
Abstract
The current global obesity epidemic has resulted in more women entering pregnancy with a body mass index in the overweight and obese range. It has been shown that offspring of obese women are at increased risk of obesity and type 2 diabetes in childhood and adult life, thus giving rise to an ‘intergenerational cycle’ of metabolic dysfunction. Importantly, studies in recent years have highlighted that the oocyte and/or early pre-implantation embryo is particularly vulnerable to the effects of maternal obesity resulting in long-lasting endocrine and metabolic effects for the offspring. Investigations into the molecular mechanisms underlying the programming of obesity and insulin resistance in liver, muscle and adipose tissue have highlighted the role of epigenetic changes within these tissues, which are recruited within the developing embryo and/or fetus. The periconceptional period is also an important period for intervention where dietary intervention in overweight/obese women is relatively more feasible. While dieting before pregnancy may have metabolic benefits for the offspring, there are however also metabolic and endocrine costs for the offspring. Thus, we need a better evidence base for the development of dietary interventions in obese women before pregnancy and around the time of conception to maximise the metabolic benefits and minimise the metabolic costs for the next generation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Overweight and obesity affect 39 % and 13 %, respectively, of adults worldwide [1]. Consequently, more women in the developed world are entering pregnancy either overweight or obese [2–4]. Obese women are more insulin resistant than their normal-weight counterparts, both before and during pregnancy [5], and this is associated with an increased risk of developing gestational diabetes mellitus (GDM) and of giving birth to a large baby with increased fat mass [5–8]. Indeed, as of 2013, 17 % of all live births were associated with hyperglycaemia in pregnancy [9]. Importantly, exposure to either maternal obesity or to impaired glucose tolerance during pregnancy is also associated with an increased risk of obesity and features of insulin resistance in childhood, adolescence and adult life [10–12]. Globally, 42 million preschool children were overweight in 2013 [13]. This suggests that exposure to maternal obesity may result in an ‘intergenerational cycle’ of obesity and insulin resistance [3, 14, 15]. There has, therefore, been significant interest in understanding the type of dietary and lifestyle interventions such as increased physical activity both before and during pregnancy, which may lead to optimal outcomes for the mother and her offspring [15, 16]. This chapter will summarise the epidemiological, clinical as well as experimental studies that have highlighted the relationship between maternal obesity with or without pre-existing GDM and type 2 diabetes mellitus (T2DM) and the later onset of obesity and insulin resistance in the offspring in childhood and adult life. Furthermore, some of the potential benefits and risks of maternal dietary restriction and weight loss will also be highlighted.
This chapter will also highlight some of the molecular mechanisms underpinning the programming of obesity, insulin resistance and T2DM specifically in insulin-sensitive tissues and what is known about the epigenetic mechanisms that are recruited within the developing embryo in the face of maternal obesity or dietary restriction and weight loss, which result in the programming of these metabolic pathways.
7.1 Maternal Obesity and Its Association with Short- and Longer-Term Pregnancy Outcomes
Maternal overnutrition and overweight or obese status has a significant impact on the health of the offspring in later life. Data from more than 200 countries between 1980 and 2008 highlight that there is a steady increase in the prevalence of obesity in every region of the world, including most countries of low and middle incomes, with the steepest rises in higher-income countries [17]. Not surprisingly, this global obesity ‘epidemic’ includes women of reproductive age with more women entering pregnancy with a BMI in the overweight (i.e. a BMI ≥ 25 kg/m2) or obese (i.e. a BMI ≥ 30 kg/m2) range [3]. The prevalence of obesity in women aged between 20 and 39 years is now around 15–28 % in women in the USA, UK and Australia [2–4, 18, 19]. In the USA, La Coursiere and colleagues found an increase in the proportion of women entering pregnancy both overweight and obese between 1991 and 2001 [20]. At present, more than 60 % of all pregnancies in the USA are in women who are either overweight or obese at conception [21, 22]. Similarly, in Australia, the prevalence of maternal overweight and obesity was 34 % in a population giving birth between 1998 and 2002 and 43 % in a population measured at their first antenatal visit between 2001 and 2005 [23, 24]. Furthermore, the incidence of childhood obesity, which strongly predicts adult obesity [25], is also increasing; an estimated 22 million children aged under 5 years are estimated to be overweight or at risk of becoming overweight worldwide [26] and 1 in 10 children (155 million) aged between 5 and 17 years are overweight [27].
Obesity imposes a number of serious risks during pregnancy including increased rates of twinning and miscarriage in early pregnancy, and maternal obesity has also led to increased rates of hypertension, pre-eclampsia and venous thromboembolism [28].
7.1.1 Maternal Obesity and the Developmental Programming of Obesity and Insulin Resistance: Evidence from Human Studies
In addition to the clinical risks conferred by obesity to the pregnant mother, maternal obesity also has longer-term consequences for the offspring including increased adiposity [5–8]. The underlying mechanisms that result in obesity in the offspring of overweight or obese women most likely are a result of a dysregulation of glucose, insulin and lipid metabolism in these offspring [29, 30]. Women who enter into pregnancy obese are more insulin resistant than their lean and overweight counterparts, particularly before pregnancy and in early gestation [5], resulting in an increased risk of developing insulin resistance and GDM [31]. Exposure of the developing fetus to maternal hyperglycaemia results in excess fetal growth; while maternal glucose freely crosses the placental barrier, there is no trans-placental transfer of maternal insulin [32]. The fetal pancreas responds to the increased glucose supply from the mother by synthesising and secreting insulin to maintain its own glucose homeostasis. Insulin acts as a fetal growth hormone promoting growth and adiposity [32]. Obese women are, therefore, at a greater risk of giving birth to a larger, heavier and fatter baby [5–8]. This has been demonstrated in various epidemiological and clinical studies, which have shown that overweight/obese women tended to have infants with an increased risk of macrosomia; defined as birth weight ≥4 kg [33–36].
Furthermore, these offspring of obese and/or diabetic mothers are not only heavier at birth but remain so throughout childhood and adult life. According to a recent systematic review, an increase in pre-pregnancy overweight/obesity in women increases the risk of having a large for gestational age (LGA) baby and these babies are subsequently at an increased risk of being overweight/obese in later life [37]. It has also been shown that the offspring of Pima Indian women with pre-existing GDM and T2DM were larger for gestational age at birth and, at every age, were heavier than the offspring of prediabetic or non-diabetic women [7, 38, 39] and that children of women with diabetes during pregnancy were on average 30 % heavier than expected for their height at 8 years of age [40]. Boney and colleagues also reported that obesity in children at 11 years of age was a strong predictor of insulin resistance [10]. Furthermore, children of obese mothers that were born LGA were at twice the risk of developing the metabolic syndrome accompanying childhood obesity at this age [10]. The impact of maternal obesity is still present in her offspring in adult life. Parsons and colleagues carried out a longitudinal study of the 1958 British birth cohort to determine the influence of birth weight on BMI at different stages of later life. BMI of the participants was measured at ages 7, 11, 16, 23 and 33 years, and they concluded that maternal weight or her BMI largely explained the association between a high birth weight and a high adult BMI of the offspring [41].
A recent longitudinal study of 421 mother–daughter pairs [42] has highlighted the concern that the increase in obesity in women entering pregnancy will, in turn, lead to propagation of an ‘intergenerational’ cycle of obesity and insulin resistance [3, 15]. Kubo et al. found that girls who were exposed to maternal GDM and hyperglycaemia in utero were at a higher risk of increased adiposity and that this risk increases if the mother was overweight/obese [42]. Moreover, they found that the risk of obesity was highest among offspring of mothers with GDM and pregravid obesity [42].
7.1.2 Maternal Obesity and the Developmental Programming of Obesity and Insulin Resistance: Evidence from Animal Studies
As most women who are obese at conception remain obese through their pregnancy, it is difficult to determine the separate or interdependent contributions of maternal pre-pregnancy BMI, gestational weight gain and glycaemic control on the metabolic outcomes for the offspring in human studies. Experimental studies in animals are, therefore, key to address these questions. Indeed, studies in both small and large animals have also provided evidence for the association between maternal obesity and subsequent programming of obesity and insulin resistance in the offspring. Studies in rodents have involved maternal consumption of either a high-fat only [43–45] or high-fat, high-sugar junk food diet, which is reflective of an obesogenic Western diet in humans [46, 47] from before pregnancy and during gestation. In some studies, the period of overnutrition was also extended to encompass lactation [45–47]. Despite the differences in diet composition, length of exposure to maternal overnutrition and whether offspring were weaned onto a standard chow or high-fat diet, these studies all found that a common outcome was increased adiposity in the offspring [43–48]. Furthermore, it has also been shown that exposure of the offspring to a high-fat diet resulted in increased adiposity specifically in the visceral fat depot [49, 50]. Visceral fat accumulation has been shown to be associated with the development of insulin resistance [51, 52]. Studies have also documented that maternal overnutrition in rodents is associated with insulin resistance in the offspring [46, 47, 49] due to altered expression of key components of the insulin signalling pathway [53] as well as poor glucose tolerance [43, 46, 47, 49], associated with a combination of β-cell dysfunction and insulin resistance [44, 49] in the offspring. Furthermore, in some cases, maternal obesity during pregnancy and lactation also resulted in obesity-induced non-alcoholic fatty liver disease (NAFLD) in the offspring, which is characterised by evidence of steatosis, liver injury, raised inflammatory cytokines and the beginning of fibrogenesis [54].
