
Abstract
Notch receptors participate in a highly conserved signaling pathway that regulates normal developmental and tissue homeostasis in a context-dependent manner. Deregulated Notch signaling is involved in numerous human diseases and recently, a substantial body of evidence has been generated in support of this pathway playing critical roles in several types of cancer. The finding that activating Notch-1 mutations are frequently found in patients suffering from T-cell acute lymphoblastic leukemia is one of the best examples, but an abnormal expression of different human Notch receptors also contributes to B-cell tumors as well as a number of solid cancers such as breast, colon, pancreas, brain, lung, skin and other tissues. Several γ-secretase inhibitors are currently being explored for their potential therapeutic applications in Notch-associated tumors. Alternative approaches involve the development of antibodies to inhibit Notch receptors, their activating ligands, or other components of the Notch pathway. In this book chapter, we review the rationale for Notch inhibition in cancer, the current state of the art, as well as potential strategies that try to target the oncogenic properties of Notch signaling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
Notch pathway was first recognized as an important developmental pathway in Drosophila in the first half of the twentieth century [1]. Several decades later, it was shown this pathway powerfully influences stem cell maintenance, differentiation and cell fate decisions [2–5]. In the last years, a number of preclinical but also clinical studies have shown that Notch signaling plays important roles in numerous human diseases, including a broad spectrum of malignancies [6–10]. Therefore, the Notch pathway has a tremendous potential as a new target in cancer therapy and there is growing evidence that synergy can result from combining Notch inhibition with already existing treatment modalities such as chemotherapy (CT), radiation and other pathway inhibitors [11–15]. This book chapter will address the current knowledge of Notch signaling in various types of hematological and solid tumors as well as potential therapies that try to target its oncogenic properties.
2.2 Notch Signaling Pathway
As introduction, it is important to consider the complex studies of this highly conserved signaling pathway. The Notch pathway is a short-range communication system in which contact between a cell expressing a membrane-associated ligand and a cell expressing a transmembrane receptor sends the receptor-expressing cell (and possibly both cells) a cell fate regulatory signal. This signal takes the form of a cascade of transcriptional regulatory events, that involves the expression of hundred if not thousands of genes, and has profound context-dependent phenotypic consequences [16].
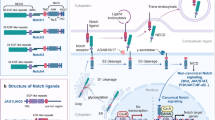
Mature Notch receptors are large single pass transmembrane noncovalent heterodimers consisting of an extracellular subunit and a transmembrane subunit. Whereas the fly genome contains only one Notch receptor, and worms have two that act redundantly [17], mammals have four paralogs (Notch-1, -2, -3, and -4) that display both redundant [18] and unique functions [19]. The extracellular domain of all Notch proteins contains 29–36 tandem epidermal growth factor (EGF) -like repeats, some of which mediate interactions with ligands. The activating interaction with ligand presented by neighboring cells (trans interactions) are mediated by repeats 11–12, whereas inhibitory interaction with ligand co-expresses in the same cell (cis interactions) are mediated by repeats 24–29 [20]. Many EGF repeats bind calcium, which plays an important role in determining the structure and affinity of Notch to its ligands [21, 22]. The EGF repeats are followed by a unique negative regulatory region (NRR) composed of three cysteine-rich Lin12-Notch repeats (LNR) and a heterodimerization domain (HD). The NRR plays a critical role in preventing receptor activation in the absence of ligand. The single transmembrane domain (TMD) is terminated by a “stop translocation” signal comprised of 3–4 Arg/Lys residues. Intracellulary, the RBPjĸ association module (RAM) domain forms a high affinity binding module of 12–20 aminoacids centered on a conserved WxP motif [23]. A long unstructured linker containing one nuclear localizing sequence (NLS) links RAM to seven ankyrin repeats (ANK domain). Following the ANK domain are an additional bipartite NLS and a loosely defined and evolutionarily divergent transactivation domain (TAD). The very C-terminus contains conserved proline/glutamic acid/serine/threonine-rich motifs (PEST), which regulate the stability of the Notch intracellular domain (NICD).
Most canonical Notch ligands are similar but smaller single-pass transmembrane proteins, that are characterized by three related structural motifs: a specialized delta/serrate/lag-2 (DSL) domain at the N-terminus, a specialized tandem of EGF repeats called the DOS domain (Delta and OSM-11-like proteins), and EGF-like repeats [24, 25]. Both the DSL and DOS domains are involved in receptor binding, with the DSL domain involved in both trans and cis interactions with Notch. Notch ligands can be classified based on the presence/absence of a cysteine-rich domain (Jagged/Serrated vs. Delta, respectively) and a DOS domain [25]. There are three Delta-like proteins (DLL-1,-3, and -4) and two Jagged proteins (JAG-1 and -2), which are differentially expressed in different cells. Other ligands have been proposed, such as the NOV (nephroblastoma-overexpressed), the neural adhesion molecule F3/contactin [26], the related NB-3 protein [27] and the EGF repeat protein DNER [28], but they have not been as well-established as Delta-like and Jagged.
Signal transduction by Notch receptors relies on a series of proteolytic cleavages. During trafficking to the cell surface, Notch is cleaved by a furin-like protease at a site termed S1 located within an unstructured loop protruding from the HD domain. As a result, mature Notch receptors are heterodimers made up of non-covalently associated extracellular and transmembrane subunits that are held together by the intrinsic stability of the HD domain [29]. Signaling is initiated when Notch receptor of one cell binds to a Notch ligand expressed on a neighbouring cell. This set in motion events that lead to the cleavage of Notch by two additional proteases. The first cleavage is carried out extracellularly by an alpha-secretase (a disentegrin and metalloprotease ADAM-10 and ADAM-17) at site 2 (S2), which is located 12–14 amino acids external to the TMD [30, 31]. In the resting state, the S2 site is deeply buried within the NRR, indicating that a substantial conformational change must precede S2 cleavage. S2 cleavage requires the endocytosis of ligand into the ligand-expressing cell, leading to speculation that mechanical forces transmitted to the NRR via endocytosis are responsible for the intramolecular movements that precede S2 cleavage [32]. The shedding of the Notch ectodomain creates a membrane-tethered intermediate, the Notch extracellular truncation (NEXT), that is cleaved within its transmembrane domain by γ-secretase, a multi-subunit protease consisting of presenilin 1 or 2, PEN-2, APH-1, and nicastrin. The ultimate cleavage at the site S3 frees the intracellular domain of Notch (NICD) from the membrane, allowing it to translocate to the nucleus [33]. Whether γ-secretase cleavage occurs at the cell surface or following endocytosis within endocytic vesicles remains controversial.
NICD contains several protein-protein interaction domains, including seven iterated ankyrin repeats (ANKs) that are needed for all known Notch functions and a so-called RAM domain. Nuclear NICD associates with the DNA-binding protein CSL (for CBF1, Suppressor of Hairless, Lag-2; also known as RBP-Jĸ) through high-affinity RAM contacts and lower-affinity ANK contacts. In humans, CBF1 is the only CSL factor identified to date. Binding of ANK to CSL creates an extended, composite surface groove that recruits scaffold proteins of the Mastermind-like (MAML)/Lag-3 family, the third component of the core Notch transcription complex (NTC). The NTC in turns interacts with chromatin modifying factors such as the histone acetyl-tranferases p300 and pCAF, as well as components of the mediator complex, to transactivate target genes. In the absence of NICD, CSL can associate with multiple proteins that suppress transcription, including multiple complexes with histone deacetylase activity and other factors with histone demethylase activity [34, 35]. For this reason, CSL has been likened to a molecular switch capable of actively suppressing or stimulating transcription, depending of the Notch activation status of a cell. Normal Notch transcription complexes are believed to have a half-life of the order of minutes, in part due to the presence of a PEST degron domain in the C-terminus of NICD that marks activated Notch for ubiquitination and proteasomal degradation, thus terminating the signal [16]. Notch target genes are numerous and include HLH-family negative transcriptional regulators of the Hes and Hey family, but also cell cycle progression genes (c-Myc, cyclin D1), antiapoptotic genes (Bcl-2), and many others yet to be discovered [36, 37].
Other aspects of the Notch pathway are also noteworthy. The Notch ligands Delta-like and Jagged are processed in a similar fashion to Notch following binding; they are cleaved also by alpha- and gamma-secretase, liberating an intracellular domain that is believed to be localized to the nucleus [38, 39]. The functions of their intracellular domains remain unknown. The likelihood of independent functions for Delta-like and Jagged proteins should be considered in the development of Notch inhibitors, as most strategies for Notch inhibition are also likely to inhibit Delta-like and Jagged. In addition, putative non-canonical pathways have been suggested but remain incompletely characterized. Among them, physical interaction of the intracellular domain of Notch-1 with the IKK signalosome, with nuclear IKKα and with p50. And the intracellular domain of Notch-3 with cytoplasmic IKKα may mediate therapeutically relevant cross-talk with nuclear factor ĸB (NF-ĸB) [40–42]. Physical interaction of the intracellular domain of Notch-1 with p85 PI3-kinase α may [43] mediate non-nuclear cross-talk with AKT, leading to survival signaling [44].
A closer look at this apparently simple signaling pathway reveals a intricate series of mechanisms that finely regulate the timing, intensity, and biological consequences of Notch signaling, and are likely to have significant therapeutic implications. Because the pathway relies on protein-protein interactions and the NICD is short-lived, a single activated Notch receptor is likely to transactivate only one target gene for a short period of time (on the order of minutes), a “design” that enables very precise temporal and quantitative control.
2.3 Notch Pathway Functions in Normal and Cancerous Cells: Rationale for Notch Inhibition in Cancer
The evolutionary conserved Notch signaling pathway functions as a mediator of short-range cell-cell communication. Numerous functions have been attributed to Notch, with some of these helping to explain its cancer-promoting effects in many tissues. However, Notch function can substantially differ and be dependent on cell type and tissue, and often the role of Notch signaling in a given tissue is unpredictable. Notch is among the most central pathways in self-maintenance of stem cells, along with Hedgehog, Wnt, and perhaps transforming growth factor-β (TGF-β). Interestingly, Notch signals select among preexisting cellular potentials; in a context dependent manner they will either promote or suppress proliferation, cell death, acquisition of specific cell fates and activation of differentiation programs throughout development and during maintenance of self-renewing adult tissues [2–5, 45–56]. Notch has been found to be critical in development of the brain, heart, vasculature, fat, hematopoietic system, gut and immune system. For example, it drives toward a glial cell fate in the central nervous system (CNS) [45] and regulates the T helper 1 versus T helper 2 decision in the immune system [4]. Notch has two main physiological roles in the intestine. One is to maintain the proliferating undifferentiated SC/progenitors acting as a gatekeeper of crypt cells and the other is to monitor binary cell fate decisions of the transient amplifying compartment, resulting in increased proliferation of absorptive enterocytes and a severe reduction of all secretory cells-goblet, enteroendocrine, and Paneth cells [5, 46]. Notch signaling, and in particular, RBP-J, is also an important regulator of pancreatic progenitor cells that have to choose between the endocrine and the acinar cell fate [47, 48]. How Notch regulates this cell fate decision is not fully understood. One possible mechanism suggested that Hes-1(hairy/enhancer of split 1) represses the expression of neurogenin3, which functions as a pro-endocrine factor, and the cell-cycle regulator p57 [49], thereby preventing progenitor cells from exiting the cell cycle and from differentiating into the endocrine lineage. In addition, some reports confirm that although Notch is not very active in homeostatic conditions, it seems to play important roles during tissue regeneration [50]. Interestingly, a physiological role for Notch signaling in the melanocyte lineage has also been demonstrated. Notch acts through Hes-1 and plays an indispensable role in the maintenance of melanocytic stem cells and melanoblasts in the epidermis. When Notch-1 and/or Notch-2 are ablated in the melanocyte lineage, a diluted initial hair pigmentation at birth and premature hair graying in subsequent hair cycles is observed [51, 52].
As referred above, Notch signaling can also induce terminal differentiation, which is accompanied with growth suppression. The skin is one of the best-studied examples of Notch exerting growth suppressive functions. The epidermis is composed of multiple layers of keratinocytes that are separated from the dermis by a basement membrane. Skin stem cells (SC) and transient amplifying cells are found within the basal cell layer of the epidermis and they are the reservoir for epidermal SC. Keratinocytes that undergo cell-cycle arrest detach from the basement membrane and move upward to form a supra-basal spinous layer and post-mitotic keratinocytes continue to migrate toward the outer surface of the skin to form the granular layer. Notch-1, Notch-2, and Notch-3 mRNA are highly expressed in the basal cell layer and to a lesser extent in the suprabasal layer of the human epidermis [53]. DLL-1 expression was shown to be highest in regions where potential SC reside which led to the suggestion that DLL-1 mediated Notch signaling induces SC to differentiate into transient amplifying cells. Cell culture experiments combined with genetic mouse studies suggest that Notch signaling induces terminal differentiation processed in keratinocytes [53, 54]. The p63 gene is important for the self-renewing properties and stratification of keratinocytes in the skin. P63 is expressed in the proliferating compartment of the skin and is downregulated as soon as keratinocytes start to differentiate. Notch-1 and p63 negatively regulate each other and thereby regulate the balance between self-renewing and differentiation [55]. In the context of cancer, p63 is often upregulated in epithelial tumors, including squamous cell carcinomas, in which Notch receptor expression is often downregulated [56]. Additional Notch-mediated mechanisms that help keratinocytes to differentiate are the induction of keratin1/10 and involucrin, as well as downregulation of integrin expression [54].
