
Abstract
It is well accepted that stress, measured by increased glucocorticoid secretion, leads to profound reproductive dysfunction. In times of stress, glucocorticoids activate many parts of the fight or flight response, mobilizing energy and enhancing survival, while inhibiting metabolic processes that are not necessary for survival in the moment. This includes reproduction, an energetically costly procedure that is very finely regulated. In the short term, this is meant to be beneficial, so that the organism does not waste precious energy needed for survival. However, long-term inhibition can lead to persistent reproductive dysfunction, even if no longer stressed. This response is mediated by the increased levels of circulating glucocorticoids, which orchestrate complex inhibition of the entire reproductive axis. Stress and glucocorticoids exhibits both central and peripheral inhibition of the reproductive hormonal axis. While this has long been recognized as an issue, understanding the complex signaling mechanism behind this inhibition remains somewhat of a mystery. What makes this especially difficult is attempting to differentiate the many parts of both of these hormonal axes, and new neuropeptide discoveries in the last decade in the reproductive field have added even more complexity to an already complicated system. Glucocorticoids (GCs) and other hormones within the hypothalamic-pituitary-adrenal (HPA) axis (as well as contributors in the sympathetic system) can modulate the hypothalamic-pituitary-gonadal (HPG) axis at all levels—GCs can inhibit release of GnRH from the hypothalamus, inhibit gonadotropin synthesis and release in the pituitary, and inhibit testosterone synthesis and release from the gonads, while also influencing gametogenesis and sexual behavior. This chapter is not an exhaustive review of all the known literature, however is aimed at giving a brief look at both the central and peripheral effects of glucocorticoids on the reproductive function.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
It is well accepted that stress, measured by increased glucocorticoid secretion, leads to profound reproductive dysfunction. In times of stress, glucocorticoids activate many parts of the fight or flight response, mobilizing energy and enhancing survival, while inhibiting metabolic processes that are not necessary for survival in the moment. This includes reproduction, an energetically costly procedure that is very finely regulated. In the short term, this is meant to be beneficial, so that the organism does not waste precious energy needed for survival. However, long-term inhibition can lead to persistent reproductive dysfunction, even after the stressor has been removed. This response is mediated by the increased levels of circulating glucocorticoids, which orchestrate complex inhibition of the entire reproductive axis. The reproductive and stress systems share a common architecture with central (hypothalamic stimulation of pituitary in both) and peripheral (gonads and adrenal respectively) components. Stress and glucocorticoids exhibits both central and peripheral inhibition of the reproductive hormonal axis. While this has long been recognized as an issue, understanding the complex signaling mechanism behind this inhibition remains somewhat of a mystery. In fact, the presenting complaint of Harvey Cushing’s first patient with the syndrome that bears his name was secondary amenorrhea. She had normal menarche at age 14, but menses ceased two years later. What makes this especially difficult is attempting to differentiate the many parts of both of these hormonal axes, and new neuropeptide discoveries in the last decade in the reproductive field have added even more complexity to an already complicated system. Glucocorticoids (GCs) and other hormones within the hypothalamic-pituitary-adrenal (HPA) axis (as well as contributors in the sympathetic system) can modulate the hypothalamic-pituitary-gonadal (HPG) axis at all levels—GCs can inhibit release of GnRH from the hypothalamus, inhibit gonadotropin synthesis and release in the pituitary, and inhibit testosterone synthesis and release from the gonads, while also influencing gametogenesis and sexual behavior. This chapter is not an exhaustive review of all the known literature however is aimed at giving a brief look at both the central and peripheral effects of glucocorticoids on the reproductive function.
Reproductive Physiology: A Primer
Reproduction in mammals is a complex and precisely regulated hormonal process that requires the coordination of both the central nervous system and the peripheral reproductive organs for successful procreation. Negative and positive feedback signals tightly regulate the reproductive hormonal axis, also known as the hypothalamo-pituitary-gonadal (HPG) axis to maintain homeostasis. Perturbations in the axis, such as those caused by stress, can therefore have profound effects on reproductive ability, as even small changes can have large effects downstream, or even stop the axis in its tracks. Much of this chapter is focused on research conducted in rodents, however there are many species and sex differences in reproductive research indicating that stress can exert a myriad of effects to inhibit reproduction.
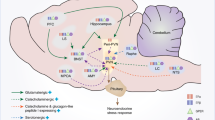
Studying the reproductive system of males offers a simpler “model” of HPG axis to begin examining reproductive physiology, due to its relative consistency over the lifetime post-puberty, as opposed to the cycling of female reproductive hormones during the post-pubertal and pre-menopausal years. Gonadotropin-releasing hormone (GnRH) is secreted from the hypothalamus in a pulsatile manner, crossing the hypophyseal portal system into the anterior pituitary. In the pituitary, GnRH stimulates the synthesis and release of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). LH and FSH circulate systemically to trigger testosterone release from the testes and gametogenesis. In turn, testosterone (T), as well and LH and FSH, negatively feedback on the axis to maintain homeostasis [1–6] (Fig. 11.1).

General schematic of hypothalamic-pituitary-gonadal axis in the rodent model
Females, however, present a much more complicated picture. Unlike males, females experience hormonal surges to trigger ovulation, a phenomenon where estrogen switches from exerting negative feedback on the axis to positive feedback [7–12]. In rats, ovulation occurs once every 4 days. Similarly to males, GnRH is released from the hypothalamus, and LH and FSH circulate to the ovaries to trigger the release of estradiol (E2) and progesterone (P) as well as development of the ovum [6, 12–14]. Estradiol from the ovaries tightly regulates the HPG axis, exerting negative feedback onto the hypothalamus to inhibit GnRH release. However, when the developing ovum is near completion, and ovulation is due to occur, there is a switch in the hormonal system. Increasing estradiol secretion from the developing follicle triggers a change from negative to positive feedback of estradiol on the hypothalamus, resulting in an increase of GnRH secretion and leading to a surge of LH secretion from the pituitary to trigger ovulation [15, 16]. However, until recently it was unknown what triggered the switch between negative and positive feedback.
Research in the last decade has shed more light on that switch during ovulation while also revealing that the axis is not nearly as simple as it appeared. There are currently two different hypotheses on what triggers the switch between negative and positive feedback in the HPG axis: The first one focuses on progesterone as the culprit, whereas the second one points to allopregnanolone. Supporting the first hypothesis, earlier research found that the estrogen surge initiates synthesis of progesterone receptors (PRs) in the hypothalamus, indicating progesterone is just as critical for ovulation as the estrogen surge [17–19]. However, studies on ovariectomized (OVX) and andrenalectomized (ADX) rats indicate that this progesterone does not come from the ovaries or adrenals, areas typical for steroid synthesis and systemic release. OVX rats given a normal dose of E2 can still exhibit LH surges [20]. Additionally, this surge can be blocked by administering trilostane, which inhibits the enzyme 3β-hydroxysteroid dehydrogenase (3β-HSD), an enzyme critical for synthesis of progesterone [20]. This data suggests that not only is pre-LH surge progesterone necessary for a successful LH surge, it is progesterone synthesized outside of the ovary and adrenals that regulates the surge. Hypothalamic progesterone synthesis, also known as neuroprogesterone, is a likely location, particularly hypothalamic astrocytes, which have been found to be the main source of neuroprogesterone in the hypothalamus, as they have all the steroidogenic enzyme machinery necessary for synthesis, regulate releasing factors and possess both types of estradiol receptors—Erα and ERβ receptors, which enable them to respond to the estradiol peak that precedes the LH surge [20–25].
While GnRH is the main coordinator of the HPG axis, secreted from the hypothalamus through the hypophyseal portal system to the anterior pituitary to stimulate release of LH, there are many factors upstream of GnRH that can impact its release, preventing successful ovulation. Two in particular, directly in the HPG axis, have been recently discovered: kisspeptin (KISS1) and gonadotropin-inhibitory hormone (GnIH) [26–34]. These two neuropeptides have opposing effects—KISS1 stimulates GnRH release from the hypothalamus [35–40], while GnIH inhibits it [30, 41–45]. A proposed mechanism of KISS1 action in the neuroprogesterone model of the LH surge states that the peak of estradiol also triggers an increase of progesterone receptors (PRs) on KISS1 neurons in the hypothalamus, which respond to the neuroprogesterone secreted from the astrocytes (activated also by estradiol to trigger the neuroprogesterone synthesis, likely through membrane estrogen receptors, specifically mERα). Neuroprogesterone, binding to the PRs on the KISS neurons, trigger the release of KISS, which then activates the GnRH neurons, leading to increased secretion of GnRH to trigger the LH surge from the pituitary [24, 46–48].
An alternative hypothesis for the switch between negative and positive feedback of E2 in the hypothalamus argues that progesterone is not the driving factor of ovulation, but it is a derivative of progesterone, allopregnanolone, that modulates GnRH release from the hypothalamus via increasing glutamate release. This in turn could act on NMDA receptors on the GnRH neurons, stimulating GnRH release [49–51]. While it is still not clear exactly what mechanism is truly responsible for positive feedback of E2 during ovulation, the many regulators of GnRH and GnRH itself are all impacted by glucocorticoids, and stress can lead to a disruption of homeostasis that results in both short-term reproductive dysfunction and long-term infertility.
Stress, whether psychological or physical, via the activation of the HPA axis, and subsequent increase in serum concentration of glucocorticoids, can inhibit the reproductive axis at every level, from the hypothalamus down to the ovaries or testes. Due to how closely regulated the HPG axis is, even small interferences in the hormonal milieu responsible for successful breeding can cause major dysfunction in the system (Fig. 11.2). The female ovulatory system discussed above, as well as GnRH afferents such as GnIH and KISS, exhibit many points at which stress can disrupt the axis and cause fertility issues. While acute stress inhibiting the reproductive axis is found to be adaptive, preventing animals from breeding when times are not optimal for raising young, chronic stress and long-term shutdown of the reproductive axis can lead to prolonged dysfunction and infertility. In addition to causing infertility, this hypogonadism can contribute to other medical conditions such as osteoporosis. We will examine how stress impacts each part of the reproductive axis individually, and how this can add up to detrimental fertility issues.