Similar observations linking maternal obesity and the development of increased adiposity and insulin resistance in the offspring have also been made in a primate model of maternal overnutrition [55]. One of the earliest studies conducted in the baboon showed that overnutrition in the pre-weaning period permanently increased offspring adiposity as a consequence of fat cell hypertrophy [56]. Sheep have also been used as a large animal model to study the impact of maternal overnutrition/obesity on the developmental programming of obesity and insulin resistance in the fetus and in the offspring. The sheep fetus is similar to the human fetus in its dependence on glucose as a major source of energy [57–59]. Moreover, both sheep and humans are precocial species, exhibit the same newborn-to-maternal weight ratios and have the same temporal pattern of fetal organ development throughout pregnancy [58, 60]. Importantly, in both sheep and humans, the appetite regulatory network develops in the hypothalamus before birth, in contrast to rodents, in which this neural network develops after birth [61]. There have been a series of studies on the effects of maternal overnutrition/obesity in the sheep from 60 days before conception and throughout pregnancy on the fetal and postnatal lamb. Similar to findings in rodent studies, maternal obesity in sheep leads to increased adiposity in the offspring in late gestation [62], which persists after birth [63]. These changes were attributed to increased gene expression and protein abundance of fatty acid (FA) and glucose transporters (GLUTs) as well as increased expression of enzymes mediating FA biosynthesis in fat depots of the offspring [62]. Maternal obesity also resulted in decreased pancreatic weight and β-cell number in the fetus in late gestation as well as in a reduction in circulating plasma insulin concentration at term [64]. Moreover, these offspring displayed upregulation of inflammatory signalling pathways in skeletal muscle, which promotes adipogenesis and downregulates myogenesis [65, 66]. This along with the increased adiposity would result in an increased risk of developing insulin resistance and T2DM in the postnatal animal. Indeed, when the offspring of obese ewes were subjected to ad libitum feeding at maturity, they showed increased appetite, growth rates and adiposity and decreased glucose tolerance and insulin sensitivity [67].
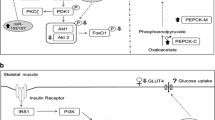
Late gestation has also been identified as a period during which exposure to maternal obesity can result in programming of obesity in the offspring. Exposure to maternal overnutrition during the last month of pregnancy resulted in an increase in fetal glucose and insulin concentrations and an upregulation of key adipogenic, lipogenic and adipokine genes, including peroxisome proliferator-activated receptor (PPAR) γ, leptin and adiponectin in perirenal adipose tissue of the sheep fetus in late gestation [68] as well as increased fasting plasma glucose concentrations and a higher relative subcutaneous fat mass in the first month of life in the postnatal lamb [69]. Furthermore, leptin expression was higher in both the perirenal and subcutaneous fat depots in the postnatal lamb of the overnourished ewe [70]. It is clear, therefore, that maternal overnutrition in late gestation results in an initial upregulation of adipogenic and lipogenic genes in the perirenal fat in fetal life followed by an upregulation in leptin expression in the perirenal and subcutaneous fat depots and the emergence of leptin resistance in the hypothalamic network which regulates appetite in postnatal life. In contrast to its effects on programming of the fat–brain axis, exposure to maternal obesity in late gestation had less impact on hepatic glucose metabolism in the offspring. Maternal overnutrition during the last month of gestation resulted in decreased hepatic expression of the mitochondrial (PEPCK-M) isoform of phosphoenolpyruvate carboxykinase both before birth and in postnatal life. There was, however, no impact on the cytosolic isoform of PEPCK (PEPCK-C), glucose-6-phosphatase, PPARγ co-activator (PGC)-1, PPARα and 11β-hydroxysteroid dehydrogenase type 1 (11βHSD1) in the postnatal lamb [71].
7.2 The ‘Intergenerational Cycle’ of Obesity and Insulin Resistance
Although there is variation between studies in the length of the feeding regimes and types of dietary challenges, studies in both small and large animals show that exposure to maternal overnutrition/obesity consistently results in a similar phenotype in the offspring including a high body fat mass and in most cases impaired insulin signalling in the liver, muscle and adipose tissue [15, 72]. Together, these findings implicate a possible ‘intergenerational cycle’ of obesity and insulin resistance (Fig. 7.1). In a recent study by Graus-Nunes et al., a maternal high-fat diet consumed before and throughout pregnancy and lactation resulted in impaired whole-body metabolism as well as altered development of the pancreas in the F1 and F2 offspring [73]. The maternal contribution to the intergenerational transmission of obesity and insulin resistance appears to be mediated, at least in part, by the transgenerational accumulation of epigenetic modifications [74]. This has led to a significant interest in the type of dietary and lifestyle interventions that could be imposed before and during pregnancy that could lead to optimal metabolic outcomes for the mother and the offspring [15, 75] (Fig. 7.1).

The intergenerational cycle of obesity and insulin resistance. Is there a role for pre-pregnancy weight loss to break this cycle?
7.3 Molecular Mechanisms Underlying the Programming of Insulin Resistance and T2DM in Insulin-Sensitive Tissues
Defects in insulin signalling itself are among the earliest indications that an individual is predisposed to the development of insulin resistance and subsequently T2DM [76, 77]. To date, however, the underlying molecular mechanisms which result in resistance to the actions of insulin including transcriptional and post-transcriptional dysregulation of genes involved in insulin signalling as well as post-translational modification(s) and degradation of their corresponding protein(s) are poorly understood [78, 79]. Furthermore, although it has been shown that maternal obesity is associated with an increased risk of obesity and insulin resistance in the offspring, the genetic and/or epigenetic modifications within insulin-sensitive tissues such as liver and skeletal muscle which contribute to the insulin-resistant phenotype remain unknown.
7.3.1 Metabolic Role of the Liver
In postnatal life, both in human and in sheep, the liver plays an essential role in carbohydrate metabolism by maintaining plasma glucose concentrations within a very narrow range over both short and long periods of time. This is partly achieved by the actions of insulin, which regulates hepatic glucose output by suppressing gluconeogenesis and glycogenolysis [80–82]. Insulin acts through a complex, highly integrated network that controls several processes downstream of the insulin receptor (IR) [77, 83]. The IR itself is found enriched in caveolae, which are flask-shaped invaginations of the plasma membrane. Structural components of caveolae are made up by the caveolin (Cav) gene family, namely Cav-1 and Cav-2, which are usually co-expressed in adipose tissue and the liver, and Cav-3, which is expressed in skeletal, cardiac and smooth muscle [84]. Localisation of the IR within caveolae serves to ensure metabolic signalling specificity downstream of it and Cav-1 also stabilises the receptor against proteasomal degradation [84].
In the presence of insulin, IR phosphorylates IRS proteins that are linked to the activation of the PI3K–Akt pathway, which is responsible for most of the metabolic actions of insulin [77]. PI3K acts to catalyse the formation of the lipid second messenger PIP3, which allows the localisation and activation of PDK1 and the subsequent activation of Akt. PDK1 is also critical for activating aPKCζ, which in the liver leads to an increase in lipid synthesis [77]. In contrast, activation of Akt results in a reduction in phosphorylation of the transcription factor FoxO1, which together with the transcriptional co-activator PGC1α plays an important role in regulating the expression of both PEPCK-C and G6Pase [80–82, 85]. PEPCK-C is mainly considered to be the rate-limiting enzyme in gluconeogenesis whereas hydrolysis of glucose-6-phosphate by G6Pase is the ‘final common pathway’ and is rate determining for the release of glucose into the circulation by gluconeogenesis and glycogenolysis [86].
The liver is also the most important site for the removal of FFA from circulating blood plasma [87, 88]. It coordinates the synthesis of FAs and the esterification of FAs to produce TGs and their subsequent packaging into VLDLs for export to adipose tissue [81, 87, 89]. The liver also regulates the rate of FA oxidation and ketogensis [81, 87, 89] and is therefore able to handle large amounts of fat without accumulating triacylglycerol and causing peripheral lipotoxicity [89].
7.3.2 Maternal Obesity Programs Molecular Changes in Hepatic Metabolic Processes in the Offspring
Shankar and colleagues found that at 21 days after birth, the offspring of rats that were overnourished from 3 weeks before until conception had increased phosphorylation of the IR and Akt, two key molecules of the insulin signalling network [90]. Furthermore, in another rodent model of metabolic programming by maternal obesity, female mice were fed a highly palatable diet high in sugar and fats for 6 weeks prior to pregnancy and throughout gestation and lactation [91]. Offspring of obese dams, which were weaned onto a control chow diet, had decreased hepatic IRS-1 abundance at 3 months of age. Moreover, hepatic phosphorylation of IRS-1 at Ser 307, which results in inhibition of insulin signalling, was also increased in these offspring [91].
In addition to defects in the insulin signalling network, increased hepatic fat deposition is also a very common feature of obesity and insulin resistance [92, 93] and NAFLD has been shown to be initiated by insulin resistance [94]. Most studies investigating the effects of maternal obesity on hepatic metabolism have examined perturbations in lipid metabolism. For example, studies investigating the impact of either a high-fat, high-sugar or high-fat only diet from before and during pregnancy and lactation on the offspring found that these mice developed a condition with marked similarity to NAFLD including increased liver TGs and hepatic fibrogenesis [54]. These offspring also had changes in gene expression, which indicated upregulation of lipogenesis, oxidative stress and inflammation [95]. Furthermore, rodent offspring exposed to maternal obesity also show impaired FA oxidation possibly due to a decrease in hepatic mitochondrial function [96]. Finally, a study by McCurdy and colleagues investigating the effect of a chronic high-fat diet on the development of fetal metabolic systems reported that maternal high-fat feeding triggered development of fatty liver and hepatic oxidative stress in the fetus, which persisted in postnatal life [97].
7.3.3 Metabolic Role of Skeletal Muscle
Skeletal muscle is the major site of postprandial glucose clearance from the circulation and accounts for up to 75 % of insulin-dependent glucose uptake [98]. The rate-limiting step for glucose clearance is the transport of glucose across the plasma membrane by facilitated diffusion of glucose through a family of specific GLUTs. GLUT-4 is the predominant glucose transporter in postnatal life, and in the postprandial state, binding of insulin to its receptor on myocytes leads to translocation of GLUT-4 to the plasma membrane, thereby permitting glucose entry into the cell [99, 100]. In the presence of insulin, IR phosphorylates IRS proteins, which then act as docking proteins for the activation of PI3K. PI3K acts to catalyse the formation of the lipid second messenger PIP3, which allows the localisation and activation of PDK1 and the subsequent activation of Akt and aPKC through phosphorylation of the Thr 308 and Thr 410 sites, respectively [100]. The positive actions of PI3K can be negatively regulated by phospholipid phosphatases, e.g. PTEN, which dephosphorylate and inactivate PIP3 [77].