Because Notch plays a critical role in many fundamental processes and in a wide range of tissues, it is not surprising that aberrant gain or loss of Notch signaling components have been directly linked to multiple human disorders, from developmental syndromes, such as the Tetralogy of Fallot, Alagille syndrome, familial aortic valve disease, spondylocostal dysostosis or syndactyly [57], to adult onset diseases such as CADASIL (cerebral autosomal dominant artheriopathy with subcortical infarcts and leukoencephalopathy), Alzheimer’s disease [58], and cancer [6–10].
As we referred above, Notch is one of the most powerful of the stem cell-promoting pathways, making it relevant for cancer given the undifferentiated/de-differentiated state of most tumor cells. The growing evidence for the “cancer stem cell” (CSC) hypothesis may make Notch a particularly exciting target in Oncology. This hypothesis states that cancers harbor a usually small subpopulation of pluripotent “CSC” or “cancer-initiating cells” that retains SC character and gives rise to the bulk population of cancer cells through a process of aberrant differentiation that recapitulates that of normal tissues. Such cells have now been isolated and cultured from leukemias, breast cancers, glioblastomas, and many other cancers. They are characterized by properties of normal SC, such as indefinite self-renewal through asymmetric cell division [59, 60] but also the ability to differentiate into cells resembling normal cell types in a given tissue, very slow proliferation rates and resistance to standard treatments such as CT and radiation, owing, in part, to overexpression of ABC export pumps and cell cycle checkpoints proteins [59, 61, 62]. Whether CSC are derived from the malignant transformation of normal tissue SC or from the “dedifferentiation” of normal non-SC is a matter of considerable debate. What seems likely is that these cells are uniquely capable of resisting anticancer agents, surviving for a long time in a nearly quiescent status and eventually cause disease recurrences and/or metastasis after apparently complete remissions. Thus, a complete eradication of them will be necessary to attain a cure. This will require targeting of pathways that participate in the survival, replication and differentiation decisions in pluripotent cells, such as the Notch pathway [59, 63–65].
Some of the impact of Notch inhibition in cancer cells results from its extensive cross-talk with several essential proteins and pathways in tumorigenesis [66–74]. Notch regulates expression of important receptor tyrosine kinases such as the epidermal growth factor receptor (EGFR) and the vascular endothelial growth factor receptor-1 (VEGFR-1) [66, 67] and also interacts with fibroblast growth factor receptor (FGFR) signaling [68]. It has been also demonstrated that Notch activity sustains the phosphatidyl inositol 3-kinase (PI3K)/Akt/mTOR pathway and is a mediator of the oncogenic function of the RAS/MAPK pathways [69–71]. In addition, Notch and the nuclear factor-ĸB (NF-ĸB) pathway are intimately related, with multiple points of interaction described [72], and the Myc oncogene is a direct target of Notch, mediating much of the oncogenic effects of Notch in T-cell malignancies [73]. In some instances, other oncogenic pathways have been shown to cross-talk with Notch or its downstream activity, as is the case for the hypoxia/hypoxia inductible factor-1α (HIF-1α) pathway [74]. It is important to note that some of the described interactions are context-dependent and do not occur in all cellular backgrounds.
The direct effects of Notch inhibition on cancer cells may vary. Since Notch activity promotes cell survival and has anti-apoptotic functions through its interaction with important anti-apoptotic pathways such as Akt, it is not surprising that Notch inhibition has most frequently been shown to trigger apoptosis in cancer cells [70, 75]. Notch inhibition has also been shown to slow cancer cell proliferation and there is some evidence indicating that there may be important roles for Notch in the cell cycle in some settings [76]. Senescence has also been linked to the Notch pathway as its mediator, Hes-1, has been shown to play a critical role in blocking senescence [77]. Finally, the interaction between cancer cells and the surrounding stroma is receiving increasing attention as a key factor in tumor progression. “Tumor stroma” includes endothelial cells, necessary for tumor angiogenesis, fibroblasts that can produce growth factors and cytokines, as well as many subtypes of immunocytes, from T cells to dendritic cells or NK cells. There is significant evidence that bidirectional intercellular communication involving Notch signals takes place between tumor cells and stromal cells in some malignancies, suggesting that targeting the Notch-ligand interaction in endothelial cells can have therapeutic implications. For example, Notch blockade can impact the angiogenesis process [78–83]. The precise mechanisms by which Notch regulates the vasculature seem to be diverse. Notch/DLL-4 signaling directly regulates angiogenic endothelial cells and Notch also seems to regulate aspects of vascular development such as arterial versus venous fate [78]. Moreover, Notch also regulates the expression of the VEGFR-1, a key receptor for vascular formation. A number of reports have shown direct antiangiogenic effects from Notch inhibition [67]. A major role for Notch in blood vessels is supported by the vascular nature of the defects in the human disease called CADASIL syndrome caused by Notch-3 mutations [51]. Recently, multiple groups have found that signaling via the Notch ligand DDL-4 regulates endothelial sprouting and its inhibition lead to disordered and unproductive endothelial growth and decreased tumor size, even in tumor with vascular endothelial growth factor (VEGF) inhibition resistance [79, 80]. In addition, in mice the knockouts of Notch-1, DDL-1 or JAG-1 are embryonic lethal due principally to vascular defects [81] and small-molecule Notch-inhibiting drugs have been shown to have potent antiangiogenic effects in cancer animal models [82, 83].
2.4 Notch Signaling in Hematologic and Solid Tumors
2.4.1 Notch Signaling in T-Cell Acute Lymphoblastic Leukemia (T-ALL)/Lymphomas
Notch-1 was discovered in 1991 through analysis of T-cell lymphoblastic leukemias/lymphomas (T-LL) with balanced (7;9) translocations [84]. The translocation breakpoint were shown to fall within Notch-1 on chromosome 9 and the T-cell receptor β (TCRβ) locus on chromosome 7 and to result in fusion of the 3’end of Notch-1 to TCRβ enhancer/promoter elements, which drive the expression of aberrant Notch-1 transcripts encoding truncated, constitutively nuclear Notch-1 polypeptides. Expression of similar forms of Notch-1 in murine hematopoietic stem cells (HSC) induces the appearance of T-LL [85, 86], whereas no tumors are observed when the same polypeptides are expressed in HSCs with genetic defects that abrogate T-cell development [87]. These studies revealed that Notch-1 had a special oncotropism for T-cell progenitors and it is both necessary and sufficient for T-cell development from HSCs [88].
The degree of Notch-1 involvement in human T-LL became apparent in 2004, when Notch-1 gain-of-function mutations were found in roughly 60 % of primary human T-LL [6]. The most common Notch-1 mutations in human T-LL consist of point substitutions or small in-frame insertions or deletions [29]. In 40–45 % of T-ALL, mutations perturb the NRR domain, leading to ligand-independent signaling or increased sensitivity to ligand. Other Notch-1 mutations cause displacement of the extracellular ADAM cleavage site away from the NRR domain, or shifting of the NNR domain away from the transmembrane domain, in both instances leading to de-regulated ADAM cleavage at the S2 site which results in ligand-independent proteolysis [29, 89]. A second type of Notch-1 mutations results in truncation of the C-terminal PEST domain, leading to increased stability of NICD and hence prolonged transcriptional activation of Notch-1 target genes [6]. Remarkably, individual tumors may harbor as many as three Notch-1 mutations, typically aligned in cis in a single allele and mutations abrogating NRR function are often found together with PEST domain mutations, indicating that T-cell transformation is driven by selection for increasingly high levels of Notch activity. Consistent with this observation, Notch-1 mutations have been detected as secondary events in T-cell subclonal populations in T-ALL patients [90]. Finally, given the strong selection for Notch-1 gain of function in T-LL and the ability of leukemic blasts to proliferate in many tissues where access to ligand is likely limited, it is likely that other mechanisms of ligand-independent Notch-1 activation remain to be discovered in human T-LL. This is particularly true of tumors that only have PEST deletions, as such mutations may have little or no effect on Notch-1 signaling in the absence of some mechanisms (e.g. an NRR mutation) that promotes Notch-1 proteolysis [6, 91].
In the context of transformed T-cell progenitors, Notch signaling induces and reinforces a programme of gene expression that supports cell growth. The most important direct target genes include c-Myc [6, 92, 93] and Hes-1 [6, 92, 94]. A key pathways activated by Notch-1 include the PI3K/Akt/mTOR, since Notch-1 signaling via Hes-1 down-regulates PTEN, an important negative regulator or PI3K/Akt signaling [71, 95, 96]. Notch-1 also up-regulates the expression of the interleukin-7 (IL7) receptor, which activates PI3K/Akt signaling in an IL7-dependent fashion. In culture systems, the growth of primary human T-LL cells is IL7-dependent, suggesting that the IL7/IL7-receptor signaling axis is an important mediator of growth [97, 98]. Notch-related T-LL is also associated with constitutive activation of NF-ĸB [40] and abnormalities of E2A [99], but the mechanistic bases for these relationships are not fully elucidated. In addition to Notch-1, developing thymocytes also express Notch-3, which appears to be a downstream target for Notch-1 [6, 92]. Transgenic mice expressing N3ICD develop T-LL, but the relative importance of Notch-3 compared with Notch-1 has been uncertain [87].
Despite the confirmed role of Notch-1 mutations in all genetic and clinical subtypes of human T-ALL, associations between mutation status and outcome have been inconsistent. While a few series have suggested that Notch-1 mutations are associated with worse outcomes [100], most have shown no association or a trend towards more favourable responses [6, 101, 102]. Although Notch signaling has been linked to resistance to glucocorticoids in studies of T-LL cell lines [103], no such association has been found in primary tumors; in fact, in some series the trend is towards better responses to glucocorticoids among tumors with Notch-1 mutations [104].
2.4.2 Notch Signaling in Solid Tumors
The Notch pathway activation is a common step in the initiation and/or progression of many different human cancers, including breast cancer [105–118], colon cancer [119–123], pancreatic cancer [124–129], medulloblastoma [130–135], melanoma and other cutaneous tumors [136–148], Ewing’s sarcoma [149], Kaposi’s sarcoma [150], osteosarcoma [151], lung cancer [152], hepatocellular carcinoma [153, 154] or ovarian cancer [155, 156].
2.4.2.1 Breast Cancer
The first evidence describing a link between aberrant Notch signaling in solid tumors came from the observation in animal studies that the integration of the mouse mammary tumor virus (MMTV) into the Notch-4 gene leads to the formation of mammary tumors [105]. Over the past years several mouse studies have establish the role of Notch signaling during the normal mammary gland development [106, 107] and correlative evidence has also been accumulated implicating this pathway in human breast cancer. Activated forms of Notch-1 and Notch-4 have been identified in several human breast cancer cell lines [8] and Notch-3 has been shown to play an important role in the proliferation of ErbB2-negative breast tumor cell lines.
The first clue that Notch might be aberrantly expressed in primary human breast cancer came from a study demonstrating increased expression of Notch-1 in four breast cancer tumors that overexpressed H-ras identifying Notch-1 as a downstream target of oncogenic H-Ras [109]. Interestingly, tumor samples expressing high levels of both Notch-1 and its ligand JAG-1 are associated with particularly low overall patient survival rates, suggesting a synergistic effect of these expression level changes on tumor progression [110]. Increased accumulation of N1ICD and Hes-1 expression in ductal carcinoma in situ (DCIS) compared with normal breast tissue also predicts a reduced time to recurrence 5 years after surgery [111]. This finding confirms that both the accumulation of NICD as a useful prognostic marker for recurrence as well as changes in the Notch signaling pathway may be associated with the progression from DCIS to invasive disease. Consistent with these observations, activated Notch signaling [8, 112] and consequent upregulation of genes that promote tumor growth [128–130] have been observed in breast cancer cell lines and primary breast cancers. In particular, Notch-4 expression, as detected by immunohistochemistry, correlated with Ki67 in infiltrating breast carcinoma of ductal or lobular histologies [112]. Activation of Notch signaling in estrogen receptor (ER)-negative breast cancer results in direct transcriptional up-regulation o the apoptosis inhibitor, and cell-cycle regulator survivin [113] and levels of Slug, a transcriptional repressor and Notch target, are elevated and correlate with increased expression of JAG-1 in human breast cancers [114].
On the other hand, Notch signaling plays a role in breast CSC and two recent studies have demonstrated that the Notch pathway is important for promoting the commitment of mammary stem cells to the luminal lineage at the expense of the myoepithelial lineage in both man and mice [116, 117]. The Notch pathway in vivo appears to be preferentially active in mammary luminal cells, with prominent expression of the active form of Notch-1 and its target genes (Hey1 and Hey2) in luminal progenitor cells. Expression of N1ICD in mammary stem cell promoted luminal cell-fate specification at the expense of the myoepithelial lineage. The constitutive overexpression of N1ICD led to specific expansion of luminal progenitors and their self-renewal, finally leading to hyperplasia and tumorigenesis [116]. In contrast to the mouse, where Notch-1 is the key determinant of luminal fate selection [117], Raouf et al. have showed that Notch-3 is critical for the restriction of bipotent progenitor cells to the luminal pathway in human breast tissue and that other Notch receptors could not substitute for this activity. Nevertheless, they showed that Notch-4 gene expression is highest in undifferentiated human clonogenic mammary progenitor cells, becoming markedly downregulated when these cells committed to the luminal lineage [116]. This finding is interesting in light of a study by Harrison et al., who have identified the activated form of the Notch-4 receptor in basal CD44+ breast CSC cell lines and in primary human samples, whereas N1ICD was observed at higher levels in the luminal cells of normal breast epithelium [118]. The differential distribution of Notch-1 and Notch-4 in basal CSC and more differentiated cells would suggest different roles for each receptor. Therefore, targeting the Notch-4 receptor specifically might be a feasible therapeutic approach. In summary, the luminal progenitor cell can be implicated as a potential cell of origin for tumors in which the Notch pathway has been activated inappropriately, leading to hyperplasia and eventually tumorigenesis.