Summary schematic of glucocorticoid and HPA axis interaction with the HPG axis and reproductive function
Glucocorticoid Effects on the Hypothalamus
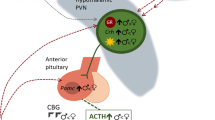
Stress causes the activation of the hypothalamic-pituitary-adrenal axis by a cascade of hormonal release: neural signals onto the hypothalamus cause the release of corticotropin-releasing hormone (CRH, previously known as CRF) to the hypophyseal portal system, via the median eminence. CRH in turn, activates cells in the pituitary to release adrenocorticotrophic hormone (ACTH) to the general circulation. ACTH stimulates cells in the adrenal cortex to synthesize and release the third hormone in this cascade of the glucocorticoid family, which in rodents is corticosterone (CORT). Stress and high glucocorticoids have a profound negative impact on the reproductive system, and within the hypothalamus both CRH and CORT affect GnRH and its afferents to inhibit reproductive success, as detailed in the next two sections. As discussed above, the timing of ovulation in female rodents is highly regulated by multiple factors controlling GnRH levels. Stress has been shown to impact each one of these factors, combining to effectively shut down the reproductive axis from the top. A difficulty in studying the effects of stress on reproduction is to accurately differentiate between the effects of CRH and the effects of CORT, both elevated during stress exposure, as well as determining specifically which level of the HPG axis is being directly affected. While it is well accepted that there are central effects of CRH directly on GnRH, completely separating those effects from downstream peripheral effects of CORT on GnRH is complex. Many manipulations of either CORT or CRH influence the other by feedback regulation, making the dissection of mechanisms even more difficult. In this section, we will try to differentiate the two to better understand how stress influences reproductive success.
Direct Regulation of GnRH by Glucocorticoids
All central influences on reproduction driven by stress converge on GnRH, and an inhibition of the GnRH signal from the hypothalamus can occur both directly on the GnRH neuron itself and indirectly via influence on GnRH afferents. While many peptides, steroids and neurotransmitters regulate the GnRH surge, stress and glucocorticoids can directly inhibit GnRH release from the hypothalamus. To regulate LH secretion from the pituitary, GnRH is released from the hypothalamus in pulses, and modulation of the pulsatile release of GnRH affects downstream gonadotropin release from the pituitary. Many types of stress have been found to affect the pulse generator of GnRH, and both CRH and CORT have been implicated in this mechanism of action [52–54]. GnRH crosses the hypophyseal portal system, a small circulatory system between the hypothalamus and the pituitary, to stimulate release of LH and FSH from the pituitary. Inhibiting the pulse generator of GnRH leads to a decrease in GnRH secretion from the hypothalamus. However, measuring GnRH release from the rodent hypothalamus is incredibly difficult, as the hypophyseal portal system is small and hard to sample from. To determine whether GC’s are inhibiting GnRH release from the hypothalamus, researchers use downstream release of LH from the pituitary as a proxy for GnRH. If stress leads to a decrease in LH release, it is assumed that this is due to a decrease in GnRH release from the hypothalamus. Intracerebroventricular (ICV) administration of CRH has been found to inhibit LH pulses in rats [55] and this response can be blocked or reversed by administration of CRH antagonists [55, 56]. It appears that this suppression is mediated in part by both CRH receptor subtypes, CRH-R1 and CRH-R2, however which receptor is predominant depends on the type of stressor utilized [57–59]. CRH axon terminals directly interact with GnRH dendrites, and in vitro studies have shown that CRH can inhibit GnRH release. Other studies have also shown that infusing CRH into the medial preoptic area of females rats lead to a 60 % decrease in GnRH release from the hypothalamus [60, 61]. Though many studies have used in vitro systems to determine how GnRH can be inhibited by high CRH and CORT, recent studies using sheep, however, which have a much larger hypophyseal portal system than rodents, have allowed researchers to show in vivo that glucocorticoids inhibit GnRH release in the hypothalamus. Prolonged corticosterone administration IV caused a drop in GnRH release in the portal system of ewes [62] supporting the long-standing theory that glucocorticoids’ central actions function to inhibit GnRH release from the hypothalamus. Release has also been inhibited in hypothalamic explants incubated with CORT and the glucocorticoid receptor agonist dexamethasone (DEX) in a dose-dependent manner [63].
Populations of GnRH neurons have been found to express both CRH receptors (CRH-R1, R2) [64] and glucocorticoid receptors (GRs) directly in both mice and rats [65, 66], indicating a method for which glucocorticoids (GCs) can act to directly inhibit GnRH synthesis and release. Glucocorticoids have also been found to inhibit GnRH transcription [67–69] and identified glucocorticoid response elements near the GnRH gene that regulate GnRH transcription. The GT1 cell line derived from GnRH-neurons, which synthesize and release GnRH [70, 71] express glucocorticoid receptors [72]. Further studies show that GCs can represses GnRH gene expression and release from these cells [73]. Studies in vivo have shown that chronic CORT treatment can inhibit GnRH expression, leading to a decrease in serum LH levels [74]. This however, had no effect on FSH levels, nor did it affect gonadotropin mRNA.
The effect of peripheral circulating glucocorticoids on GnRH and consequently the LH surge is highly variable and likely dependent on the severity and length of the stressor. In different acute stress studies glucocorticoids are shown to exert a full spectrum of effects on the LH surge that range from complete inhibition, to little or no effect to a positive activator of LH release. It emphasizes the importance of comparing stress paradigms and understanding how different stressors trigger the HPA axis. Acute stress tends to provide variable results on many different outcomes—life history, age and gender can influence this response greatly. In contrast, chronic stress has been consistently shown to inhibit the LH surge and ovulation in the literature, indicating that while short-term stressors may exert variable effects on reproduction, likely due to the type of stressor, long-term stress reliably causes reproductive dysfunction.
Indirect Regulation of GnRH by Glucocorticoids
Glucocorticoids may also exert an inhibitory influence on reproduction and GnRH output by influencing hormones upstream of GnRH, adding another level of control over reproduction. Two hormones in particular have been recently identified upstream of GnRH that respond directly to stress, affecting GnRH synthesis and secretion from the hypothalamus. Kisspeptin (KISS1) and gonadotropin-inhibitory hormone (GnIH), mentioned earlier, have opposing effects on GnRH. KISS1 is an activator of GnRH and plays a critical role in the maintenance of the GnRH pulse generator in the hypothalamus as well as the estrogen surge responsible for ovulation. Many different types of stressors, including exposure to the endotoxin lipopolysaccharide, acute hypoglycemia, immobilization (restraint) stress and social isolation, lead to downregulation of KISS1 and it’s receptor KISS1R in the population of kisspeptin neurons in the median pre-optic area (mPOA) of the hypothalamus, as well as decreases in KISS1 expression in the arcuate nucleus, leading to downstream decreases in LH secretion [75–77]. Kisspeptin neurons have been shown to express both CRH-R and GR, implying that they could potentially respond directly to increase in either hormones following a stress stimulus [78]. These changes can transmit downstream into inhibition of the GnRH pulse generator, which has been shown to rely on kisspeptin input, and ultimately translate to downstream inhibition of LH secretion from the pituitary.
A subset of neurons in the hypothalamus expressing kisspeptin project directly to GnRH neurons [79–82], and has been implicated in modulation of the GnRH pulse generator. These neurons, known as the KNDy neurons, express two other neuropeptides, neurokinin B and dynorphin, and strongly respond to CORT. This subgroup offers yet another pathway in which stress via glucocorticoids can indirectly inhibit the reproductive axis. Dynorphin, an endogenous opioid in the brain signals through the kappa-subtype of the opioid receptor (KOR) in the hypothalamus, and administering dynorphin to female rats has been shown to inhibit LH pulses from the pituitary [83]. Stress has been shown to increase dynorphin release [84] which could lead to downstream inhibition of LH [85].
Gonadotropin-inhibitory hormone (GnIH) is another hormone upstream of GnRH that is regulated by stress to inhibit reproduction. GnIH was originally discovered in birds [30] and a mammalian orthologue, Rfamide-related peptide-3 (RFRP3) has since been identified in many species including rats, mice, hamsters and humans [26, 27, 86]. RFRP3 inhibits the GnRH pulse generator, decreasing GnRH release from the hypothalamus and leading to decreased gonadotropin secretion from the pituitary [41, 45, 87]. Both acute and chronic immobilization stress in male rats were found to increase RFRP3 mRNA and peptide levels in the hypothalamus and adrenalectomy blocked this effect, revealing that this is due to circulating glucocorticoid levels [88]. This increase in RFRP3 by glucocorticoids led to downstream inhibition of LH release from the pituitary. In vitro studies utilizing a cell line derived from RFRP3-ergic neurons has shown that RFRP3 neurons express the glucocorticoid receptor (GR) and also possess two glucocorticoid response elements (GREs) at the RFRP promoter region, further evidence pointing to a direct regulation of RFRP3 neurons by glucocorticoids [89, 90]. RFRP3 neurons in mammals can directly inhibit GnRH pulses from the hypothalamus, a response enhanced by high glucocorticoids after stress. In female rats, research has shown that high glucocorticoids after chronic stress can lead to an increase in RFRP3 levels in all stages of the estrous cycle and that this increase is sustained for at least one more estrous cycle after the stress has ceased. This increase leads to downstream reproductive dysfunction with fewer copulatory events in females exposed to chronic stress, lower pregnancy rates and decreased litter sizes [91]. This new research into the effect of glucocorticoids on RFRP3 levels and long-term reproductive dysfunction in female rodents could shed light on a more detailed mechanism of chronic inhibition of the HPG axis and reproductive success by glucocorticoids.
These are only a small sample of the possible indirect mechanisms of glucocorticoids on GnRH secretion. Much is still not well understood about the mechanism of action of glucocorticoids on GnRH levels, especially since there have been a slew of novel discoveries of peptides upstream of GnRH that increases the complexity of the regulation of reproductive neural systems by glucocorticoids. The difficulty in measuring GnRH levels from rodents has also made it difficult to come to generate conclusions about the mechanism of action of GCs on GnRH.
Glucocorticoid Effects in the Pituitary
While many of the stress effects on GnRH can lead to downstream pituitary dysfunction, glucocorticoids also directly influence reproduction at the level of the pituitary. This inhibition can happen via many different mechanisms, including modulating the sensitivity of the pituitary to changes in GnRH secretion, a decrease in synthesis of the gonadotropes LH and FSH, as well as decreasing the secretion of LH and FSH from the pituitary. However, the effects of glucocorticoids directly on the pituitary in secretion and synthesis are highly variable, indicating that the type of stressor and duration is very important when discussing this. This variability may stem from only looking at a part of the inhibition, such as focusing on just LH secretion as the output, rather than examining the mechanisms of synthesis and responsiveness of the pituitary itself.
