Expression and significance of Notch1, Jagged1 and VEGF in human non-small cell lung cancer. Zhong nan da xue xue bao. Yi xue ban= Journal of Central South University. Medical Sciences, 32(6), 1031–1036.
Andersen, S., et al. (2011). Correlation and coexpression of HIFs and NOTCH markers in NSCLC. Anticancer Research, 31(5), 1603–1606.
Mariscal, J., et al. (2016). Molecular profiling of circulating tumour cells identifies notch1 as a principal regulator in advanced non-small cell lung cancer. Scientific Reports, 6, 37820.
Chen, C.-Y., et al. (2017). Expression of notch gene and its impact on survival of patients with resectable non-small cell lung cancer. Journal of Cancer, 8(7), 1292.
Sherry, S. T., et al. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311.
Bollig-Fischer, A., et al. (2015). Racial diversity of actionable mutations in non–small cell lung cancer. Journal of Thoracic Oncology, 10(2), 250–255.
Lee, S. Y., et al. (2017). A functional polymorphism in DTX1 gene of notch pathway predicts the prognosis of surgically resected non-small cell lung cancer. AACR Proceedings: AACR annual meeting 2017; Washington, DC http://cancerres.aacrjournals.org/content/77/13_Supplement/5727.short.
Quan, X., et al. (2017). Single nucleotide polymorphism rs3124599 in Notch1 is associated with the risk of lung cancer in northeast Chinese non-smoking females. Oncotarget, 8(19), 31180.
Xu, K., Moghal, N., & Egan, S. E. (2012). Notch signaling in lung development and disease. In Notch signaling in embryology and Cancer (pp. 89–98). Springer, New York, NY.
Zheng, Y., et al. (2013). A rare population of CD24+ ITGB4+ Notch hi cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell, 24(1), 59–74.
Arasada, R. R., et al. (2014). EGFR blockade enriches for lung cancer stem–like cells through Notch3-dependent signaling. Cancer Research, 74(19), 5572–5584.
Justilien, V., et al. (2012). Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One, 7(4), e35040.
Lefort, K., & Dotto, G. P. (2004). Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Seminars in Cancer Biology, 14 (5), 374–386. Academic Press.
Lowell, S., et al. (2000). Stimulation of human epidermal differentiation by Delta–Notch signalling at the boundaries of stem-cell clusters. Current Biology, 10(9), 491–500.
Rangarajan, A., et al. (2001). Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. The EMBO Journal, 20(13), 3427–3436.
Li, L., Sheehan, C., & Ross, J. (2009). Notch signaling in non small cell lung cancers (NSCLC) is associated with squamous differentiation and favorable clinical outcome. Laboratory Investigation. Nature Publishing Group 75 varick st, 9TH FLR, New York, NY 10013-1917 USA.
Schwartz, A. G., et al. (2007). Reproductive factors, hormone use, estrogen receptor expression and risk of non–small-cell lung cancer in women. Journal of Clinical Oncology, 25(36), 5785–5792.
Schwartz, A. G., et al. (2005). Nuclear estrogen receptor β in lung cancer: Expression and survival differences by sex. Clinical Cancer Research, 11(20), 7280–7287.
Skov, B. G., Fischer, B. M., & Pappot, H. (2008). Oestrogen receptor β over expression in males with non-small cell lung cancer is associated with better survival. Lung Cancer, 59(1), 88–94.
Zhang, M., et al. (2016). Does Notch play a tumor suppressor role across diverse squamous cell carcinomas? Cancer Medicine, 5(8), 2048–2060.
Wael, H., et al. (2014). Notch1 signaling controls cell proliferation, apoptosis and differentiation in lung carcinoma. Lung Cancer, 85(2), 131–140.
Zheng, Q., et al. (2007). Notch signaling inhibits growth of the human lung adenocarcinoma cell line A549. Oncology Reports, 17(4), 847–852.
Baumgart, A., et al. (2010). ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non–small cell lung cancer. Cancer Research, 70(13), 5368–5378.
Meng, X., & Yu, J. (2012). Implementation of hypoxia measurement into lung cancer therapy. Lung Cancer, 75(2), 146–150.
Graves, E. E., et al. (2010). Hypoxia in models of lung cancer: Implications for targeted therapeutics. Clinical Cancer Research, 16(19), 4843–4852.