Activation of Akt acts to phosphorylate and inhibit AS160, which is involved in the regulation of glucose uptake through the redistribution of GLUT-4 from intracellular vesicles to the plasma membrane [101, 102]. This ultimately leads to an increase in glucose transport into the cell [101, 102]. Similarly, aPKCs have also been shown to play a role in insulin-stimulated glucose uptake and GLUT-4 translocation in adipocytes and muscle [99]. In skeletal muscle, glucose is utilised to generate energy via glycolysis and is also converted to glycogen for storage [103]. Akt is also involved in the regulation of glycogen synthesis through the actions of the serine/threonine kinase GSK3 [77, 104, 105]. GSK3 consists of two highly homologous isoforms, GSK3α and GSK3β, and acts to phosphorylate and inactivate GS [104, 105]. In resting cells, GSK3 activity is high, but on stimulation, GSK3 is inactivated through phosphorylation; GSK3α is phosphorylated at Ser 21 and GSK3β at the equivalent residue, Ser 9 [104, 105]. There is evidence from both human and animal studies that defects in these downstream components of the insulin signalling pathway are present in the insulin-resistant state [100, 104].
7.3.4 Maternal Obesity Programs Molecular Changes in Metabolic Processes in Skeletal Muscle of the Offspring
Although there is a wealth of evidence showing the association between maternal obesity and the subsequent insulin-resistant phenotype in the offspring, there is a paucity of studies that have been carried out to try and determine the molecular basis of this relationship. One study by Shelley and colleagues observed that 3-month-old mice that were exposed to maternal obesity before and during gestation and lactation had decreased abundance of IRS-1. IRS-1 is required for the activation of PI3K, which is needed for the phosphorylation and activation of Akt and subsequent glucose uptake. These mice also had decreased abundance of the catalytic subunit of PI3K, p110β, as well as decreased phosphorylated Akt [53].
Studies in sheep have produced similar results although different molecules in the insulin signalling cascade appear to be affected. A study by Yan and colleagues where ewes were overnourished from 60 days before conception and throughout pregnancy reported that there were defects in insulin signalling in skeletal muscle of the adult offspring at the receptor level, which is in contrast to rodent studies that showed impaired post-receptor signalling [53, 106]. Similar to rodents, however, these lambs also had increased phosphorylated IRS-1 abundance and decreased phosphorylated Akt abundance at 22 months of age [106].
7.3.5 Metabolic Role of Adipose Tissue
Most energy reserves are stored in adipocytes as triacylglycerol (TAG), which arises from two major processes: uptake of free fatty acids (FFA) from plasma or de novo lipogenesis from non-lipid precursors such as glucose [107]. Insulin plays a key role in the latter as it stimulates glucose uptake into adipose tissue. Glucose is transported into adipocytes by facilitated diffusion through a family of specific GLUTs. GLUT-4 is redistributed from intracellular vesicles to the plasma membrane in response to insulin as it is in skeletal muscle via activation of the IR [99, 100]. Furthermore, insulin also plays an important role in uptake, esterification and storage of FFA in adipocytes [108]. Insulin stimulates the activity of lipoprotein lipase LPL, which generates FFA for TAG synthesis by enabling the release of FFA from lipoproteins and is the important first step in TAG synthesis [107]. Insulin also suppresses the activity of hormone-sensitive lipase (HSL), which is expressed in white and brown adipose tissue and is a principal regulator of FFA release from adipose tissue [107].
Adipose tissue also secretes a number of adipokines, e.g. leptin and adiponectin, which play a key role in the development of insulin resistance associated with increased adiposity [109]. For example, obese patients with insulin resistance/T2D have reduced adiponectin [110]. Adiponectin also regulates hepatic glucose production through its actions on gluconeogenic genes [111]. Another adipokine, leptin, acts as a circulating signal of fat mass [112]. Indeed, there is a direct relationship between cord blood concentrations of leptin at delivery and birth weight or neonatal adiposity both in normal pregnancies [113, 114] and in pregnancies complicated by maternal diabetes [115].
7.3.6 Maternal Obesity Programs Molecular Changes in Metabolic Processes in Adipose Tissue of the Offspring
Borengasser and colleagues have investigated the early effects of programming that occur prior to the emergence of adiposity and weight gain in 21-day-old offspring of dams that were overfed from before pregnancy [21, 96]. They found that these offspring had increased expression of adipogenic, lipogenic and adipokine (leptin and adiponectin) genes in white adipose tissue (WAT) [21]. Furthermore, the increased abundance of adipogenic proteins PPARγ, CCAAT-enhancer-binding protein (C/EBP)-α and C/EBP-β resulted ultimately in increased adipocyte differentiation, which was present in the offspring of obese dams at 21 days of age and persisted at 100 days of age [21]. In addition to these changes, insulin signalling was also upregulated in WAT of the offspring. This was characterised by increased protein abundance of the IR, GSK3α/β and increased GLUT-4 gene expression [21]. Phosphorylation of Akt following acute insulin stimulation was also approximately 1.9-fold greater in these offspring [21].
Interestingly, these findings associated with insulin signalling are in contrast to those of Fernandez-Twinn et al. in their study to dissect out the effects of maternal diet-induced obesity on offspring insulin resistance that were independent of the increased adiposity [72]. Using a mouse model of maternal diet-induced obesity with a diet rich in fat and simple sugars representative of a Western human diet, they found that the young mice of the obese dams displayed impaired adipose tissue insulin signalling [72]. These mice had reduced IR, IRS-1, the regulatory and catalytic subunit of PI3K, Akt1 and Akt2 protein abundance in WAT [72]. These findings suggest that although exposure to maternal obesity contributes to an obese phenotype, the adipose tissue of these offspring develops resistance to the actions of insulin even before the appearance of increased adiposity. What is also interesting is that, taken together, both these studies highlight the fact that adipose tissue is exquisitely sensitive to programming by maternal obesity and that the differences in timing, duration and type of maternal over-feeding can produce contrasting changes prior to the onset of obesity in the offspring. Exposure to pre-pregnancy obesity appears to program increased insulin signalling in WAT of the offspring, whereas when the period of exposure to maternal obesity is extended to encompass both gestation and lactation, there appears to be a switch to decreased insulin signalling.
7.4 Exposure to Maternal Obesity During Gestation and the Epigenetic Origins of Obesity
A number of recent studies in rodents have investigated the impact of a maternal high-fat diet throughout gestation on epigenetic changes in the adipose tissue and liver of the offspring [74, 116, 117]. Transcriptomic changes in the study by Borengasser et al. outlined in the previous section have been shown to be associated with alterations in DNA methylation of CpG sites and CpG island shores, which are proximal to developmentally important genes including C/EBP-β [21]. Changes in adiponectin and leptin expression in adipose tissue of offspring exposed to maternal high-fat diet were found to be due to alterations in both acetylation and methylation of histone H3K9 within the adiponectin promoter and changes in methylation of histone H4K20 within the leptin promoter [116]. The offspring of high-fat fed dams also showed increased hepatic expression of the cytosolic isoform of the gluconeogenic gene, phosphoenolpyruvate carboxykinase, which was attributed to histone modifications associated with transcriptional activation [117]. Importantly, the effects of maternal high-fat feeding appear to be transgenerational. The F2 offspring derived from both grand-maternal and maternal obesity appeared to be extremely susceptible to developing obesity due to the transgenerational accumulation of epigenetic modifications including histone methylation, which then contribute to an increase in lipogenesis [74].
Recent evidence has also identified microRNAs (miRNAs), a class of small (~22 nt), non-coding RNAs, as important regulators of insulin signalling through its role in post-transcriptional gene regulation either by cleavage or translational repression of their specific target mRNAs [79, 118–120]. In their study described above, Fernandez-Twinn and colleagues showed that the decreased IRS-1 protein abundance in WAT of offspring exposed to maternal diet-induced obesity before and during pregnancy and lactation was linked to increased expression of miR-126 [72]. He and colleagues have also shown in a rat model of T2DM that expression of all members of the miR-29 family (miR-29a, miR-29b and miR-29c) was upregulated in liver, adipose tissue and skeletal muscle of Goto-Kakizaki rats [118]. The authors then mimicked insulin resistance in 3 T3-L1 adipocytes in order to determine the molecular mechanisms involved in the regulation of insulin signalling by the mir-29 family. They found that there was an increase in expression of miR-29a and miR-29b as well as a parallel decrease in the abundance of insulin signalling molecules, in particular, Ser 473 phospho-Akt, which resulted in a subsequent decrease in glucose uptake [118]. The mir-103/mir-107 family have also emerged as regulators of hepatic insulin signalling in two models of insulin-resistant, obese mice. Specifically, Trajkovski and colleagues have shown that miR-103/miR-107 were among the five most upregulated miRNAs in the livers of insulin-resistant obese mice [120]. Cav-1 carries a seed match to miR-103/miR-107 in its 3’UTR and is, therefore, its direct target. Consequently, Cav-1 expression was decreased after overexpression of miR-107 in the liver of mice.
Finally, Jordan and colleagues have shown that expression of hepatic miR-143 was increased in db/db mice as well as mice exposed to a high-fat diet. They also showed in a transgenic mouse model of miR-143 overexpression that these mice had impaired glucose metabolism through the induction of insulin resistance as shown by a decreased in phosphorylation of Akt [79].