Synthesis of LH and FSH is a highly regulated process involving not just GnRH levels, but steroid gonadal hormones as well [92, 93]. LH and FSH are glycoprotein heterodimers that consist of a common glycoprotein alpha-subunit (aGSU) and a specific β-subunit for either FSH or LH (LHβ and FSHβ) [94]. As rat and mice gonadotrope cells express GRs [95], it is likely that GR transcriptional regulation influences the synthesis of these subunits, both the specific β-subunits as well as the common alpha subunit to also regulate expression levels. Breen et.al. found that both daily immobilization stress and CORT administration led to reduced LH-β mRNA levels, as well as decreased LH release from the pituitary in vivo. They also showed in vitro, using a gonadotope cell line, Lβ T2 that CORT decreases GnRH-induced increase in LHβ mRNA levels as well as identified the LHβ promoter regions that are CORT and DEX responsive [96].
Similar to the effects seen in the hypothalamus, acute stress leads to variable results. Several studies utilizing multiple types of acute stressors on gonadotropin synthesis and release found that acute stress stimulated the HPG axis, leading to increased levels of LH, prolactin (PRL) and FSH in the plasma [97, 98]. Others have found though that acute administration of glucocorticoids can reduce the LH peak in females [99, 100] and injection of CRH peripherally has been shown to inhibit ovulation and block the LH surge completely [54]. Baldwin and Sawyer found that an acute injection of DEX early in the estrous cycle of rats can delay the onset of ovulation. Administration of LH though, in addition to DEX, can recover the ovulatory event, indicating that this delay of the estrous cycle is due to DEX inhibiting the LH surge necessary for ovulation, rather than a problem within the ovary [101]. There is some debate over timing of sample collection—some studies see an initial stimulation of LH but an inhibitory response later [102]. Collu et.al. found that after the first 15 min of an acute stressor, female rats exhibited an increase in LH plasma levels, however after 6 h of immobilization stress LH plasma levels in the females had dropped below control levels, supporting the theory that the acute stimulation of the HPG axis is only transitory [102]. Differences in the stressors, duration, sample collection and timing of the experiment may explain the differences found in LH release from the pituitary after acute stress and so it is hard to make concrete conclusions about acute stress and pituitary function in reproduction.
Chronic immobilization stress, on the other hand, has been shown to reliably reduce LH levels in both males and females [102–110]. One such study found that chronic immobilization stress reduced plasma levels of LH, and prolactin (PRL) with no change in FSH. However, pituitary levels of LH and FSH protein increased, showing that synthesis and secretion are not always matched up-it appears in this study, chronic stress did not influence synthesis, but inhibited release in some way [109]. This difference is critical when comparing studies—most studies do not look at both synthesis and secretion simultaneously, which may explain differences in findings.
Glucocorticoids can also change the responsiveness of the pituitary to GnRH secretion. This may occur via regulation of the GnRH receptor in the pituitary, however other studies have shown that this can also occur independently of changes in the GnRH receptor [111]. Baldwin (1979) found that stress affects estrogen feedback onto the pituitary, making it less responsive to GnRH secretion [112]. GnRH binding to its receptor on the gonadotropes is necessary to induce synthesis of aGSU, LHβ and FSHβ subunits, as well as stimulate the dimerization of the subunits for successful synthesis and release from the gonadotrope cells. GnRH receptors appear to be transcriptionally regulated by GR [113, 114].
This section focused predominately on the influence CORT and GR has on LH synthesis and secretion. CORT has been shown to also decrease FSH mRNA levels as well in some studies [110, 115], however, there is extreme variability in the FSH response in regards to stress. FSH secretion levels are rarely affected by stress, especially acute stress, showing little to no change in most studies looking at it [110, 116]. This likely is due to other regulatory signals on FSH beyond corticosterone. Many studies, in fact, show increases in FSHβ (beta) mRNA post-stress, both after immobilization stress and corticosterone or cortisol administration in many species in both acute and chronic studies, sometimes accompanied by decreases in LHβ or with no change in LH at all [96, 111, 117–119]. This also happens with little to no change in actual FSH secretion, so it is hard to draw conclusions on how stress regulates FSH levels.
In summary, based on current literature it seems impossible to draw clear answers about glucocorticoid inhibition of reproduction at the level of the pituitary. The range of studies utilizing different species, different sexes, a variety of stressors as well as different lengths of time of the stressors themselves is likely part of the cause of the confusion. Some studies show that acute stress increases the pituitary gonadotropins while some find decreases in gonadotropins. Some studies show changes in the synthesis of the gonadotropins, but no change in release and others find that release is altered with no change in synthesis. Much research is still to be done in this area to fully elucidate how glucocorticoids affect the pituitary directly in terms of reproduction.
Glucocorticoids and the Gonads
The final component of the HPG axis, the gonads, is yet another level in which GCs regulate the HPG axis. In the gonads, GCs can act to inhibit many critical steps to complete the reproductive process. Corticosterone can inhibit steroidogenesis, inhibiting the synthesis of testosterone (T), estrogen (E2) and progesterone (P), as well as directly inhibiting the release of these steroids from the gonads. GCs modulate the expression of the LH-receptor (LHR) on the gonads, changing how the gonads respond to LH and leading to downstream effects on steroids. GCs can also regulate gametogenesis, the development of mature sperm and ovum, to inhibit reproduction at the levels of the gamete. These effects can all be completed in the absence of influences from the hypothalamus and pituitary, emphasizing how profoundly stress can influence reproduction.
Glucocorticoid Effects in the Testes
Research has shown that GR is localized in several different cell populations within the testes, including importantly the Leydig cells, which is where steroidogenesis occurs within the testes, as well as in the Sertoli cells, primary spermatocytes and the epididymis [121–123]. This indicates that GRs can regulate not only steroidogenesis and the release of T, but spermatogenesis as well, either through affecting the primary population of cells or affecting the last steps of maturation in the epididymis. Both acute and chronic stress experiments have shown that high GCs inhibits testosterone secretion, spermatogenesis and libido [124–128] as expected. This effect is due specifically to circulating GC levels in the blood and action via GR because ACTH treatment in adrenalectomized animals fails to replicate these findings [129]. Some studies show that this decreased testosterone release can occur either via downregulation of the LH receptor in Leydig cells [130] or through inhibition of the enzymes necessary for testosterone biosynthesis [125, 126, 128, 129, 131–133]. Overall, these changes result in decreased testosterone synthesis and release from the gonads.
Glucocorticoids may also impact spermatogenesis, as GRs are present on the primary spermatocytes as well as within the epididymis. High glucocorticoids have been found to induce testicular germ cell apoptosis [134, 135] as well as Leydig cell apoptosis [136], which has a profound inhibitory influence on male reproductive abilities. Chronic stress has been shown to also decrease the number of spermatids within the testis [137], and in humans it has been shown that chronic stress leads to decreased sperm numbers, likely through a combination of the above responses [138]. Expression of GR in all these spermatogenic area indicates that glucocorticoids can act directly on the testes to regulate sperm production. Stress and high levels of GCs likely inhibit reproduction both indirectly and directly at the level of the gonads, with decreased secretion of LH from the pituitary decreasing testosterone release, and direct inhibition of testosterone synthesis and sperm production by GCs.
Glucocorticoid Effects in the Ovaries
The role of glucocorticoids within the ovary is somewhat more complicated than it is within the testes. Rather than a straight inhibitory role of GCs on ovarian function, some GC effects are actually beneficial to the ovaries and are necessary for maintenance of the follicular development pathway. During each cycle, many follicles are activated for development within the ovaries, however not all fully develop to maturity and it appears GCs are an active part in that selection process. This is necessary for normal ovarian function, but likely is very finely controlled, and high stress may tip the balance between a “good” level of GCs and a “maladaptive” level that leads to ovarian dysfunction.
A way in which the ovaries control levels of GCs through follicular development during the female estrous or menstrual cycle is via expression of the enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD). 11β-HSD is a family of enzymes responsible for catalyzing the conversion of inactive cortisone to cortisol or vice versa to regulate glucocorticoid exposure. 11β-HSD1 activates cortisol predominately, however the reaction is bidirectional, and 11β-HSD1 can also inactivate cortisol. 11β-HSD2 on the other hand unidirectionally inactivates cortisol, converting it back to cortisone [139, 140]. Researchers have identified that many of the cells within the ovaries, including the follicles and corpus luteum, express 11β-HSD1, 11β-HSD2 and GR [141–145], indicating that there are possibly many regulatory effects of glucocorticoids on follicular development and ovarian function. Interestingly, the ovaries differentially regulate 11β-HSD1 and HSD2 throughout the cycle. 11β-HSD2, which inactivates GCs, is highly expressed in developing follicles in the ovary, while 11β-HDS1, which activates GCs, is highly expressed in follicles that have been luteinized, meaning they have been activated by an LH surge and ready for an ovulatory event [141, 146, 147]. This indicates that the ovaries upregulate 11β-HSD2 while developing in order to inactivate GCs present in the ovary while the follicles are maturing in order to enhance development, but choose to activate circulating GCs once the follicle is released for ovulation. These activated and functional GCs may act as an anti-inflammatory response triggered by the rupturing of the ovarian surface epithelium during ovulation [148, 149]. These two examples show how GCs are likely necessary for normal function of the ovaries, however their levels are tightly regulated via variability in expression of the 11β-HSD1 and 2. These enzymes are actually manipulated via gonadotropin signals from the pituitary, with LH controlling expression of 11β-HSD1 expression (thus activating GCs during ovulation). This regulation via gonadotropins provides a mechanism through which excess GCs could influence enzymatic regulation of GCs. As these two enzymes are so narrowly regulated during the ovarian cycle, stress and high GC secretion from the adrenals can easily dysregulate these signals and cause profound fertility problems in both ovarian function during ovulation and uterine function during fertilization, implantation and pregnancy.
