
Abstract
The significance of glycosphingolipids and glycoproteins is discussed in their relation to normal aging and pathological aging, aging with diseases. Healthy myelin that looks stable is found to be gradually degraded and reconstructed throughout life for remodeling. An exciting finding is that myelin P0 protein is located in neurons and glycosylated in aging brains. In pathological aging, the roles of glycosphingolipids and glycoproteins as risk factors or protective agents for Alzheimer’s and Parkinson’s diseases are discussed. Intensive studies have been performed aiming to remove the risks from and to restore the functional deficits of the brain. Some of them are expected to be translated to therapeutic means.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Aging
- Alzheimer’s disease
- Parkinson’s disease
- Ganglioside
- Glycoprotein
- Cholinergic
- Remyelination
- Sialylcholesterol
- Warfarin
19.1 Preface
Aging is defined as the detrimental process of a body composed of somatic cells after cessation of reproductive activity. The body as a short-term vehicle for passing on the genes to the next generation is evolutionally disposable as based on the disposable soma theory of aging (Kirkwood 1977). The theory means that the body must take natural courses to death. Individuals, however, do not always fulfill their maximum life spans, but frequently suffer from various diseases injurious to their health. In this sense, aging is classified into two types, aging without disease, physiological aging and aging with disease, pathological aging. In this article, glycoconjugate changes in the brain that occur during physiological aging are described first, followed by discussion of their possible participation in the pathophysiology and pathogenesis of age-related neurological diseases. Kobata (2011) introduced the word “glycogerontology” for the field covering the roles of glycoconjugates in relation to gerontology and geriatrics. The physiology and pathology of glycosphingolipids (GSLs) and glycoproteins, will be reviewed here with special reference to their roles in glycogerontology.
19.2 Glycoconjugates in Aging Brains
19.2.1 Age-Related Changes of Glycosphingolipids in the Brain
GSLs are composed of neutral and acidic glycolipids, and the latter are divided into two groups, sulfatides and sialic acid-containing GSLs or gangliosides. The sialyl-oligosaccharide structures of gangliosides show great diversity as a result of the combination of neutral oligosaccharides of various chain lengths and different numbers of sialic acids attached to the core neutral sugars (Ando 1983) as shown in Fig. 19.1. Because of the complexity in their carbohydrate structure, it is hypothesized that gangliosides play crucial roles in cellular events (Cantù et al. 2011; Ohmi et al. 2012; Yu et al. 2012), and as a result they have attracted the most attention of any of the GSLs from researchers in aging research. The supposition is that alterations in the concentration and composition of gangliosides in aging neural membranes should be correlated with age-related functional changes in the brain.

Pathways for formation of sulfatide and gangliosides. Transferases are typed in bold-italic. Cer ceramide, CST galactosylceramide sulfotransferase, Gal galactose, GalNAc N-acetylgalactosamine, Glc glucose, GalT-I galactosyltransferase I, GalT-II galactosyltransferase II, GalT-III galactosyltransferase III, GlcT glucosyltransferase, SA sialic acid. ST-I, II, III, IV, V, and VI correspond to sialyltransferases I (GM3 synthase), II (GD3 synthase), III, IV, V, and VI, respectively
Age-related compositional changes of gangliosides in the human brain were first reported by (Suzuki 1965). In the frontal cortices, GD1a, which is predominant in new-born brains, decreases with age. GM1 also tends to decrease. On the other hand, GD1b and GT1b both increase and by age 30 reach adult levels which normally remain essentially constant until a person is near 90 years of age. These trends in ganglioside pattern changes were supported by the data reported by Segler-Stahl et al. (1983). Similarly, gangliosides GD1b, GT1b, and GQ1b in b-series gangliosides (Fig. 19.1) were shown to increase with age, while GM1 and GD1a decrease (Svennerholm et al. 1989). As GM1 is the major component of myelin (Ledeen and Yu 1982), b-series gangliosides in neural components other than myelin may continue to increase to reduce the relative contents of GM1 in the brain after the period of active myelination. Alternatively, the decrease of GM1 in advanced age may indicate some disintegration of myelin structure. Disruption of the myelin sheath has been reported to occur in aged brain (Peters et al. 2001). The myelin components were shown to never be stable but to be dynamically turned over in physiological aging (Ando et al. 2003). Probably as the consequence of the remodeling of the composition of myelin, cognitive declines with aging were hypothesized to correlate with the altered white matter tracts (Aine et al. 2011; Schulze et al. 2011).
19.2.2 Age-Related Changes of Glycosphingolipids in Synapses
The brain is composed of neurons, glias, and blood vessels. The ganglioside distribution patterns were shown biochemically to be distinct for neural cell types as well as their subcellular fractions (Ando 1983). Immunohistochemical identification of the regional distribution of major gangliosides in the rat brain (Kotani et al. 1993), indicated that a specific ganglioside or set of gangliosides might be responsible for particular neuronal functions. To elucidate their biological roles, it will be important to obtain information on the ganglioside contents and composition of particular cells and subcellular components. In respect to neurons, nerve endings or synapses are functional elements for neurotransmission, and gangliosides in synaptic membranes are thought to affect efficiency of acetylcholine release (Ando 2012; Ando et al. 2004; Tanaka et al. 1997) and long-term potentiation at synapses (Furuse et al. 1998; Wieraszko and Seifert 1986). Waki et al. (1994) developed excellent methods for quantitative isolation of gangliosides from membrane preparations and for their accurate quantitation using gas chromatography–mass spectrometry. Using this method the ganglioside content and composition of mouse brain synaptic plasma membranes was shown to remain constant from adult to senescence. This result seems to be reasonable. Functional units such as synaptic plasma membranes may retain the structure required for their function until senescence, even though the density of the units decreases with age resulting in the changes in ganglioside content and composition observed in tissues or whole brains as described in Sect. 19.2.1.
During the juvenile period of brain development, the content of GD1a was shown to increase and then decrease in the human frontal cortex (Suzuki 1965). GD1a found in cerebral microsomal fractions may be considered a marker for dendritic arborization (Yusuf and Dickerson 1978). Synaptic membrane preparations from mouse brains showed high concentrations of GD1a after birth and reduced levels of GD1a at 6 months of age (Waki et al. 1994). The transient increase in GD1a seemed to coincide with the temporary increase in synaptic density in infant brains (Huttenlocher 1979), and its decrease might correspond to synapse elimination during brain development. GD1a levels then remained constant throughout adult life. These observations indicate that GD1a expression may be related to arborization and synaptogenesis that occur in the initial formation of the neuronal network.
19.2.3 Age-Related Changes in Myelin GSLs
Many studies have examined the effects of age on various brain regions. Most of them, however, were focused on specific areas and the data seemed to be too fragmented to make valid comparisons. Upon comparing results obtained for the composition of 16 automatically segmented measures of the human brain Walhovd et al. (2005) found distinct age changes in different brain structures. Two representative age responses observed were that the volume of white matter showed a curvilinear relationship with age, while the volume of gray matter was reduced in a linear fashion. In the case of cerebral white matter, the volume increased early in life, and decreased during senescence following the steady-state adult period. The main components of white matter are axons and myelin sheaths. While no changes were found in the diameters of axons with age, the numbers of myelin lamellae increased in aged monkeys (Peters et al. 2001). These morphometric observations (Peters et al. 2001; Walhovd et al. 2005) indicate that some changes may continuously take place in myelin components across one’s life span.
Age-related changes in the glycosphingolipids composition of white matter were determined for the human brain (Svennerholm et al. 1994). The three major GSLs, cerebrosides, sulfatides, and gangliosides were found to decrease (μmole per gram tissue weight) with advancing age starting at ~20 years. The same group published somewhat different age-related changes for these glycolipids in separate papers. Cerebroside content in white matter remained constant till 90 years and then sharply declined (Svennerholm et al. 1991). Another paper reported that GM1 in white matter increase in the early developmental age until 1 year and remained at a constant level in the adult (Vanier et al. 1971). These discrepancies might come from their research designs in which glycoconjugates were quantified based on fresh tissue weights. Cellular composition and even tissue water content may change with aging. To avoid such effects analyses should be performed on distinct components of the brain and GSL content related to dry weight.
Norton and Poduslo (1973) isolated myelin fractions from rat brains and found age-related changes in lipid composition along with myelination. The rate of accumulation of cerebroside in the brain paralleled that of myelin. The content of cerebroside in myelin increased during the active myelinogenesis period till 30 days of age, and then remained at constant until rats were 425 days. On the other hand, the content of sulfatide in myelin increased more slowly than that of cerebroside even during active myelinogenesis, and continued to increase gradually during adult life. Sulfatide is produced from cerebroside by the action of galactosylceramide sulfotransferase (Fig. 19.1), and the enzyme is thought to remain active in the remodeling of mature myelin. The biological function of sulfatide was revealed using a mouse model incapable of synthesizing it (Ishibashi et al. 2002). The sulfatide-deficient mice were normal at birth, developed neurological deficits after 6 weeks of life, and survived to >1 year of age. Immunohistochemical studies indicated that sulfatide was not necessary for initial cluster formation, but was essential for proper localization of axonal proteins such as Na+ and K+ channels as well as the maintenance of these proteins around nodes.