2.4.2.2 Colon Cancer
It has become evident that the accurate coordination of both the Notch and the Wnt signals controls intestinal epithelial cell fate decisions and it is essential in normal intestinal development and consequently it may play an important role in intestinal tumorigenesis [119, 120]. Indeed, tracing cells in which Notch-1 was activated, or detecting expression of the Notch target gene Hes-1 [3], indicated uniform Notch-1 activation in adenomas of adenomatous polyposis coli (APC) mice as well as in human colon cancer cell lines and primary human colon cancer tissue samples [121] implying that Notch and Wnt signaling are simultaneously active in the proliferating adenoma cells. Reedijk et al. provide correlative evidence of active Notch signaling in human adenocarcinomas [122]. Gene expression of both Jagged ligands, Notch-1, LFNG, and Hes-1 was detected by in situ hybridization and shown to be at comparable or greater levels than normally observed in cells of the crypt base. In another study performed by Fre et al. the interplay between the two signaling pathways was assessed in vivo by modulating Notch activity in mice carrying either a loss- or a gain-of-function mutation of Wnt signaling [123]. The proliferative effect of active Notch signaling has on early intestinal progenitors requires Wnt signaling, whereas its influence on intestinal differentiation appears Wnt independent. This synergy was also observed in human intestinal adenomas. The analysis of Hes-1 expression in human colon cancer samples showed that 12 out of 15 polyps of both sporadic and hereditary low-grade adenomas present strong nuclear Hes-1 expression, whereas Hes-1 is either not detected or expressed at low levels in human adenocarcinomas. Similarly, Hey1, HeyL, and the Notch ligands JAG-1 and JAG-2 were expressed at higher levels in human adenomas than carcinomas. These observations warrant the conclusion that elevated Notch signaling in benign adenomas may contribute to the initiation of colorectal cancer. On the other hand high Notch signals in adenomas could be interpreted to maintain a tumor suppressive function, whereas it seems to be dispensable at later stages of colon cancer development.
2.4.2.3 Pancreatic Cancer
There is increasing evidence that link Notch to the development and/or progression of pancreatic cancer. First, multiple Notch receptors, ligands, and downstream target genes have been shown to be expressed in early metaplastic lesions (known as pancreatic intraepithelial neoplasms, PanIN) as well as in pancreatic adenocarcinoma tissue of mice and humans [124, 125], indicating that Notch expression might be an early event in the development of pancreatic cancer. In addition, Notch could possibly cooperate with other key pathways in pancreatic tumorigenesis. For example, TGF-α-induced EGF receptor signaling is frequently found in pancreatic cancers and transgenic overexpression of TGF-α in mice also results in acinar to ductal metaplasia and correlated with increased Notch signaling [125]. The K-ras protooncogene is mutated in most pancreatic adenocarcinoma but also in early stage lesions indicating that activating K-ras mutations occur at an early stage during pancreatic carcinogenesis [126]. Thus, simultaneous expression of NICD with an oncogenic form of K-ras in either pancreatic progenitors or mature acinar cells resulted in the development of PanIN lesions at time points where expression of NICD or K-ras alone did not lead to such lesions. These results strongly suggest that Notch and K-ras synergize and can cooperate to initiate pancreatic carcinogenesis in animal models [127]. Finally, experimental data on murine pancreatic adenocarcinoma strongly suggest that Notch signaling can also promote progression from PanIN lesion to pancreatic adenocarcinoma and therefore could represent a therapeutic target [128, 129].
Although these studies are very encouraging, many issues remain to be resolved. These include elucidation of the role Notch plays in pancreatic cancer and whether interference with Notch influences disease outcome, how do Notch receptors and ligands get re-expressed during the development of early PanIN lesions or during their progression to pancreatic is also unclear, as is the question of which Notch target genes are activated during this process and what is their role.
2.4.2.4 Medulloblastoma
Multiple signaling pathways, which are involved in regulating neural stem cells, are also aberrantly activated in medulloblastoma, such as the sonic Hedgehog and the Wnt pathways [130, 131]. Analysis of primary medulloblastoma tumor samples also revealed increased mRNA expression of Notch-2 but not Notch-1. In 15 % of the examined tumors increased Notch-2 expression levels correlated with Notch-2 gene amplification suggesting that it may play a more important role for this neoplasm compared to the other Notch receptor family members [132, 133]. Moreover, increased Hes-1 expression correlated with poor patient survival prognosis [134].
Additional evidence that Notch signaling is involved in medulloblastoma is derived from experiments trying to interfere with the Notch cascade. Pharmacological inhibition of Notch activation or using soluble Delta ligands or small interfering RNAs (siRNAs) approaches induced apoptosis and led to pronounced reduction of viable cells in medulloblastoma cell lines and/or primary explant cultures [134, 135]. Reciprocal gain-of-function studies overexpressing N2ICD promoted cell proliferation, and tumor growth in xenotransplantation experiments. Somewhat surprisingly, similar experiments using N1ICD resulted in growth inhibition [134], which suggests that both Notch receptor exhibit very distinct functions, in contradiction with the in vivo finding [135]. How the N2ICD growth promoting function differs from a N1ICD mediated growth inhibitory function is unknown and needs further investigation.
2.4.2.5 Skin Tumors
Recent studies suggest that activation of the Notch signaling pathway is important to preserve the melanocyte SC (MSC) and may also play a role in melanoma progression. Microarray profiling comparing the gene expression pattern of normal melanocytes to human melanomas revealed up-regulation of Notch receptors, ligands, and downstream target genes. Notch-2 and Hey1 mRNA were overexpressed in melanoma cells compared to nevi and normal melanocytes [136]. Furthermore, JAG-2 mRNA is upregulated in highly invasive melanoma cell lines [137]. Massi et al. have also found that the expression of Notch-1 and Notch-2, as well as Notch ligands, was upregulated in human “dysplastic nevi” and melanomas as compared with common melanocytic nevi [138]. These results suggest that the activation of Notch may represent an early event in melanocytic tumor growth leading to the hypothesis that up-regulation of Notch signaling may sustain tumor progression. The oncogenic effect of Notch-1 on primary melanoma cells was mediated by β-catenin, which was upregulated following Notch-1 activation [9]. Moreover, the oncogenic effect of activated Notch-1 is at least partially mediated through regulation of the MAPK and PI3K-Akt pathways and hyperactivated PI3K-Akt signaling results in the upregulation of Notch-1 through NF-κβ activity [139, 140]. In addition, Notch-1 signaling enhances tumor cell adhesion and increases N-cadherin expression, a cell adhesion protein whose expression is highly correlated with melanoma progression and metastasis [140].
Pinnix et al. have recently provided good evidence that deregulation of Notch signaling activity plays a specific role in promoting a transformed phenotype in human melanocytes and has defined the importance of Notch signaling in human melanoma [141]. Through analysis of a large panel of cell lines and patient lesions they could show that Notch receptors 1, 2, and 4 are overexpressed particularly when compared against primary melanocytes or normal human skin and that ectopic N1ICD expression resulted in the loss of E-cadherin expression and up-regulation of MCAM, two well-characterized events in melanoma development. These findings suggest that Notch signaling plays a specific role in promoting the transformed phenotype in human melanocytes and acts as a driving force in melanocyte transformation. Nonetheless, genetic loss-of-function analyses in established melanoma models need to be performed in order to convincingly demonstrate that Notch activation is an obligate event necessary for melanoma development and/or tumor progression.
In non-melanoma skin tumors, data suggesting that Notch has tumor suppressive activities in the skin is mostly derived from genetic mouse studies and correlative expression studies of human skin lesions. Conditional inactivation of several signaling components of the Notch cascade including Notch-1, Notch-1-Notch-2-Notch-3 concomitantly, RBP-J, and Presinilin1 and 2 in mouse skin results in hyperproliferation of the skin, hair loss, and epidermal cyst formation within less than 4 weeks [142, 143]. Skin tumors in mice lacking Notch-1 develop only after a long latency period (approximately 12 months), but removal of additional Notch components accelerates time to tumor onset to as early as ~70 days in mice heterozygous for Notch-2 and lacking Notch-1 and Notch-3 [144]. The spontaneous tumors of these mice are papillomas with a subset progressing to heavily vascularized basal cell carcinoma-like tumors and a few squamous cell like tumors. The long latency of tumor onset in Notch-1 deficient mice suggest that loss of Notch-1 signaling on its own is not sufficient to develop skin tumors. During the latency period additional mutations are likely to accumulate thus sufficiently deregulating growth leading to tumor development [144]. In this scenario Notch would cooperate with additional oncogenic mutations and thereby contribute to tumor development.
The genetic mouse data seem to be consistent with observations in human skin cancer. Human basal cell carcinomas exhibit downregulated Notch-1, Notch-2, and JAG-1 expression [53]. Moreover, reduced expression of Notch-1, Notch-2, and Hes-1 was shown in a panel of human oral and skin squamous cell carcinoma cell lines, as well as in surgically excised squamous cell carcinomas from patients. In addition, suppression of Notch signaling in primary human keratinocytes that express an activated form of the Ras gene is sufficient to cause aggressive squamous cell carcinomas in xenograft models [145], as seen with mouse keratinocytes [146]. Downregulation of Notch-1 expression in human skin tumors may be linked to compromised p53 function, a regulator of Notch-1 expression [147, 148]. Interestingly, a similar link between suppression of Notch and p53 activity was reported for Ewing’s sarcoma [149], possibly indicating that such a mechanism could be conserved in tissues where Notch has growth suppressive functions.
Although the skin is clearly the best-studied organ system in which Notch exerts tumor suppressive properties, much more work needs to be done to map all the tissues where Notch loss may promote cancer. The prostate [157], small cell lung cancer [158] and hepatocellular carcinoma [154] are all locations where loss of Notch signaling may promote dysplasia.
2.5 Therapeutic Approaches to Modulating Notch Signaling
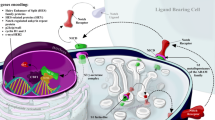
For targeting purposes, some features of the Notch pathway have unique relevance. First, the fact that the Notch pathway members has not enzymatic activity and signaling cascade triggered by Notch-ligand interactions does not include and enzymatic amplification step means that “signal intensity” can be modulated very precisely by cellular regulatory mechanisms. As a result, the downstream effects of Notch activation are exquisitely dose dependent [159]. This means that complete shutdown of the pathway may not always be necessary to achieve a therapeutic effect. A second important feature is that the intracellular half-life of the active form of Notch, NICD, is generally very short, in the order of minutes, as we referred above, though it may be longer in transformed cells [109]. The Notch signal is essentially a short pulse of gene regulation [159], which implies that sustained inhibition may not always be necessary and that intermittent inhibition may be successful. A third key feature is that the effects of Notch are remarkably context dependent. This means that Notch signals can be used for different purposes in different cell types and that systemic inhibition of Notch signaling is likely to have a multitude of effects in different cell types. Therefore, for therapeutic purposes it necessary to determine whether there is a level (or timing) of Notch inhibition that is sufficient to attain efficacy in disease control without causing intolerable adverse events (Fig. 2.1).

Notch pathway. Alternative therapies to inhibit Notch pathway
There is definitely a potential therapeutic benefit for targeting Notch in cancers including CSC depletion, tumor angiogenesis reduction, differentiation induction, and even cell death [59]. The efficacy of targeting Notch in cancer will vary within cancer types; even within the same cancer, targeting Notch may result in different effects on tumor subpopulations [11–13, 59]. Several inhibitory strategies are currently tested in preclinical setting as well as in clinical trials, including the following:
-
1.
Various GSI of different selectivity and efficacy, all preventing the S3 cleavage and thereby activation of all Notch receptors.
-
2.
Inhibitory antibodies (Abs) against individual Notch receptors and ligands with the aim to block specific receptor-ligand interactions.
-
3.
Receptor specific inhibitory Abs masking the S2 cleavage site, thereby blocking ADAM-protease-mediated cleavage of the receptors.
-
4.
Novel stapled peptides blocking the formation of a functional NICD-MAML transcription complex of unknown selectivity and low efficacy.
Some on the more promising strategies, with their potential advantages and disadvantages, are discussed in detail below.
2.5.1 GSIs
GSIs were first developed as potential therapies to treat and/or prevent Alzheimer’s disease, as the amyloid precursor protein (APP) implicated in this illness is also cleaved by the gamma-secretase [160, 161]. The off-target effects of the first-generation GSIs on Notch signaling immediately suggested that these agents might prove particularly useful in treating Notch-related tumors and they represent the pioneering class of Notch inhibitors both in the laboratory and in the clinic.
There are numerous GSIs (Table 2.1) commercially available for research and a number of chemical structures have been used as the basis for these compounds. The most commonly used is a modified di- or tri-peptide, usually with one to two aromatic hydrocarbon rings included. This has yield hydrophobic compounds which are cell-permeable and that act as reversible inhibitors of γ-secretase. In the laboratory, the most widely employed is DAPT and another frequently-used compound in the structurally similar Lilly GSI L685, 458. A structurally different compound which is also available preclinically is compound E (PF-03044014). Other classes of GSIs includes diazepine-type structures, with DBZ (dibenzazepine) as an example, agents based on an isocoumarin foundation, such as JLK6, that can bind and inhibit γ-secretase irreversibly or potent agents with a sulfonamide core, such as Compound 18 [64, 134, 172]. The specificity, selectivity, and dosing strategies of GSIs have been improving steadily over the last years. They have the advantage of relative ease of administration and oral bioavailability. In general, small molecules can be dosed more precisely than Abs because of their relatively short biological half-life and simpler dose-response relationships. An additional potential advantage is the fact that a single agent can block the activation of all four Notch homologues since they all depend on γ-secretase. Even though there are at least six different γ-secretase complexes in humans, and subtype-specific inhibitors might be developed, Notch appears to be a substrate for each of these complexes.