7.5 The Periconceptional Period Is a Critical Window for the Development of Postnatal Obesity
Currently, it appears that for obese women, pre-pregnancy BMI rather than gestational weight gain is associated with an increased risk of pre-eclampsia, GDM and the delivery of a macrosomic infant [121]. It has also been reported that siblings born to women who had undergone bariatric surgery for the treatment of severe obesity had a lower BMI and obesity risk than their siblings who were born prior to maternal surgery and weight loss [122]. Moreover, previous studies have shown that even in women who are ovulating regularly, increased BMI correlates with reduced conception rates [34, 123], suggesting that obesity affects critical periconceptional events [124]. The very earliest stages of embryo growth are primarily controlled by the quality of the oocyte, also known as oocyte developmental competence. Clinically, oocyte quality has been directly assessed in only three studies with the most recent and larger study finding that although there was no difference in the number of follicles aspirated, the number of mature oocytes was significantly reduced in morbidly obese women [125].
An experimental study in mice by Minge and colleagues, which investigated the impact of maternal obesity on oocyte and early embryo development, showed that when cultured in vitro oocytes from obese mice exhibited slower development to the four- to eight-cell stage and through to the blastocyst stage. These blastocysts also have reduced level of mitochondrial DNA and these changes were still present in the fetus at 14.5 days even though the blastocysts were transferred to normal-weight surrogates [126]. Furthermore, they also showed that these negative effects of maternal obesity can be improved by treatment with insulin sensitisers administered around the time of conception. These results indicate that maternal obesity as well as her peripheral insulin sensitivity during the periconceptional period is an important determinant of the developmental outcomes of the offspring [127]. This indicates that the periconceptional period, which includes some or all of the following early developmental stages: oocyte maturation and follicular development, i.e. pre-pregnancy events, conception and embryo/blastocyst growth up until implantation [15], is therefore a critical window for the development of postnatal obesity in the offspring.
7.5.1 An ovine Model of Maternal Obesity in the Periconceptional Period and Programming of Later Obesity In the Offspring
In order to determine whether exposure to maternal obesity in the periconceptional period alone has any specific impact on adiposity and metabolic function in the offspring, we have developed an embryo transfer model in sheep [128–132]. In this model, non-pregnant donor ewes were either overnourished or normally nourished for at least 4 months before conception. One week after conception, single embryos were transferred from obese or normal weight ‘donor’ ewes to non-obese ‘recipient’ ewes, which were maintained on a control diet for the remainder of pregnancy [128–132]. Thus, exposure to a high nutrient environment encompassed oocyte maturation, follicular development, conception and growth of the early pre-implantation embryo. This model is unique in that exposure of the offspring to maternal obesity is confined strictly to the periconceptional period.
Donor ewes that were overnourished during the periconceptional period were heavier than ewes on the control level of nutrition at 4 weeks before conception [131], and these ewes also had an obese phenotype at conception [131] as determined by their body condition score [133]. Furthermore, similar to obese humans [134], donor ewes that were overnourished also had increased plasma insulin but not plasma glucose concentrations [131].
7.5.2 Impact of Exposure to Obesity in the Periconceptional Period on Fat Mass in the Offspring
There is a sex-specific effect of exposure to maternal obesity during the periconceptional period on the body fat mass of lambs at 4 months of age [131]. Female but not male lambs, conceived in obese ewes, had an increased total fat mass. Specifically, the greatest impact of maternal periconceptional obesity appeared to be on the visceral fat depots, i.e. the perirenal and omental fat depots, in these female lambs [131]. Interestingly, the weights of these depots were also higher in female compared to male lambs [131]. There was also a significant relationship between total fat mass of female lambs at 4 months of age and the weight of donor ewes at conception [131].
Furthermore, an investigation of the expression of genes that regulate the differentiation and the development of adipose tissue as well as the storage of lipids in the perirenal, omental and subcutaneous fat depots of these lambs found that the increased adiposity in female lambs was not due to changes in expression of the adipogenic, lipogenic and adipokine genes PPARγ, glyceraldehyde 3-phosphate dehydrogenase, lipoprotein lipase, leptin and adiponectin [131]. Further studies on the role of the insulin signalling and other key metabolic pathways which regulate lipogenesis within these fat depots are required.
7.5.3 Impact of Exposure to Obesity in the Periconceptional Period on Insulin Signalling in the Liver and Muscle in the Offspring
Follow-up studies have also been carried out to determine whether exposure to maternal obesity during the periconceptional period may result in the molecular features of insulin resistance in the offspring either as a consequence of the impact of increased adiposity on insulin-sensitive tissues such as the liver and muscle [135, 136] or as a consequence of the programming of specific changes in the abundance of insulin signalling molecules in these tissues of metabolic importance [137–139].
While exposure to maternal obesity around the time of conception resulted in an increase in body fat mass in female lambs, we found that there were programmed changes in gene expression and protein abundance of key insulin signalling molecules in the liver and to a more limited extent in skeletal muscle of both male and female lambs conceived in obese ewes [128, 129]. There was a decreased hepatic abundance of the insulin receptor as well as phosphorylated Akt (Ser 473) and FoxO1 (Thr 24) in the young offspring of obese ewes. Interestingly, however, there was a paradoxical effect on the expression of key factors which regulate hepatic gluconeogenesis; expression of 11βHSD1, PEPCK-C and PEPCK-M was decreased in lambs exposed to maternal obesity [129]. These contrasting changes suggest that there are distinct mechanisms involved, which are programmed by maternal obesity during the periconceptional period and which impact hepatic insulin signalling and gluconeogenic factors separately. Findings in skeletal muscle of the offspring, however, showed that exposure to maternal obesity during the periconceptional period did not appear to impact directly on the early components of the insulin signalling pathway. Instead, this exposure resulted in specific changes in the abundance of molecules downstream of Akt [128]. Taken together, these results suggest that exposure to maternal obesity during the periconceptional period acts directly, independently of increased adiposity, to program changes within the insulin signalling pathway in the liver and skeletal muscle.
7.5.4 Impact of Exposure to Obesity in the Periconceptional Period on Hepatic Fatty Acid Metabolism in the Offspring
In addition to defects in the insulin signalling network, increased hepatic fat deposition is also a common feature of obesity and insulin resistance [92, 93]. Indeed, maternal obesity both before and throughout pregnancy has been shown to result in alterations in hepatic FA oxidation and lipogenesis in the offspring [54, 97]. In addition to changes in the abundance of hepatic insulin signalling molecules, we also found that exposure to maternal obesity during the periconceptional period resulted in downregulation of hepatic PGC1-α and PPARα and also resulted in a compensatory increase in the abundance of AMP-activated protein kinase (AMPK) α1 and α2, which may initially limit the impact of these changes on intra-hepatic FA oxidation in the young offspring [130]. Furthermore, hepatic expression of sterol regulatory element-binding protein 1, a key lipogenic gene, was also increased in these lambs. It is possible that with ageing and/or exposure to a high-caloric diet, these offspring may be susceptible to hepatic lipid accumulation and steatosis [130].
7.5.5 Exposure to Maternal Obesity During the Periconceptional Period and the Epigenetic Origins of Obesity
During the early stages of development, the differentiation and development of different cell types is regulated by epigenetic mechanisms, which play a role in modulating chromatin architecture [140]. Furthermore, epigenetic regulation plays a key role in conferring phenotype plasticity, which allows organisms to adapt their gene expression and function in response to the environment [141]. Each cell type, therefore, has its own epigenetic signature which reflects genotype, developmental history and environmental influences. This is ultimately reflected in the phenotype of the cell and organism [142]. Early embryogenesis in mammals is a critical period for the establishment of the epigenome [143]; during the period between conception and implantation, there is de-methylation of the genome followed by a wave of re-methylation shortly after implantation [142]. This period, therefore, represents a critical window in development during which the embryo is vulnerable to environmental and/or nutritional cues that disrupt the establishment of epigenetic marks such as DNA methylation, histone modification and miRNAs [144]. Importantly, although the genome of an individual is largely stable, the epigenome has the potential to be reversibly modified by exposure to a range of nutritional and environmental factors [145]. Parental nutrition has been shown to permanently influence metabolism of the offspring through epigenetic mechanisms and these changes also appear to be stable and transgenerational [146].
Using the embryo transfer model described above [128–132], we found that exposure of the oocyte/early embryo to maternal obesity resulted in upregulation of hepatic expression of miR-29b, miR-103 and miR-107 [129]. Expression of these miRNAs has been shown to be related to decreased insulin signalling in adipocytes [118] and liver [120] and is increased in murine models of obesity and T2DM [79, 118, 120].
Since defects in insulin signalling are among the earliest indicators that an individual is predisposed to the development of insulin resistance and T2DM [77], our results suggest that miRNAs may be potential epigenetic regulators that are sensitive to programming by the metabolic and nutritional environment associated with maternal obesity specifically during the periconceptional period. MiRNAs could, therefore, play a key role in the transduction of the metabolic consequences of maternal obesity from the mother to the offspring.
7.6 Weighing Up the Benefits and Costs of Maternal Dietary and Lifestyle Interventions in Obese Mothers for Their Offspring
Clinical and experimental studies have provided clear evidence that exposure to a maternal obesogenic environment from before pregnancy and around the time of conception has long-lasting metabolic consequences for the offspring. There has been a growing focus, therefore, on nutritional health of women in the periconceptional period and on what weight loss interventions can be safely introduced in overweight/obese women seeking to become pregnant [15]. There is, however, now more than ever a need for a better understanding of both the benefits and the possible negative effects of maternal dietary and lifestyle interventions in obese mothers for their offspring.