Yu and Iqbal (1979) found that gangliosides GM1 and GM4 were concentrated in the myelin fraction isolated from the human brain. Since myelin constitutes the bulk of the oligodendrocyte plasma membranes, GM1 and GM4 may be synthesized in the cell bodies and incorporated into myelin. This leads to the question of why the ganglioside composition of myelin is quite dissimilar to that of parent oligodendrocytes whose ganglioside pattern is complex and rather similar to that of neurons. Saito and Yu (1992) performed an in vitro experiment in which incubation of the myelin fraction obtained from young rats with sialidase was found to produce a ganglioside pattern similar to that observed in in vivo maturation of myelin. They suggested that myelin-associated sialidase may play a role in the processing of gangliosides in myelin membranes. This is an interesting hypothesis in light of another suggesting a role for the sialidase-GM1 interaction in stabilization of the multilamellar structure of myelin sheaths (Saito and Yu 1993). The activities of membrane-bound sialidase were determined for synaptic plasma membranes and nuclear membranes, and their age-related changes examined. The enzyme activities decreased by one-sixth in synaptic plasma membranes (Saito et al. 1995) and by one-third in nuclear membranes (Saito et al. 2002) in old animals.
In my laboratory, the lipid composition of myelin fractions isolated from mouse brains at different ages was examined (Ando 1985). Age-related changes of cerebroside and sulfatide were a little different from those reported for rats (Norton and Poduslo 1973). Our data showed that the contents of cerebroside and sulfatide as expressed per dry weight of myelin both remained at near constant levels from weaning to old age (20–820 days of age), and increased, almost suddenly, at the oldest old age of 1,055 days. With respect to gangliosides in mouse myelin, the content continued to increase from young to old age, and suddenly decreased at the last stage of life. The changes seen in myelin GSLs at the extreme old age may indicate occurrence of an abrupt metabolic disruption in myelin. Similar age-changes in myelin glycolipids at the final stage of life were also reported for human brains (Walhovd et al. 2005; Svennerholm et al. 1991).
To understand the age-related changes in myelin composition metabolic turnover rates for myelin components were measured (Ando et al. 2003). Myelin has a tightly compacted multi-membrane structure containing the least volume of cytosole among membranes, and was not expected to be actively metabolized, so short-term labeling experiments seemed inappropriate. In long-term monitoring studies, depending upon precursors used, the turnover rates of radioisotope-labeled myelin components were reported to vary 6–167 days as half-lives for phosphatidylcholine (Sun and Sun 1979). These big discrepancies may reflect the fact that different tracers recycle at different rates. Our in vivo deuterium-labeling method was shown to eliminate the contribution of the recycling or reuse of labels so that more reliable turnover rates of myelin components could be determined (Ando et al. 2003). Turnover rates of myelin components as half-lives were calculated from decay curves of initially labeled molecules. In fact, very long half-lives of up to 1 year could be measured. Individual components of myelin in the mouse brain were found to be metabolized at separate rates, and their turnover rates were affected differently by aging. Turnover rates of GSLs were calculated from the incorporation rates or disappearance rates of their labeled neutral sugar moieties. Cerebroside and GM1 appeared to be rapidly incorporated into myelin in infant brains as were cholesterol and phospholipids, and their turnover rates varied with aging, decreasing during young and adult periods and rebounding in the senescence stage. The accelerated metabolism of myelin in old age may explain the curious compositional changes observed at the last stage of a mouse’s life (Ando 1985) as well as in human brains (Walhovd et al. 2005; Svennerholm et al. 1991). The metabolism of GM1 appeared to be composed of two compartments in myelin, one with a short half-life of 52 days and another with a long half-life of 131 days, which correspond to rapidly and slowly exchanging pools, respectively (Ando et al. 2003). The concept that metabolic turnover of membrane components occurs continuously in mature myelin and the finding that turnover rates change with age may provide a better understanding of the mechanism underlying myelin aging.
19.2.4 Alterations in the Lipid Portions of Glycosphingolipids with Aging
GSLs are inserted into the outer layer of neural membranes through their ceramide portions. Ceramide is composed of a fatty acid linked to sphingosine via an amide linkage. It is known that the ceramide portion plays a number of biological roles. It may control the interaction of the saccharide portion with external ligands (Kannagi et al. 1982), and regulate the aggregative properties and surface dynamics of the GSLs (Yohe et al. 1976). In a model system using synaptosomes and liposomes increasing concentrations of gangliosides in the membranes were shown to increase membrane fluidity or decrease membrane microviscosity (Ando et al. 1986). Gangliosides may affect membrane physicochemical characteristics such as fluidity through the homophilic interaction of the sialylsaccharides and the heterophilic interaction of the saccharides with other ligands, and further through the lipidic interaction of their ceramide portions with other lipids in membranes. Recently, Furukawa’s group (Ohmi et al. 2012) showed, using mice expressing mutant ganglioside synthases, that glycolipid-enriched microdomain/rafts architecture was destroyed by ganglioside deficiency. Others generated mutant mice defective in their ability to synthesize ceramide containing very long-chain fatty acids (C22-C24) due to ablation of ceramide synthase 2 (Ben-David et al. 2011). The mice had reduced levels of both nonhydroxy-C22-C24- and 2-hydroxy-C22-C24-galactosylceramide, and developed brain lesions. Myelin degeneration and detachment occurred in the brain. The mice also exhibited abnormal motor behavior and histological abnormalities such as vacuolization and astrogliosis.
Age-related changes in the ceramide portion of GSLs were studied in order to learn more about the probable physiological roles of ceramides. Changes in fatty acid composition were studied using samples from human brains (Mansson et al. 1978; Svennerholm and Ställberg-Stenhagen 1968). The adult fatty acid composition of cerebrosides and sulfatides in regard to degree of unsaturation and total percentage of C22-C26 acids was reached at 2 years of age, but the percentage of odd-numbered fatty acids continued to increase up to about 10–15 years. Fatty acid changes occurred in cerebrosides first and in sulfatides later. The delay in sulfatides may reflect the fact that cerebrosides are precursors in the synthesis of sulfatides (Norton and Poduslo 1973). Stearic acid was reported to be the major component in the ceramide portions of gangliosides in human brains (Mansson et al. 1978). The proportion of stearic acid was found to decrease from about 94 to 86 % in both GM1 from white matter and GT1 from the cerebral cortex upon aging. These data were obtained from studies of three brains, one from a 77 year old (Svennerholm and Ställberg-Stenhagen 1968), one from a 71 year old and one from an 89 year old (Mansson et al. 1978).
Long-chain bases (LCBs) or sphingosines in the ceramide portion of GSLs in the brain are composed of two major species, d18:1 and d20:1. The compositional changes in LCB with aging were first observed in human brains (Mansson et al. 1978). The averaged values for the molar proportion of d20:1 were shown to increase from about 20 to 70 % with advancing age. Age-related changes in the LCBs of gangliosides were studied in detail using rat forebrains (Palestini et al. 1990). The d18:1 LCB, predominant at 3 days (91–96 %), diminished with age and at 2 years was 73, 65, 61, 59, and 45 % of the total for GD1a, GM1, GT1b, GD1b, and GQ1b, respectively. The content of d20:1 LCB, low at birth (4–9 %), increased with age in all gangliosides and at 2 years 27–55 % of the total. Molecular species of all gangliosides carrying d18:1 LCB were virtually devoid of C20 fatty acid. Analysis of the ceramide portions of gangliosides isolated from synaptosomes and myelin fractions of rat brains of different ages indicated that the fatty acid composition did not undergo appreciable changes (Palestini et al. 1993). Large age-related changes in LCB composition were observed in all gangliosides in both synaptosomal and myelin fractions. The steady increase in the proportion of d20:1 LCB observed in the two subcellular fractions appeared to coincide with the age-related changes reported for gangliosides isolated from rat whole brains.
Sugiura et al. (2008) provided a good answer for the question of whether the different molecular species would show different distribution patterns in the brain. Using imaging mass spectrometry, they found that gangliosides containing d18:1 or d20:1 LCB were differentially distributed in mouse brain. While the d18:1-species was widely distributed throughout the frontal brain, the d20:1-species selectively localized along the entorhinal-hippocampal projections, especially in the molecular layer of the dentate gyrus. The finding of developmental- and age-related accumulation of the d20:1 species in the hippocampal formation provided evidence that changes in the ganglioside molecular species may contribute to the process of brain aging.
19.2.5 Age-Related Changes in the Carbohydrate Structure of Glycoproteins
The glycan moieties of glycoproteins are known to play crucial roles not only in modulating the property of the stem glycoproteins, but also in regulating various molecular recognition processes (Kobata 1992). In the developing brain, nervous system glycans have been implicated as important mediators of adhesive interactions among neural cells (Schachner and Martini 1995). Age-related changes in glycoproteins in the central nervous system (CNS) have been infrequently documented in the literature, but evidence for them has been summarized in review articles (Kobata 2011; Sato and Endo 2010). The myelin glycoprotein P0 is one that has been studied.
Originally using a goat anti-P0 antibody and immunofluorescence, the myelin P0 glycoprotein was found to be located exclusively in the myelin of peripheral nerves, but not in CNS myelin (Ishaque et al. 1980). Subsequently, Endo’s group (Sato et al. 1999) was able to show that P0 was present in the spinal cord of the rat. They further demonstrated by immunohistochemical and immunocytochemical analyses that CNS neuronal cells expressed P0 (Sato and Endo 2000). To explore whether any age-related changes occurred in the glycans of CNS P0, glycoproteins obtained from the brain or spinal cord of 9-week old and 29-month old rats were separated by electrophoresis and stained with Lens culinaris agglutinin (Sato et al. 1999). This lectin specifically binds to the P0 glycan. While the glycoprotein patterns of spinal cords showed marked differences between the two age groups, samples from brains did not. Nonglycosylated P0 molecules present in the young spinal cord were replaced with glycosylated ones during aging. As it was reported that the glycan moiety of P0 plays an important role in cell–cell adhesion (Yazaki et al. 1992), the appearance of glycosylated P0 may function in the remodeling of neural structures that occurs with aging. A study on age-related changes in the glycan structure of peripheral nerve myelin glycoprotein P0 indicated that P0 from adult rats contained high-mannose and/or hybrid-type oligosaccharides not seen in P0 from 5-day-old nerves (Brunden 1992).