2.5.1.1 GSI for Notch-Targeted Cancer Therapeutics: T-ALL and Other Hematologic Malignancies
Given the well-documented role of overactive Notch signaling in T-ALL [6, 29, 89–99], many of the first studies to explore the potential efficacy of GSI-based cancer treatments focused on T-ALL human cell lines and mouse xenografts models.
In an early study, five human T-ALL cell lines were found to undergo G0/G1 cell cycle arrest, reduction in cell proliferation, and increased apoptosis following treatment with Compound E [6], findings that were subsequently replicated with additional T-ALL cell lines [14, 15, 173–175]. Specific inhibition of Notch signaling was demonstrated by reduced NICD levels and transcriptional downregulation of Notch-1-responsive genes. Compound E was also shown to enhance the sensitivity of T-ALL cell lines to other agents, including dexamethasone and imatinib [14]. Similar effects on G0/G1 cell cycle arrest and apoptosis were also observed with the cyclic sulfonamide GSI MRK-003 for three T-ALL cell lines [176]. Nevertheless, these studies also revealed that only a subset of T-ALL cell lines responded positively to GSI therapy [6, 177] and mixed results were also reported with rodent xenografts models of T-ALL using various GSIs [15, 163]. For example, the GSI PF-03084014 was found to exert robust antitumor effects in six Notch-1-driven T-ALL xenografts [15], and MRK-003 similarly downregulated Notch signaling, inducing apoptosis and causing complete tumor regression in mouse xenografts of thirteen different human T-ALL lines [163]. However, the evaluation of the GSI RO4929097 in a panel of mouse xenografts representing several different human cancers revealed no effect on two T-ALL xenografts or six precursor-B ALL xenografts, although tumor growth delays were observed for other xenografts tested, particularly for osteosarcoma [168]. One likely factor that could explain these incongruent outcomes in T-ALL cell lines and xenograft models is that a variety of different GSI and dosing regimens have been employed in the studies performed to date, making it difficult to compare the results. Indeed, different T-ALL cell lines and xenografts are exquisitely sensitive to different dosing regimens. For instance, in one T-ALL xenograft study comparing seven different MRK-003 dosing regimens, high doses administered on a 3-days-on/4-days-off, weekly, or bimonthly schedule were effective, but moderately lower doses administered on similar schedules were ineffective in conferring antitumor protection [163]. In a second mouse xenograft study with PF-03084014, much stronger antitumor efficacy was observed for a 7-days-on/7-days-off dosing schedule compared to a 3-days-on/4-days-off dosing schedule [15]. These results highlight the necessity of carefully evaluating GSI dose levels, dosing schedules, and therapeutic windows to determine the optimal design of clinical trials for candidate GSI agents. A second factor contributing to the differential response of T-ALL tumors to GSI therapy is that these tumors are genetically heterogeneous. Several T-ALL cell lines that exhibit high levels of NICD are resistant to GSI treatment were found to harbor mutations in the FBW7 gene, which encodes an F-box ubiquitin ligase required for NICD degradation by the proteosome [177]. The FBW7 mutations abrogate its binding to the substrates, thus allowing NICD to evade its normal down-modulation. A significant percentage (8.1 %) of primary T-ALL isolates were also found to harbor FBW7 mutations, illustrating the tendency of T-ALL cells to acquire secondary mutations under selective pressure for continued tumor growth. Mutational inactivation of the PTEN tumor suppressor gene has also been documented in T-ALL cell lines with GSI resistance, resulting in hyperactive PI3K/Akt/mTOR signaling that confers this resistance by bypassing the requirements for Notch signaling during leukemic clone growth [71]. As these studies indicate, it is of great importance to identify biomarkers and perform tumor genotyping in order to predict which molecular subtypes of T-ALL and other cancers are more likely to obtain benefit with GSI-based treatments.
A phase I clinical trial of MK-0752, which involved 10 patients with relapsed or refractory T-ALL, six of whom had activating Notch1 mutations, proved disappointing [159]. The orally administered daily dose (300 mg/m2 per day) had sever gastrointestinal toxicities (diarrhea, fatigue and cough), and patients were on the trial for a median of 11 days. The dose and daily schedule were clearly too toxic. Evaluation of Notch1 levels in peripheral blood mononuclear cells did not correlate with gain-of-function Notch1 mutations or clinical benefit. Several phase I studies are currently underway to evaluate different GSI (MK-0752, RO4929097, MRK-003, PF-03084014) for the treatment of T-ALL.
On the other hand, emerging evidence indicates that GSIs might also prove useful for treating hematologic tumors of B-cell origin. Many B-cell tumor lines have been found to be sensitive to GSIs, providing a rationale for further testing in animal models and ultimately in human clinical trials. DAPT treatment causes a significant reduction in cell proliferation for Hodkin’s lymphoma and multiple myeloma cells, and GSI-I, GSI-XII and DAPT inhibit growth and induce apoptosis of large B-cell lymphoma and Burkitt’s lymphoma cells [173, 178]. Contradictory results have been obtained with precursor-B ALL cell lines, with one study reporting no significant effect of RO4929097 on six different cell lines [168], while another study found that GSI-I induces apoptosis in precursor-B ALL cell lines and primary lymphoblasts, and blocked or delayed engraftment in 50 % of precursor-B ALL mouse xenografts [179]. Furthermore, MRK003 treatment induced caspase-dependent apoptosis and inhibited proliferation of multiple myeloma and non-Hodkin lymphoma cell lines and patient cells. Examination of signaling events after treatment showed time-dependent decrease in levels of the notch intracellular domain, Hes1 and c-Myc. MRK003 downregulated cyclin D1, Bcl-Xl and Xiap levels in non-Hodkin lymphoma cells and p21, Bcl-2 and Bcl-Xl in multiple myeloma cells. In addition, MRK003 caused an upregulation of pAkt, indicating crosstalk with the PI3K/Akt pathway [164]. A complicating question is that unlike truly γ-secretases-specific GSIs, some GSI compounds including GSI-I and –XII also target the proteasome, leading to the suggestion that simultaneous inhibition of both γ-secretase and the proteosome is required for the pro-apoptotic activities of some GSIs in B-cell tumors [179]. Finally, GSI can also modulate the Notch-dependent interactions between B cells and stromal osteoblasts, osteoclasts and fibroblasts. GSI treatment ameliorates the stromal cell-mediated drug resistance of multiple myeloma cells in vitro and enhanced the antitumor activities of melphalan and doxorubicin in a murine multiple myeloma model [75]. Therefore, GSI-based therapies might prove broadly applicable for B-cell neoplasias despite the complexities of Notch signaling and its crosstalk with other pathways in this class of hematologic tumors.
2.5.1.2 GSI-Based Therapies for Solid Tumors
Numerous studies using human cancer cell lines and xenografts models have established the potential utility of GSI-based therapies for solid tumors [13, 64, 75, 83, 128, 162, 165, 168, 169, 180–183]. For instance, the in vitro and in vivo properties of PF-03084014 have been investigated in a panel of breast cancer xenografts models [162]. In vitro, PF-03084014 exhibited activity against tumor cell migration, endothelial cell tube formation, and mammosphere formation. In vivo, apoptosis, anti-proliferation, reduced tumor cell self-renewal ability, impaired tumor vasculature, and decreased metastasis activity after the treatment of PF-03084014 was also observed. PF-03084014 treatment displayed significant antitumor activity in 10 of the 18 breast xenograft models [180]. However, the antitumor efficacy in most of them did not correlate with the in vitro antiproliferation results in the corresponding cell lines, suggesting the critical involvement of tumor microenvironment during Notch activation. In the tested breast xenograft models, the baseline expressions of the Notch receptors, ligands, and the cleaved Notch-1 failed to predict the antitumor response to PF-03084014, whereas several Notch pathway target genes, including Hey2, Hes-4, and Hes-3 were strong predictors of response. In addition, RO4929097 has shown to downregulated the Notch target genes Hes-1, Hey1, and HeyL in inflammatory breast cancer cells. However, the putative self-renewal mammosphere formation assay efficiency was increased with the drug [181]. Authors further showed that RO429097 inhibits normal T-cell synthesis of some inflammatory cytokines, including TNF-α and interleukin-8 (IL-8) production in the microenvironment. The therapeutic effect of GSI in K-rasG12V-driven non-small cell lung cancer (NSCLC) has also been shown in mice carrying autochthonous NSCLCs [182]. Treated carcinomas present reduced Hes-1 levels and reduced phosphorylated ERK without changes in phosphorylated MEK. Mechanistically, Hes-1 directly binds to and represses the promoter of DUSP1, encoding a dual phosphatase that is active against phospho-ERK. Accordingly, GSI treatment upregulates DUSP1 and decreases phospho-ERK. A recently published study has analyzed a panel of human pancreatic cancer cell lines as well as patient-derived pancreatic cancer xenografts to determine their responsiveness to MRK-003, a potent and selective GSI [165]. Pretreatment of pancreatic cancer cells with MRK-003 in cell culture significantly inhibited the subsequent engraftment in immunocompromised mice. MRK-003 monotherapy significantly blocked tumor growth in 5 of 9 (56 %) pancreatic cancer xenografts. A combination of MRK-003 and gemcitabine showed enhanced antitumor effects compared with gemcitabine in 4 of 9 (44 %) pancreatic adenocarcinoma xenografts, reduced tumor cell proliferation, and induced both apoptosis and intratumoral necrosis. Gene expression analysis of untreated tumors indicated that upregulation of NF-κB pathway components was predictive of sensitivity to MRK-003, whereas upregulation in B-cell receptor signaling and nuclear factor erythroid-derived 2-like 2 pathway correlated with response to the combination of MRK-003 with gemcitabine. Finally, RO4929097 reduces the tumor initiating potential of human melanoma cell lines by decreasing the levels of Notch transcriptional target Hes-1. RO4929097 also decreased tumor volume and blocked the invasive growth pattern of metastatic melanoma cell lines in vivo [169]. Another GSI, MRK-003 has also shown to reduce growth and cell invasion in uveal melanoma cell lines [183].
Several phase I clinical trials with GSIs have also been performed in patients with solid tumors [184–187]. The primary aims of these studies were to determine a maximum-tolerated dose (MTD) for phase II dosing, assess safety, and examine potential antitumor efficacy, as well as pharmacokinetic (PK) and pharmacodynamic (PD) end points. In the study by Krop et al. [166], MK-0752 was re-investigated in 103 patients under three schedules and at flat doses: schedule A with continuous, once-per-day dosing; schedule B with dosing on 3 days of 7; and schedule C with once-per-week dosing. Both schedules A and B evaluated two cohorts before dose-limiting diarrhea occurred with schedule A and unexpected dose-limiting fatigue occurred with schedule B. Dose expansions at the lower dose of 450 mg on both schedules indicated unacceptable toxicity; therefore, a weekly dosing schedule was pursued. However, by the time it was decided that neither schedule A nor B was worth pursuing, a total of 38 patients had already been treated in the expansion cohort. It is unclear why schedule A or B was re-investigated when the dose and the schedules were clearly too toxic in the previously reported MK-0752 study in patients with T-ALL. Not surprisingly, identical toxicities were reported in both studies. On schedule C (once-per-week dosing), 65 patients were treated in eight cohorts. Significant inhibition of Notch signaling was observed with the 1,800–4,200-mg weekly dose levels, confirming target engagement at those doses. Clinical benefit was observed, with one objective complete response and an additional 10 patients with stable disease longer than 4 months were observed among patients with high-grade gliomas. Although this drug has been reported to penetrate the CNS in mouse models, how effectively this occurs in patients is unknown. A second study with MK-0752 has been performed in 23 children with refractory or recurrent CNS malignancies [167]. MK-0752 was administered once daily for 3 consecutive days of every 7 days at escalating dosages starting at 200 mg/m2. Interestingly, no clinical responses or durable stable disease has been reported. Perhaps, this reflects lower daily dosing and generally low plasma levels, below which CNS penetration was observed in the animal models.
Tolcher et al. have evaluated three schedules of oral RO4929097 in 110 patients with refractory locally advanced or metastatic solid tumors [170]. In schedule A, 58 patients were treated for 3 days on and 4 days off for the first 2 weeks, followed by a week of rest. In schedule B, 47 patients were treated for the first 7 consecutive days of a 3-week cycle. Despite multiple dose escalations, an MTD (using a classic 3 +3 design) could not be defined for either schedule A or B. In fact, dose escalation was halted at a dose of 270 mg on schedule A and 135 mg on schedule B because PK evidence indicated CYP3A4 autoinduction at doses greater than 24 mg on schedule A and 18 mg on schedule B, with a decline in plasma concentrations with continued dosing. Treatment was well tolerated at the doses of both schedules A and B. Tumor responses included one partial response in a patient with colorectal adenocarcinoma with neuroendocrine features, one mixed response (stable disease) in a patient with sarcoma, one nearly complete FDG-PET response in a patient with melanoma and prolonged stable disease in several other tumor types.
The activity of RO4929097 has recently been tested in a phase II trial including 37 patients with metastatic colorectal cancer who had received at least two prior lines of systemic CT. Patients were treated at the dose of 20 mg daily, 3 days on and 4 days off continuously. No objective radiographic responses were observed and only six patients had stable disease as their best response. Median progression free survival (PFS) was 1.8 months and median overall survival (OS) was 6 months, which suggests that RO4929097 at the study dose has minimal single agent activity in this malignancy [171].