Le, Q.-T., et al. (2006). An evaluation of tumor oxygenation and gene expression in patients with early stage non–small cell lung cancers. Clinical Cancer Research, 12(5), 1507–1514.
Graves, E. E., Maity, A., & Le, Q.-T. (2010). The tumor microenvironment in non–small-cell lung cancer. Seminars in Radiation Oncology, 20(3), 156–163. WB Saunders.
Mees, G., et al. (2009). Molecular imaging of hypoxia with radiolabelled agents. European Journal of Nuclear Medicine and Molecular Imaging, 36(10), 1674–1686.
Vikram, D. S., Zweier, J. L., & Kuppusamy, P. (2007). Methods for noninvasive imaging of tissue hypoxia. Antioxidants & Redox Signaling, 9(10), 1745–1756.
Chen, Y., et al. (2007). Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Research, 67(17), 7954–7959.
Gustafsson, M. V., et al. (2005). Hypoxia requires notch signaling to maintain the undifferentiated cell state. Developmental Cell, 9(5), 617–628.
Eliasz, S., et al. (2010). Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene, 29(17), 2488–2498.
Han, Y. H., et al. (2008). Antimycin A as a mitochondrial electron transport inhibitor prevents the growth of human lung cancer A549 cells. Oncology Reports, 20(3), 689–693.
Goodwin, J., et al. (2017). The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nature Communications, 8, 15503.
Peiris-Pagès, M., et al. (2016). Cancer stem cell metabolism. Breast Cancer Research, 18(1), 55.
Mazzone, M., et al. (2010). Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proceedings of the National Academy of Sciences, 107(11), 5012–5017.
Miele, L., Golde, T., & Osborne, B. (2006). Notch signaling in cancer. Current Molecular Medicine, 6(8), 905–918.
Bachireddy, P., Rakhra, K., & Felsher, D. (2012). Immunology in the clinic review series; focus on cancer: Multiple roles for the immune system in oncogene addiction. Clinical & Experimental Immunology, 167(2), 188–194.
Rakhra, K., et al. (2010). CD4+ T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell, 18(5), 485–498.
Odunsi, K., & Old, L. J. (2007). Tumor infiltrating lymphocytes: Indicators of tumor-related immune responses. Cancer Immunity., 7(3). http://cancerimmunolres.aacrjournals.org/content/canimmarch/7/1/3.full-text.pdf.
Bailis, W., et al. (2013). Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity, 39(1), 148–159.
Xu, H., et al. (2012). Notch-RBP-J signaling regulates the transcription factor IRF8 to promote inflammatory macrophage polarization. Nature Immunology, 13(7), 642–650.
Xu, J., et al. (2015). NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. The Journal of Clinical Investigation, 125(4), 1579.
Sauma, D., et al. (2012). Notch signalling regulates cytokine production by CD8+ and CD4+ T cells. Scandinavian Journal of Immunology, 75(4), 389–400.
Kassner, N., et al. (2010). Cutting edge: Plasmacytoid dendritic cells induce IL-10 production in T cells via the Delta-like-4/Notch axis. The Journal of Immunology, 184(2), 550–554.
Yvon, E. S., et al. (2003). Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood, 102(10), 3815–3821.
Delaney, C., et al. (2005). Dose-dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood, 106(8), 2693–2699.
Dorsch, M., et al. (2002). Ectopic expression of Delta4 impairs hematopoietic development and leads to lymphoproliferative disease. Blood, 100(6), 2046–2055.
Koch, U., et al. (2008). Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. The Journal of Experimental Medicine, 205(11), 2515–2523.
Huang, Y., et al. (2011). Resuscitating cancer immunosurveillance: Selective stimulation of DLL1-Notch signaling in T cells rescues T cell function and inhibits tumor growth. Cancer Research, 71(19), 6122–6131.
Shanker, A., et al. (2014). Cancer therapy by resuscitating Notch immune surveillance. Journal for Immunotherapy of Cancer, 2(Suppl 1), O1.
Ladi, E., et al. (2005). The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. The Journal of Cell Biology, 170(6), 983–992.
Amsen, D., Antov, A., & Flavell, R. A. (2009). The different faces of Notch in T-helper-cell differentiation. Nature Reviews. Immunology, 9(2), 116–124.
Keerthivasan, S., Suleiman, R., Lawlor, R., Roderick, J., Bates, T., Minter, L., … & Miele, L. (2011). Notch signaling regulates mouse and human Th17 differentiation. The Journal of Immunology, 1003658.