Following the finding of expression of P0 in the mammalian CNS, its cellular localization in rat spinal cord was examined by immunohistochemical and immunocytochemical methods using a polyclonal anti-P0 antibody (Sato and Endo 2000). Nissl staining-positive motor neurons and sensory neurons showed strong reactivity with the antibody. The neuronal localization of P0 was confirmed by double immunofluorescence labeling using the anti-P0 antibody and an anti-neurofilament monoclonal antibody. Further analysis indicated that neurons expressing P0 in the spinal cord of 30 month-old rats had a different morphology than those of 14 week-old rats. The number of neurons stained in the old rats was about 80 % of that in the young rats, while the average size of neurons in the old rats was about 62 % of that in young rats. These observations indicate that both cell number and average size of neurons in the spinal cord decreased with age supporting the hypothesis that the activity and survival of the neurons might be regulated or changed by age-related changes in glycosylation of P0.
Endo’s lab (Sato et al. 2006), using 2D-electrophresis using Concanavalin A staining, surveyed the contents of cytosolic glycoproteins in rat cerebral cortices and found several spots increased in the aged brains. The glycoprotein that was most prominently increased was identified as cathepsin D. It was detected in the cytosolic fractions of aged rats, but not in those of young adult rats. Cathepsin D in the microsomal fractions did not show age-related changes. The increase of cytosolic cathepsin D during aging was not due to disruption of the lysosomal membrane, because other lysosomal enzymes did not increase in the cytosolic fractions. The level of cathepsin D transcripts in aged rats was 1.6 times higher than in the young adults. Cathepsin D is known to digest neurofilaments and tau (Bednarski and Lynch 1996) which are components of the cytoskeleton of neuronal cells. The enhanced expression of cathepsin D may facilitate cytoskeletal degradation leading to morphological changes and functional loss of neurons in aged brains.
Sialic acid is present in both glycoproteins and gangliosides. It attaches to the nonreducing terminals of sugar chains by α2-3, α2-6, or α2-8 ketosidic linkage. A homopolymer of α2-8-linked sialic acid moieties (polysialic acid, PSA) is found on neural cell adhesion molecules (NCAM), and is known to modulate the adhesion property of NCAM and to regulate neurite outgrowth and cell migration (Brusés and Rutishauser 1998). PSA-NCAM (embryonic form of NCAM) was shown to be present in adult brain regions where neuronal regeneration occurred but its expression decreased during aging (Seki and Arai 1991). PSA-NCAM expression is lost after the cessation of neuronal cell migration and synapse formation, but can be retained by axons capable of synaptic remodeling.
The histochemical distribution of sialyl α2-3 galactose and sialyl α2-6 galactose was examined in the rat hippocampus using Maackia amurensis lectin to label the former, and Sambucus sieboldiana for the latter, and electron microscopy to visualize them (Sato et al. 2001). Both lectins stained the plasma membranes of pyramidal cells and synapses. The staining intensity by both the lectins of synapses was reduced in 30-month-old rats. In addition to staining glycoproteins, the Maackia amurensis lectin could also bind all of the series of gangliosides possessing sialyl α2-3 galactose linkages. The Sambucus sieboldiana lectin could bind the α-series of gangliosides possessing sialyl α2-6 galactose. Expression of sialyl α2-6 galactose decreased in the aged brain.
19.3 Glycoconjugates and Age-Related Diseases
19.3.1 Gangliosides and the Pathology of Alzheimer’s Disease
An interesting question addressed by Mizutani’s group was whether the histopathological changes that occur in Alzheimer’s brains were the results of accelerated aging (Mizutani and Kasahara 1997; Yamada et al. 1998). In advancing physiological or normal aging, morphometric measurements of brain volumes in comparison with intracranial volumes showed a very slow progression of brain atrophy and an insignificant correlation between the rate of atrophy and age. In contrast, similar measurements with Alzheimer’s brains revealed intensive atrophy occurred (Yamada et al. 1998). The hippocampal atrophy observed in Alzheimer’s brains was distinct from that seen in control, nondemented brains (Mizutani and Kasahara 1997). In the hippocampal formation of control brains, no atrophy was observed in the hippocampus and parahippocampus. In fact, in all cases with Alzheimer’s disease the stratum lacunosum-radiatum was decreased in thickness, and this was accompanied by loss of myelin and fibrillary gliosis. Interestingly, the numbers of senile plaques and neurofibrillary tangles (NFTs) and the degrees of neuronal loss in the pyramidal layer varied from case to case. The severer degeneration in the parahippocampus including the entorhinal cortex than in the hippocampus indicates that degeneration of the entorhinal cortex may induce degeneration of the hippocampus because the perforant pathway in the hippocampus originates from the entorhinal cortex. Entorhinal cortex dysfunction was detected in early Alzheimer’s disease by positron emission tomography (Eustache et al. 2001). In regard to cognitive dysfunction, the concept of “synaptic pathology,” in which synaptic loss is the major correlate of cognitive deficits, was proposed by Terry et al. (1991; Masliah and Terry 1993), and supported by others (Blennow et al. 1996; Heinonen et al. 1995; Scheff et al. 1993). Based on the observations made using Alzheimer’s brains, a dementia animal model was successfully generated by inducing synapse-specific lesions in the entorhinal cortex (Ando et al. 2002). This supported the synaptic pathology hypothesis.
Alzheimer’s disease is an age-related disorder, but is not the result of advanced normal aging. The progressive cognitive dysfunction seen in Alzheimer’s disease is characterized by the accumulation of senile plaques and NFTs, and the degeneration of neurons in brain regions such as the hippocampal formation. A recent review of the literature on the correlation of Alzheimer’s neuropathologic changes with cognitive status stresses that the severity of cognitive impairment correlates best with the burden of neocortical NFTs (Nelson et al. 2012). NFTs are related to neuronal loss. In contrast, from the standpoint of the pathogenesis of Alzheimer’s disease, generation of amyloid β (Aβ)-peptides is thought to be at the beginning of a cascade that leads to the disease. Recent studies indicate that assembly of Aβ-peptides into Aβ-oligomers or protofibrils, can cause cognitive declines by disrupting synaptic function (Dahlgren et al. 2002; Matsumura et al. 2011; O’Nuallian et al. 2010). Wild type Aβ monomers assemble first into protofibrils and then amyloid fibrils to form senile plaques. Aβ-peptides deposit in diffuse plaques, the earliest stage of senile plaques. Yanagisawa et al. (1995) isolated the diffuse plaque fraction as a “light Aβ” from Alzheimer’s disease and Down’s syndrome brains and found that Aβ associated with diffuse plaques bound to ganglioside GM1 in a noncovalent fashion. Yanagisawa’s group hypothesized that GM1-bound Aβ acted as a seed in the initiation of amyloid fibril formation (Kakio et al. 2001). Another in vitro experiment showed that Aβ selectively bound to membranes containing GM1 and that no Aβ binding was observed with GM1-free membranes (Choo-Smith et al. 1997). Studies of brains from monkeys indicated that GM1-Aβ was formed in early endosomes and transported to late endosomes for degradation (Kimura and Yanagisawa 2007). It is thought that impaired recycling in the endosome pathway results in accumulation of GM1-Aβ in endosomes and that its subsequent transport to the cell surface where it acts as a seed for amyloid fibril formation. Furthermore, it was shown that a toxic soluble Aβ assembly was formed in a GM1 dependent manner through incubation of soluble Aβ with neuronal membranes prepared from aged mouse brains (Yamamoto et al. 2007). These observations may help to define the mechanism underlying the plaque-independent neuronal death seen in Alzheimer’s disease.
19.3.2 Expression of Unusual Gangliosides in the Alzheimer’s Brain Indicative of Brain Plasticity
Expression of c-series gangliosides in Alzheimer’s brains was reported by our laboratory (Takahashi et al. 1991). Since the c-series of gangliosides with trisialyl residues were structurally identified (Ando and Yu 1979), the metabolic pathway for their synthesis from GT3 (Fig. 19.1) was established (Yu et al. 2004). The c-series gangliosides were found to be expressed in embryonic brains and designated as fetal antigens (Hirabayashi et al. 1988). Anti-c-series ganglioside antibodies were found to label neuritic elements composing senile plaques as well as perivascular amyloid deposits (Takahashi et al. 1991). Positive staining with the antibodies was not observed in brains of nondemented individuals, except for those containing a small number of senile plaques. NFTs were immunolabeled with the monoclonal antibody A2B5, which had been shown to recognize the c-series ganglioside, GQ1c (Emory et al. 1987). Subsequently A2B5 was shown to react with gangliosides other than those in the c-series, and with sulfatides (Majocha et al. 1989) and even with glycoproteins having α2,8-trisialic acid units (Inoko et al. 2010). In this context, our specific anti-c-series ganglioside antibodies definitely demonstrated the presence of the fetal gangliosides in Alzheimer’s brains. This finding was strengthened by the evidence that another fetal antigen, microtubule-associated protein 5 (MAP5), colocalized with c-series gangliosides in NFTs (Takahashi et al. 1991). The expression of fetal antigens such as c-series gangliosides and MAP5 may indicate that neuronal regeneration (cell proliferation and sprouting) occurs along with neuronal degeneration in the Alzheimer’s brain. Evidence for the regeneration in the Alzheimer’s brain was also provided by the observation of increased dendritic sprouting and arborization (Probst et al. 1983; Scheibel and Tomiyasu 1978). These findings are consistent with the concept that neuronal regeneration does occur while degeneration proceeds (Lopez-Toledano and Shelanski 2004; Uchida 2010). As the brain has innate plasticity, it is possible that development of synapses achieved by various interventions may help protect the brain from degenerative insults and to recover from their damage (Ando 2012).