In the ovaries, high amounts of GCs, surpassing the amount that is typically inactivated by 11β-HSD2, can suppress LH function and inhibit estrogen release and synthesis [131, 146, 150]. Studies both in vivo and in vitro have shown that GCs can influence not only LH response, but also inhibit transcription of the enzymes necessary for steroid biosynthesis, critically inhibiting p450 aromatase, necessary for conversion of testosterone to estrogen. In rat granulosa cell cultures, FSH triggers the increase of aromatase activity, promoting estrogen synthesis for ovulation. Administration of both CORT and DEX inhibited this FSH-induced increase, however stimulated progesterone synthesis and did not inhibit pre-existing aromatase function. This indicates that GCs act to inhibit induction of aromatase activity specifically, not necessarily affecting granulosa cell function as a whole [151]. Glucocorticoid treatment was also found to decrease LH receptor in cultured granulosa cells [152], indicating that GCs can act directly on the ovarian cells to decrease FSH-stimulated functions, including aromatase activity and LH receptor binding.
Interestingly, GC effects on oocyte maturation appear to be species dependent. Studies in humans and pig have shown that GCs can inhibit meiotic development in the oocytes [153, 154], however studies in sheep and mice have shown no effect of GCs on final oocyte maturation [155, 156]. However a recent study in mice showed that high levels of CRH in the cytoplasm of the ovaries due to restraint stress induced ovarian apoptosis, decreasing follicular development independent of GR. This increase of CRH was acting on thecal cells in the ovary, decreasing testosterone and estrogen levels and increasing progesterone, creating a hormonal imbalance between estrogen and progesterone that led to decreased oocyte success [157]. These differences are likely due to problems intrinsic to in vitro models that utilize only the ovarian granulosa cells. In addition, another in vivo study in mice utilizing predatory stress found that while high GCs did not affect oocyte maturation, blastocyst formation was significantly decreased in these mice, showing that GCs may have a stronger effect on embryo development or the oocyte potential for fertilization, rather than maturation of the oocytes in general [158]. The next section will explore GCs effects on pregnancy and fertilization more closely.
The role of glucocorticoid function in the ovary is incredibly complex and narrowly regulated. The actions of GCs are regulated via differential transcription of the two 11β-HSD enzymes, transcription of which is controlled through gonadotropin release form the pituitary. Glucocorticoids are critical for maintenance of ovarian function, involved in functional apoptosis of follicles to maintain normal follicular development, as well as its anti-inflammatory role necessary for ovulation to occur. However, high stress can tip the scales from functional to dysfunctional, overwhelming the ability of 11β-HSD to regulate GC levels and causing ovarian problems ranging from a decreased responsiveness to LH levels, decreased synthesis of estrogen due to inhibition of aromatase release, and potentially inhibiting the final step of oocyte maturation.
Glucocorticoids, Implantation and Pregnancy Success
Even if an ovum can be successfully developed in times of stress, and the HPG axis still functional enough to trigger ovulation, GCs can still act to influence the uterus to prevent successful implantation and completion of pregnancy. Glucocorticoids typically act in opposition to estrogenic actions, and this becomes increasingly critical in implantation. For successful implantation of a blastocyst, progesterone and estrogen regulate uterine cell proliferation, and are necessary for the changes in both the blastocyst and uterine epithelium for successful adhesion. Glucocorticoids inhibit estradiol-stimulated uterine growth and decreases estrogen receptor concentrations in the uterus [159–162].
Pregnancy itself requires a delicate immune balance and regulation of the maternal immune cells in order for survival of both the fetus and the mother. It is suggested that the high levels of progesterone (P) released from the corpus luteum of the ovary after ovulation and sustained by the placenta throughout pregnancy help regulate the mothers immune system. There is some research indicating that membrane-bound progesterone receptors act to inhibit maternal T-cell during pregnancy [163, 164]. This combines with a series of other downstream immune events that allows the maternal immune system to accept the foreign fetus and expression of progesterone and related progesterone factors such as progesterone-induced blocking factor (PIBF) continues to increase through pregnancy. High levels of Th1 cytokines in mice have been shown to be abortogenic, and progesterone during pregnancy binding to progesterone receptors have been shown to release PIBF, which in turn decreases natural killer (NK) cells in the uterus and induces Th2 cytokine development, changing the balance towards an anti-abortive immune response [163–167]. However studies using restraint stress in rodents has found that stress early in pregnancy leads to decreased embryo success, showing higher abortion rates in the mice and smaller litter sizes [168–170]. Wiebold et.al. found that this was due to decreases in corpora lutea, lower levels of serum progesterone and fewer implantation sites [168]. In humans, circadian cortisol levels are suppressed in early pregnancy, and women who have been found to have high morning cortisol levels in the first weeks of pregnancy were more likely to experience spontaneous abortions [130]. This however appears to be specific to the peri-implantation time, as studies have not found that circadian levels of cortisol are indicative of likelihood of a miscarriage later in the first trimester [131]. Human data on the subject is inconclusive, but the immunosuppressive effects of high cortisol, as well GC’s influence on decreasing progesterone and PIBF release support the idea that stress can have a profound influence on early miscarriage rates.
Glucocorticoids play a significant role in pregnancy maintenance, opposing estrogen’s ability to ready the uterus for implantation and inhibiting progesterone’s anti-abortive immune response. While much of research into this focuses on stress during the pregnancy itself, there could be long-term effects of stress prior to the pregnancy that could affect pregnancy success as well, maybe via long-term inhibition of progesterone.
Conclusion: Stress and Its Many Effects on Reproductive Ability
Physiologically, glucocorticoids exert many effects on surrounding cells and are necessary for life. Within normal ranges, GCs regulate homeostasis and are critical for our stress response. In times of stress, high levels of GCs shut down physiological processes not relevant for survival in that time, including reproduction. GCs and the HPA axis can act upon every level of the HPG axis, both directly and indirectly inhibiting gonadotropin release from the pituitary and exerting direct effects on the gonads. Stress and high GCs decrease the release of GnRH from the hypothalamus, either by directly inhibiting GnRH pulses or inhibiting upstream regulators of GnRH release. This can lead to downstream decreases in LH release form the pituitary, however GCs can also directly inhibit the synthesis and release of gonadotropins from the anterior pituitary. The decrease of LH and sometimes FSH from the pituitary can decrease steroid release form the gonads, and circulating GCs can also act directly on the gonads to inhibit the transcription of enzymes necessary for gonadal steroid biosynthesis. There are sex and species differences in all these responses. It is a complex and confusing field, however new techniques utilizing cell-specific knockdowns of GR and/or other peptides involved in this response can help clarify the more specific roles of GCs and reproductive dysfunction. This becomes increasingly important as we find that infertility rates continue to increase in humans, likely due to high stress exposure in day-to-day lives. Understanding the molecular mechanisms behind how stress impairs fecundity and reproductive success, especially in females, is critical to helping improve fertility rates.
References
Handa RJ, Weiser MJ. Gonadal steroid hormones and the hypothalamo-pituitary-adrenal axis. Front Neuroendocrinol. 2014;35(2):197–220. doi:10.1016/j.yfrne.2013.11.001.
Jennes L, Conn PM. Gonadotropin-releasing hormone and its receptors in rat brain. Front Neuroendocrinol. 1994;15(1):51–77. doi:10.1006/frne.1994.1003.
King JC, Tobet SA, Snavely FL, Arimura AA. LHRH immunopositive cells and their projections to the median eminence and organum vasculosum of the lamina terminalis. J Comp Neurol. 1982;209(3):287–300. doi:10.1002/cne.902090307.
Levine JE, Bauer-Dantoin AC. Neuroendocrine regulation of the luteinizing hormone-releasing hormone pulse generator in the rat. Recent Prog Horm Res. 1991;47:97–151.
Moenter SM, Anthony DeFazio R, Pitts GR, Nunemaker CS. Mechanisms underlying episodic gonadotropin-releasing hormone secretion. Front Neuroendocrinol. 2003;24(2):79–93. doi:10.1016/S0091-3022(03)00013-X.
Haisenleder DJ, Dalkin AC, Ortolano GA, Marshall JC, Shupnik MA. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128(1):509–17. doi:10.1210/endo-128-1-509.
Sarkar DK, Chiappa SA, Fink G, Sherwood NM. Gonadotropin-releasing hormone surge in pro-oestrous rats. Nature. 1976;264(5585):461–3. doi:10.1038/264461a0.
Park O-K, Ramirez VD. Spontaneous changes in LHRH release during the rat estrous cycle, as measured with repetitive push-pull perfusions of the pituitary gland in the same female rats. Neuroendocrinology. 1989;50(1):66–72. doi:10.1159/10.1159/000125203.
Marshall JC, Griffin ML. The role of changing pulse frequency in the regulation of ovulation. Hum Reprod. 1993;8 Suppl 2:57–61. http://www.ncbi.nlm.nih.gov/pubmed/8276970. Accessed 27 May 2014.
Marshall JC, Dalkin AC, Haisenleder DJ, Griffin ML, Kelch RP. GnRH pulses—the regulators of human reproduction. Trans Am Clin Climatol Assoc. 1993;104:31–46. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2376610&tool=pmcentrez&rendertype=abstract. Accessed 27 May 2014.
Herbison AE. Estrogen positive feedback to gonadotropin-releasing hormone (GnRH) neurons in the rodent: the case for the rostral periventricular area of the third ventricle (RP3V). Brain Res Rev. 2008;57(2):277–87. doi:10.1016/j.brainresrev.2007.05.006.
Chazal G, Faudon M, Gogan F, Laplante E. Negative and positive effects of oestradiol upon luteinizing hormone secretion in the female rat. J Endocrinol. 1974;61(3):511–2. doi:10.1677/joe.0.0610511.
Shupnik MA. Gonadotropin gene modulation by steroids and gonadotropin-releasing hormone. Biol Reprod. 1996;54(2):279–86. doi:10.1095/biolreprod54.2.279.
Knobil E. The neuroendocrine control of the menstrual cycle. Recent Prog Horm Res. 1980;36:53–88.
Baird DT, McNeilly AS. Gonadotrophic control of follicular development and function during the oestrous cycle of the ewe. J Reprod Fertil Suppl. 1981;30:119–33. http://europepmc.org/abstract/MED/6300383. Accessed 2 Sept 2014.
Legan SJ, Karsch FJ. A daily signal for the LH surge in the rat. Endocrinology. 1975;96(1):57–62. doi:10.1210/endo-96-1-57.
Ferin M, Tempone A, Zimmering PE, Van de Wiele RL. Effect of antibodies to 17beta-estradiol and progesterone on the estrous cycle of the rat. Endocrinology. 1969;85(6):1070–8. doi:10.1210/endo-85-6-1070.