Sierra, R. A., et al. (2014). Rescue of Notch 1 signaling in antigen-specific CD8+ T cells overcomes tumor-induced T cell suppression and enhances immunotherapy in cancer. Cancer Immunology Research, 2(8), 800–811.
Radtke, F., et al. (1999). Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity, 10(5), 547–558.
Pear, W. S., et al. (1996). Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. The Journal of Experimental Medicine, 183(5), 2283–2291.
Biktasova, A. K., et al. (2015). Multivalent forms of the Notch ligand DLL-1 enhance antitumor T cell immunity in lung cancer and improve efficacy of EGFR targeted therapy. Cancer Research: p. canres. 1154.2014.
Mathieu, M., et al. (2013). Notch signaling regulates PD-1 expression during CD8+ T-cell activation. Immunology and Cell Biology, 91(1), 82–88.
Srivastava, M., et al. (2015). Dual targeting of delta-like ligand 4 (DLL4) and programmed death 1 (PD1) inhibits tumor growth and generates enhanced long-term immunological memory. Cancer Research, 75(15 Suppl), 255–255.
Tran, I. T., et al. (2013). Blockade of individual Notch ligands and receptors controls graft-versus-host disease. The Journal of Clinical Investigation, 123(4), 1590–1604.
Fasnacht, N., et al. (2014). Specific fibroblastic niches in secondary lymphoid organs orchestrate distinct Notch-regulated immune responses. Journal of Experimental Medicine, 211(11), 2265–2279.
Rizzo, P., et al. (2008). Rational targeting of Notch signaling in cancer. Oncogene, 27(38), 5124–5131.
Ranganathan, P., Weaver, K. L., & Capobianco, A. J. (2011). Notch signalling in solid tumours: A little bit of everything but not all the time. Nature Reviews Cancer, 11(5), 338–351.
Paris, D., et al. (2005). Inhibition of angiogenesis and tumor growth by β and γ-secretase inhibitors. European Journal of Pharmacology, 514(1), 1–15.
Maraver, A., et al. (2012). Therapeutic effect of γ-secretase inhibition in Kras G12V-driven non-small cell lung carcinoma by derepression of DUSP1 and inhibition of ERK. Cancer Cell, 22(2), 222–234.
Hayashi, I., et al. (2012). Neutralization of the γ-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene, 31(6), 787–798.
Ridgway, J., et al. (2006). Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature, 444(7122), 1083–1087.
Yan, M., et al. (2010). Chronic DLL4 blockade induces vascular neoplasms. Nature, 463(7282), E6–E7.
Wu, Y., et al. (2010). Therapeutic antibody targeting of individual Notch receptors. Nature, 464(7291), 1052–1057.
Nickoloff, B. J., Osborne, B. A., & Miele, L. (2003). Notch signaling as a therapeutic target in cancer: A new approach to the development of cell fate modifying agents. Oncogene, 22(42), 6598–6608.
Kopan, R., & Ilagan, M. X. G. (2004). γ-secretase: Proteasome of the membrane? Nature Reviews Molecular Cell Biology, 5(6), 499–504.
Maetzel, D., et al. (2009). Nuclear signalling by tumour-associated antigen EpCAM. Nature Cell Biology, 11(2), 162–171.
Takebe, N., Nguyen, D., & Yang, S. X. (2014). Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacology & Therapeutics, 141(2), 140–149.
Konishi, J., et al. (2010). Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene, 29(4), 589–596.
Kaur, G., et al. (2016). Bromodomain and hedgehog pathway targets in small cell lung cancer. Cancer Letters, 371(2), 225–239.
Ambrogio, C., et al. (2016). Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nature Medicine, 22(3), 270.
Gold, K. A., et al. (2013). A phase I/II trial combining erlotinib with gamma secretase inhibitor RO4929097 in advanced non-small cell lung cancer (NSCLC). Journal of Clinical Oncology. American Society of Clinical Oncology 2318 MILL ROAD, STE 800, ALEXANDRIA, VA 22314 USA.
Luistro, L., et al. (2009). Preclinical profile of a potent γ-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Research, 69(19), 7672–7680.
De Strooper, B., Iwatsubo, T., & Wolfe, M. S. (2012). Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine, 2(1), a006304.