GSI-based Notch inhibition is now being evaluated for some solid tumors in preclinical and clinical trials that are currently underway. In fact, the Cancer Therapy Evaluation Program now has several trials that are readdressing the issue of optimal dose and schedule (A Phase I Study of Various Administration Schedules of RO4929097 With Multi-Parameter Assessment [Biomarkers, Pharmacokinetics, Pharmacodynamics] in Patients With Advanced Solid Cancers; and the Randomized Drug Interaction Study of RO4929097 for Advanced Solid Tumors). Several studies are in fact using schedules not even tested in phase I studies, such as daily low doses of drugs (15 mg per day) so as to avoid autoinduction (Phase IB/II Study of GDC-0449 [NSC 747691] in Combination With RO4929097, a Gamma-Secretase Inhibitor [GSI] in Advanced/Metastatic Sarcomas). All of these issues might have been avoided by examining the tumors of the patients. It is thus quite clear that it is essential to examine pharmacology in real time. As newer biologic and small molecule inhibitors are investigated, we will continue to encounter agents for which an MTD cannot be readily defined. In this context, pathway inhibition in matched-pair tumor samples will be absolutely critical to help define the minimal biologically effective dose and optimize the drug development process [188].
Preclinical and clinical trials evaluating GSIs in cancer are summarized in Table 2.1. Table 2.3 showed ongoing clinical trials with these compounds.
2.5.2 Notch Immunotherapy: Antibody Inhibitors of Notch Activity
An alternative approach for inhibiting Notch signaling is immunotherapy using antibodies directed against Notch, its Delta/Jagged ligands, or other components of the pathway (Table 2.2). One potential advantage of Abs inhibitors is their specificity, allowing specific members of the pathway to be targeted with high affinity, potentially limiting mechanism-based toxicity caused by global inhibition of Notch signaling [11–13]. One situation in which a specific biologic may be preferable is in malignancies where a particular mutated or otherwise deregulated Notch paralogue is known to be the primary oncogenic event or if the target has a relatively restricted expression pattern compared to other pathway members. Abs, however, are large molecules, though the delivery/access to cancer cells could be a main difficulty. For certain cancers such as brain tumors, local delivery may be an option, but for most metastatic cancers it is necessary to have efficient systemic distribution. Thus, inhibitory Abs of Notch may be most easily applied toward hematopoietic malignancies or for antiangiogenic purposes. Other potential disadvantages of biologics in this setting include their generally complex dose-response curves in vivo and their long biological half-lives. If intermittent inhibition of Notch signaling is desirable to minimize adverse events, using an mAb that will remain in circulation for days or weeks may prove challenging in terms of regimen designs. Of course, the biological half-lives of mAbs can be modulated by recombinant engineering or generation of F(ab)2 s, F(ab)s or even single chain Fvs [11, 13].
Inhibitory Abs directed against Notch ligands, including DLL-1 and DLL-4 have been developed. As mentioned above, Notch signaling via the ligand DLL-4 was reported by multiple groups to suppress angiogenic sprouting by endothelial cells and DLL-4 overexpression is found in tumor vasculature and in tumor cells to activate Notch signaling [79, 80]. Studies targeting blood vessel formation employing blocking Abs to DLL-4 revealed substantial tumor growth reduction in cancer cell line-based xenograft models [80]. The antitumor effect was shown to be the result of deregulated angiogenesis characterized by increasing sprouting in endothelial tip cells leading to chaotic and dysfunctional vasculature in the tumor. Thus, inhibiting DLL-4 disrupts productive angiogenesis in a different way from traditional antiangiogenic therapies causing hyperproliferation of tumor vessels that leads to a reduction in tumor growth. Importantly, this occurred even in cancer models that were resistant to VEGF Abs, an established and powerful antiangiogenic approach in cancer therapy [79, 80, 195–197]. This has prompted an aggressive effort to develop DDL-4 Abs for clinical usage. A land mark study by Hoey and colleagues demonstrated that blocking DLL-4 signaling inhibits tumor growth through multiple mechanisms, including a reduction in CSC frequency. In addition to the previously described effect on deregulating angiogenesis, they showed that selectively inhibiting DLL-4 signaling in human tumor cells with a humanized anti-hDLL4 21M18 Ab leads to a decrease in colon tumor growth, a delay in tumor recurrence after chemotherapeutic treatment, and a decrease in the percentage of tumorigenic cells. In a second study, the combination of specific DLL-4 Notch blockade and ionizing radiation impairs tumor growth in human colorectal carcinoma and human head and neck xenografts by promoting non-functional tumor angiogenesis and extensive tumor necrosis, independent of tumor DLL-4 expression [198]. Fischer et al. tested the efficacy of anti-DLL4 antibodies in KRAS mutant tumors in a panel of early passage colon tumor xenograft models derived from patients. It was efficacious against both wild-type and mutant KRAS colon tumors as a single agent and in combination with irinotecan [199]. Further analysis of mutant KRAS tumors indicated that the anti-DLL4/irinotecan combination produced a significant decrease in colon cancer stem cell frequency while promoting apoptosis in tumor cells. Following those preclinical studies providing evidence of antitumor activity, phase I clinical trials of the use of two different anti-DLL4 human mAbs- REGN421 and OMP-21M18- in treatment of solid tumors are currently in progress. A recent report, however, has raised some important safety concerns in the use of blocking DLL-4 chronically [200]. The authors showed that prolonged DLL-4 blockade using a rat model resulted in severe disruption of normal tissue homeostasis, caused pathological activation of endothelial cells and ultimately led to the development of vascular/endothelial cell-based tumors resembling hemangioblastoma in skin, heart, and lung. If this adverse event is borne out by others, it may present a major obstacle to the usage of DLL-4 Abs in clinic.
Blocking Abs to Notch or its ligands may serve not only antiangiogenic functions but also directly inhibit cancer cells. A growing number of reports have described the development of Abs that specifically antagonize the Notch paralogues Notch-1, 2 and 3. Some of these Abs seem to work by recognizing and stabilizing the extracellular NRR of Notch that undergoes a conformational change upon ligand bindings to facilitate ADAM protease cleavage at the S2 site [201–204]. This raises the interesting prospect that Abs could fine-tune Notch activity, increasing or attenuating signaling by individual Notch family members by different mechanisms. Several in vitro studies on human tumor lines indicate that they are able to inhibit oncogenic Notch signaling, albeit not as potently as cell-penetrating, small molecule GSIs [203]. One interesting report has emerged in which anti-NRR Abs were developed that specifically block activity of either Notch-1 or Notch-2 [202]. The Notch-1 anti-NRR showed good antitumor effects, but without the gut toxicity associated with combined Notch-1 and Notch-2 inhibition. Sharma et al. have developed the mAb 602.101, which specifically recognizes Notch-1, inhibited ligand-dependent expression of downstream target genes of Notch such as Hes-1, Hes-5, and HEY-L in the breast cancer cell line MDA-MB-231 [204]. The mAb also decreased cell proliferation and induced apoptotic cell death. Furthermore, exposure to this Ab reduced CD44(Hi)/CD24(Low) subpopulation in MDA-MB-231 cells, suggesting a decrease in the cancer stem-like cell subpopulation. This was confirmed by showing that exposure to the Ab decreased the primary, secondary, and tertiary mammosphere formation efficiency of the cells. The Ab also modulated expression of genes associated with stemness and epithelial-mesenchymal transition. In a third study, Abs to Notch-3 were also reported that can either block or stimulate receptor signaling [201]. These findings provide insights into the mechanisms of Notch autoinhibition and activation and pave the way for the further development of specific antibody-based modulators of the Notch receptors, which are likely to be of utility in a wide range of experimental and therapeutic settings. A number of Notch-targeting Abs (NNR-1, NNR-1, NNR-3) are currently being evaluated in preclinical studies, and the anti-Notch mAb OMP-59R5 is under phase I clinical trial evaluation (Table 2.3).
MAbs could also be used to target γ-secretase itself, an approach that has received little attention due to the availability of highly effective GSIs. Nevertheless, human γ-secretase is heterogeneous, with at least six different complexes possible due to differential usage of either presenilin-1 or −2 as well as Aph-1Aς, Aph-1AL or Aph-1B, and targeted inhibition of particular γ-secretase subtypes could potentially be beneficial in some therapeutic contexts. In a recent published study, a novel mAb A5226A against the extracellular domain component Nicastrin has been shown to inhibit γ-secretase activity by competing with substrate binding, and to interfere with proliferation of T-ALL cell lines and tumor growth in T-ALL mouse xenografts [42].
Preclinical trials evaluating antibodies inhibitors of Notch activity are summarized on Table 2.2.
2.5.3 Peptide-Based Approaches
The transcriptional effector complex downstream of Notch has also been targeted using a different-based approach. A recent publication has converted the peptide MAML1 transcription factor-based inhibitors into a drug-like molecule able to target the Notch/CSL transcription complex [194]. The crystal structure of Notch/MAML1/CSL identifies a nearly continuous stretch of α-helices at the interface of the three proteins. Moellering et al. hypothesized that a helical peptide mimetic might be able to compete for binding to NICD with full-length MAML1 and therefore inhibit transcriptional activation of Notch-targeted genes [205]. The researchers designed a series of six stapled α-helices peptides derived from MAML1, thus named for covalent backbone bonds stabilizing the helix. The stapled peptide was also more resistant to protease recognition and degradation and it was actively taken up by cells and entered the nucleus, where they can target the transcriptional process. In vitro cell culture studies confirmed that one peptide, SAHM1, prevented MAML1 from binding to the NICD-CSL complex, blocked the expression of Notch-1 target genes, and reduced proliferation of human T-ALL cell lines. The inhibitory effect was confirmed in a murine model of T-ALL reducing tumor burden significantly compared with vehicle. This strategy holds promise and, in principle, it could be applied to other components of the Notch pathway, including ADAM10/17 proteases or Notch glycosylation enzymes, although an important consideration in the extent to which such agents might also disrupt other cellular processes that depend upon the same enzymes.
2.5.4 Combinatorial Therapies Involving Notch Inhibition
As is becoming clear for many targeted inhibitors in cancer, Notch inhibition may be best not as solitary therapy but in combination with other agents. Such combinations will be made possible only through a thorough understanding of cross-talk between Notch and other developmental and non-developmental pathways that may play roles in specific malignancies. Given that deregulated Notch signaling plays an ancillary role in many cancers that are primarily caused by malfunction of other signaling pathways and cell growth mechanisms and the growing body of evidence demonstrating that Notch inhibitors sensitize to more standard treatments such as chemotherapy and radiotherapy [206–208], a promising approach is to combine Notch inhibition with other chemotherapeutic agents that target these other pathways. In T-ALL cell lines harboring both Notch-1 mutations and Abl1 fusions, certain combinatorial treatment regimens using GSIs with the kinase inhibitor imatinib have demonstrated synergistic antitumor effects [14]. Real et al. achieved a promising breakthrough using the combination of a GSI with dexametasone in glucocorticoid-resistant tumor cell lines [103]. Moreover, dexamethasone counteracts lethal gut toxicity induced by the GSI and the authors outline how the combination therapy induces apoptosis in T-ALL cell lines, primary human T-ALL cells, and in xenografts of such T-ALL cell lines in mice to a much greater extent than either dexamethasone or the GSI alone. A second study has shown similar conclusions. Combination treatment of the GSI PF-03084014 with glucocorticoids induced a synergistic antileukemic effect in human T-ALL cell lines and primary human T-ALL patient samples. Mechanistically, PF-03084014 plus glucocorticoid treatment induced increased transcriptional upregulation of the glucocorticoid receptor and glucocorticoid target genes. Glucocorticoid treatment effectively reversed PF-03084014-induced gastrointestinal toxicity via inhibition of goblet cell metaplasia [209]. Synergistic effects were not observed, however, when GSIs were combined with etoposide, methotrexate, vincistine or l-asparaginase [14]. In multiple myeloma, combined inhibition of Notch using GSI-XII treatment and Bcl-2/Bcl-xL using the small molecule ABT-737 resulted in synergistic cytotoxicity in cell lines and mouse xenografts models [209]. Enhanced antimyeloma effects have also been observed for combinations involving GSI-XII and bortezomib in cell lines and primary bone marrow isolates [210].
The utility of combining GSIs with conventional chemotherapy, hormonotherapy or targeted agents has also been confirmed for solid tumors [211–219]. In breast cancer, GSIs such as LY-411,575 and MRK-003 were found to prevent or reduce ErbB-2-positive tumors recurrence when combined with lapatinib or trastuzumab in cancer xenografts, and partially reversed trastuzumab resistance in refractory tumors [211]. Notch signaling is prominently regulated by Her2/Neu and trastuzumab-induced inhibition of ErbB-2 leads to Notch-1 activation [212], which in turns activates PI3K/Akt/mTOR signaling [70, 213], a tumor-promoting event that is attenuated by GSI treatment in some ErbB-2-positive breast cancer lines [69, 212]. In addition, a newly discovered feedback between Notch and the ER-α [112, 137, 214] supports combining Notch inhibitors with anti-estrogens and this combination is being investigated in ongoing clinical trials. An unexpected but potentially useful observation was that co-treatment with tamoxifen greatly alleviated the intestinal toxicity of orally administered GSIs, suggesting that this combination may be not only more effective but also safer than single GSI treatment. Oxaliplatin-induced activation of Notch-1 signaling in metastatic colorectal cancer is reduced by simultaneous GSI treatment, resulting in enhanced tumor sensitivity to oxaliplatin [215]. Additionally, the combination of PF-03084014 and irinotecan may be effective in reducing tumor recurrence in a colorectal cancer preclinical explants model in those tumors exhibiting elevated levels of the Notch pathway [216]. Synergistic anti-tumor effects have also been documented recently for the GSI MRK-003 together with rapamycin in pancreatic cancer, which was attributed to enhanced inhibition of the PI3K/Akt/mTOR pathway by the combined therapy [217].