19.3.3 Functional Significance of α-Series Gangliosides in Alzheimer’s Disease
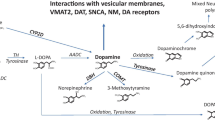
Whittaker and his colleagues (Richardson et al. 1982) found that cholinergic-specific antigens in the Chol-1 family, comprised of Chol-1α, Chol-1β, and Chol-1γ, were gangliosides. We isolated and characterized two molecular species corresponding to Chol-1α, termed GT1aα and GQ1bα as illustrated in Fig. 19.2 (Ando et al. 1992; Hirabayashi et al. 1992). The “α” in the abbreviations is assigned to the unique branching structure of a sialic acid residue linked α2-6 to the N-acetylgalactosamine in the gangliotetraose backbone (Taki et al. 1986). Immunostaining indicated that Chol-1α was present in cholinergic nuclei such as the septal nucleus (Irie et al. 1994). To examine the involvement of gangliosides in the function of cholinergic synapses, a series of monoclonal antibodies against gangliosides were tested for their ability to suppress release of acetylcholine. Only the anti-Chol-1α antibody, GGR-41, affected acetylcholine release (Ando et al. 2004). To ascertain its mechanism of action, choline uptake and acetylcholine synthesis were measured using synaptosomes in the presence of GGR-41. Both choline uptake and acetylcholine synthesis were inhibited in a dose-dependent manner by GGR-41. When Chol-1α was added to a synaptosomal fraction, it accelerated high affinity choline uptake into synaptosomes and this resulted in enhancement of acetylcholine synthesis. These observations led to the question of whether Chol-1α gangliosides participated physiologically in the cognitive function of the brain. To answer this, GGR-41 was continuously infused into the rat brain septal area in order to disrupt the septohippocampal cholinergic pathway (Ando et al. 2004). This resulted in a reduction in the learning ability of the rats similar to that seen in rats given mecamylamine, a nicotinic cholinergic receptor antagonist. Memory retention was also severely impaired in rats infused with GGR-41. Chol-1α is thought to localize with nicotinic acetylcholine receptors because it was originally found in torpedo electric organs composed of pure nicotinic nerve terminals (Richardson et al. 1982). In sum, Chol-1α functions to accelerate acetylcholine turnover in cholinergic synapses and serve as a cholinergic marker.

Synthetic pathways for α-series gangliosides (Chol-1α antigens). Dashed lines indicate sialylation reactions catalyzed by sialyltransferase VII that catalyzes formation of sialyl 2-6 N-acetylgalactosamine linkages. Alpha-series of gangliosides marked by *1, *2, and *3 were characterized as Chol-1α by Irie et al. (1996), Ando et al. (1992) and Hirabayashi et al. (1992), respectively
Expression of Chol-1α antigens is known to be developmentally regulated as they appear at the time of cholinergic synapse formation in the rat brain (Derrington and Borroni 1990). Neurogenesis and neuronal regeneration are enhanced in cholinergic lesioned brains (Ho et al. 2009) and by environmental stimulation in aged brains (Nakamura et al. 1999). Expression of fetal c-series gangliosides in Alzheimer’s brains may also reflect neuronal plastic responses (Section 19.3.2). To examine possible alterations in ganglioside metabolism in relation to Alzheimer’s disease, animal models with disrupted ganglioside biosynthesis have been developed (Ariga et al. 2010, 2011, 2013; Bernardo et al. 2009; Oikawa et al. 2009). Ganglioside changes were analyzed in brains from double transgenic (Tg) mice that coexpressed amyloid precursor protein with the Swedish mutation and presenilin-1 with a deletion of exon 9 (Ariga et al. 2010). No significant changes were detected in the concentration and composition of major gangliosidesin brains from the double-Tg mice. In contrast, expression of cholinergic gangliosides such as GT1aα and GQ1bα (Chol-1α antigens) increased. Their increased expression may reflect cholinergic neuronal regeneration response to damages induced by Aβ. The above double-Tg mice were cross-bred with GD3S(St8sia1)-/- mice to generate mice deficient in GD3-synthase responsible for synthesis of b-series gangliosides (Fig. 19.1) (Bernardo et al. 2009). In the triple-Tg brains all the b-series gangliosides including GD3 were absent, while GM1 and GD1a were increased. Surprisingly, triple-Tg mice showed memory performance similar to that of wild-type control and GD3S−/− mice, while the double-Tg mice exhibited cognitive impairments. Consistent with normalized cognition, Aβ plaques were almost eliminated. These results indicate that b-series gangliosides may be one of the causes of Aβ accumulation. Remarkably in the triple-Tg brain, the concentration of GT1aα was elevated, while no expression of GQ1bα was observed. Thus, the elevated cholinergic ganglioside GT1aα may contribute to memory retention (Ariga et al. 2013). The double-Tg mice expressing human amyloid precursor protein having the Swedish and London mutations were crossbred with GM2-synthase knockout mice (Oikawa et al. 2009). The mutant mice expressing a large amount of GM3 but not GM1 showed a remarkable increase of Aβ deposition in vascular tissues (amyloid angiopathy). This observation may indicate another mechanism exists for Aβ deposition than that covered by the hypothesis that GM1-bound Aβ is involved in amyloid fibril formation (Kakio et al. 2001) (Sect. 19.3.1). These results indicate that the significance of gangliosides relevant to the physiology and pathology of the brain cannot be deduced simply from studies of the loss-of-function or gain-of-function of each ganglioside. Further comprehensive studies are needed.
19.3.4 Glycoproteins: In Connection with Alzheimer’s Disease
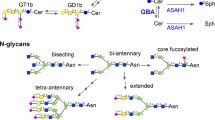
Core N-glycosylation and N-glycans were reported to modulate the synthesis of amyloid precursor protein (Pahlsson et al. 1992), suggesting that N-glycosylation status might affect the metabolic pathway of amyloid precursor protein (APP). Analysis of the N-glycan structures determined of mutant amyloid precursor proteins (Swedish type and London type) produced by transfected C17 cells showed that they contained higher contents of bisecting N-acetylglucosamine (GlcNAc) residues than normal APP (Akasaka-Manya et al. 2008). To examine the reason for overexpression of the bisecting structure, expression of N-acetylglucosaminyltransferase III (GnT-III)-mRNA (GnT-III is the enzyme responsible for synthesizing bisecting GlcNAc residues) (Fig. 19.3) was measured and found to be increased in Alzheimer’s brains (Akasaka-Manya et al. 2010). Incubation of Neuro2a cells with Aβ42 increased GnT-III gene expression. In a separate setting, Neuro2a cells transfected with a GnT-III expression vector downregulated Aβ production. Fiala et al. (2007) reported that blood monocytes exposed to Aβ peptides upregulated transcription of GnT-III and increased Aβ clearance by phagocytosis. Thus, upregulation of GnT-III appears to exert two effects: inhibition of Aβ production in neurons and enhancement of its clearance by monocytes. Both responses can be protective against the further progression of Alzheimer’s disease.

Bisecting GlcNAc residues in N-glycans synthesized by N-acetylglucosaminyltransferase III (GnT-III)
The possible mechanisms by which bisecting GlcNAc residues could reduce Aβ production include the following (Akasaka-Manya et al. 2010): (1) Addition of bisecting GlcNAc may affect the conformation of the APP, thereby inducing changes in its susceptibility to α-, β-, and/or γ-secretases. (2) The increase of bisecting GlcNAc on the APP leads to changes in its N-glycan structure such as diminished degrees of elongation, branching, and sialylation. (3) Bisecting GlcNAc affects trafficking of the APP and, as a result, its susceptibility to secretases. This is supported by the report that localization and trafficking of APP is affected by its glycan modifications (McFarlane et al. 1999). (4) The bisecting GlcNAc directly affects secetase activities. Support for this possibility was provided the observations that (a) enzyme activities of α- and β-secretases were significantly increased and decreased, respectively, in GnT-III-transfected Neuro2a cells (Akasaka-Manya et al. 2010); (b) Western blot analysis indicated that changes in N-glycan structures were present in the secretases (TACE and BACE). These findings may account for changes in secretase activities and resultant inhibition of Aβ formation.
19.3.5 Glucocerebrosidase Gene Mutations as a Risk Factor for Parkinson’s Disease
Some patients with Gaucher disease were found to develop parkinsonism (Mitsui et al. 2009; Rhouma et al. 2012; Sunwoo et al. 2011). Gaucher disease is an autosomal recessive, lysosomal storage disease caused by mutations in the β-glucocerebrosidase gene (Beutler and Grabowski 1995). Glucocerebrosidase (GBA) is a lysosomal enzyme that catalyzes the hydrolysis of glucocerebroside to ceramide and glucose. Homozygous mutations in the GBA that affect its activity result in accumulation of glucocerebroside in various tissues. Heterozygous loss of function mutations at the GBA locus are a potent risk factor for Parkinson’s disease (PD) (Lwin et al. 2004) as evidenced by the finding of multiple cases of parkinsonism among Gaucher disease carriers (Goker-Alpan et al. 2004).