Yan, M. (2011). Therapeutic promise and challenges of targeting DLL4/NOTCH1. Vascular Cell, 3, 17.
Li, D., et al. (2014). The notch ligand JAGGED1 as a target for anti-tumor therapy. Frontiers in Oncology, 4, 254.
Alketbi, A., & Attoub, S. (2015). Notch signaling in cancer: Rationale and strategies for targeting. Current Cancer Drug Targets, 15(5), 364–374.
Chiorean, E. G., et al. (2015). A phase I first-in-human study of enoticumab (REGN421), a fully human Delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clinical Cancer Research, 21(12), 2695–2703.
Brunner, A., et al. (2016). Effects of anti-DLL4 treatment on non-small cell lung cancer (NSCLC) human xenograft tumors. AACR. (http://cancerres.aacrjournals.org/content/76/14_Supplement/4652.short) Proceedings: AACR 107th Annual Meeting 2016; April 16–20, 2016; New Orleans, LA.
Globenewswire.com. (2015). Oncomed presents demcizumab data from phase 1B clinical trial in non-small cell lung cancer patients at the European lucg cancer conference. Available from: http://globenewswire.com/news-release/2015/04/16/725132/10129223/en/OncoMed-Presents-Demcizumab-Data-From-Phase-1b-Clinical-Trial-in-Non-Small-Cell-Lung-Cancer-Patients-at-the-European-Lung-Cancer-Conference.html?print=1.
Rudin, C. M., et al. (2017). Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. The Lancet Oncology, 18(1), 42–51.
Gordon, W. R., et al. (2007). Structural basis for autoinhibition of Notch. Nature Structural & Molecular Biology, 14(4), 295–300.
Li, K., et al. (2008). Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. Journal of Biological Chemistry, 283(12), 8046–8054.
Aste-Amézaga, M., et al. (2010). Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One, 5(2), e9094.
Daniel, D. B., Rudin, C. M., Hart, L., Spigel, D. R., Edelman, M. J., Goldschmidt, J., Bordoni, R., et al. (2017). 1530PDResults of a randomized, placebo-controlled, phase 2 study of tarextumab (TRXT, anti-Notch2/3) in combination with etoposide and platinum (EP) in patients (pts) with untreated extensive-stage small-cell lung cancer (ED-SCLC). Annals of Oncology, 28(suppl_5).
OncoMed Pharmaceuticals, I. (2017). OncoMed’s phase 2 trial of tarextumab in small cell lung cancer does not meet endpoints. In Company also announces discontinuation of brontictuzumab phase 1b study. OncoMed Pharmaceuticals, Inc: Online.
Chiang, A., Mclaughlin, J., Pietanza, M. C., Spira, A., Jotte, R., Gadgeel, S., Mita, A. et al. (2015). NOTCH3 protein expression and outcome in small cell lung Cancer (SCLC) and therapeutic targeting with Tarextumab (anti-NOTCH 2/3). Journal of Thoracic Oncology, 10(9), S361. New York, NY: Elsevier Science Inc.
Davis, S. L., et al. (2013). A first-in-human phase I study of the novel cancer stem cell (CSC) targeting antibody OMP-52M51 (anti-Notch1) administered intravenously to patients with certain advanced solid tumors. In Proceedings of the AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics.
Liu, Z., et al. (2011). Notch1 loss of heterozygosity causes vascular tumors and lethal hemorrhage in mice. The Journal of Clinical Investigation, 121(2), 800.
Pamela Munster, S. G. E., Patnaik, A., Shields, A., Tolcher, A. W., Davis, S. L., Heymach, J. V., Xu, L., Kapoun, A. M., Faoro, L., Dupont, J., & Ferrarotto, R. (2015). Safety and preliminary efficacy results of a first-in-human phase I study of the novel cancer stem cell (CSC) targeting antibody brontictuzumab (OMP-52M51, anti-Notch1) administered intravenously to patients with certain advanced solid tumors. [Poster] [cited 2018 January 24 2018]. Available from: http://posters.omed.s3.amazonaws.com/2015_N1_solid_tumor_triple_meeting.pdf.
Geles, K. G., et al. (2015). Therapeutic targeting the NOTCH3 receptor with antibody drug conjugates. Cancer Research, 75(15 Suppl), 1697–1697.
Mizugaki, H., et al. (2012). γ-Secretase inhibitor enhances antitumour effect of radiation in Notch-expressing lung cancer. British Journal of Cancer, 106(12), 1953–1959.