Inhibitors of the PI3K-AKT-mTOR pathway may also be useful in combination with Notch inhibitors, and there is evidence that this strategy may reverse resistance to GSI in T-ALL that carry PTEN inactivating mutations [71]. Whether this strategy can be successful in other cancers characterized by PTEN loss is unclear. The complex cross talk between Notch and NF-ĸB suggests that, at least in some circumstances, drugs that inhibit NF-ĸB activity directly or indirectly could be successfully combined with Notch inhibitors [40, 218, 219]. As DLL-4 mAb appear to be effective independently of VEGF, they may be useful in combination with agents that block the VEGF pathway such as bevacizumab [78–81]. Finally, a particularly interesting treatment option is the combination of Notch inhibitors with inhibitors of other key stem cell pathways. For example, recent results show potent anti-cancer effects from combining a Notch-inhibiting agent and a Hedgehog pathway inhibitor in glioblastoma stem cell lines [220]. Hedgehog-inhibitor plus GSI combinations are also being investigated in ongoing clinical trials in breast cancer.
2.5.5 Future Approaches for Notch Inhibition
2.5.5.1 Other Potential Approaches to Small-Molecule Inhibitors
At the theoretical level, it could be possible to generate small-molecule inhibitors of Notch that act at different levels. Whereas attention has focused only on γ-secretase as a vulnerable point in Notch processing, it may also be feasible to use α-secretase inhibitors (ASI). The α-secretase enzymes that cleave Notch are thought to be ADAM-7 and ADAM-10 [221], and inhibitors that block both these ADAMs have been developed [222]. There may be theoretical advantages of an ASI over a GSI; for example, an ASI would not have to enter the cell to act. The process of testing ASIs as Notch inhibitors in cancer is currently underway.
While the inhibition of an enzymatic activity is typically the most common strategy to block a protein or pathway, examples are beginning to emerge of the potential druggability of protein-protein interactions. This strategy has been shown in reports in which small-molecule agents were derived to interrupt the interaction of the fusion protein EWS-Fli 1 with the RNA helicase RHA [223] or the p53/MDM2 interaction [224]. A number of protein-protein interactions in the Notch pathway would be logical targets for disruption, including Notch-Notch ligand, NICD-CBF1 transcription factor, or NICD-MAML.
Another promising approach relies on the discovery that the γ-secretase cleavage of Notch occurs not at the cell membrane but in the acidic endosomes [225]. A number of agents with the potential to interfere with endosomal acidification have been screened for their ability to reduce Notch activity. The Na+/H+ antiporter Monensin emerged as a potent Notch inhibitor [226].
2.5.5.2 Genetic Strategies
Genetic strategies for Notch inhibition may also find limited application in cancer therapy, particularly for hematopoietic malignancies or localized tumors, such as in lung or brain. One potential option could consist of delivery of a gene or pseudogene encoding a Notch-inhibiting peptide or protein. A dominant-negative form of MAML has been used in vitro to inhibit canonical Notch signaling via CBF1 [227] and other genes known to down-regulate Notch could also serve this function, such as the Numb/Numb-like or FBXW-7 genes [228]. Agents that up-regulate the expression of these endogenous Notch-inhibiting genes could be another way of blocking Notch activity.
Delivery of RNA interference represents a similar strategy for Notch-inhibiting cancer therapy, but possibly one with more potential for clinical success. Similarly to Notch-inhibiting genes, delivery remains one of the main problems in developing such strategies, but it is relatively less challenging to deliver small oligonucleotides than its whole genes. Either siRNAs or endogenous or artificial microRNAs could be generated. Each microRNA targets numerous genes and in general each gene is targeted by more than one microRNA. MicroRNAs thus offer the potential to simultaneously target more than one gene of interest, though the target genes may not be suppressed as efficiently as by siRNAs. For example, the microRNA miR-326 has been shown to target both Notch-1 and Notch-2 and to decrease Notch activity [229]. The tumor-suppressive micro-RNA miR-34a has also been shown to target Notch-1 and Notch-2 and microRNA-206 has targeted Notch-3. In some cases, transfecting these microRNAs has demonstrated not only to decrease Notch activity but also to kill cancer cells, as in the case of the miR-326 and glioblastoma cells [229]. In addition, viral or liposomal vectors have been developed for genetic strategies such as RNA interference. Recent studies suggest that cancer cells have been shown to shed large amounts of microvesicles that can transmit cytoplasmic contents to nearby cells and that siRNAs or microRNAs can be transferred in this fashion to suppress gene expression in those cells. Thus, even if a limited percentage of cancer cells is transfected with a therapeutic vector, the transfected cancer cells may “share” with neighboring untransfected cancer cells to obtain benefit. It remains an open question, however, whether siRNAs or microRNAs would be successful agents for Notch inhibition and cancer therapy.
2.5.6 Potential Risks of Notch Inhibition
Different strategies for Notch inhibition in cancer may also pose potential significant risk and side effects. As with the initial evaluations of GSIs for the treatment of Alzheimer’s disease, studies involving first-generation GSIs in T-ALL and other tumors found that this therapy fails to distinguish individual Notch receptors, inhibits other signaling pathways and cause significant systemic toxicity, attributed to dual inhibition of Notch-1 and 2 [203]. One of the most common adverse events is severe diarrhea. This is likely an on-target side effect of Notch inhibition, given that Notch drives gastrointestinal precursor cells toward an epithelial fate and away from a secretory cell fate; therefore, chronic Notch inhibition in the gut causes metaplastic conversion of proliferative cells in intestinal crypts and adenomas into secretory globet cells [3, 5, 46]. Secretory diarrhea showed its potential to be a dose-limiting toxicity in all these trials and it is likely one that will be problematic for any systematically-delivered Notch inhibitor. However, it has been found that intermittent rather than continuous oral administration of GSIs (e.g. giving a week off each month) greatly ameliorates the intestinal toxicity, presumably because it allows at least some intestinal stem cells to correctly differentiate as enterocytes. Corticosteroid treatment may also help to minimize the gut toxicity of Notch inhibition while maintaining anti-tumor efficacy [103]. Other adverse effects of systemic GSI treatment in mice include reversible thymic suppression, reversible hair depigmentation, hair loss or reversible hyperkeratosis. Hair loss in dose-escalation experiments is an indication that a toxic dose has been reached and is associated with diarrhea and weight loss. GSI are not significantly myelotoxic, making such a potential application at least theoretically feasible.
There has also been concern about other two theoretical risks of long-term Notch inhibition have been posited. The first one is the potential for damage or ablate the normal adult stem cell populations in various organs, which possible impact is difficult to determine, but could include anything from hematopoietic collapse to subtle cognitive dysfunction. No signs of such toxicities have been uncovered in the earliest clinical trials, but the dosing was relatively short in those studies. The second potential risk may be even more concerning, as it involves an increased incidence of certain cancers. Whereas Notch plays an oncogenic role in most tissues, it acts as a tumor suppressor in some, such as B lymphocytes, neuroendocrine lung cells and certain skin cells [146, 158]. For example, loss of Notch signaling in the skin causes a barrier defect that causes local inflammation, predisposing to transformation, hyperproduction of thymic stromal lymphopoietin, and systemic immunological disturbances [142, 143]. Thus long-term Notch inhibition may increase the risk of cancers in these cellular compartments, though this has not yet been demonstrated.
2.6 Conclusions and Future Perspectives
Notch is a critical pathway in stem cell maintenance, development and cancer [2–5]. It has been shown to be important in numerous hematologic and solid malignancies [6–10, 105–156], and its potential utility against “tumor stem cells” makes it a particularly high-value target [59, 63–65]. Despite last years have seen rapid advances in the development of therapeutic approaches to treat cancers and other diseases associated with dysfunctional Notch signaling, patients are not yet routinely treated by deliberately targeting the Notch pathway aside from clinical trials. Most Notch-directed therapies involve the use of GSIs, that have long been used in basic and preclinical investigation [13–15, 64, 75, 83, 128, 162, 165, 168, 169, 180–183] as Notch inhibitors and they have recently begun clinical trials in cancer patients [166, 167, 170, 171, 184–191, 195–197] (Table 2.3). Other more selective inhibitors of Notch and Notch ligands, such as DLL-4 Abs, are in preclinical or early clinical development and may show great promise against not only cancer cells but also tumor angiogenesis [79, 80, 184–187, 189–197]. Optimism for Notch should be tempered somewhat by adverse events such as gastrointestinal toxicity that are beginning to be observed in clinical trials and other problems from long-term Notch inhibition remain to be discovered [142, 143, 145, 157, 184, 186, 223, 224]. Promising results have also been obtained through less common approaches, such as using Notch 1 ectodomain expression to inhibit tumor growth and angiogenesis [226], inhibiting the ADAM metalloproteases that perform key activating Notch cleavages [227], expressing dominant-negative fragments of MAML [215, 228] and expressing DLL-1 and JAG-1 fusion proteins to modulate Notch signaling [229]. Although all these strategies show great potential for realistic therapeutic intervention of Notch signaling in the future, they also highlight the need to identify groups of patients and/or subtypes of cancers who are most likely to benefit from Notch inhibitors. To that end, it remains to be determined which cancers and specific subtypes are characterized by active Notch signaling, what specific roles are performed by different components of the Notch signaling pathway in a given tumor, and whether global or selective Notch modulation is most desirable. Another key point is the study of biomarkers on Notch-associated cancers to understand other cellular events and signaling pathways interactions that contribute to tumor progression, thereby guiding the selection of the most effective treatment, which in many cases will involve GSIs or Notch immunotherapy in combination with other chemotherapeutic or targeted agents.
References
Dexter JS (1914) The analysis of a case of continuous variation in Drosophila by a study of its linkage interactions. Am Nat 48:712–758
Kumano K, Chiba S, Kunisato A et al (2003) Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 18:699–711
Van Es JH, van Gijn ME, Riccio O et al (2005) Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435:959–963
Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA (2004) Instruction of distinct CD4 T helper cell fates by different Notch ligands on antigen-presenting cells. Cell 117:515–526
Fre S, Huyghe M, Mourikis P et al (2005) Notch signals control the fate of immature progenitor cells in the intestine. Nature 435:964–968
Weng AP, Ferrando AA, Lee W et al (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306:269–271
Purow BW, Haque RM, Noel MW et al (2005) Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 65:2353–2363
Stylianou S, Clarke RB, Brennan K (2006) Aberrant activation of Notch signalling in human breast cancer. Cancer Res 66:1517–1525
Balint K, Xiao M, Pinnix CC et al (2005) Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest 115:3166–3176
Haruki N, Kawaguchi KS, Eichenberger S et al (2005) Dominant-negative Notch3 receptor inhibits mitogen-activated protein kinase pathway and the growth of human lung cancers. Cancer Res 65:3555–3561
Purow B (2012) Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol 727:305–319
Groth C, Fortini ME (2012) Therapeutic approaches to modulating Notch signaling: current challenges and future prospects. Semin Cell Dev Biol 23:465–472
Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L (2008) Rational targeting of Notch signaling in cancer. Oncogene 27:5124–5131
De Keersmaecker K, Lahortiga I, Mentens N et al (2008) In vitro validation of γ-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica 93:533–542
Wei P, Walls M, Qiu M et al (2010) Evaluation of selective γ-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther 9:1618–1628
Kopan R, Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanisms. Cell 137:216–233
Fitzgerald K, Wilkinson HA, Greenwald I (1993) Glp-1 can substitute for lin-12 in specifying cell fate decisions in Caenorhabditis elegans. Development 119:1019–1027
Krebs LT, Iwai N, Nonaka S et al (2003) Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes Dev 17:1207–1212
Cheng HT, Kim M, Valerius MT et al (2007) Notch 2, but not Notch 1, is required for proximal fate acquisition in the mammalian nephron. Development 134:801–811
De Celis JF, Bray SJ (2000) The Abruptex domain of Notch regulates negative interactions between Notch, its ligands and Fringe. Development 127:1291–1302
Cordle J, Redfieldz C, Stacey M et al (2008) Localization of the delta-like-1-binding site in human Notch-1 and its modulation by calcium affinity. J Biol Chem 283:11785–11793
Raya A, Kawakami Y, Rodriguez-Esteban C et al (2004) Notch activity acts as a sensor for extracellular calcium during vertebrate left-right determination. Nature 427:121–128
Lubman OY, Ilagan MX, Kopan R, Barrick D (2007) Quantitative dissection of the Notch: CSL interaction: insights into the Notch-mediated transcriptional switch. J Mol Biol 365:577–589