An international collaborative study was done to ascertain the frequency of GBA mutations in ethnically diverse patients with PD (Sidransky et al. 2009). Sixteen centers participated, including five from the Americas, six from Europe, two from Israel, and three from Asia. Two GBA mutations, L444P and N370S, were found in 15.3 % of Ashkenazi Jewish patients with PD, and in 3.2 % of non-Ashkenazi patients indicating a strong association between GBA mutations and PD. When a complete sequence analysis of the variants was carried out for a large cohort of European, mostly French, patients with PD (Lesage et al. 2011), the results revealed that carrier frequency in the non-Ashkenazi Jewish populations was 7 %, much higher than that, the 3.2 %, reported by the above 16 centers’ study (Sidransky et al. 2009). These results indicate that limited screening might miss more than half of the mutant alleles. They also showed that GBA mutations were significantly more frequent (odds ratio = 6.98, 95 % confidence interval 2.54–19.21; p = 0.00002) in PD than in controls. After the 16 centers’ study, resequencing of GBA was performed for Japanese patients with PD. The frequency of pathogenic variants in the heterozygous state was shown to be 9.4 % in Parkinson’s patients compared to 0.37 % in controls (odds ratio, 28.0). Mutations in the GBA gene are hypothesized to accelerate the pathogenesis of PD (Clark et al. 2007). Support for this was provided by the observation that GBA carriers had a 2.5 year earlier age of onset of PD compared to noncarriers. Although genetic research in the past delineated many mutations that cause PD, such as those in genes encoding E3 ubiquitin ligase parkin (PARK2), leucine-rich repeat kinase 2 (LRRK2), and alpha-synuclein (Bras et al. 2008), research on GBA mutations supports the idea that they are the most common genetic risk factor for PD (Lesage et al. 2011; Neumann et al. 2009).
To address the question of what deleterious effects are caused by GBA mutations, cerebral metabolic activity was assessed in carriers of the GBA mutation both with and without parkinsonism using positron emission tomography (Kono et al. 2010). All GBA mutation carriers had significantly decreased glucose metabolic rates in the supplemental motor area (cortex region anterior to the primary motor cortex), and the carriers with parkinsonism showed additional hypometabolism. The hypometabolism in the cortex region may be related to the clinical manifestation of parkinsonism. Measurement of GBA activity showed it decreased significantly in brains of Parkinson’s patients carrying heterozygous GBA mutation (Gegg et al. 2012). The greatest deficiency was found in the substantia nigra (58 % decrease) known to be affected in PD. In PD the brain loses dopaminergic neurons which results in severe reduction in the dopamine content of the striatum (Hornykiewicz 1966). Protein levels of GBA were also reduced, indicating that the lowered expression of the enzyme as well as its decreased activity could contribute to its deficiency (Gegg et al. 2012). Immunofluorescence studies on brain tissues from patients with PD associated with GBA mutations showed that the enzyme was present in 75 % of Lewy bodies while GBA-positive Lewy bodies were found in only 4 % of the subjects without mutations (Goker-Alpan et al. 2010).
Possible mechanisms for the link between GBA gene mutations and PD have been speculated. Mutations in α-synuclein are known to result in aberrant aggregation of the protein, which is associated with neuronal death. The aggregated polymers are proposed to be necessary prerequisites for Lewy body formation seen in PD (Ishizawa et al. 2003). Functional loss of GBA in primary cultured neurons was found to compromise lysosomal protein degradation, cause accumulation of α-synuclein, and result in neurotoxicity through aggregation-dependent mechanisms (Mazulli et al. 2011). These results indicate that increased glucosylceramide, the GBA substrate, directly influences amyloid formation of α-synuclein. Increased glucosylceramide due to a deficiency of GBA is speculated to disrupt the membrane binding of α-synuclein, enhancing its aggregation in the cytoplasm (DePaolo et al. 2009). Another theory about the relationship of GBA to PD involves its interference with the clearance of mutated proteins. Since most mutations in GBA are missense mutations, the protein likely becomes aberrantly folded and as a result undergoes parkin-mediated poly-ubiquitination and subsequent proteasome-mediated degradation. The presence of mutant GBA might cause build-up of other parkin substrates, causing endoplasmic reticulum-stress and eventual apoptosis of the neurons (Westbroek et al. 2011). Finally, a physical linkage between α-synuclein and GBA was verified using immunoprecipitation and immunofluorescence (Yap et al. 2011).
19.3.6 Gangliosides: In Relation to the Pathogenesis of Parkinson’s Disease
The symptoms of PD are manifested after dopaminergic innervation of the striatum is lost as a result of degeneration of dopaminergic neurons in the substantia nigra (Hornykiewicz 1966). Over two decades ago GM1 was hypothesized to act as a neurotrophic factor for dopaminergic neurons (Schneider et al. 1992). A comprehensive review on degenerative diseases and the therapeutic potentials of gangliosides are described in Chap. 20. A causative aspect of reduced gangliosides in PD is partly discussed here. Recently Using a genetic mouse model of Parkinson’s disease in which major gangliosides were depleted Ledeen’s group (Wu et al. 2011) proved that GM1 was involved in the pathogenesis of parkinsonism. The knockout mice were generated by disrupting the B4galnt1 gene for GM2/GD2 synthase thereby eliminating synthesis of GM2, GD2, and all gangliotetraose gangliosides. The B4galnt1−/− mice manifested clinical parkinsonism and the pathological loss of dopaminergic neurons in the substantia nigra. The symptoms of parkinsonism were largely attenuated by administration of a GM1 analogue, LIGA-20, developed by Costa’s group (Manev et al. 1990). This finding supports the hypothesis that GM1 is involved in the pathogenesis of PD. Ledeen’s group (Wu et al. 2012) also found that heterozygous mice with one defective allele for the B4galnt1 gene displayed virtually the same degree of parkinsonism as the knockout mice. Interestingly, the levels of GM1 and GD1a, a-series gangliosides (Fig. 19.1), decreased in the heterozygous mice by 43 % and 46 %, respectively, while those of b-series gangliosides did not significantly decrease. Loss of dopaminergic neurons was evident in the heterozygous mice, and dopamine levels in the striatum decreased progressively as the animals aged. Treatment with LIGA-20 increased the number of tyrosine hydroxylase-positive neurons in the substantia nigra and levels of dopamine in the striatum, indicating recovery of dopaminergic neurons. Combined these observations indicate that the heterozygous mouse model carrying the mutant B4galnt1 gene will be quite useful in future studies of PD.
In human PD, the number of dopaminergic neurons in the substantia nigra, identified by staining for tyrosine hydroxylase, decreased by 40 % compared to that in controls (Wu et al. 2012). Quantification of dopaminergic neurons expressing GM1 in 11 Parkinson’s patients revealed a noticeable GM1 deficiency: 19.7 % of dopaminergic neurons in the patients compared to 61.8 % in controls. This provides an interesting parallel to the deficit of GM1 observed in the heterozygous mouse model discussed above. GM1 staining with cholera toxin B-FITC (specific for GM1) was significantly decreased in both nondopaminergic neurons as well as dopaminergic neurons in Parkinson’s brain sections, indicating the widespread abnormal expression of gangliosides in the diseased brain. Not addressed is whether the decreased GM1 is the cause for or result of PD. So far no mutations of genes involved in ganglioside metabolism have been identified as risk factors for PD.
Anti-GM1 ganglioside antibodies have been found in the sera of patients with neurological diseases such as lower motor neuron syndromes (Pestronk et al. 1990; Sunwoo et al. 2011), amyotrophic lateral sclerosis (Pestronk et al. 1988; Rhouma et al. 2012), and Alzheimer’s (Chapman et al. 1988; Saito et al. 2002). In an early survey of a limited number of parkinsonian demented patients high titers of anti-GM1 antibodies were found (Saito et al. 2002). Subsequently Zappia et al. (Zappia et al. 2002) studied a large group of PD patients, most of whom had a tremor-dominant form of the disease, and found that more than one-quarter had sera with increased levels of IgM anti-GM1 antibodies. It is hypothesized that increased anti-GM1 antibodies could affect the function of ganglioside GM1 in dopaminergic neurons. Anti-ganglioside immune responses are speculated to contribute to axonal damage via a T cell-mediated mechanism in multiple sclerosis (Pender et al. 2003; McFarlane et al. 1999). When patients with lower motor neuron syndromes and high serum titers of IgM anti-GM1 antibodies were subjected to repeated plasma exchanges (Pestronk et al. 1994; Beutler and Grabowski 1995), removal of the antibodies brought about progressive improvement in muscle strength. These findings indicate that anti-ganglioside antibodies may act as a risk factor for neuronal dysfunction or disease pathogenesis.
19.4 Strategies for Anti-Aging and Prevention of Age-Related Diseases
Studies of glycoconjugates expression and aging have provided much information (Sects. 19.2 and 19.3), and some may provide the basis for development of future therapeutics. Possibilities include use of the knowledge for inducing remyelination, clearance of Aβ from the brain, and enhancement of innate neuronal plasticity.