7.6.1 The Benefits of Maternal Dietary Restriction and Weight Loss: Breaking the ‘Intergenerational Cycle’ of Obesity and Insulin Resistance
Previous studies have suggested that childhood obesity may be prevented by normalising body composition and nutrition and improving the general health of young women of childbearing age before becoming pregnant, thereby preventing the prevalence of the ‘intergenerational cycle of obesity’ and the serious co-morbidities associated with obesity [147]. Other studies have focused on limiting gestational weight gain through both physical activity and the reduction of dietary intake [148–150] on the premise that a healthy, active pregnancy may help to minimise the cycle [151]. Although most studies showed favourable weight-related outcomes indicating that interventions can help pregnant and postpartum women manage their weight, knowledge gaps still remain regarding the benefits and potential harm associated with dietary and lifestyle interventions for overweight and obese pregnant women and their offspring [148–151].
Furthermore, as traditional ‘diet and exercise’ approaches do not often achieve robust and sustained weight loss [152], surgical approaches have also emerged as an effective albeit complex strategy to promote durable weight loss, improve insulin resistance and improve or reverse T2DM especially in morbidly obese women [152, 153]. A number of studies have found a decreased prevalence of obesity in the offspring of mothers who underwent maternal surgical weight loss prior to pregnancy [154, 155]. Furthermore, a recent study by Guénard and colleagues found that the improved cardiometabolic risk profiles of offspring born after maternal weight loss surgery were associated with epigenetic changes including differential methylation patterns of glucoregulatory genes [156].
In contrast to human studies, there have been relatively few experimental animal studies, which have investigated whether maternal dietary restriction and/or exercise are able to reverse the outcomes associated with maternal obesity. We have shown in the sheep that dietary restriction and weight loss experienced by the mother in the periconceptional period alone is able to ablate the effects of a high maternal pre-pregnancy weight on offspring adiposity [131]. Furthermore, in a study where the nutritional intake of female rats that were previously on a high-fat diet was ‘restricted’ to normal chow from 1 month prior to conception and throughout pregnancy and lactation, dietary ‘restriction’ was able to normalise fat mass, serum triglycerides, leptin and insulin levels in 3-week-old male offspring [157]. Interestingly, these changes occurred even though these dams that were exposed to dietary ‘restriction’ remained heavier than their control counterparts at conception [157], indicating that in this instance, it is the metabolic response to dietary restriction rather than a lower maternal body weight, which confers metabolic benefits in the offspring. Studies investigating the impact of exercise from before and/or throughout pregnancy in rodents have found that exercise is able to ablate the increase in plasma leptin and triglycerides caused by exposure to maternal obesity [158]. Expression of key genes involved in glucose and lipid metabolism as well as markers of inflammation was also reduced to control levels in skeletal muscle and adipose tissue of offspring from dams that underwent voluntary exercise during pregnancy [159].
7.6.2 The Metabolic and Endocrine Costs of Maternal Dietary Restriction and Weight Loss
While maternal dieting before pregnancy has metabolic benefits, there are also potential metabolic and endocrine costs for the offspring. Studies of people born at the time of the Dutch famine in 1944–1945 have shown that exposure to undernutrition during both early and mid-pregnancy in a population that was previously well nourished was associated with a reduction in glucose tolerance and increased insulin concentration at age 50 and 58 [160]. Furthermore, experimental evidence in sheep has shown that nutritional restriction imposed in ewes with a normal body across both the periconceptional and early gestation periods (from 60 days before until 30 days after conception) has an adverse impact on the glucose–insulin axis of the offspring in postnatal life [161]. This impaired glucose tolerance also persists in the adult offspring [162]. It is not known, however, whether a similar period of dietary restriction and weight loss imposed on obese ewes will also have consequences for the metabolic health of the offspring.
We have also shown that in most instances dietary restriction and weight loss in the periconceptional period were unable to ablate the effects of maternal obesity on the abundance of insulin signalling proteins in both the liver [129] and skeletal muscle [128] or on signalling molecules involved in hepatic lipid metabolism in the postnatal lamb [130]. Instead, exposure to dietary restriction in either normal-weight or obese ewes resulted in a downregulation of a different subset of insulin signalling proteins within both liver and skeletal muscle compared to offspring exposed to maternal obesity. Furthermore, the reduced abundance of some insulin signalling molecules were conserved both in liver and skeletal muscle of offspring exposed to maternal dietary restriction during the periconceptional period [128, 129]. This suggests that the mechanism involved is one that is able to impact tissues that arise from different cell lineages as the liver and skeletal muscle arise from the endoderm and mesoderm, respectively [163]. This leads us to ask the question of whether the oocyte/early embryo is most sensitive to perturbations during the period encompassing 1 month before and 1 week after conception. Moreover, since the specification of different cell types is regulated by epigenetic mechanisms such as histone or DNA modification, which can modulate chromatin architecture [140], it is possible that changes in the abundance of insulin signalling molecules in liver and skeletal muscle of the offspring are due to the recruitment of epigenetic mechanisms within the developing embryo. Indeed, studies which have investigated the impact of maternal undernutrition during the periconceptional period in sheep have found that changes in the abundance of insulin signalling molecules in liver and skeletal muscle were inversely associated with changes in expression of specific miRNAs in these metabolic organs [164, 165].
It has also been shown in sheep that moderate dietary restriction imposed during the periconceptional period results in an increase in fetal arterial blood pressure and in an earlier activation of the fetal pre-partum cortisol surge [166, 167]. Furthermore, dietary restriction in both normal weight and obese ewes during the periconceptional period has also been shown to result in an enhanced cortisol response to stress in female lambs at 3–4 months of age [132]. Investigation into the possible mechanism(s) underlying this observation found that both male and female lambs from normal-weight ewes that were exposed to dietary restriction during the periconceptional period had a loss of DNA methylation within the differentially methylated region (DMR) of H19/IGF2 in the adrenal gland [132]. Moreover, these lambs had increased activation of the downstream components of the intra-adrenal renin–angiotensin system through an increase in the abundance of angiotensin-converting enzyme and angiotensin type 1 receptor in the adrenal cortex [168]. Interestingly, Heijmans and colleagues have shown that individuals whose mother was exposed to the Dutch Hunger Winter famine during the periconceptional period had lower methylation of the IGF2 DMR measured in their blood six decades later [169].
7.7 Summary and Conclusions
There is, therefore, solid evidence that exposure to maternal obesity or to impaired glucose tolerance in utero programs an increased risk of obesity and features of insulin resistance in the offspring, thus potentially fuelling an ‘intergenerational cycle’ of obesity and insulin resistance. Importantly, it appears that the oocyte and/or early pre-implantation embryo is particularly vulnerable to the effects of maternal obesity, resulting in long-lasting endocrine and metabolic effects for the offspring. Furthermore, the impact of maternal obesity on different insulin-sensitive tissues may be programmed independently rather than as a result of the indirect effects of increased adiposity. Investigations into the molecular mechanisms underlying the programming of obesity and insulin resistance in liver, muscle and adipose tissue have highlighted the role of epigenetic changes within these tissues, which are recruited within the developing embryo and/or fetus (Fig. 7.2).

The impact of maternal obesity during the periconceptional on different insulin-sensitive tissues in the offspring may be programmed independently rather than as a result of the indirect effects of increased adiposity. Epigenetic changes within these tissues may be the conduit through which a life of metabolic vulnerability is programmed in tissues of metabolic importance
Finally, it is clear that weight loss achieved through dietary restriction in overweight/obese women prior to and around the time of conception may not be the optimal intervention to break the ‘intergenerational cycle’ of obesity and insulin resistance. Furthermore, the longer-term effects of maternal exercise in overweight/obese mothers during the pre-conception period on the offspring remain to be determined. These findings highlight the need for a better evidence base for the development of dietary interventions in obese women before pregnancy which maximise the metabolic benefits and minimise the metabolic costs for the offspring.