Labhsetwar AP. Role of estrogens in ovulation: a study using the estrogen-antagonist, I.C.I. 46,474. Endocrinology. 1970;87(3):542–51. doi:10.1210/endo-87-3-542.
Chappell PE, Levine JE. Stimulation of gonadotropin-releasing hormone surges by estrogen. I. Role of hypothalamic progesterone receptors. Endocrinology. 2000;141(4):1477–85. doi:10.1210/endo.141.4.7428.
Micevych P, Sinchak K, Mills RH, Tao L, LaPolt P, Lu JKH. The luteinizing hormone surge is preceded by an estrogen-induced increase of hypothalamic progesterone in ovariectomized and adrenalectomized rats. Neuroendocrinology. 2003;78(1):29–35. doi:10.1159/000071703.
Kuo J, Hamid N, Bondar G, Prossnitz ER, Micevych P. Membrane estrogen receptors stimulate intracellular calcium release and progesterone synthesis in hypothalamic astrocytes. J Neurosci. 2010;30(39):12950–7. doi:10.1523/JNEUROSCI.1158-10.2010.
Micevych P, Soma KK, Sinchak K. Neuroprogesterone: key to estrogen positive feedback? Brain Res Rev. 2008;57(2):470–80. doi:10.1016/j.brainresrev.2007.06.009.
Micevych PE, Chaban V, Ogi J, Dewing P, Lu JKH, Sinchak K. Estradiol stimulates progesterone synthesis in hypothalamic astrocyte cultures. Endocrinology. 2007;148(2):782–9. doi:10.1210/en.2006-0774.
Micevych P, Sinchak K. The neurosteroid progesterone underlies estrogen positive feedback of the LH surge. Front Endocrinol (Lausanne). 2011;2:90. doi:10.3389/fendo.2011.00090.
Chaban VV, Lakhter AJ, Micevych P. A membrane estrogen receptor mediates intracellular calcium release in astrocytes. Endocrinology. 2004;145(8):3788–95. doi:10.1210/en.2004-0149.
Ubuka T, Inoue K, Fukuda Y, et al. Identification, expression, and physiological functions of Siberian hamster gonadotropin-inhibitory hormone. Endocrinology. 2012;153(1):373–85. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1434.
Ubuka T, Morgan K, Pawson A, et al. Identification of human GnIH homologs, RFRP-1 and RFRP-3, and the cognate receptor, GPR147 in the human hypothalamic pituitary axis. PLoS One. 2009;4(12):1334–9. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1233.
Ubuka T, Lai H, Kitani M, et al. Gonadotropin-inhibitory hormone identification, cDNA cloning, and distribution in rhesus macaque brain. J Comp Neurol. 2009;517:841–55. doi:10.1002/cne.22191.
Ukena K, Iwakoshi E, Minakata H, Tsutsui K. A novel rat hypothalamic RFamide-related peptide identified by immunoaffinity chromatography and mass spectrometry. FEBS Lett. 2002;512(1–3):255–8. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1299.
Tsutsui K, Saigoh E, Ukena K, et al. A novel avian hypothalamic peptide inhibiting gonadotropin release. Biochem Biophys Res Commun. 2000;275(2):661–7. doi:10.1006/bbrc.2000.3350.
De Roux N, Genin E, Carel J-C, Matsuda F, Chaussain J-L, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100(19):10972–6. doi:10.1073/pnas.1834399100.
Thompson EL, Patterson M, Murphy KG, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol. 2004;16(10):850–8. doi:10.1111/j.1365-2826.2004.01240.x.
Gottsch ML, Cunningham MJ, Smith JT, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145(9):4073–7. doi:10.1210/en.2004-0431.
Navarro VM, Castellano JM, Fernández-Fernández R, et al. Effects of KiSS-1 peptide, the natural ligand of GPR54, on follicle-stimulating hormone secretion in the rat. Endocrinology. 2005;146(4):1689–97. doi:10.1210/en.2004-1353.
Dhillo WS, Chaudhri OB, Patterson M, et al. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. 2005;90(12):6609–15. doi:10.1210/jc.2005-1468.
Li X-F, Kinsey-Jones JS, Cheng Y, et al. Kisspeptin signalling in the hypothalamic arcuate nucleus regulates GnRH pulse generator frequency in the rat. Tena-Sempere M, ed. PLoS One. 2009;4(12):e8334. doi:10.1371/journal.pone.0008334.
Maeda K-I, Ohkura S, Uenoyama Y, et al. Neurobiological mechanisms underlying GnRH pulse generation by the hypothalamus. Brain Res. 2010;1364:103–15. doi:10.1016/j.brainres.2010.10.026.
Roseweir AK, Kauffman AS, Smith JT, et al. Discovery of potent kisspeptin antagonists delineate physiological mechanisms of gonadotropin regulation. J Neurosci. 2009;29(12):3920–9. doi:10.1523/JNEUROSCI.5740-08.2009.
Messager S, Chatzidaki EE, Ma D, et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci U S A. 2005;102(5):1761–6. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1232.
Millar RP, Roseweir AK, Tello JA, et al. Kisspeptin antagonists: unraveling the role of kisspeptin in reproductive physiology. Brain Res. 2010;1364:81–9. doi:10.1016/j.brainres.2010.09.044.
Pineda R, Garcia-Galiano D, Sanchez-Garrido MA, et al. Characterization of the inhibitory roles of RFRP3, the mammalian ortholog of GnIH, in the control of gonadotropin secretion in the rat: in vivo and in vitro studies. Am J Physiol Endocrinol Metab. 2010;299(1):E39–46. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1269.
Khan AR, Kauffman AS. The role of kisspeptin and RFamide-related peptide-3 neurones in the circadian-timed preovulatory luteinising hormone surge. J Neuroendocrinol. 2012;24(1):131–43. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1435.
Clarke I, Smith J, Henry B, et al. Gonadotropin-inhibitory hormone is a hypothalamic peptide that provides a molecular switch between reproduction and feeding. Neuroendocrinology. 2012;95(4):305–16. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1596.
Wu M, Dumalska I, Morozova E, van den Pol AN, Alreja M. Gonadotropin inhibitory hormone inhibits basal forebrain vGluT2-gonadotropin-releasing hormone neurons via a direct postsynaptic mechanism. J Physiol. 2009;587(7):1401. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1234.
Kriegsfeld LJ, Gibson EM, Williams WP, et al. The roles of RFamide-related peptide-3 in mammalian reproductive function and behaviour. J Neuroendocrinol. 2010;22(7):692–700. doi:10.1111/j.1365-2826.2010.02031.x.
Clarkson J, d’Anglemont de Tassigny X, Moreno AS, Colledge WH, Herbison AE. Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J Neurosci. 2008;28(35):8691–7. doi:10.1523/JNEUROSCI.1775-08.2008.
Sinchak K, Wagner EJ. Estradiol signaling in the regulation of reproduction and energy balance. Front Neuroendocrinol. 2012;33(4):342–63. doi:10.1016/j.yfrne.2012.08.004.
Christensen A, Bentley GE, Cabrera R, et al. Hormonal regulation of female reproduction. Horm Metab Res. 2012;44(8):587–91. doi:10.1055/s-0032-1306301.
Giuliani FA, Yunes R, Mohn CE, Laconi M, Rettori V, Cabrera R. Allopregnanolone induces LHRH and glutamate release through NMDA receptor modulation. Endocrine. 2011;40(1):21–6. doi:10.1007/s12020-011-9451-8.
Sim JA, Skynner MJ, Herbison AE. Direct regulation of postnatal GnRH neurons by the progesterone derivative allopregnanolone in the mouse. Endocrinology. 2001;142(10):4448–53. doi:10.1210/endo.142.10.8451.
el-Etr M, Akwa Y, Fiddes RJ, Robel P, Baulieu EE. A progesterone metabolite stimulates the release of gonadotropin-releasing hormone from GT1-1 hypothalamic neurons via the gamma-aminobutyric acid type A receptor. Proc Natl Acad Sci U S A. 1995;92(9):3769–73. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=42043&tool=pmcentrez&rendertype=abstract. Accessed 27 May 2014.
Breen KM, Karsch FJ. New insights regarding glucocorticoids, stress and gonadotropin suppression. Front Neuroendocrinol. 2006;27(2):233–45. doi:10.1016/j.yfrne.2006.03.335.
Li XF, Knox AMI, O’Byrne KT. Corticotrophin-releasing factor and stress-induced inhibition of the gonadotrophin-releasing hormone pulse generator in the female. Brain Res. 2010;1364:153–63. doi:10.1016/j.brainres.2010.08.036.
Rivier C, Vale W. Influence of corticotropin-releasing factor on reproductive functions in the rat. Endocrinology. 1984;114(3):914–21. doi:10.1210/endo-114-3-914.
Cates PS, Li XF, O’Byrne KT. The influence of 17beta-oestradiol on corticotrophin-releasing hormone induced suppression of luteinising hormone pulses and the role of CRH in hypoglycaemic stress-induced suppression of pulsatile LH secretion in the female rat. Stress. 2004;7(2):113–8. doi:10.1080/1025389042000218988.
Bowe JE, Li XF, Kinsey-Jones JS, Brain SD, Lightman SL, O’Byrne KT. The role of corticotrophin-releasing hormone receptors in the calcitonin gene-related peptide-induced suppression of pulsatile luteinising hormone secretion in the female rat. Stress. 2008;11(4):312–9. doi:10.1080/10253890701801448.
Li XF, Bowe JE, Kinsey-Jones JS, Brain SD, Lightman SL, O’Byrne KT. Differential role of corticotrophin-releasing factor receptor types 1 and 2 in stress-induced suppression of pulsatile luteinising hormone secretion in the female rat. J Neuroendocrinol. 2006;18(8):602–10. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p701.
Li XF, Bowe JE, Lightman SL, O’Byrne KT. Role of corticotropin-releasing factor receptor-2 in stress-induced suppression of pulsatile luteinizing hormone secretion in the rat. Endocrinology. 2005;146(1):318–22. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p710.
Kinsey‐Jones J, Li X, Knox A, et al. Corticotrophin‐releasing factor alters the timing of puberty in the female rat. J Neuroendocrinol. 2010;22(2):102–109. file:///Users/annageraghty/Documents/Papers/2010/Kinsey‐Jones/Journal of Neuroendocrinology 2010 Kinsey‐Jones.pdf.