19.4.1 Myelin Repair and Remyelination
The fact that the volume of white matter decreases in senescence (Walhovd et al. 2005) indicates that enhanced demyelination occurs in aging brains and more severely in demyelinating disease brains (Zhang et al. 2011). Cognitive declines appear to correlate with altered myelination of nerve tracts (Aine et al. 2011; Schulze et al. 2011). While disruption of mature myelin is going on, remyelination takes place (Peters and Sethares 2003). This is supported by observations indicating that the number of myelin lamellae increase (Peters et al. 2001) and intermodal lengths of myelin get shorter with advancing age (Bowley et al. 2010). To enhance myelin repair or remyelination following demyelination, promoters of remyelination have been examined as potential therapeutics. The age-associated inefficiency of remyelination (Shields et al. 1999) is known to be due to impairment of both recruitment and differentiation of oligodendrocyte progenitor cells (OPC) (Sim et al. 2002). Myelin synthesis is preceded by downregulation of OPC differentiation inhibitors such as PSA-NCAM (Shen et al. 2008). This downregulation is epigenetically controlled by recruitment of histone deacetylases to promoter regions, indicating that efficient remyelination requires deacetylation of nucleosomal histones. Epigenetic control of remyelination is potentially an important therapeutic target. Another promising intervention to enhance remyelination is hormonal stimulation of OPC (Calzà et al. 2010; Fernandez et al. 2004). Thyroid hormone was shown to regulate proliferation and differentiation of OPC and induce myelinating oligodendrocytes. Actually, systematic administration of thyroid hormone enhanced myelination in an experimental allergic encephalomyelitis model (Fernandez et al. 2004) as well as in a cuprizone-induced demyelination model (Franco et al. 2008). Using an in vivo deuterium labeling method (Ando et al. 2003) thyroxine was found to enhance incorporation of ganglioside GM1 into mature myelin of adult mice (Ando et al. 1984).
19.4.2 Acceleration of Aβ Clearance
Aging is the major risk factor for neurodegenerative diseases such as Alzheimer’s disease and PD (Sect. 19.3) with accumulation of aberrant precipitates of amyloid-β peptide in brains from the former and α-synuclein in those from the latter. Clearance of those toxic compounds from the brain is driven by an active transport system at the blood–brain barrier (BBB) and blood–cerebrospinal fluid barrier. In the transport system at the barriers, the receptor low-density lipoprotein receptor-related protein 1 takes up the toxic compounds at the abluminal sites of capillary endotherial cells, and P-glycoprotein (P-gp) at the luminal sites of the cells excretes them into the blood stream. Decreased function of P-gp was observed in Alzheimer’s patients (Vogelgesang et al. 2004) and in Parkinson’s patients (Bartels et al. 2008). The expression of P-gp decreases in the BBB with age (Silverberg et al. 2010). As the reduced clearance of amyloid-β across the BBB is thought to enhance amyloid accumulation, a decrease in P-gp could be a risk factor for developing Alzheimer’s disease. In this context, development of a model to upregulate expression of P-gp and enhance clearance of amyloid-β could provide a new method for treating Alzheimer’s disease. Support for this idea was provided by both in vitro (Abuznait et al. 2011) and in vivo studies (Hartz et al. 2010).
Serum amyloid P (SAP) is also thought to be involved in the pathogenesis of Alzheimer’s disease. SAP, a member of the pentraxin family, is a glycoprotein secreted by the liver into the blood stream. The main functions of SAP are to recognize carbohydrates, nuclear substances, and amyloid fibrils (Agrawal et al. 2009). SAP does not cross the BBB, however, neurons produce it in the brain, and its production is upregulated in Alzheimer’s brains (Yasojima et al. 2000). SAP is assumed to contribute to the pathogenesis of Alzheimer’s disease by binding to amyloid plaques and entering neurons to induce apoptotic cell death (Ulbanyi et al. 2003). Although the hallmarks of Alzheimer’s disease include amyloid plaques and neurofibrillary tangles, a significant population of individuals who have plaques and tangles have no signs of cognitive impairment. Crawford et al. (2012) measured SAP levels in the hippocampus and cerebral cortex of post mortem samples obtained from Alzheimer’s patients and nondemented individuals with Alzheimer’s neuropathology, and found that the latter had no significant difference in SAP levels compared to normal controls while the former had increased SAP levels. The lack of dementia in individuals with Alzheimer’s neuropathology and low levels of SAP, a marker of inflammation, may be due to reduced inflammatory responses. From the therapeutic point of view, reduction of the increased serum SAP might be beneficial for Alzheimer’s patients. Pepys et al. (2002) invented a drug, R-1-[6- [R-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl] pyrrolidine-2-carboxylic acid that is a competitive inhibitor of SAP binding to amyloid fibrils. Kolstoe et al. (2009) administered it to patients with probable Alzheimer’s disease for 12 weeks, and found that the SAP concentrations in serum and cerebrospinal fluid were significantly reduced. Their trial was too short to provide measurable neurological or cognitive effects.
19.4.3 Gene-Transfer of Enzymes Responsible for Glycoconjugates
Loss of function mutations at the GBA locus is a known risk factor for PD (Lwin et al. 2004) (see Sect. 19.3.5). Sardi et al. (2011) developed mouse models of Gaucher disease including homozygote (Gba1D409V/D409V) and heterozygotes (Gba1D409V/+ and Gba1+/−) that showed loss of GBA activity. GBA activity was reduced in homozygous mice (19 % of that of wild type) and heterozygous (59 % for Gba1D409V/+and 54 % for Gba1+/−). Adeno-associated virus-mediated expression of exogenous GBA in the hippocampus of homozygous mice brought about remarkable amelioration of both the neuropathology and memory deficits. Sardi et al. (2013) developed a transgenic mouse model overexpressing A53T α-synuclein and using it found that augmented striatal expression of exogenous GBA in the mice reduced the levels of both cytosolic soluble α-synuclein and membrane-associated striatal α-synuclein.
The observation that PSA-NCAM (see Sect. 19.2.5) was expressed concurrently with sprouting of injured or axotomized Purkinje cells in glial scars (Dusart et al. 1999) led to interrogation of whether induction of PSA expression in damaged tissues in adult CNS could enhance neuronal regeneration. To test the therapeutic potential of PSA, a viral vector encoding polysialyltransferase was employed to transfect scar astrocytes and sustain expression of high levels of PSA (El Maarouf et al. 2006). This resulted in substantial growth of axonal processes through the spinal injury site. Induced expression of PSA may be a promising strategy for promoting neural tissue repair. Similar genetic manipulations using viral vectors encoding GnT-III (see Sect. 19.3.4) could be applied to Alzheimer’s brains to inhibit amyloid β production and to enhance the clearance of amyloid β as previously suggested (Akasaka-Manya et al. 2010). A possible alternative would be to use the curcumin derivative, bisdemethoxycurcumin found to induce expression of GnT-III (Fiala et al. 2007).
19.4.4 Ganglioside Analogues and Environmental Effectors of Neuronal Plasticity
Gangliosides, sialic acid-containing GSLs, are important mediators of neuronal function, age-related neuronal dysfunction, and Alzheimer’s pathology. Studies have shown that gangliosides may mimic nerve growth factor neurotrophic activity thereby restoring cholinergic parameters (Cuello et al. 1989; Ferrari et al. 1995; Mutoh et al. 1995; Rabin and Mocchetti 1995). The beneficial effects of ganglioside GM1 were shown in the experimental models of glutamate neurotoxicity (Favaron et al. 1988; Vaccarino et al. 1987; Wu et al. 2005), brain and spinal cord lesions (Geisler et al. 1991; Oderfeld-Nowak et al. 1993), and cerebral ischemia (Seren et al. 1990). Although gangliosides have been hypothesized to serve as neuroprotective agents capable of reducing brain and spinal cord damages in disease conditions, they were proven to attenuate neuronal apoptosis induced by serum deprivation, ionomycin, or cyclosporine A in an in vitro system (Ryu et al. 1999). However, gangliosides did not attenuate neuronal necrosis induced by exposure of cultured cortical cells to excitotoxins, and even increased necrosis induced by oxidative stress. Thus, gangliosides seem to rescue neurons from becoming apoptotic.
Two decades ago, GM1 was reported to activate the TrkA (tropomyosin-related kinase A) receptor (Ferrari et al. 1995; Mutoh et al. 1995; Rabin and Mocchetti 1995). Later it was shown to activate TrkC by inducing release of neurotrophin-3 (Rabin et al. 2002). In contrast to GM1 which induces neurotrophin-3 but not BDNF, a synthetic analogue of GM1, LIGA20 (Manev et al. 1990), was found to increase secretion of BDNF by stimulating TrkB in NIH-3T3 fibroblasts. When the same amount of GM1 and LIGA20 were given orally to rats, the concentration of LIGA20 in the brain was 50-fold higher than that of GM1 (Polo et al. 1994). This may account for the superior potency of LIGA20 compared to natural GM1.
LIGA20 is similar in structure of lyso-GM1 (Manev et al. 1990). Although LIGA20 appears to be membrane permeable, it was hypothesized that smaller molecules would be more suitable for transportation across the BBB. To address this sialylcholesterol was synthesized (Sato et al. 1987). Effects of the two stereoisomers, α- and β-sialylcholesterols, on cholinergic synapses were examined (Tanaka and Ando 1996) to determine whether they could ameliorate age-related decrements in synaptic functions observed in studies of mouse brain synaptosomes (Tanaka et al. 1996). While both isomers enhanced acetylcholine release from synaptosomes, their underlying mechanisms differed. Alpha-sialylcholesterol increased depolarization-induced influx of calcium ions to enhance acetylcholine release, while not affecting choline uptake. On the other hand, β-sialylcholesterol activated high-affinity choline uptake, resulting in enhancement of acetylcholine synthesis followed by augmentation of acetylcholine release. The β-isomer had no effect on calcium ion influx. These results imply that the two isomers of sialylcholesterol modulate the synaptic functions in different ways, and that their effects on cholinergic synaptic transmission are synergistic. The results also indicate that the actions of the synthetic sialylcholesterols on synapses mimic those of naturally occurring gangliosides (Ando et al. 1998). The action of the α-isomer on calcium channels appears to be similar to that observed using ganglioside GQ1b. In contrast, the β-isomer activation of the choline transporter was similar to the effect of the Chol-1α ganglioside (see Sect. 19.3.3). The similarity of the effects of both gangliosides and sialylcholesterols on synaptic action is summarized in a review (Ando 2012).