References
WHO. Global Health Observatory (GHO) data: Overweight and obesity (8 Jan 2016). Available from: http://www.who.int/gho/ncd/risk_factors/overweight_text/en/
Flegal KM, Carroll MD, Ogden CL, Johnson CL (2002) Prevalence and trends in obesity among US adults, 1999-2000. JAMA 288(14):1723–1727
Kumanyika SK, Obarzanek E, Stettler N, Bell R, Field AE, Fortmann SP et al (2008) Population-based prevention of obesity: the need for comprehensive promotion of healthful eating, physical activity, and energy balance: a scientific statement from American Heart Association Council on Epidemiology and Prevention, Interdisciplinary Committee for prevention (formerly the expert panel on population and prevention science). Circulation 118(4):428–464
Ogden CL, Yanovski SZ, Carroll MD, Flegal KM (2007) The epidemiology of obesity. Gastroenterology 132(6):2087–2102
Catalano PM, Ehrenberg HM (2006) The short- and long-term implications of maternal obesity on the mother and her offspring. BJOG 113(10):1126–1133
Catalano PM (2003) Obesity and pregnancy—the propagation of a viscous cycle? J Clin Endocrinol Metabol 88(8):3505–3506
Pettitt DJ, Nelson RG, Saad MF, Bennett PH, Knowler WC (1993) Diabetes and obesity in the offspring of Pima Indian women with diabetes during pregnancy. Diabetes Care 16(1):310–314
Silverman BL, Rizzo TA, Cho NH, Metzger BE (1998) Long-term effects of the intrauterine environment: the northwestern university diabetes in pregnancy center. Diabetes Care 21(Suppl 2):B142–B149
Federation ID (2013) IDF diabetes Atlas, 6th edn. International Diabetes Federation, Brussels
Boney CM, Verma A, Tucker R, Vohr BR (2005) Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 115(3):e290–e296
Dorner G, Plagemann A (1994) Perinatal hyperinsulinism as possible predisposing factor for diabetes mellitus, obesity and enhanced cardiovascular risk in later life. Horm Metab Res 26(5):213–221
Whitaker RC (2004) Predicting preschooler obesity at birth: the role of maternal obesity in early pregnancy. Pediatrics 114(1):e29–e36
WHO. 10 Facts on obesity (8 Jan 2016). Available from: http://www.who.int/features/factfiles/obesity/facts/en/index2.html
Zhang S, Rattanatray L, McMillen IC, Suter CM, Morrison JL (2011) Periconceptional nutrition and the early programming of a life of obesity or adversity. Prog Biophys Mol Biol 106(1):307–314
Zhang S, Rattanatray L, Morrison JL, Nicholas LM, Lie S, McMillen IC (2011) Maternal obesity and the early origins of childhood obesity: weighing up the benefits and costs of maternal weight loss in the periconceptional period for the offspring. Exp Diabetes Res 2011
Oteng-Ntim E, Varma R, Croker H, Poston L, Doyle P (2012) Lifestyle interventions for overweight and obese pregnant women to improve pregnancy outcome: systematic review and meta-analysis. BMC Med 10:47
Finucane MM, Stevens GA, Cowan MJ, Danaei G, Lin JK, Paciorek CJ et al (2011) National, regional, and global trends in body-mass index since 1980: systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9·1 million participants. Lancet 377(9765):557–567
Poston L, Harthoorn LF, Van Der Beek EM (2011) Obesity in pregnancy: implications for the mother and lifelong health of the child. A consensus statement. Pediatr Res 69(2):175–180
Statistics ABo (2007) Australian social trends. Canberra
LaCoursiere DY, Bloebaum L, Duncan JD, Varner MW (2005) Population-based trends and correlates of maternal overweight and obesity, Utah 1991-2001. Am J Obstet Gynecol 192(3):832–839
Borengasser SJ, Zhong Y, Kang P, Lindsey F, Ronis MJ, Badger TM et al (2013) Maternal obesity enhances white adipose tissue differentiation and alters genome-scale DNA methylation in male rat offspring. Endocrinology 154(11):4113–4125
Flegal KM, Carroll MD, Kit BK, Ogden CL (2012) Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999-2010. JAMA 307(5):491–497
Athukorala C, Rumbold AR, Willson KJ, Crowther CA (2010) The risk of adverse pregnancy outcomes in women who are overweight or obese. BMC Pregnancy Childbirth 10:56
Callaway LK, Prins JB, Chang AM, McIntyre HD (2006) The prevalence and impact of overweight and obesity in an Australian obstetrics population. Med J Aust 184(2):56–59
Singh AS, Mulder C, Twisk JWR, Van Mechelen W, Chinapaw MJM (2008) Tracking of childhood overweight into adulthood: a systematic review of the literature. Obes Rev 9(5):474–488
Kelishadi R (2007) Childhood overweight, obesity, and the metabolic syndrome in developing countries. Epidemiol Rev 29(1):62–76
Rooney K, Ozanne SE (2011) Maternal over-nutrition and offspring obesity predisposition: targets for preventative interventions. Int J Obes (Lond) 35(7):883–890
Ryan D (2007) Obesity in women: a life cycle of medical risk. Int J Obes (Lond) 31(Suppl 2):S3–S7
Lawlor DA (2013) The society for social medicine John Pemberton Lecture 2011. Developmental overnutrition-an old hypothesis with new importance. Int J Epidemiol 42(1):7–29
Lawlor DA, Relton C, Sattar N, Nelson SM (2012) Maternal adiposity—a determinant of perinatal and offspring outcomes? Nat Rev Endocrinol 8(11):679–688
Catalano PM, Kirwan JP, Haugel-de Mouzon S, King J (2003) Gestational diabetes and insulin resistance: role in short- and long-term implications for mother and fetus. J Nutr 133(5 Suppl 2):1674S–1683S
Freinkel N (1980) Of pregnancy and progeny. Diabetes 29(12):1023–1035
Bergmann RL, Richter R, Bergmann KE, Plagemann A, Brauer M, Dudenhausen JW (2003) Secular trends in neonatal macrosomia in Berlin: influences of potential determinants. Paediatr Perinat Epidemiol 17(3):244–249
Jensen DM, Damm P, Sørensen B, Mølsted-Pedersen L, Westergaard JG, Ovesen P et al (2003) Pregnancy outcome and prepregnancy body mass index in 2459 glucose-tolerant Danish women. Am J Obstet Gynecol 189(1):239–244
May R (2007) Prepregnancy weight, inappropriate gestational weight gain, and smoking: relationships to birth weight. Am J Hum Biol 19(3):305–310
Sebire NJ, Jolly M, Harris JP, Wadsworth J, Joffe M, Beard RW et al (2001) Maternal obesity and pregnancy outcome: a study of 287 213 pregnancies in London. Int J Obes (Lond) 25(8):1175–1182
Yu Z, Han S, Zhu J, Sun X, Ji C, Guo X (2013) Pre-pregnancy body mass index in relation to infant birth weight and offspring overweight/obesity: a systematic review and meta-analysis. PLoS One 8(4)
Petitt DJ, Baird HB, Aleck KA (1983) Excessive obesity in offspring of Pima Indian women with diabetes during pregnancy. N Engl J Med 308(5):242–245
Pettitt DJ, Bennett PH, Knowler WC (1985) Gestational diabetes mellitus and impaired glucose tolerance during pregnancy. Long-term effects on obesity and glucose tolerance in the offspring. Diabetes 34(Suppl 2):119–122
Silverman BL, Rizzo T, Green OC, Cho NH, Winter RJ, Ogata ES et al (1991) Long-term prospective evaluation of offspring of diabetic mothers. Diabetes 40(Suppl 2):121–125
Parsons TJ, Power C, Manor O (2001) Fetal and early life growth and body mass index from birth to early adulthood in 1958 British cohort: longitudinal. Br Med J 323(7325):1331–1335
Kubo A, Ferrara A, Windham GC, Greenspan LC, Deardorff J, Hiatt RA et al (2014) Maternal hyperglycemia during pregnancy predicts adiposity of the offspring. Diabetes Care 37(11):2996–3002
Buckley AJ, Keserü B, Briody J, Thompson M, Ozanne SE, Thompson CH (2005) Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism 54(4):500–507
Cerf ME, Williams K, Nkomo XI, Muller CJ, Du Toit DF, Louw J et al (2005) Islet cell response in the neonatal rat after exposure to a high-fat diet during pregnancy. Am J Physiol Regul Integr Comp Physiol 288(5):R1122–R1128
Guo F, Catherine Jen KL (1995) High-fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol Behav 57(4):681–686
Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C et al (2009) Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 52(6):1133–1142
Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EHJM et al (2008) Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 51(2):383–392
Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJJ, Badger TM (2008) Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 294(2):R528–R538
Taylor PD, McConnell J, Khan IY, Holemans K, Lawrence KM, Asare-Anane H et al (2005) Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am J Physiol Regul Integr Comp Physiol 288(1):R134–R139
Volpato AM, Schultz A, Magalhães-da-Costa E, Correia MLDG, Águila MB, Mandarim-de-Lacerda CA (2012) Maternal high-fat diet programs for metabolic disturbances in offspring despite leptin sensitivity. Neuroendocrinology 96:272–284
Frayn KN (2000) Visceral fat and insulin resistance—Causative or correlative? Br J Nutr 83(Suppl 1):S71–S77
Yamashita S, Nakamura T, Shimomura I, Nishida M, Yoshida S, Kotani K et al (1996) Insulin resistance and body fat distribution: contribution of visceral fat accumulation to the development of insulin resistance and atherosclerosis. Diabetes Care 19(3):287–291
Shelley P, Martin-Gronert MS, Rowlerson A, Poston L, Heales SJR, Hargreaves IP et al (2009) Altered skeletal muscle insulin signaling and mitochondrial complex II-III linked activity in adult offspring of obese mice. Am J Physiol Regul Integr Comp Physiol 297(3):R675–R681
Oben JA, Mouralidarane A, Samuelsson AM, Matthews PJ, Morgan ML, McKee C et al (2010) Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol 52(6):913–920
McMillen IC, Adam CL, Muhlhausler BS (2005) Early origins of obesity: programming the appetite regulatory system. J Physiol 565(1):9–17
Lewis DS, Bertrand HA, McMahan CA (1986) Preweaning food intake influences the adiposity of young adult baboons. J Clin Invest 78(4):899–905
DiGiacomo JE, Hay WW Jr (1990) Effect of hypoinsulinemia and hyperglycemia on fetal glucose utilization. Am J Physiol Endocrinol Metab 259(4):E506–E512
Ford SP, Tuersunjiang N (2013) Maternal obesity: how big an impact does it have on offspring prenatally and during postnatal life? Expert Rev Endocrinol Metab 8(3):261–273
Hay WW Jr, DiGiacomo JE, Meznarich HK, Hirst K, Zerbe G (1989) Effects of glucose and insulin on fetal glucose oxidation and oxygen consumption. Am J Physiol Endocrinol Metab 256(6):E704–E713