Ikezawa, Y., et al. (2017). Inhibition of Notch and HIF enhances the antitumor effect of radiation in Notch expressing lung cancer. International Journal of Clinical Oncology, 22(1), 59–69.
Purow, B. W., et al. (2008). Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis, 29(5), 918–925.
Jin, S., et al. (2008). Notch signaling regulates platelet-derived growth factor receptor-β expression in vascular smooth muscle cells. Circulation Research, 102(12), 1483–1491.
Yuan, X., et al. (2014). Notch signaling and EMT in non-small cell lung cancer: Biological significance and therapeutic application. Journal of Hematology & Oncology, 7(1), 87.
Tammela, T., et al. (2008). Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature, 454(7204), 656–660.
Funahashi, Y., et al. (2010). Notch regulates the angiogenic response via induction of VEGFR-1. Journal of Angiogenesis Research, 2(3), 2.
Espinosa, L., et al. (2002). p65-NFκB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. Journal of Cell Science, 115(6), 1295–1303.
Wang, Z., et al. (2010). Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer, 1806(2), 258–267.
Mori, M., et al. (2015). Notch3-Jagged signaling controls the pool of undifferentiated airway progenitors. Development, 142(2), 258–267.
Ranganathan, P., et al. (2011). Hierarchical phosphorylation within the ankyrin repeat domain defines a phosphoregulatory loop that regulates Notch transcriptional activity. Journal of Biological Chemistry, 286(33), 28844–28857.
Alamgeer, M., et al. (2013). Cancer stem cells in lung cancer: Evidence and controversies. Respirology, 18(5), 757–764.
Leung, E. L.-H., et al. (2010). Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One, 5(11), e14062.
Eramo, A., et al. (2008). Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death & Differentiation, 15(3), 504–514.
Bertolini, G., et al. (2009). Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proceedings of the National Academy of Sciences, 106(38), 16281–16286.
Chen, Y.-C., et al. (2008). Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One, 3(7), e2637.
Jiang, F., et al. (2009). Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Molecular Cancer Research, 7(3), 330–338.
Shi, Y., et al. (2012). The side population in human lung cancer cell line NCI-H460 is enriched in stem-like cancer cells. PLoS One, 7(3), e33358.
Sullivan, J. P., et al. (2010). Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Research, 70(23), 9937–9948.
Okudela, K., et al. (2012). Expression of the potential cancer stem cell markers, CD133, CD44, ALDH1, and β-catenin, in primary lung adenocarcinoma—their prognostic significance. Pathology International, 62(12), 792–801.
Wang, Z., et al. (2014). Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Research, 74(21), 6364–6374.
Garcia-Heredia, J. M., et al. (2017). The cargo protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the Notch pathway by abducting NUMB. Clinical Cancer Research, 23(14), 3871–3883.
Liu, Y.-P., et al. (2013). Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Research, 73(1), 406–416.
Rosell, R., et al. (2017). OA10.03 YAP-NOTCH and STAT3 signaling rebound as a compensatory response to gefitinib or osimertinib treatment in EGFR mutant lung cancer. Journal of Thoracic Oncology, 12(1), S281–S282.
Kelly, K., et al. (2008). Phase III trial of maintenance gefitinib or placebo after concurrent chemoradiotherapy and docetaxel consolidation in inoperable stage III non-small-cell lung cancer: SWOG S0023. Journal of Clinical Oncology, 26(15), 2450–2456.
Goss, G. D., et al. (2010). A phase III randomized, double-blind, placebo-controlled trial of the epidermal growth factor receptor inhibitor gefitinb in completely resected stage IB-IIIA non-small cell lung cancer (NSCLC): NCIC CTG BR.19. Journal of Clinical Oncology, 28(18s), abstr LBA7005.
Acknowledgments
We would like to thank Joseph Amann, Yung-Mae Yao, Susan Cole, and Rajeswara Arasada for the critical review of this manuscript. We would like to thank Mikhail Dikov for providing a schematic of Notch’s role in T cell maturation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
Sinicropi-Yao, S.L., Koenig, M.J., Carbone, D.P. (2018). Notch in Lung Cancer. In: Miele, L., Artavanis-Tsakonas, S. (eds) Targeting Notch in Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-8859-4_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-8859-4_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-8857-0
Online ISBN: 978-1-4939-8859-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)