Neuronal plasticity, a crucial requirement for adapting to new environments, developing neuronal integrity, and restoring damaged neural architecture, are central themes in basic and clinical neuroscience. Age-related deterioration in brain functions or damage caused by neurodegenerative diseases or various insults can be partly restored by innate neuronal plasticity. Stimulation of plasticity can be considered the ultimate strategy for maintaining brain function during aging and for its efficient recovery from neuronal damage. Synaptic plasticity was enhanced as revealed by increased synaptogenesis and cognitive improvement when aged rats were put in an enriched environment (Ando 2012; Saito et al. 1994). In a mouse model of PD, physical activity and environmental enrichment stimulated oligodendrocyte differentiation in the substantia nigra, again indicating their positive effect on cellular plasticity (Klaissle et al. 2012). Neuronal plasticity is thought to be regulated by at least two groups of molecules: neurotrophins and adhesion molecules. The effect of activity on plasticity may be explained by the observation that expression of BDNF and NCAM were upregulated in the spinal cords of rats given exercise (Macias et al. 2002). Drugs can also affect neuronal plasticity. Warfarin is routinely prescribed for patients with atrial fibrillation or cerebral infarction, but since it is also a vitamin K antagonist and may cause vitamin K-deficiency it must be used with care. The first indication that vitamin K was needed by the nervous system was the observation by Sundaram and Lev (1988) that young mice fed warfarin had decreased brain concentrations of sulfatide due to reduced activity of galactocerebroside sulfotransferase, needed for its synthesis (Fig. 19.1). Lifelong intake of a low-vitamin K diet was shown to impair cognitive performance by old rats. They also had low levels of gangliosides in the pons medulla and midbrain (Carrié et al. 2011). Sulfatide contents were not different between high- and low-dosed groups in old rats. In humans, fetal exposure to warfarin is known to induce warfarin embryopathy identified by symptoms such as blindness and mental retardation due to optic and cerebral atrophies (Hall et al. 1980). Patients with early stage Alzheimer’s disease were found to have significantly lower intakes of vitamin K compared to age- and gender-matched controls (Presse et al. 2008). With regard to brain plasticity or responsiveness, both positive and negative environmental risk factors such as enriched environment and warfarin must be considered.
19.5 Concluding Remarks
In Sect. 19.2, age-related changes in glycoconjugates, mostly GSLs and glycoproteins, in the central nervous system are summarized.
In Section 3, new findings on the pathogenesis of age-related neurodegenerative diseases such as Alzheimer’s and Parkinson’s are discussed. Ganglioside GM1 may function as a seed for amyloid fibril formation, and the expression of Chol-1α gangliosides in the Alzheimer’s model mouse brain is thought to be a compensatory response to restore cholinergic function. Evidence suggests that mutations in the gene for GBA and partial deficits in a-series gangliosides may contribute to PD.
In section 4, possible strategies for anti-aging and the prevention of age-related dementia are discussed, including those to (1) enhance remyelination, (2) enhance clearance of amyloid-β, and (3) express the GBA gene. To ameliorate the functional deficits in aging and diseased brains, ganglioside-analogues, LIGA20 and sialylcholesterols, have been tested. Alpha- and β-stereoisomers of the latter are found to activate calcium ion channels and high-affinity choline uptake at presynapses, respectively. Finally, environmental factors (eg physical activity and drugs) affecting neuronal plasticity in relation to glycoconjugates are discussed.
Abbreviations
- APP:
-
Amyloid precursor protein
- Aβ:
-
Amyloid beta
- BBB:
-
Blood–brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- CBA:
-
Glucocerebrosidase
- CNS:
-
Central nervous system
- GnT-III:
-
N-acetylglucosaminyltransferase III
- LCB:
-
Long-chain base
- MAP5:
-
Microtubule-associated protein 5
- NCAM:
-
Neural cell adhesion molecule
- NFTs:
-
Neurofibrillary tangles
- OPC:
-
Oligodendrocyte precursor cell
- PD:
-
Parkinson’s disease
- P-gp:
-
P-glycoprotein
- PSA:
-
Polysialic acid
- SAP:
-
Serum amyloid P
References
Abuznait AH, Cain C, Ingram D, Burk D, Kaddoumi A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta-amyloid: investigation of P-glycoprotein as a novel therapeutic target for Alzheimer’s disease. J Pharm Pharmacol. 2011;63:1111–8.
Agrawal A, Singh PP, Bottazzi B, Garlanda C, Montovani A. Pattern recognition by pentraxins. Adv Exp Med Biol. 2009;653:98–116.
Aine CJ, Sanfratello L, Adair JC, Knoefel JE, Caprihan A, Stephen JM. Development and decline of memory functions in normal, pathological and healthy successful aging. Brain Topogr. 2011;24:323–39.
Akasaka-Manya K, Manya H, Sakurai Y, Wojczyk BS, Spitalnik SL, Endo T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj J. 2008;25:775–86.
Akasaka-Manya K, Manya H, Sakurai Y, Wojczyk BS, Kozutsumi Y, Sato Y, et al. Protective effect of-glycan bisecting GlcNAc residues on β–amyloid production in Alzheimer’s disease. Glycobiology. 2010;20:99–106.
Ando S. Review: gangliosides in the nervous system. Neurochem Int. 1983;5:507–37.
Ando S. Biochemistry of brain aging. Nihon Rinsho. 1985;43:1399–403 (in Japanese).
Ando S. Neuronal dysfunction with aging and its amelioration. Proc Jpn Acad Ser B Phys Biol Sci. 2012;88:266–82.
Ando S, Yu RK. Isolation and characterization of two isomers of brain tetrasialogangliosides. J Biol Chem. 1979;254:12224–9.
Ando S, Tanaka Y, Ono Y, Kon K. Incorporation rate of GM1 ganglioside into mouse brain myelin: effect of aging and modification by hormones and other compounds. Adv Exp Med Biol. 1984;174:241–8.
Ando S, Tanaka Y, Kon K. Membrane aging of the brain synaptosomes with special reference to gangliosides. In: Tettamanti G et al., editors. Gangliosides and neural plasticity. Padva: Liviana Press; 1986. p. 23–30.
Ando S, Hirabayashi Y, Kon K, Inagaki F, Tate S, Whittaker VP. A trisialoganglioside containing a sialyl-α2,6-N-acetylgalactosamine residue is a cholinergic-specific antigen, Chol-1α. J Biochem. 1992;111:287–90.
Ando S, Tanaka Y, Waki H, Kon K, Iwamoto M, Fukui F. Gangliosides and sialylcholesterol as modulators of synaptic functions. Ann N Y Acad Sci. 1998;845:232–9.
Ando S, Kobayashi S, Waki H, Kon K, Fukui F, Tadenuma T, et al. Animal model of dementia induced by entorhinal damage and partial restoration of cognitive deficits by BDNF and carnitine. J Neurosci Res. 2002;70:519–27.
Ando S, Tanaka Y, Toyoda Y, Kon K. Turnover of myelin lipids in aging brain. Neurochem Res. 2003;28:5–13.
Ando S, Tanaka Y, Kobayashi S, Fukui F, Iwamoto M, Waki H, et al. Synaptic function of cholinergic-specific Chol-1α ganglioside. Neurochem Res. 2004;29:857–67.
Ariga T, Yanagisawa M, Wakada C, Ando S, Buccafusco JJ, McDonald MP, et al. Ganglioside metabolism in a transgenic mouse model of Alzheimer’s disease: expression of Chol-1α antigens in the brain. ASN Neuro. 2010;2:e00044.
Ariga T, Wakada C, Yu RK. The pathological roles of ganglioside metabolism in Alzheimer’s disease: effect of gangliosides on neurogenesis. Int J Alzheimers Dis. 2011;2011:193618.
Ariga T, Itokazu Y, McDonald MP, Hirabayashi Y, Ando S, Yu RK. Brain gangliosides of a transgenic mouse model of Alzheimer’s disease with deficiency in GD3-synthase: expression of elevated levels of a cholinergic-specific ganglioside, GT1aα. ASN Neuro. 2013;5:141–8.
Bartels AL, Willemsen ATM, Kortekaas R, de Jong BM, de Vries R, de Klerk O, et al. Decreased blood–brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm. 2008;115:1001–9.
Bednarski E, Lynch G. Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. J Neurochem. 1996;67:1846–55.
Ben-David O, Pewzner-Jung Y, Brenner O, Laviad EL, Kogot-Levin A, Weissberg I, et al. Encephalopathy caused by ablation of very long acyl chain ceramide synthesis may be largely due to reduced galactosylceramide levels. J Biol Chem. 2011;286:30022–33.
Bernardo A, Harrison FE, McCord M, Zhao J, Bruchey A, Davies SS, et al. Elimination of GD3 synthase improves memory and reduces amyloid-β plaque load in transgenic mice. Neurobiol Aging. 2009;30:1777–91.
Beutler E, Grabowski G. Gaucher disease. In: Scriver CR, Beaudet al, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. New York, NY: McGraw-Hill; 1995. p. 2641–70.
Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer’s disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the apo E4 allele. J Neural Transm. 1996;103:603–18.
Bowley MP, Cabral H, Rosene DL, Peters A. Age changes in myelinated nerve fibers of the cingulate bundle and corpus callosum in the rhesus monkey. J Comp Neurol. 2010;518:3046–64.
Bras J, Singleton A, Cookson MR, Hardy J. Potential role of ceramide metabolism in Lewy body disease. FEBS Lett. 2008;275:5767–73.