Cordle J, Johnson S, Tay JZ et al (2008) A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol 15:849–857
Komatsu H, Chao MY, Larkins-Ford J et al (2008) OSM-11 facilitates LIN-12 Notch signaling during C. elegans vulval development. PLoS Biol 6:e196
Hu QD, Ang BT, Karsak M et al (2003) F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell 115:163–175
Cui XY, Hu QD, Tekaya M et al (2004) NB-3/Notch1 pathway via Deltex1 promotes neural progenitor cell differentiation into oligodendrocytes. J Biol Chem 279:25858–25865
Saito SY, Takeshima H (2006) DNER as key molecule for cerebellar maturation. Cerebellum 5:227–231
Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC (2006) Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol 26:4642–4651
van Tetering G, van Diest P, Verlaan I et al (2009) Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem 284:31018–31027
Bozkulak EC, Weinmaster G (2009) Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol 29:5679–5695
Gordon WR, Roy M, Vardar-Ulu D et al (2009) Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood 113:4381–4390
Wolfe MS, Kopan R (2004) Intramembrane proteolysis: theme and variations. Science 305:1119–1123
Weng AP, Lau A (2005) Notch signaling in T-cell acute lymphoblastic leukemia. Future Oncol 1:51–519
Sakano D, Kato A, Parikh N et al (2010) BCL6 canalizes Notch-dependent transcription, excluding Mastermind-like1 from selected target genes during left-right patterning. Dev Cell 18:450–462
Liefke R, Oswald F, Alvarado C et al (2010) Histone demethylase KDM5A is an integral part of the core Notch-RBP-J repressor complex. Genes Dev 24:590–601
Devgan V, Mammucari C, Millar SE, Brisken C, Dotto GP (2005) P21WAF1/Cip1 is a negative transcriptional regulator of Wnt4 expression downstream of Notch1 activation. Genes Dev 19:1485–1495
Bland CE, Kimberly P, Rand MD (2003) Notch-induced proteolysis and nuclear localization of the Delta ligand. J Biol Chem 278:13607–13610
LaVoie MJ, Selkoe DJ (2003) The Notch ligands, jagged and delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem 278:34427–34437
Vilimas T, Mascarenhas J, Palomero T et al (2007) Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med 13:70–77
Song LL, Peng Y, Yun J et al (2008) Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene 27:5833–5844
Hao L, Rizzo P, Osipo C et al (2010) Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene 29:201–213
Sade H, Krishna S, Sarin A (2004) The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Biol Chem 279:2937–2944
Perumalsamy LR, Nagala M, Sarin A (2010) Notch-activated signaling cascade interacts with mitochondrial remodeling proteins to regulate cell survival. Proc Natl Acad Sci U S A 107(15):6882–6887
Morrison SJ, Perez SE, Quiao Z et al (2000) Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 101:499–510
Riccio O, van Gijn ME, Bezdek AC et al (2008) Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27(Kip1) and p57(Kip2). EMBO Rep 9:377–383
Nakhai H, Siveke JT, Klein B et al (2008) Conditional ablation of Notch signaling in pancreatic development. Development 135:2757–2765
Cras-Meneur C, Li L, Kopan R, Permutt MA (2009) Presenilins, Notch dose control the fate of pancreatic endocrine progenitors during a narrow developmental window. Genes Dev 23:2088–2101
Georgia S, Soliz R, Li M, Zhang P, Bhushan A (2006) p57 and Hes1 coordinate cell cycle exit with self-renewal of pancreatic progenitors. Dev Biol 298:22–31
Sivek JT, Lubeseder-Martellato C, Lee M et al (2008) Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 134:544–555
Kumano K, Masuda S, Sata M et al (2008) Both Notch1 and Notch2 contribute to the regulation of melanocyte homeostasis. Pigment Cell Melanoma Res 21:70–78
Schouwey K, Delmas V, Larue L et al (2007) Notch1 and Notch2 receptors influence progressive hair graying in a dose-dependent manner. Dev Dyn 236:282–289
Thelu J, Rossio P, Favier B et al (2002) Notch signalling is linked to epidermal cell differentiation level in basal cell carcinoma psoriasis and wound healing. BMC Dermatol 2:7
Rangarajan A, Talora C, Okuyama R et al (2001) Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J 20:3427–3436
Nguyen BC, Lefort K, Mandinova A et al (2006) Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev 20:1028–1042
Westfall MD, Pietenpol JA (2004) p63: molecular complexity in development and cancer. Carcinogenesis 25:857–864
Garg V, Muth AN, Ransom JF et al (2005) Mutations in NOTCH1 cause aortic valve disease. Nature 21:180–184
Louvi A, Arboleda-Velasquez JF, Artavanis-Tsakonas S (2006) CADASIL: a critical look at a Notch disease. Dev Neurosci 28:5–12
Pannuti A, Foreman K, Rizzo P et al (2010) Targeting Notch to target cancer stem cells. Clin Cancer Res 16:3141–3152
Eyler CE, Risch JN (2008) Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 26:2839–2845
Bao S, Wu Q, McLendon RE et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760
Liu G, Yuan X, Zeng Z et al (2006) Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 5:67
Alison MR, Islam S, Lim SM (2009) Number crunching in the cancer stem cell market. Breast Cancer Res 11:302
Fan X, Matsui W, Khaki L et al (2006) Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res 66:7445–7452
Sikandar SS, Pate KT, Anderson S et al (2010) NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res 70:1469–1478
Purow BW, Sundaresan TK, Burdick MJ et al (2008) Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis 29:918–92535
Funahashi Y, Shawber CJ, Vorontchikhina M et al (2010) Notch regulates the angiogenic response via induction of VEGFR-1. J Angiogenes Res 2:3
Yoon K, Nery S, Rutlin ML et al (2004) Fibroblast growth factor receptor signaling promotes radial glial identity and interacts with Notch1 signaling in telencephalic progenitors. J Neurosci 24:9497–9506
Efferson CL, Winkelmann CT, Ware C et al (2010) Downregulation of Notch pathway by a γ-secretase inhibitor attenuates AKT/mammalian target of rapamycin signaling and glucose uptake in an ERBB2 transgenic breast cancer model. Cancer Res 70:2476–2484
Meurette O, Stylianou S, Rock R et al (2009) Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res 69:5015–5022
Palomero T, Sulis ML, Cortina M et al (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med 13:1203–1210
Espinosa L, Santos S, Ingles-Esteve J et al (2002) p65-NFkappaB synergizes with Notch to activate transcription by triggering cytoplasmic translocation of the nuclear receptor corepressor N-CoR. J Cell Sci 115:1295–1303
Weng AP, Millholland JM, Yashiro-Ohtani Y et al (2006) c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 20:2096–2109
Gustafsson MV, Zheng X, Pereira T et al (2005) Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell 9:617–628
Nefedova Y, Sullivan DM, Bolick SC et al (2008) Inhibition of Notch signaling induces apoptosis of myeloma cells and enhances sensitivity to chemotherapy. Blood 27:6164–6174
Ishikawa Y, Onoyama I, Nakayama KI et al (2008) Notch-dependent cell cycle arrest and apoptosis in mouse embryonic fibroblasts lacking Fbxw7. Oncogene 27:6164–6174
Sang L, Coller HA, Roberts JM (2008) Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science 321:1095–1100
Hirashima M, Suda T (2006) Differentiation of arterial and venous endothelial cells and vascular morphogenesis. Endothelium 13:137–145
Hellstrom M, Phng LK, Hofmann JJ et al (2007) Dll4 signaling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445:776–780
Noguera-Troise I, Daly C, Papadopoulos NJ et al (2006) Blockade of Dll4 inhibits tumour growth by promoting nonproductive angiogenesis. Nature 444:1032–1037
Xue Y, Gao X, Lindsell CE et al (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8:723–730
Masuda S, Kumano K, Suzuki T et al (2009) Dual antitumor mechanisms of Notch signaling inhibitor in a T-cell acute lymphoblastic leukemia xenograft model. Cancer Sci 100:2444–2450
Luistro L, He W, Smith M et al (2009) Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 69:7672–7680
Ellisen LW, Bird J, West DC et al (1991) TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66:649–661
Aster J, Pear W, Hasserjian R et al (1994) Functional analysis of the TAN-1 gene, a human homolog of Drosophila notch. Cold Spring Harb Symp Quant Biol 59:125–136
Pear WS, Aster JC, Scott ML et al (1996) Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med 183:2283–2291
Bellavia D, Campese AF, Checquolo S et al (2002) Combined expression of pTalpha and Notch3 in T cell leukemia identifies the requirement of preTCR for leukemogenesis. Proc Natl Acad Sci U S A 99:3788–3793
Radtke F, Wilson A, Stark G et al (1999) Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10:547–558
Sulis ML, Williams O, Palomero T et al (2008) NOTCH1 extracellular juxtamembrane expansión mutations in T-ALL. Blood 112:733–740
Mansour MR, Duke V, Foroni L et al (2007) Notch-1 mutations are secondary events in some patients with T-cell acute lymphoblastic leukemia. Clin Cancer Res 13:6964–6969
Chiang MY, Xu L, Shestova O et al (2008) Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest 118:3181–3194
Palomero T, Lim WK, Odom DT et al (2006) NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A 103:18261–18266
Li X, Gounari F, Protopopov A et al (2008) Oncogenesis of T-ALL and nonmalignant consequences of overexpressing intracellular NOTCH1. J Exp Med 205:2851–2861
Dudley DD, Wang HC, Sun XH (2009) Hes1 potentiates T cell lymphomagenesis by up-regulating a subset of notch target genes. PLoS One 4:e6678
Cullion K, Draheim KM, Hermance N et al (2009) Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 113:6172–6181
Chan SM, Weng AP, Tibshirani R et al (2007) Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood 110:278–286
Gonzalez-Garcia S, Garcia-Peydro M, Martin-Gayo E et al (2009) CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7R{alpha} gene expression in early human thymopoiesis and leukemia. J Exp Med 206:779–791
Armstrong F, Brunet de la Grange P, Gerby B et al (2009) NOTCH is a key regulator of human T-cell acute leukemia initiating cell activity. Blood 113:1730–1740
Reschly EJ, Spaulding C, Vilimas T et al (2006) Notch1 promotes survival of E2A-deficient T cell lymphomas through pre-T cell receptor-dependent and -independent mechanisms. Blood 107:4115–4121
Zhu YM, Zhao WL, Fu JF et al (2006) NOTCH1 mutations in T-cell acute lymphoblastic leukemia: prognostic significance and implication in multifactorial leukemogenesis. Clin Cancer Res 12:3043–3049
Asnafi V, Buzyn A, Le Noir S et al (2009) NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood 113:3918–3924
Larson Gedman A, Chen Q, Kugel Desmoulin S et al (2009) The impact of NOTCH1, FBW7 and PTEN mutations on prognosis and downstream signaling in pediatric T-cell acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Leukemia 23:1417–1425
Real PJ, Tosello V, Palomero T et al (2009) Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med 15:50–58
Breit S, Stanulla M, Flohr T et al (2006) Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108:1151–1157
Gallahan D, Kozak C, Callahan R (1987) A new common integration region (int-3) for mouse mammary tumor virus on mouse chromosome 17. J Virol 61:218–220
Buono KD, Robinson GW, Martin C et al (2006) The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol 293:565–580
Raafat A, Lawson S, Bargo S et al (2009) Rbpj conditional knockout reveals distinct functions of Notch4/Int3 in mammary gland development and tumorigenesis. Oncogene 28:219–230
Yamaguchi N, Oyama T, Ito E et al (2008) NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res 68:1881–1888
Weijzen S, Rizzo P, Braid M et al (2002) Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med 8:979–986
Reedijk M, Odorcic S, Chang L et al (2005) High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 65:8530–8537
Farnie G, Clarke RB, Spence K et al (2007) Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J Natl Cancer Inst 99:616–627
Rizzo P, Miao H, D’Souza G et al (2008) Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res 68:5226–5235
Lee CW, Raskett CM, Prudovsky I, Altieri DC (2008) Molecular dependence of estrogen receptor-negative breast cancer on a notch-survivin signaling axis. Cancer Res 68:5273–5281
Leong KG, Niessen K, Kulic I et al (2007) Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med 204:2935–2948
Rustighi A, Tiberi L, Soldano A et al (2009) The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol 11:133–142
Raouf A, Zhao Y, To K et al (2008) Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell 3:109–118
Bouras T, Pal B, Vaillant F et al (2008) Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 3:429–441
Harrison H, Farnie G, Howell SJ et al (2010) Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res 70:709–718
Nakamura T, Tsuchiva K, Watanabe M et al (2007) Crosstalk between Wnt and Notch signaling in intestinal epithelial cell fate decision. J Gastroenterol 42:705–710
Kim HA, Koo BK, Cho JH et al (2012) Notch1 counteracts WNT/β-catenin signaling through chromatin modification in colorectal cancer. J Clin Invest 122:3245–3259
Fernández-Majada V, Aguilera C, Villanueva A et al (2007) Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci U S A 104:276–281
Reedijk M, Odorcic S, Zhang H et al (2008) Activation of Notch signaling in human colon adenocarcinoma. Int J Oncol 33:1223–1229
Fre S, Pallavi SK, Huyghe M et al (2009) Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc Natl Acad Sci U S A 106:6309–6314
Büchler P, Gazdhar A, Schubert M et al (2005) The Notch signaling pathway is related to neurovascular progression of pancreatic cancer. Ann Surg 242:791–800
Miyamoto Y, Maitra A, Ghosh B et al (2003) Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 3:565–576
Habbe N, Shi G, Meguid RA et al (2008) Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A 105:18913–18918
De La O JP, Emerson LL, Goodman JL et al (2008) Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 105:18907–18912
Plentz R, Park JS, Rhim AD et al (2009) Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136:1741–1749
Mazur PK, Einwächter H, Lee M et al (2010) Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 107:13438–13443
Clifford SC, Lusher ME, Lindsey JC et al (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–2670
Lee Y, Kawagoe R, Sasai K et al (2007) Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 26:6442–6447