Anthony RV, Scheaffer AN, Wright CD, Regnault TR (2003) Ruminant models of prenatal growth restriction. Reprod Suppl 61:183–194
Mühlhäusler BS, McMillen IC, Rouzaud G, Findlay PA, Marrocco EM, Rhind SM et al (2004) Appetite regulatory neuropeptides are expressed in the sheep hypothalamus before birth. J Neuroendocrinol 16(6):502–507
Long NM, Rule DC, Zhu MJ, Nathanielsz PW, Ford SP (2012) Maternal obesity upregulates fatty acid and glucose transporters and increases expression of enzymes mediating fatty acid biosynthesis in fetal adipose tissue depots. J Anim Sci 90(7):2201–2210
Ford SP, Zhang L, Zhu M, Miller MM, Smith DT, Hess BW et al (2009) Maternal obesity accelerates fetal pancreatic β-cell but not α-cell development in sheep: prenatal consequences. Am J Physiol Regul Integr Comp Physiol 297(3):R835–R843
Zhang L, Long NM, Hein SM, Ma Y, Nathanielsz PW, Ford SP (2011) Maternal obesity in ewes results in reduced fetal pancreatic β-cell numbers in late gestation and decreased circulating insulin concentration at term. Domest Anim Endocrinol 40(1):30–39
Du M, Yan X, Tong JF, Zhao J, Zhu MJ (2010) Maternal obesity, inflammation, and fetal skeletal muscle development. Biol Reprod 82(1):4–12
Tong JF, Yan X, Zhu MJ, Ford SP, Nathanielsz PW, Du M (2009) Maternal obesity downregulates myogenesis and beta-catenin signaling in fetal skeletal muscle. Am J Physiol Endocrinol Metab 296(4):E917–E924
Long NM, George LA, Uthlaut AB, Smith DT, Nijland MJ, Nathanielsz PW et al (2010) Maternal obesity and increased nutrient intake before and during gestation in the ewe results in altered growth, adiposity, and glucose tolerance in adult offspring. J Anim Sci 88(11):3546–3553
Mühlhäusler BS, Duffield JA, McMillen IC (2007) Increased maternal nutrition stimulates peroxisome proliferator activated receptor-γ, adiponectin, and leptin messenger ribonucleic acid expression in adipose tissue before birth. Endocrinology 148(2):878–885
Mühlhäusler BS, Adam CL, Findlay PA, Duffield JA, McMillen IC (2006) Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J 20(8):1257–1259
Mühlhäusler BS, Duffield JA, McMillen IC (2007) Increased maternal nutrition increases leptin expression in perirenal and subcutaneous adipose tissue in the postnatal lamb. Endocrinology 148(12):6157–6163
Rattanatray L, Muhlhausler BS, Nicholas LM, Morrison JL, McMillen IC (2014) Impact of maternal overnutrition on gluconeogenic factors and methylation of the phosphoenolpyruvate carboxykinase promoter in the fetal and postnatal liver. Pediatr Res 75(1-1):14–21
Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes DE, Piekarz A, Ferland-McCollough D et al (2014) Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab 3(3):325–333
Graus-Nunes F, Dalla Corte Frantz E, Lannes WR, da Silva Menezes MC, Mandarim-de-Lacerda CA, Souza-Mello V (2015) Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 31(2):380–387
Li J, Huang J, Li JS, Chen H, Huang K, Zheng L (2012) Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol 56(4):900–907
Adamo KB, Ferraro ZM, Goldfield G, Keely E, Stacey D, Hadjiyannakis S et al (2013) The maternal obesity management (MOM) trial protocol: a lifestyle intervention during pregnancy to minimize downstream obesity. Contemp Clin Trials 35(1):87–96
Kahn CR (2003) Knockout mice challenge our concepts of glucose homeostasis and the pathogenesis of diabetes. Exp Diabesity Res 4(3):169–182
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: Insights into insulin action. Nat Rev Mol Cell Biol 7(2):85–96
Deshmukh A, Salehzadeh F, Metayer-Coustard S, Fahlman R, Nair KS, Al-Khalili L (2009) Post-transcriptional gene silencing of ribosomal protein S6 kinase 1 restores insulin action in leucine-treated skeletal muscle. Cell Mol Life Sci 66(8):1457–1466
Jordan SD, Krüger M, Willmes DM, Redemann N, Wunderlich FT, Brönneke HS et al (2011) Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 13(4):434–448
Nordlie RC, Foster JD, Lange AJ (1999) Regulation of glucose production by the liver. Annu Rev Nutr 19:379–406
Postic C, Dentin R, Girard J (2004) Role of the liver in the control of carbohydrate and lipid homeostasis. Diabetes Metab 30(5):398–408
Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J et al (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413(6852):131–138
Braccini L, Ciraolo E, Morello F, Lu X, Hirsch E (2009) PI3K signaling: a crossroads of metabolic regulation. Expert Rev Endocrinol Metab 4(4):349–357
Cohen AW, Combs TP, Scherer PE, Lisanti MP (2003) Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab 285(6):E1151–E1160
Krebs M, Roden M (2005) Molecular mechanisms of lipid-induced insulin resistance in muscle, liver and vasculature. Diabetes Obes Metab 7(6):621–632
Massillon D, Barzilai N, Chen W, Hu MZ, Rossetti L (1996) Glucose regulates in vivo glucose-6-phosphatase gene expression in the liver of diabetic rats. J Biol Chem 271(17):9871–9874
Bell AW (1979) Lipid metabolism in liver and selected tissues and in the whole body of ruminant animals. Prog Lipid Res 18(3):117–164
Bergman EN, Havel RJ, Wolfe BM, Bohmer T (1971) Quantitative studies of the metabolism of chylomicron triglycerides and cholesterol by liver and extrahepatic tissues of sheep and dogs. J Clin Invest 50(9):1831–1839
Azhar S, Kelley G (2007) PPARα: its role in the human metabolic syndrome. Future Lipidol 2(1):31–53
Shankar K, Kang P, Harrell A, Zhong Y, Marecki JC, Ronis MJJ et al (2010) Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology 151(6):2577–2589
Martin-Gronert MS, Fernandez-Twinn DS, Poston L, Ozanne SE (2010) Altered hepatic insulin signalling in male offspring of obese mice. J Dev Orig Health Dis 1(3):184–191
Angulo P (2002) Medical progress: nonalcoholic fatty liver disease. N Engl J Med 346(16):1221–1231
Marchesini G, Brizi M, Blanchi G, Tomassetti S, Bugianesi E, Lenzi M et al (2001) Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 50(8):1844–1850
Targher G, Bertolini L, Padovani R, Rodella S, Tessari R, Zenari L et al (2007) Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 30(5):1212–1218
Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC et al (2009) Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 50(6):1796–1808
Borengasser SJ, Lau F, Kang P, Blackburn ML, Ronis MJJ, Badger TM et al (2011) Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS One 6(8):e24068
McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE et al (2009) Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 119(2):323–335
Klip A, Paquet MR (1990) Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care 13(3):228–243
Farese RV (2002) Function and dysfunction of aPKC isoforms for glucose transport in insulin-sensitive and insulin-resistant states. Am J Physiol Endocrinol Metab 283(1):E1–E11
Zierath JR, Krook A, Wallberg-Henriksson H (2000) Insulin action and insulin resistance in human skeletal muscle. Diabetologia 43(7):821–835
Alkhateeb H, Chabowski A, Glatz JFC, Gurd B, Luiken JJFP, Bonen A (2009) Restoring AS160 phosphorylation rescues skeletal muscle insulin resistance and fatty acid oxidation while not reducing intramuscular lipids. Am J Physiol Endocrinol Metab 297(5):E1056–E1066
Cartee GD, Wojtaszewski JFP (2007) Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab 32(3):557–566
Petersen KF, Shulman GI (2002) Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol 90(Suppl 5):11G–18G
Henriksen EJ, Dokken BB (2006) Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr Drug Targets 7(11):1435–1441
Kockeritz L, Doble B, Patel S, Woodgett JR (2006) Glycogen synthase kinase-3—An overview of an over-achieving protein kinase. Curr Drug Targets 7(11):1377–1388
Yan X, Huang Y, Zhao JX, Long NM, Uthlaut AB, Zhu MJ et al (2011) Maternal obesity-impaired insulin signaling in sheep and induced lipid accumulation and fibrosis in skeletal muscle of offspring. Biol Reprod 85(1):172–178
Lafontan M (2008) Advances in adipose tissue metabolism. Int J Obes (Lond) 32(Suppl 7):S39–S51
Gross DN, Van Den Heuvel APJ, Birnbaum MJ (2008) The role of FoxO in the regulation of metabolism. Oncogene 27(16):2320–2336
Antuna-Puente B, Feve B, Fellahi S, Bastard JP (2008) Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab 34(1):2–11
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S et al (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8(11):1288–1295
Zhou H, Song X, Briggs M, Violand B, Salsgiver W, Gulve EA et al (2005) Adiponectin represses gluconeogenesis independent of insulin in hepatocytes. Biochem Biophys Res Commun 338(2):793–799
Ahima RS, Flier JS (2000) Leptin. Annu Rev Physiol 62:413–437
Matsuda J, Yokota I, Iida M, Murakami T, Naito E, Ito M et al (1997) Serum leptin concentration in cord blood: relationship to birth weight and gender. J Clin Endocrinol Metabol 82(5):1642–1644
Shekhawat PS, Garland JS, Shivpuri C, Mick GJ, Sasidharan P, Pelz CJ et al (1998) Neonatal cord blood leptin: Its relationship to birth weight, body mass index, maternal diabetes, and steroids. Pediatr Res 43(3):338–343
Tapanainen P, Leinonen E, Ruokonen A, Knip M (2001) Leptin concentrations are elevated in newborn infants of diabetic mothers. Horm Res 55(4):185–190
Masuyama H, Hiramatsu Y (2012) Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 153(6):2823–2830
Strakovsky RS, Zhang X, Zhou D, Pan YX (2011) Gestational high fat diet programs hepatic phosphoenolpyruvate carboxykinase gene expression and histone modification in neonatal offspring rats. J Physiol 589(11):2707–2717
He A, Zhu L, Gupta N, Chang Y, Fang F (2007) Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol 21(11):2785–2794
Pandey AK, Verma G, Vig S, Srivastava S, Srivastava AK, Datta M (2011) MiR-29a levels are elevated in the db/db mice liver and its overexpression leads to attenuation of insulin action on PEPCK gene expression in HepG2 cells. Mol Cell Endocrinol 332(1–2):125–133
Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M et al (2011) MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474(7353):649–653