Rivier C, Rivest S. Effect of stress on the activity of the hypothalamic-pituitary-gonadal axis: peripheral and central mechanisms. Biol Reprod. 1991;45(4):523–32. http://www.ncbi.nlm.nih.gov/pubmed/1661182. Accessed 9 Sept 2013.
Rivest S, Rivier C. Central mechanisms and sites of action involved in the inhibitory effects of CRF and cytokines on LHRH neuronal activity. Ann N Y Acad Sci. 1993;697(1 Corticotropin):117–41. doi:10.1111/j.1749-6632.1993.tb49928.x.
Oakley AE, Breen KM, Clarke IJ, Karsch FJ, Wagenmaker ER, Tilbrook AJ. Cortisol reduces gonadotropin-releasing hormone pulse frequency in follicular phase ewes: influence of ovarian steroids. Endocrinology. 2009;150(1):341–9. doi:10.1210/en.2008-0587.
Calogero AE, Burrello N, Bosboom AM, Garofalo MR, Weber RF, D’Agata R. Glucocorticoids inhibit gonadotropin-releasing hormone by acting directly at the hypothalamic level. J Endocrinol Invest. 1999;22(9):666–70. http://www.ncbi.nlm.nih.gov/pubmed/10595829. Accessed 28 May 2014.
Jasoni CL, Todman MG, Han S-K, Herbison AE. Expression of mRNAs encoding receptors that mediate stress signals in gonadotropin-releasing hormone neurons of the mouse. Neuroendocrinology. 2005;82(5–6):320–8. doi:10.1159/000093155.
Ahima RS, Harlan RE. Glucocorticoid receptors in LHRH neurons. Neuroendocrinology. 1992;56(6):845–50. http://www.ncbi.nlm.nih.gov/pubmed/1369593. Accessed 27 May 2014.
Dondi D, Piccolella M, Messi E, et al. Expression and differential effects of the activation of glucocorticoid receptors in mouse gonadotropin-releasing hormone neurons. Neuroendocrinology. 2005;82(3–4):151–63. doi:10.1159/000091693.
DeFranco DB, Attardi B, Chandran UR. Glucocorticoid receptor-mediated repression of GnRH gene expression in a hypothalamic GnRH-secreting neuronal cell line. Ann N Y Acad Sci. 1994;746:473–5. http://www.ncbi.nlm.nih.gov/pubmed/7825918. Accessed 28 May 2014.
Tellam DJ, Perone MJ, Dunn IC, et al. Direct regulation of GnRH transcription by CRF-like peptides in an immortalized neuronal cell line. Neuroreport. 1998;9(14):3135–40. doi:10.1097/00001756-199810050-00003.
Tellam DJ, Mohammad YN, Lovejoy DA. Molecular integration of hypothalamo-pituitary-adrenal axis-related neurohormones on the GnRH neuron. 2011. http://www.nrcresearchpress.com/doi/abs/10.1139/o00-060#.U4Z5rlhdX-Y. Accessed 29 May 2014.
Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI. Immortalization of hypothalamic GnRH by genetically targeted tumorigenesis. Neuron. 1990;5(1):1–10. doi:10.1016/0896-6273(90)90028-E.
Wetsel WC, Mellon PL, Weiner RI, Negro-Vilar A. Metabolism of pro-luteinizing hormone-releasing hormone in immortalized hypothalamic neurons. Endocrinology. 1991;129(3):1584–95. doi:10.1210/endo-129-3-1584.
Chandran UR, Attardi B, Friedman R, Dong KW, Roberts JL, DeFranco DB. Glucocorticoid receptor-mediated repression of gonadotropin-releasing hormone promoter activity in GT1 hypothalamic cell lines. Endocrinology. 1994;134(3):1467–74. doi:10.1210/endo.134.3.8119188.
Attardi B, Tsujii T, Friedman R, et al. Glucocorticoid repression of gonadotropin-releasing hormone gene expression and secretion in morphologically distinct subpopulations of GT1-7 cells. Mol Cell Endocrinol. 1997;131(2):241–55. http://www.ncbi.nlm.nih.gov/pubmed/9296383. Accessed 28 May 2014.
Gore A, Attardi B, DeFranco D. Glucocorticoid repression of the reproductive axis: effects on GnRH and gonadotropin subunit mRNA levels. Mol Cell Endocrinol. 2006;256(1-2):40–8. Papers
Kinsey‐Jones J, Li X, Knox A, et al. Down‐regulation of hypothalamic kisspeptin and its receptor, Kiss1r, mRNA expression is associated with stress‐induced suppression of luteinising hormone secretion in the female rat. J Neuroendocrinol. 2009;21(1):20–29. file:///Users/annageraghty/Documents/Papers/2009/Kinsey‐Jones/Journal of Neuroendocrinology 2009 Kinsey‐Jones-1.pdf.
Iwasa T, Matsuzaki T, Murakami M, et al. Decreased expression of kisspeptin mediates acute immune/inflammatory stress-induced suppression of gonadotropin secretion in female rat. J Endocrinol Invest. 2008;31(7):656–9. http://europepmc.org/abstract/MED/18787387. Accessed 28 May 2014.
Grachev P, Li XF, O’Byrne K. Stress regulation of kisspeptin in the modulation of reproductive function. Adv Exp Med Biol. 2013;784:431–54. doi:10.1007/978-1-4614-6199-9_20.
Takumi K, Iijima N, Higo S, Ozawa H. Immunohistochemical analysis of the colocalization of corticotropin-releasing hormone receptor and glucocorticoid receptor in kisspeptin neurons in the hypothalamus of female rats. Neurosci Lett. 2012;531(1):40–5. doi:10.1016/j.neulet.2012.10.010.
Grachev P, Li XF, Hu MH, et al. Neurokinin B signaling in the female rat: a novel link between stress and reproduction. Endocrinology. 2014;155(7):2589–601. doi:10.1210/en.2013-2038.
Goodman RL, Hileman SM, Nestor CC, et al. Kisspeptin, neurokinin B, and dynorphin act in the arcuate nucleus to control activity of the GnRH pulse generator in ewes. Endocrinology. 2013;154(11):4259–69. doi:10.1210/en.2013-1331.
Okamura H, Tsukamura H, Ohkura S, Uenoyama Y, Wakabayashi Y, Maeda K. Kisspeptin and GnRH pulse generation. Adv Exp Med Biol. 2013;784:297–323. doi:10.1007/978-1-4614-6199-9_14.
Wakabayashi Y, Yamamura T, Sakamoto K, Mori Y, Okamura H. Electrophysiological and morphological evidence for synchronized GnRH pulse generator activity among Kisspeptin/neurokinin B/dynorphin A (KNDy) neurons in goats. J Reprod Dev. 2013;59(1):40–8. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3943231&tool=pmcentrez&rendertype=abstract. Accessed 28 May 2014.
Bowe JE, Li XF, Kinsey-Jones JS, et al. Calcitonin gene-related peptide-induced suppression of luteinizing hormone pulses in the rat: the role of endogenous opioid peptides. J Physiol. 2005;566(Pt 3):921–8. doi:10.1113/jphysiol.2005.085662.
Nabeshima T, Katoh A, Wada M, Kameyama T. Stress-induced changes in brain Met-enkephalin, Leu-enkephalin and dynorphin concentrations. Life Sci. 1992;51(3):211–7. http://www.ncbi.nlm.nih.gov/pubmed/1352028. Accessed 28 May 2014.
Petraglia F, Vale W, Rivier C. Opioids act centrally to modulate stress-induced decrease in luteinizing hormone in the rat. Endocrinology. 1986;119(6):2445–50. doi:10.1210/endo-119-6-2445.
Ukena K, Iwakoshi E, Minakata H, Tsutsui K. A novel rat hypothalamic RFamide-related peptide identified by immunoaffinity chromatography and mass spectrometry. FEBS Lett. 2002;512(1–3):255–8. http://www.ncbi.nlm.nih.gov/pubmed/11852091. Accessed 9 Sept 2013.
Sari IP, Rao A, Smith JT, Tilbrook AJ, Clarke IJ. Effect of RF-amide-related peptide-3 on luteinizing hormone and follicle-stimulating hormone synthesis and secretion in ovine pituitary gonadotropes. Endocrinology. 2009;150(12):5549–56. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1243.
Kirby ED, Geraghty AC, Ubuka T, Bentley GE, Kaufer D. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. Proc Natl Acad Sci U S A. 2009;106(27):11324–9. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1273.
Lee Son Y, Ubuka T, Narihiro M, et al. Molecular basis for the activation of gonadotropin-inhibitory hormone gene transcription by corticosterone. Endocrinology. 2014;155(5):1817–26. doi:10.1210/en.2013-2076.
Gojska NM, Belsham DD. Glucocorticoid receptor-mediated regulation of Rfrp (GnIH) and Gpr147 (GnIH-R) synthesis in immortalized hypothalamic neurons. Mol Cell Endocrinol. 2014;384(1–2):23–31. doi:10.1016/j.mce.2013.12.015.
Geraghty A, Muroy S, Zhao S, Bentley G, Kriegsfeld L, Kaufer D. Chronic stress causes an increase in RFRP expression and leads to reproductive dysfunction in the adult female rat. [abstract]. In: 2013 Neuroscience Meet Plan, Society for Neuroscience; 2013.
Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138(3):1224–31. doi:10.1210/endo.138.3.4968.
Vale W, Rivier C, Brown M. Regulatory peptides of the hypothalamus. Annu Rev Physiol. 1977;39:473–527. doi:10.1146/annurev.ph.39.030177.002353.
Pierce JG, Parsons TF. Glycoprotein hormones: structure and function. Annu Rev Biochem. 1981;50:465–95. doi:10.1146/annurev.bi.50.070181.002341.
Kononen J, Honkaniemi J, Gustafsson JA, Pelto-Huikko M. Glucocorticoid receptor colocalization with pituitary hormones in the rat pituitary gland. Mol Cell Endocrinol. 1993;93(1):97–103. http://www.ncbi.nlm.nih.gov/pubmed/8319836. Accessed 28 May 2014.
Breen KM, Thackray VG, Hsu T, Mak-McCully RA, Coss D, Mellon PL. Stress levels of glucocorticoids inhibit LHβ-subunit gene expression in gonadotrope cells. Mol Endocrinol. 2012;26(10):1716–31. doi:10.1210/me.2011-1327.