Gaiano N, Fishell G (2002) The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci 25:471–490
Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME (2001) Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron 31:557–568
Fan X, Mikolaenko I, Elhassan I et al (2004) Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 64:7787–7793
Hallahan AR, Pritchard JI, Hansen S et al (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64:7794–7800
Hoek K, Rimm DL, Williams KR et al (2004) Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res 64:5270–5282
Gutgemann A, Golob M, Müller S, Buettner R, Bosserhoff AK (2001) Isolation of invasion-associated cDNAs in melanoma. Arch Dermatol Res 293:283–290
Massi D, Tarantini F, Franchi A et al (2006) Evidence for differential expression of Notch receptors and their ligands in melanocytic nevi and cutaneous malignant melanoma. Mod Pathol 19:246–254
Bedogni B, Warneke JA, Nickoloff BJ, Giaccia AJ, Powell MB (2008) Notch1 is an effector of Akt and hypoxia in melanoma development. J Clin Invest 118:3660–3670
Liu ZJ, Xiao M, Balint K et al (2006) Notch1 signaling promotes primary melanoma progression by activating mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt pathways and up-regulating N-cadherin expression. Cancer Res 66:4182–4190
Pinnix CC, Herlyn M (2007) The many faces of Notch signaling in skin-derived cells. Pigment Cell Res 20:458–465
Demehri S, Liu Z, Lee J et al (2008) Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol 6:e1232
Vauclair S, Nicolas M, Barrandon Y, Radtke F (2005) Notch1 is essential for postnatal hair follicle development and homeostasis. Dev Biol 284:184–193
Demehri S, Turkoz A, Kopan R (2009) Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 16:55–66
Lefort K, Dotto GP (2004) Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin Cancer Biol 14:374–386
Nicolas M, Wolfer A, Raf K et al (2003) Notch1 functions as a tumor suppressor in mouse skin. Nat Genet 33:416–421
Lefort K, Mandinova A, Ostano P et al (2007) Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev 21:562–577
Mandinova A, Lefort K, Tommasi di Vignano A et al (2008) The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J 27:1243–1254
Ban J, Bennani-Baiti IM, Kauer M et al (2008) EWS-FLI1 suppresses NOTCH-activated p53 in Ewing’s sarcoma. Cancer Res 68:7100–7109
Gasperini P, Espigol-Frigole G, McCormick PJ et al (2012) Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res 72:1157–1169
Li Y, Zhang J, Ma D et al (2012) Curcumin inhibits proliferation and invasión of osteosarcoma cells through inactivation of Notch-1 signalling. FEBS J 279:2247–2259
Yu S, Sun J, Zhang J et al (2013) Aberrant expression and association of VEGF and Dll4/Notch pathway molecules under hypoxia in patients with lung cancer. Histol Histopathol 28:277–284
Strazzabosco M, Fabris L (2012) Notch signaling in hepatocellular carcinoma: guilty in association! Gastroenterology 143:1430–1434
Wang XD, Leow CC, Zha J et al (2006) Notch signaling is required for normal prostatic epithelial cell proliferation and differentiation. Dev Biol 290:66–80
Chen X, Thiaville MM, Chen L et al (2012) Defining NOTCH3 target genes in ovarian cancer. Cancer Res 72:2294–2303
Chen X, Stoeck A, Lee SJ, Shih IM, Wang MM, Wang TL (2010) Jagged expression regulated by Notch3 and Wnt/β-catenin signaling pathways in ovarian cancer. Oncotarget 1:210–218
Sriuranpong V, Borges MV, Strock CL et al (2002) Notch signaling induces rapid degradation of achaete-scute homolog 1. Mol Cell Biol 22:3129–3139
Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF (2012) Downregulation of the Notch signaling pathway inhibits hepatocellular carcinoma cell invasion by inactivation of matrix metalloproteinase-2 and −9 and vascular endothelial growth factor. Oncol Rep 28:874–882
Deangelo D, Stone R, Silverman L et al (2006) A phase I clinical trial of the Notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J Clin Oncol 24:6585 [Abstract]
Tolia A, De Strooper B (2009) Structure and function of γ-secretase. Semin Cell Dev Biol 20:211–218
Wolfe MS, Citron M, Diehl TS, Xia W, Donkor IO, Selkoe DJ (1998) A substrate-based difluoro ketone selectivity inhibits Alzheimer’s γ-secretase activity. J Med Chem 41:6–9
Suwanjunee S, Wongchana W, Palaga T (2008) Inhibition of γ-secretase affects proliferation of leukemia and hepatoma cell lines through Notch signaling. Anticancer Drugs 19:477–486
Tammam J, Ware C, Efferson C et al (2009) Down-regulation of the Notch pathway mediated by a γ-secretase inhibitor induces anti-tumour effects in mouse models of T-cell leukaemia. Br J Pharmacol 158:1183–1195
Ramakrishnan V, Ansell S, Haug J et al (2012) MRK003, a γ-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodkin’s lymphoma. Leukemia 26:340–348
Mizuma M, Rasheed ZA, Yabuuchi S et al (2012) The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. Mol Cancer Ther 11:1999–2009
Krop I, Demuth T, Guthrie T et al (2012) Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol 30:2307–2313
Fouladi M, Stewart CF, Olson J et al (2011) Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 29:3529–3534
Kolb EA, Gorlick R, Keir ST et al (2012) Initial testing (stage 1) by the pediatric preclinical testing program of RO4929097, a γ-secretase inhibitor targeting Notch signaling. Pediatr Blood Cancer 58(5):815–8
Huynh C, Poliseno L, Segura MF et al (2011) The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One 6:e25264
Tolcher AW, Messersmith WA, Mikulski SM et al (2012) Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory or locally advanced solid tumors. J Clin Oncol 30:2348–2353
Strosberg JR, Yeatman T, Weber J et al (2012) A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer 48:997–1003
Lewis SJ, Smith AL, Neduvelil JG et al (2005) A novel series of potent gamma-secretase inhibitors based on a benzobicyclo[4.2.1]nonane core. Bioorg Med Chem Lett 15:373–378
Palomero T, Barnes KC, Real PJ et al (2006) CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to γ-secretase inhibitors. Leukemia 20:1279–1287
O’Neil J, Calvo J, McKenna K et al (2006) Activating Notch1 mutations in mouse models of T-ALL. Blood 107:781–785
Kogoshi H, Sato T, Koyama T, Nara N, Tohda S (2007) γ-secretase inhibitors suppress the growth of leukemia and lymphoma cells. Oncol Rep 18:77–80
Lewis HD, Leveridge M, Strack PR et al (2007) Apoptosis in T cell acute lymphoblastic leukemia cells after cell cycle arrest induced by pharmacological inhibition of Notch signaling. Chem Biol 14:209–219
O’Neil J, Grim J, Strack P et al (2007) FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to γ- secretase inhibitors. J Exp Med 204:1813–1824
Tohda S, Sato T, Kogoshi H, Fu L, Sakano S, Nara N (2006) Establishment of a novel B-cell lymphoma cell line with suppressed growth by γ -secretase inhibitors. Leuk Res 30:1385–1390
Meng X, Matlawska-Wasowska K, Girodon F et al (2011) GSI-I (Z-LLNle-CHO) inhibits γ-secretase and the proteosome to trigger cell death in precursor-B acute lymphoblastic leukemia. Leukemia 25:1135–1146
Zhang CC, Pavlicek A, Zhang Q et al (2012) Biomarker and pharmacological evaluation of the γ-secretase inhibitor PF-03084014 in breast cancer models. Clin Cancer Res 18:5008–5019
Debeb BG, Cohen EN, Boley K et al (2012) Pre-clinical studies of Notch signaling inhibitor RO4929097 in inflammatory breast cancer cells. Breast Cancer Res Treat 134:495–510
Maraver A, Fernández-Marcos PJ, Herranz D et al (2012) Therapeutic effect of γ-secretase inhibition in KrasG12V-driven non-small cell lung carcinoma by depression of DUSP1 and inhibition of ERK. Cancer Cell 22:222–234
Asnaghi L, Ebrahimi KB, Schreck KC et al (2012) Notch signaling promotes growth and invasion in uveal melanoma. Clin Cancer Res 18:654–665
Yan M, Callahan CA, Beyer JC et al (2010) Chronic DLL4 blockade induces vascular neoplasms. Nature 463:E6–E7
Li K, Li Y, Wu W et al (2008) Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem 283:8046–8054
Wu Y, Cain-Hom C, Choy L et al (2010) Therapeutic antibody targeting of individual Notch receptors. PLoS One 5:e9094
Aste-Amézaga M, Zhang N, Lineberger JE et al (2010) Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One 5:e9094
Gounder MM, Schwartz GK (2012) Moving forward one Notch at a time. J Clin Oncol 30:2291–2293
Hoey T, Yen WC, Axelrod F et al (2009) DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 5:168–177
Liu SK, Bham SA, Fokas E et al (2011) Delta-like ligand 4-notch blockade and tumor radiation response. J Natl Cancer Inst 103:1778–1798
Fischer M, Yen WC, Kapoun AM et al (2011) Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res 71:1520–1525
Sharma A, Paranjape AN, Rangarajan A, Dighe RR (2012) A monoclonal antibody against human Notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells. Mol Cancer Ther 11:77–86
Hayashi I, Takatori S, Urano Y et al (2012) Neutralization of the γ-secretase activity by monoclonal antibody against extracellular domain of nicastrin. Oncogene 31:787–798
Moellering RE, Cornejo M, Davis TN et al (2009) Direct inhibition of the NOTCH transcription factor complex. Nature 462:182–188
Ridgway J, Zhang G, Wu Y et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444:1083–1087
Thurston G, Noguera-Troise I, Yancopoulos GD (2007) The Delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer 7:327–331
Li JL, Sainson RC, Oon CE et al (2011) DLL4-Notch signaling mediates tumor resistance to ant-VEGF therapy in vivo. Cancer Res 71:6073–6083
Wang J, Wakeman TP, Lathia JD et al (2010) Notch promotes radioresistance of glioma stem cells. Stem Cells 28:17–28
Samon JB, Castillo-Martín M, Hadler M et al (2012) Preclinical analysis of the γ-secretase inhibitor PF-03084014 in combination with glucorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer Ther 11:1565–1575
Li M, Chen F, Clifton N et al (2010) Combined inhibition of Notch signaling and Bcl-2/Bcl-xL results in synergistic antimyeloma effect. Mol Cancer Ther 9:3200–3209
Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, Nefedova Y (2011) γ-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr) 34:545–551
Pandya K, Meeke K, Clementz AG et al (2011) Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br J Cancer 105:796–806
Osipo C, Patel P, Rizzo P (2008) ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene 27:5019–5032
Kelly AP, Finlay DK, Hinton HJ et al (2007) Notch-induced T cell development requires phosphoinositide-dependent kinase 1. EMBO J 26:3441–3450
Meng RD, Shelton CC, LI YM et al (2009) γ-secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res 69:573–582
Arcaroli JJ, Powell RW, VArella-Garcia M et al (2012) ALDH + tumor-initiating cells exhibiting gain in NOTCH1 gene copy number have enhanced regrowth sensitivity to a γ-secretase inhibitor and irinotecan in colorectal cancer. Mol Oncol 6:370–381
Vo K, Amarasinghe B, Washington K, González A, Berlin J, Dang TP (2011) Targeting Notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer 10:138
Osipo C, Golde TE, Osborne BA, Miele LA (2008) Off the beaten pathway: the complex cross talk between Notch and NF-kappaβ. Lab Invest 88:11–17
Schwarzer R, Dörken B, Jundt F (2012) Notch is an essential upstream regulator of NF-ĸβ and is relevant for survival of Hodkin and Reed-Sternberg cells. Leukemia 26:806–813
Hartmann D, de Strooper B, Serneels L et al (2002) The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 11:2615–2624
Zhou BB, Peyton M, He B et al (2006) Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non small cell lung cancer. Cancer Cell 10:39–50
Erkizan HV, Kong Y, Merchant M et al (2009) A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med 15:750–756
Vassilev LT, Vu BT, Graves B et al (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848
Vaccari T, Lu H, Kanwar R et al (2008) Endosomal entry regulates Notch receptor activation in Drosophila melanogaster. J Cell Biol 180:755–762
Sweeney C, Morrow D, Birney YA et al (2004) Notch1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jĸ. FASEB J 18:1421–1423
Maillard I, Weng AP, Carpenter AC et al (2004) Mastermind critically regulates Notch-mediated lymphoid cell fate decisions. Blood 104:1696–1702
Tsunematsu R, Nakayama K, Oike Y et al (2004) Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem 279:9417–9423
Kefas B, Comeau L, Floyd DH et al (2009) The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J Neurosci 29:15161–15168
Li Y, Guessous F, Zhang Y et al (2009) MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes. Cancer Res 69:7569–7576
Song G, Zhang Y, Wang L (2009) MicroRNA-206 targets notch3, activates apoptosis and inhibits tumor cell migration and focus formation. J Biol Chem 284:31921–31927
Skog J, Wurdinger T, van Rijn S et al (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 10:1470–1476
Yuan A, Farber EL, Rapoport AL et al (2009) Transfer of microRNAs by embryonic stem cell microvesicles. PLoS One 4:e4722
Garber K (2007) Notch emerges as new cancer drug target. J Natl Cancer Inst 99:1284–1285
Wong GT, Manfra D, Poulet FM et al (2004) Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 279:12876–12882
Zweidler-McKay PA, He Y, Xu L et al (2005) Notch signaling is a potent inducer of growth arrest and apoptosis in a wide range of B-cell malignancies. Blood 106:3898–3906
Funahashi Y, Hernandez SL, Das I et al (2008) A Notch1 ectodomain construct inhibits endothelial Notch signaling, tumor growth, and angiogenesis. Cancer Res 68(68):4727–4735
Moss ML, Stoeck A, Yan W, Dempsey PJ (2008) ADAM10 as a target for anti-cancer therapy. Curr Pharm Biotechnol 9:2–8
Weng AP, Nam Y, Wolfe MS et al (2003) Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of Notch signaling. Mol Cell Biol 23:655–664
Elyaman W, Bradshaw EM, Wang Y et al (2007) Jagged 1 and Delta1 differentially regulate the outcome of experimental autoimmune encephalomyelitis. J Immunol 179:5990–5998
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Custodio, A., Barriuso, J. (2014). What Is the Meaning of Notch Pathway and How Can We Selectively Do the Targeting?. In: Grande, E., Antón Aparicio, L. (eds) Stem Cells in Cancer: Should We Believe or Not?. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8754-3_2
Download citation
DOI: https://doi.org/10.1007/978-94-017-8754-3_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8753-6
Online ISBN: 978-94-017-8754-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)