Nohr EA, Vaeth M, Baker JL, Sørensen TIA, Olsen J, Rasmussen KM (2008) Combined associations of prepregnancy body mass index and gestational weight gain with the outcome of pregnancy. Am J Clin Nutr 87(6):1750–1759
Smith J, Cianflone K, Biron S, Hould FS, Lebel S, Marceau S et al (2009) Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metabol 94(11):4275–4283
Van Der Steeg JW, Steures P, Eijkemans MJC, Habbema JDF, Hompes PGA, Burggraaff JM et al (2008) Obesity affects spontaneous pregnancy chances in subfertile, ovulatory women. Hum Reprod 23(2):324–328
Robker RL (2008) Evidence that obesity alters the quality of oocytes and embryos. Pathophysiology 15(2):115–121
Dokras A, Baredziak L, Blaine J, Syrop C, VanVoorhis BJ, Sparks A (2006) Obstetric outcomes after in vitro fertilization in obese and morbidly obese women. Obstet Gynecol 108(1):61–69
Wu LL, Russell DL, Wong SL, Chen M, Tsai TS, St John JC et al (2015) Mitochondrial dysfunction in oocytes of obese mothers: transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 142(4):681–691
Minge CE, Bennett BD, Norman RJ, Robker RL (2008) Peroxisome proliferator-activated receptor-α agonist rosiglitazone reverses the adverse effects of diet-induced obesity on oocyte quality. Endocrinology 149(5):2646–2656
Nicholas LM, Morrison JL, Rattanatray L, Ozanne SE, Kleemann DO, Walker SK et al (2013) Differential effects of exposure to maternal obesity or maternal weight loss during the periconceptional period in the sheep on insulin signalling molecules in skeletal muscle of the offspring at 4 months of age. PLoS One 8(12):e84594
Nicholas LM, Rattanatray L, Maclaughlin SM, Ozanne SE, Kleemann DO, Walker SK et al (2013) Differential effects of maternal obesity and weight loss in the periconceptional period on the epigenetic regulation of hepatic insulin-signaling pathways in the offspring. FASEB J 27(9):3786–3796
Nicholas LM, Rattanatray L, Morrison JL, Kleemann DO, Walker SK, Zhang S et al (2014) Maternal obesity or weight loss around conception impacts hepatic fatty acid metabolism in the offspring. Obesity 22(7):1685–1693
Rattanatray L, MacLaughlin SM, Kleemann DO, Walker SK, Muhlhausler BS, McMillen IC (2010) Impact of maternal periconceptional overnutrition on fat mass and expression of adipogenic and lipogenic genes in visceral and subcutaneous fat depots in the postnatal lamb. Endocrinology 151(11):5195–5205
Zhang S, Rattanatray L, MacLaughlin SM, Cropley JE, Suter CM, Molloy L et al (2010) Periconceptional undernutrition in normal and overweight ewes leads to increased adrenal growth and epigenetic changes in adrenal IGF2/H19 gene in offspring. FASEB J 24(8):2772–2782
Russel AJF, Doney JM, Gunn RG (1969) Subjective assessment of body fat in live sheep. J Agric Sci 97:723–729
Catalano PM, Nizielski SE, Shao J, Preston L, Qiao L, Friedman JE (2002) Downregulated IRS-1 and PPARγ in obese women with gestational diabetes: relationship to FFA during pregnancy. Am J Physiol Endocrinol Metab 282(3):E522–E533
Greenfield JR, Campbell LV (2004) Insulin resistance and obesity. Clin Dermatol 22(Spec iss 4):289–295
Savage DB, Petersen KF, Shulman GI (2007) Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev 87(2):507–520
Li X, Monks B, Ge Q, Birnbaum MJ (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature 447(7147):1012–1016
Ozanne SE, Jensen CB, Tingey KJ, Storgaard H, Madsbad S, Vaag AA (2005) Low birthweight is associated with specific changes in muscle insulin-signalling protein expression. Diabetologia 48(3):547–552
Ozanne SE, Olsen GS, Hansen LL, Tingey KJ, Nave BT, Wang CL et al (2003) Early growth restriction leads to down regulation of protein kinase C zeta and insulin resistance in skeletal muscle. J Endocrinol 177(2):235–241
Isagawa T, Nagae G, Shiraki N, Fujita T, Sato N, Ishikawa S et al (2011) DNA methylation profiling of embryonic stem cell differentiation into the three germ layers. PLoS One 6(10):e26052
Duque-Guimarães DE, Ozanne SE (2013) Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol Metab 24(10):525–535
Morgan HD, Santos F, Green K, Dean W, Reik W (2005) Epigenetic reprogramming in mammals. Hum Mol Genet 14(Spec. iss. 1):R47–R58
Jiménez-Chillarón JC, Díaz R, Martínez D, Pentinat T, Ramón-Krauel M, Ribó S et al (2012) The role of nutrition on epigenetic modifications and their implications on health. Biochimie 94(11):2242–2263
Margueron R, Reinberg D (2010) Chromatin structure and the inheritance of epigenetic information. Nat Rev Genet 11(4):285–296
Semple RK, Chatterjee VK, O’Rahilly S (2006) PPAR gamma and human metabolic disease. J Clin Invest 116(3):581–589
Waterland RA, Travisano M, Tahiliani KG, Rached MT, Mirza S (2008) Methyl donor supplementation prevents transgenerational amplification of obesity. Int J Obes (Lond) 32(9):1373–1379
Kral JG (2004) Preventing and treating obesity in girls and young women to curb the epidemic. Obes Res 12(10):1539–1546
Birdsall KM, Vyas S, Khazaezadeh N, Oteng-Ntim E (2009) Maternal obesity: a review of interventions. Int J Clin Pract 63(3):494–507
Dodd JM, Crowther CA, Robinson JS (2008) Dietary and lifestyle interventions to limit weight gain during pregnancy for obese or overweight women: a systematic review. Acta Obstet Gynecol Scand 87(7):702–706
Ronnberg AK, Nilsson K (2010) Interventions during pregnancy to reduce excessive gestational weight gain: a systematic review assessing current clinical evidence using the grading of recommendations, assessment, development and evaluation (GRADE) system. BJOG 117(11):1327–1334
Adamo KB, Ferraro ZM, Brett KE (2012) Can we modify the intrauterine environment to halt the intergenerational cycle of obesity? Int J Environ Res Public Health 9(4):1263–1307
Patti ME (2013) Reducing maternal weight improves offspring metabolism and alters (or modulates) methylation. Proc Natl Acad Sci USA 110(32):12859–12860
Dalfrà MG, Busetto L, Chilelli NC, Lapolla A (2012) Pregnancy and foetal outcome after bariatric surgery: a review of recent studies. J Matern Fetal Neonatal Med 25(9):1537–1543
Barisione M, Carlini F, Gradaschi R, Camerini G, Adami GF (2012) Body weight at developmental age in siblings born to mothers before and after surgically induced weight loss. Surg Obes Relat Dis 8(4):387–391
Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S et al (2006) Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years. Pediatrics 118(6):e1644–e1649
Guénard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl MC (2013) Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci USA 110(28):11439–11444
Zambrano E, Martínez-Samayoa PM, Rodríguez-González GL, Nathanielsz PW (2010) Dietary intervention prior to pregnancy reverses metabolic programming in male offspring of obese rats. J Physiol 588(10):1791–1799
Vega CC, Reyes-Castro LA, Bautista CJ, Larrea F, Nathanielsz PW, Zambrano E (2013) Exercise in obese female rats has beneficial effects on maternal and male and female offspring metabolism. Int J Obes 39(4):712–719
Raipuria M, Bahari H, Morris MJ (2015) Effects of maternal diet and exercise during pregnancy on glucose metabolism in skeletal muscle and fat of weanling rats. PLoS One 10(4):e0120980
De Rooij SR, Painter RC, Phillips DIW, Osmond C, Michels RPJ, Godsland IF et al (2006) Impaired insulin secretion after prenatal exposure to the Dutch famine. Diabetes Care 29(8):1897–1901
Smith NA, McAuliffe FM, Quinn K, Lonergan P, Evans ACO (2010) The negative effects of a short period of maternal undernutrition at conception on the glucose-insulin system of offspring in sheep. Anim Reprod Sci 121(1–2):94–100
Todd SE, Oliver MH, Jaquiery AL, Bloomfield FH, Harding JE (2009) Periconceptional undernutrition of ewes impairs glucose tolerance in their adult offspring. Pediatr Res 65(4):409–413
Lee RSF, Depree KM, Davey HW (2002) The sheep (Ovis aries) H19 gene: genomic structure and expression patterns, from the preimplantation embryo to adulthood. Gene 301(1–2):67–77
Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE et al (2014) Impact of embryo number and maternal undernutrition around the time of conception on insulin signaling and gluconeogenic factors and microRNAs in the liver of fetal sheep. Am J Physiol Endocrinol Metab 306(9):E1013–E1024
Lie S, Morrison JL, Williams-Wyss O, Suter CM, Humphreys DT, Ozanne SE et al (2014) Periconceptional undernutrition programs changes in insulin-signaling molecules and microRNAs in skeletal muscle in singleton and twin fetal sheep. Biol Reprod 90(1):5
Edwards LJ, McMillen IC (2002) Periconceptional nutrition programs development of the cardiovascular system in the fetal sheep. Am J Physiol Regul Integr Comp Physiol 283(3):R669–R679
Edwards LJ, McMillen IC (2002) Impact of maternal undernutrition during the periconceptional period, fetal number, and fetal sex on the development of the hypothalamo-pituitary adrenal axis in sheep during late gestation. Biol Reprod 66(5):1562–1569
Zhang S, Morrison JL, Gill A, Rattanatray L, MacLaughlin SM, Kleemann D et al (2013) Dietary restriction in the periconceptional period in normal-weight or obese ewes results in increased abundance of angiotensin-converting enzyme (ACE) and angiotensin type 1 receptor (AT1R) in the absence of changes in ACE or AT1R methylation in the adrenal of the offspring. Reproduction 146(5):443–454
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES et al (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA 105(44):17046–17049
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 The American Physiological Society
About this chapter
Cite this chapter
Nicholas, L.M., McMillen, I.C. (2016). The Impact of Maternal Obesity and Weight Loss During the Periconceptional Period on Offspring Metabolism. In: Green, L., Hester, R. (eds) Parental Obesity: Intergenerational Programming and Consequences. Physiology in Health and Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-6386-7_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6386-7_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-6384-3
Online ISBN: 978-1-4939-6386-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)