Armario A, Lopez-Calderon A, Jolin T, Balasch J. Response of anterior pituitary hormones to chronic stress. The specificity of adaptation. Neurosci Biobehav Rev. 1986;10(3):245–50. doi:10.1016/0149-7634(86)90011-4.
Lopez-Calderon A, Gonzalez-Quijano MI, Tresguerres JAF, Ariznavarreta C. Role of LHRH in the gonadotrophin response to restraint stress in intact male rats. J Endocrinol. 1990;124(2):241–6. doi:10.1677/joe.0.1240241.
Blake CA. Effects of “stress” on pulsatile luteinizing hormone release in ovariectomized rats. Proc Soc Exp Biol Med. 1975;148(3):813–5. http://www.ncbi.nlm.nih.gov/pubmed/165534. Accessed 29 May 2014.
Kamel F, Kubajak CL. Modulation of gonadotropin secretion by corticosterone: interaction with gonadal steroids and mechanism of action. Endocrinology. 1987;121(2):561–8. doi:10.1210/endo-121-2-561.
Baldwin DM, Sawyer CH. Effects of dexamethasone on LH release and ovulation in the cyclic rat. Endocrinology. 1974;94(5):1397–403. doi:10.1210/endo-94-5-1397.
Collu R, Taché Y, Ducharme J. Hormonal modifications induced by chronic stress in rats. J Steroid Biochem. 1979;11(1):989–1000. doi:10.1016/0022-4731(79)90042-6.
Vreeburg JT, de Greef WJ, Ooms MP, van Wouw P, Weber RF. Effects of adrenocorticotropin and corticosterone on the negative feedback action of testosterone in the adult male rat. Endocrinology. 1984;115(3):977–83. doi:10.1210/endo-115-3-977.
Taché Y, Ducharme JR, Charpenet G, Haour F, Saez J, Collu R. Effect of chronic intermittent immobilization stress on hypophyso-gonadal function of rats. Acta Endocrinol (Copenh). 1980;93(2):168–74. http://www.ncbi.nlm.nih.gov/pubmed/7376788. Accessed 28 May 2014.
Briski KP, Sylvester PW. Differential impact of naltrexone on luteinizing hormone release during single versus repetitive exposure to restraint stress. Psychoneuroendocrinology. 1992;17(2–3):125–33. http://www.ncbi.nlm.nih.gov/pubmed/1332097. Accessed 28 May 2014.
Li XF, Edward J, Mitchell JC, et al. Differential effects of repeated restraint stress on pulsatile lutenizing hormone secretion in female Fischer, Lewis and Wistar rats. J Neuroendocrinol. 2004;16(7):620–7. doi:10.1111/j.1365-2826.2004.01209.x.
Briski KP, Sylvester PW. Effects of repetitive daily acute stress on pituitary LH and prolactin release during exposure to the same stressor or a second novel stress. Psychoneuroendocrinology. 1987;12(6):429–37. http://www.ncbi.nlm.nih.gov/pubmed/3441582. Accessed 28 May 2014.
Sakakura M, Takebe K, Nakagawa S. Inhibition of luteinizing hormone secretion induced by synthetic LRH by long-term treatment with glucocorticoids in human subjects. J Clin Endocrinol Metab. 1975;40(5):774–9. doi:10.1210/jcem-40-5-774.
Du Ruisseau P, Taché Y, Brazeau P, Collu R. Effects of chronic immobilization stress on pituitary hormone secretion, on hypothalamic factor levels, and on pituitary responsiveness to LHRH and TRH in female rats. Neuroendocrinology. 1979;29(2):90–9. http://www.ncbi.nlm.nih.gov/pubmed/116141. Accessed 9 Sept 2013.
Rivier C, Vale W. Effect of the long-term administration of corticotropin-releasing factor on the pituitary-adrenal and pituitary-gonadal axis in the male rat. J Clin Invest. 1985;75(2):689–94. doi:10.1172/JCI111748.
Suter DE, Schwartz NB, Ringstrom SJ. Dual role of glucocorticoids in regulation of pituitary content and secretion of gonadotropins. Am J Physiol. 1988;254(5 Pt 1):E595–600. http://ajpendo.physiology.org/content/254/5/E595.abstract. Accessed 20 May 2014.
Baldwin DM. The effect of glucocorticoids on estrogen-dependent luteinizing hormone release in the ovariectomized rat and on gonadotropin secretin in the intact female rat. Endocrinology. 1979;105(1):120–8. doi:10.1210/endo-105-1-120.
Maya-Núñez G, Conn PM. Transcriptional regulation of the GnRH receptor gene by glucocorticoids. Mol Cell Endocrinol. 2003;200(1–2):89–98. doi:10.1016/S0303-7207(02)00419-7.
Kotitschke A, Sadie-Van Gijsen H, Avenant C, Fernandes S, Hapgood JP. Genomic and nongenomic cross talk between the gonadotropin-releasing hormone receptor and glucocorticoid receptor signaling pathways. Mol Endocrinol. 2009;23(11):1726–45. doi:10.1210/me.2008-0462.
Bronson FH. Establishment of social rank among grouped male mice: relative effects on circulating FSH, LH, and corticosterone. Physiol Behav. 1973;10(5):947–51. doi:10.1016/0031-9384(73)90065-6.
Du Ruisseau P, Taché Y, Brazeau P, Colin R. Effects of chronic immobilization stress on pituitary hormone secretion, on hypothalamic factor levels, and on pituitary responsiveness to LHRH and TRH in female rats. Neuroendocrinology. 1979;29(2):90–9. doi:10.1159/000122910.
Ringstrom SJ, Schwartz NB. Differential effect of glucocorticoids on synthesis and secretion of luteinizing hormone (LH) and follicle stimulating hormone (FSH). J Steroid Biochem. 1987;27(1–3):625–30. doi:10.1016/0022-4731(87)90362-1.
Ringstrom SJ, McAndrews JM, Rahal JO, Schwartz NB. Cortisol in vivo increases FSH beta mRNA selectively in pituitaries of male rats. Endocrinology. 1991;129(5):2793–5. doi:10.1210/endo-129-5-2793.
Thackray VG, McGillivray SM, Mellon PL. Androgens, progestins, and glucocorticoids induce follicle-stimulating hormone beta-subunit gene expression at the level of the gonadotrope. Mol Endocrinol. 2006;20(9):2062–79. doi:10.1210/me.2005-0316.
Smals AG, Kloppenborg PW, Benraad TJ. Plasma testosterone profiles in Cushing’s syndrome. J Clin Endocrinol Metab. 1977;45(2):240–5. doi:10.1210/jcem-45-2-240.
Whirledge S, Cidlowski JA. Glucocorticoids, stress, and fertility. Minerva Endocrinol. 2010;35(2):109–25. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3547681&tool=pmcentrez&rendertype=abstract. Accessed 20 May 2014.
Schultz R, Isola J, Parvinen M, et al. Localization of the glucocorticoid receptor in testis and accessory sexual organs of male rat. Mol Cell Endocrinol. 1993;95(1–2):115–20. doi:10.1016/0303-7207(93)90036-J.
Silva EJR, Queiróz DBC, Honda L, Avellar MCW. Glucocorticoid receptor in the rat epididymis: expression, cellular distribution and regulation by steroid hormones. Mol Cell Endocrinol. 2010;325(1–2):64–77. doi:10.1016/j.mce.2010.05.013.
Bernier M, Gibb W, Collu R, Ducharme JR. Effect of glucocorticoids on testosterone production by porcine Leydig cells in primary culture. Can J Physiol Pharmacol. 1984;62(9):1166–9. doi:10.1139/y84-195.
Orr T. Effects of restraint stress on plasma LH and testosterone concentrations, Leydig cell LH/HCG receptors, and in vitro testicular steroidogenesis in adult rats. Horm Behav. 1990;24(3):324–41. doi:10.1016/0018-506X(90)90013-N.
Orr T. Role of glucocorticoids in the stress-induced suppression of testicular steroidogenesis in adult male rats. Horm Behav. 1992;26(3):350–63. doi:10.1016/0018-506X(92)90005-G.
Cumming DC, Quigley ME, Yen SS. Acute suppression of circulating testosterone levels by cortisol in men. J Clin Endocrinol Metab. 1983;57(3):671–3. doi:10.1210/jcem-57-3-671.
Marić D, Kostić T, Kovačević R. Effects of acute and chronic immobilization stress on rat Leydig cell steroidogenesis. J Steroid Biochem Mol Biol. 1996;58(3):351–5.
Saez JM, Morera AM, Haour F, Evain D. Effects of in vivo administration of dexamethasone, corticotropin and human chorionic gonadotropin on steroidogenesis and protein and DNA synthesis of testicular interstitial cells in prepuberal rats. Endocrinology. 1977;101(4):1256–63. doi:10.1210/endo-101-4-1256.
Bambino TH, Hsueh AJ. Direct inhibitory effect of glucocorticoids upon testicular luteinizing hormone receptor and steroidogenesis in vivo and in vitro. Endocrinology. 1981;108(6):2142–8. doi:10.1210/endo-108-6-2142.
Hales DB, Payne AH. Glucocorticoid-mediated repression of P450scc mRNA and de novo synthesis in cultured Leydig cells. Endocrinology. 1989;124(5):2099–104. doi:10.1210/endo-124-5-2099.
Payne AH, Sha LL. Multiple mechanisms for regulation of 3 beta-hydroxysteroid dehydrogenase/delta 5––delta 4-isomerase, 17 alpha-hydroxylase/C17-20 lyase cytochrome P450, and cholesterol side-chain cleavage cytochrome P450 messenger ribonucleic acid levels in primary cultures of mouse Leydig cells. Endocrinology. 1991;129(3):1429–35. doi:10.1210/endo-129-3-1429.
Martin LJ, Tremblay JJ. Glucocorticoids antagonize cAMP-induced Star transcription in Leydig cells through the orphan nuclear receptor NR4A1. J Mol Endocrinol. 2008;41(3):165–75. doi:10.1677/JME-07-0145.
Sasagawa I, Yazawa H, Suzuki Y, Nakada T. Stress and testicular germ cell apoptosis. 2009. http://informahealthcare.com/doi/abs/10.1080/014850101753145924. Accessed 20 May 2014.
Yazawa H. Apoptosis of testicular germ cells induced by exogenous glucocorticoid in rats. Hum Reprod. 2000;15(9):1917–20. doi:10.1093/humrep/15.9.1917.
Gao H-B, Tong M-H, Hu Y-Q, Guo Q-S, Ge R, Hardy MP. Glucocorticoid induces apoptosis in rat Leydig cells. 2013. http://press.endocrine.org/doi/abs/10.1210/endo.143.1.8604?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub=pubmed. Accessed 20 May 2014.
Almeida SA, Petenusci SO, Anselmo-Franci JA, Rosa-e-Silva AAM, Lamano-Carvalho TL. Decreased spermatogenic and androgenic testicular functions in adult rats submitted to immobilization-induced stress from prepuberty. Braz J Med Biol Res. 1998;31(11):1443–8. doi:10.1590/S0100-879X1998001100013.
Zorn B, Auger J, Velikonja V, Kolbezen M, Meden-Vrtovec H. Psychological factors in male partners of infertile couples: relationship with semen quality and early miscarriage. Int J Androl. 2008;31(6):557–64. doi:10.1111/j.1365-2605.2007.00806.x.
Seckl JR. 11Beta-hydroxysteroid dehydrogenase in the brain: a novel regulator of glucocorticoid action? Front Neuroendocrinol. 1997;18(1):49–99. doi:10.1006/frne.1996.0143.
Seckl JR, Walker BR. Minireview: 11beta-hydroxysteroid dehydrogenase type 1—a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142(4):1371–6. doi:10.1210/endo.142.4.8114.
Tetsuka M. Expression of 11 beta-hydroxysteroid dehydrogenase, glucocorticoid receptor, and mineralocorticoid receptor genes in rat ovary. Biol Reprod. 1999;60(2):330–5. doi:10.1095/biolreprod60.2.330.
Benediktsson R, Yau JLW, Brett LP, Cooke BE, Edwards CRW, Seckl JR. 11β-Hydroxysteroid dehydrogenase in the rat ovary: high expression in the oocyte. J Endocrinol. 1992;135(1):53–58. http://www.scopus.com/inward/record.url?eid=2-s2.0-0026757114&partnerID=tZOtx3y1.
McDonald SE, Henderson TA, Gomez-Sanchez CE, Critchley HOD, Mason JI. 11Beta-hydroxysteroid dehydrogenases in human endometrium. Mol Cell Endocrinol. 2006;248(1–2):72–8. doi:10.1016/j.mce.2005.12.010.
Michael AE, Evagelatou M, Norgate DP, et al. Isoforms of 11β-hydroxysteroid dehydrogenase in human granulosa-lutein cells. Mol Cell Endocrinol. 1997;132(1–2):43–52. doi:10.1016/S0303-7207(97)00118-4.
Schreiber JR, Nakamura K, Erickson GF. Rat ovary glucocorticoid receptor: identification and characterization. Steroids. 1982;39(5):569–84. doi:10.1016/0039-128X(82)90057-5.
Michael A, Cooke B. A working hypothesis for the regulation of steroidogenesis and germ cell development in the gonads by glucocorticoids and 11β-hydroxysteroid dehydrogenase (11βHSD). Mol Cell Endocrinol. 1994;100(1–2):55–63. doi:10.1016/0303-7207(94)90279-8.
Albiston AL, Smith RE, Krozowski ZS. Changes in the levels of 11β-hydroxysteroid dehydrogenase mRNA over the oestrous cycle in the rat. J Steroid Biochem Mol Biol. 1995;52(1):45–8. doi:10.1016/0960-0760(94)00154-E.
Hillier SG. Molecular biology of the female reproductive system. Amsterdam: Elsevier; 1994. p. 1–37. doi:10.1016/B978-0-08-091819-8.50005-9.
Hillier S, Tetsuka M. An anti-inflammatory role for glucocorticoids in the ovaries? J Reprod Immunol. 1998;39(1–2):21–7. doi:10.1016/S0165-0378(98)00011-4.
Michael AE, Pester LA, Curtis P, Shaw RW, Edwards CR, Cooke BA. Direct inhibition of ovarian steroidogenesis by cortisol and the modulatory role of 11 beta-hydroxysteroid dehydrogenase. Clin Endocrinol (Oxf). 1993;38(6):641–4. http://www.ncbi.nlm.nih.gov/pubmed/8334750. Accessed 29 May 2014.
Hsueh A. Glucocorticoid inhibition of FSH-induced estrogen production in cultured rat granulosa cells. Steroids. 1978;32(5):639–48. doi:10.1016/0039-128X(78)90074-0.
Schoonmaker JN, Erickson GF. Glucocorticoid modulation of follicle-stimulating hormone-mediated granulosa cell differentiation. Endocrinology. 1983;113(4):1356–63. doi:10.1210/endo-113-4-1356.
Yang J-G. Effects of glucocorticoids on maturation of pig oocytes and their subsequent fertilizing capacity in vitro. Biol Reprod. 1999;60(4):929–36. doi:10.1095/biolreprod60.4.929.
Jimena P, Castilla JA, Peran F, et al. Adrenal hormones in human follicular fluid. Eur J Endocrinol. 1992;127(5):403–6. doi:10.1530/acta.0.1270403.
González R, Ruiz-León Y, Gomendio M, Roldan ERS. The effect of glucocorticoids on ERK-1/2 phosphorylation during maturation of lamb oocytes and their subsequent fertilization and cleavage ability in vitro. Reprod Toxicol. 2010;29(2):198–205. doi:10.1016/j.reprotox.2009.10.009.
Andersen CY. Effect of glucocorticoids on spontaneous and follicle-stimulating hormone induced oocyte maturation in mouse oocytes during culture. J Steroid Biochem Mol Biol. 2003;85(2–5):423–7. doi:10.1016/S0960-0760(03)00190-0.
Liang B, Wei D-L, Cheng Y-N, et al. Restraint stress impairs oocyte developmental potential in mice: role of CRH-induced apoptosis of ovarian cells. Biol Reprod. 2013;89(3):64. doi:10.1095/biolreprod.113.110619.
Liu Y-X, Cheng Y-N, Miao Y-L, et al. Psychological stress on female mice diminishes the developmental potential of oocytes: a study using the predatory stress model. PLoS One. 2012;7(10), e48083. doi:10.1371/journal.pone.0048083.
Rabin DS. Glucocorticoids inhibit estradiol-mediated uterine growth: possible role of the uterine estradiol receptor. Biol Reprod. 1990;42(1):74–80. doi:10.1095/biolreprod42.1.74.
Bever AT, Hisaw FL, Velardo JT. Inhibitory action of desoxycorticosterone acetate, cortisone acetate, and testosterone on uterine growth induced by estradiol-17beta. Endocrinology. 1956;59(2):165–9. doi:10.1210/endo-59-2-165.
Johnson DC, Dey SK. Role of Histamine in implantation: dexamethasone inhibits estradiol-induced implantation in the rat. Biol Reprod. 1980;22(5):1136–1141. http://www.biolreprod.org/content/22/5/1136.abstract. Accessed 26 May 2014.
Zhao L-H, Cui X-Z, Yuan H-J, et al. Restraint stress inhibits mouse implantation: temporal window and the involvement of HB-EGF, estrogen and progesterone. PLoS One. 2013;8(11), e80472. doi:10.1371/journal.pone.0080472.
Chien EJ, Liao C-F, Chang C-P, et al. The non-genomic effects on Na+/H+-exchange 1 by progesterone and 20alpha-hydroxyprogesterone in human T cells. J Cell Physiol. 2007;211(2):544–50. doi:10.1002/jcp.20962.
Szekeres-Bartho J, Barakonyi A, Miko E, Polgar B, Palkovics T. The role of gamma/delta T cells in the feto-maternal relationship. Semin Immunol. 2001;13(4):229–33. doi:10.1006/smim.2000.0318.
Arck P, Hansen PJ, Mulac Jericevic B, Piccinni MP, Szekeres-Bartho J. Progesterone during pregnancy: endocrine-immune cross talk in mammalian species and the role of stress. Am J Reprod Immunol (New York, NY 1989). 2007;58(3):268–279. papers://cf7c60b8-94a1-4c79-88e4-6c57345fd583/Paper/p1437.
Szekeres-Bartho J, Wegmann TG. A progesterone-dependent immunomodulatory protein alters the Th1/Th2 balance. J Reprod Immunol. 1996;31(1–2):81–95. http://www.ncbi.nlm.nih.gov/pubmed/8887124. Accessed 29 May 2014.
Polgar B, Kispal G, Lachmann M, et al. Molecular cloning and immunologic characterization of a novel cDNA coding for progesterone-induced blocking factor. J Immunol. 2003;171(11):5956–63. http://www.ncbi.nlm.nih.gov/pubmed/14634107. Accessed 29 May 2014.
Wiebold JL, Stanfield PH, Becker WC, Hillers JK. The effect of restraint stress in early pregnancy in mice. Reproduction. 1986;78(1):185–92. doi:10.1530/jrf.0.0780185.
Joachim R, Zenclussen AC, Polgar B, et al. The progesterone derivative dydrogesterone abrogates murine stress-triggered abortion by inducing a Th2 biased local immune response. Steroids. 2003;68(10–13):931–40. doi:10.1016/j.steroids.2003.08.010.
Blois SM, Joachim R, Kandil J, et al. Depletion of CD8+ cells abolishes the pregnancy protective effect of progesterone substitution with dydrogesterone in mice by altering the Th1/Th2 cytokine profile. J Immunol. 2004;172(10):5893–9. doi:10.4049/jimmunol.172.10.5893.
Kalinka J, Szekeres-Bartho J. The impact of dydrogesterone supplementation on hormonal profile and progesterone-induced blocking factor concentrations in women with threatened abortion. Am J Reprod Immunol. 2005;53(4):166–71. doi:10.1111/j.1600-0897.2005.00261.x.
Raghupathy R, Al Mutawa E, Makhseed M, Azizieh F, Szekeres-Bartho J. Modulation of cytokine production by dydrogesterone in lymphocytes from women with recurrent miscarriage. BJOG. 2005;112(8):1096–101. doi:10.1111/j.1471-0528.2005.00633.x.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Geraghty, A.C., Kaufer, D. (2015). Glucocorticoid Regulation of Reproduction. In: Wang, JC., Harris, C. (eds) Glucocorticoid Signaling. Advances in Experimental Medicine and Biology, vol 872. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2895-8_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2895-8_11
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2894-1
Online ISBN: 978-1-4939-2895-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)