Brunden KR. Age-dependent changes in the oligosaccharide structure of the major myelin glycoprotein, P0. J Neurochem. 1992;58:1659–66.
Brusés JL, Rutishauser U. Reguration of neural cell adhesion molecule polysialylation: evidence for nontranscriptional control and sensitivity to an intracellular pool of calcium. J Cell Biol. 1998;140:1177–86.
Calzà L, Fernandez M, Giardino L. Cellular approaches to central nervous system remyelination stimulation: thyroid hormone to promote myelin repair via endogenous stem and precursor cells. J Mol Endocrinol. 2010;44:13–23.
Cantù L, Del Favero E, Sonnino S, Prinetti A. Gangliosides and the multiscale modulation of membrane structure. Chem Phys Lipids. 2011;164:796–810.
Carrié I, Bélanger E, Portoukalian J, Rochford J, Ferland G. Lifelong low-phylloquinone intake is associated with cognitive impairments in old rats. J Nutr. 2011;141:1495–501.
Chapman J, Sela BA, Wertman E, Michaelson DM. Antibodies to ganglioside GM1 in patients with Alzheimer’s disease. Neurosci Lett. 1988;86:235–40.
Choo-Smith L-P, Garzon-Rodriguez W, Glabe CG, Surewicz WK. Acceleration of amyloid fibril formation by specific binding of Aβ-(1-40) peptide to ganglioside-containing membrane vesicles. J Biol Chem. 1997;272:22987–90.
Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations on the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology. 2007;69:1270–7.
Crawford JR, Bjorklund NL, Taglialatela G, Gomer RH. Brain serum amyloid P levels are reduced in individuals that lack dementia while having Alzheimer’s disease neuropathology. Neurochem Res. 2012;37:795–801.
Cuello AC, Garofalo L, Kenigsberg RL, Maysinger D. Gangliosides potentiate in vivo and in vitro effects of nerve growth factor on central cholinergic neurons. Proc Natl Acad Sci U S A. 1989;86:2056–60.
Dahlgren KN, Manelli AM, Stine Jr WB, Baker LK. Oligomeric and fibrillar species of amyloid- β peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–53.
DePaolo J, Goker-Alpan O, Samaddar T, Lopez G, Sidransky E. The association between mutations in the lysosomal protein glucocerebrosidase and parkinsonism. Mov Disord. 2009;24:1571–8.
Derrington EA, Borroni E. The developmental expression of the cholinergic-specific antigen Chol-1 in the central and peripheral nervous system.of the rat. Dwvelop. Brain Res. 1990;52:131–40.
Dusart I, Morel MP, Wehrlé R, Sotelo C. Late axonal sprouting of injured Purkinje cells and its temporal correlation with permissive changes in the glial scar. J Comp Neurol. 1999;408:399–418.
El Maarouf A, Petridis AK, Rutishauser U. Use of polisialic acid in repair of the central nervous system. Proc Natl Acad Sci U S A. 2006;103:16989–94.
Emory CR, Ala TA, Frey WH. Ganglioside monoclonal antibody (A2B5) labels Alzheimer’s neurofibrillary tangles. Neurology. 1987;37:768–72.
Eustache F, Desgranges B, Giffard B, de la Sayette V, Barom J-C. Entorhinal cortex disruption causes memory deficit in early Alzheimer’s disease as shown by PET. Neuroreport. 2001;12:683–5.
Favaron M, Manev H, Alho H, Bertolino M, Ferret B, Guidotti A, et al. Gangliosides prevent glutamate and kinate neurotoxicity in primary neuronal cultures of neonatal rat cerebellum and cortes. Proc Natl Acad Sci U S A. 1988;85:7351–5.
Fernandez M, Giuliani A, Pirondi S, D’Intino G, Giardino L, Aloe L, et al. Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci U S A. 2004;101:16363–8.
Ferrari G, Anderson B, Stephens B, Kaplan D, Greene L. Prevention of apoptotic neuronal death by GM1 ganglioside. involvement of Trk neurotrophin receptors. J Biol Chem. 1995;270:3074–80.
Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, et al. Innate immunity and transcription of MGAT-III and toll-like receptors in Alzheimer’s disease patients are improved by bisdemetoxycurcumin. Proc Natl Acad Sci U S A. 2007;104:12849–54.
Franco PG, Silverstroff L, Soto EF, Pasquini JM. Thyroid hormones promote differentiation of oligodendrocyte progenitor cells and improve remyelination after cuprizone-induced demyelination. Exp Neurol. 2008;212:458–67.
Furuse H, Waki H, Kaneko K, Fujii S, Miura M, Sasaki H, et al. Effect of the mono- and tetra-sialogangliosides, GM1 and GQ1b, on long-term potentiation in the CA1 hippocampal neurons of the guinea pig. Exp Brain Res. 1998;123:307–14.
Gegg ME, Burke D, Heales SJR, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol. 2012;72:455–63.
Geisler FH, Dorsey FC, Coleman WP. Recovery of motor function after spinal-cord injury: a randomized placebo-controlled trial with GM-1 ganglioside. N Engl J Med. 1991;324:1829–38.
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, Mclnerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–40.
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120:641–9.
Hall JG, Pauli RM, Wilson KM. Maternal and fetal sequelae of anticoagulation during pregnancy. Am J Med. 1980;68:122–40.
Hartz AMS, Miller DS, Bauer B. Restoring blood–brain barrier P-glycoprotein reduces brain amyloid-β in a mouse model of Alzheimer’s disease. Mol Pharmacol. 2010;77:715–23.
Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljarvi L, Koivisto E, et al. Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer’s disease. Neuroscience. 1995;64:375–84.
Hirabayashi Y, Hirota M, Matsumoto M, Tanaka H, Obata K, Ando S. Development changes of C-series polysialogangliosides in chick brains revealed by mouse monoclonal antibodies M6704 and M7103 with different epitope specificities. J Biochem. 1988;104:973–9.
Hirabayashi Y, Nakao T, Irie F, Whittaker VP, Kon K, Ando S. Structural characterization of a novel cholinergic neuron-specific ganglioside in bovine brain. J Biol Chem. 1992;267:12973–8.
Ho NF, Han SP, Dawe GS. Effect of voluntary running on adult hippocampal neurogenesis in cholinergic lesioned mice. BMC Neurosci. 2009;10:57.
Hornykiewicz O. Metabolism of brain dopamine in human parkinsonism: Neurochemical and clinical aspects. In: Costa E, Yahr MD, editors. Biochemistry and pharmacology of the brain ganglia. New York, NY: Raven; 1966. p. 171–81.
Huttenlocher PR. Synaptic density in human frontal cortex—developmental changes and effects of aging. Brain Res. 1979;163:195–205.
Inoko E, Nishimura Y, Tanaka H, Takahashi T, Furukawa K, Kitajima K, et al. Developmental stage-dependent expression of an alpha 2,8-trisialic acid unit on glycoproteins in mouse brain. Glycobiology. 2010;20:916–28.
Irie F, Hashikura T, Tai T, Seyama Y, Hirabayashi Y. Distribution of cholinergic neuron-specific gangliosides (GT1aα and GQ1bα) in the rat central nervous system. Brain Res. 1994;665:161–6.
Irie F, Kurono S, Li YT, Seyama Y, Hirabayashi Y. Isolation of three novel cholinergic neuron-specific gangliosides from bovine brain and their vitro syntheses. Glycoconj J. 1996;13:177–86.
Ishaque A, Roomi MW, Szymanska I, Kowalski S, Eylar EH. The P0 glycoprotein of peripheral nerve myelin. Can J Biochem. 1980;58:913–21.
Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, et al. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci. 2002;22:6507–14.
Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW. Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol. 2003;62:389–97.
Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, and endogenous seed for Alzheimer amyloid. J Biol Chem. 2001;276:24985–90.
Kannagi R, Nudelmann E, Hakomori SI. Possible role of ceramide in defining structure and function of membrane glycolipids. Proc Natl Acad Sci U S A. 1982;79:3470–4.
Kimura N, Yanagisawa K. Endosomal accumulationof GM1 ganglioside-bound amyloid β-protein in neurons of aged monkey brains. Neuroreport. 2007;18:1669–73.
Kirkwood TB. Evolution of aging. Nature. 1977;270:301–4.
Klaissle P, Lesemann A, Huehnchen P, Hermann A, Storch A, Steiner B. Physical activity and environmental enrichment regulate the generation of neural precursors in the adult mouse substantia nigra in a dopamine-dependent manner. BMC Neurosci. 2012;13:132.
Kobata A. Structures and functions of the sugar chains of glycoproteins. Eur J Biochem. 1992;209:483–501.
Kobata A. Glycobiology in the field of gerontology (glycogerontology). Adv Exp Med Biol. 2011;705:411–29.
Kolstoe SE, Ridha BH, Bellotti V, Wang N, Robinson CV, Crutch SJ, et al. Molecular dissection of Alzheimer’s disease neuropathology by depletion of serum amyloid P component. Proc Natl Acad Sci U S A. 2009;106:7619–23.
Kono S, Ouchi Y, Terada T, Ida H, Suzuki M, Miyajima H. Functional brain imaging in glucocerebrosidase mutation carriers with and without parkinsonism. Mov Disord. 2010;25:1823–9.
Kotani M, Kawashima I, Ozawa H, Terashima T, Tai T. Differential distribution of major gangliosides in rat central nervous system detected by specific monoclonal antibodies. Glycobiology. 1993;3:137–46.
Ledeen RW, Yu RK. Gangliosides: structure, isolation, and analysis. Meth Enzymol. 1982;83:139–92.
Lesage S, Anheim M, Condroyen C, Pollak P, Durif F, Dupuits C, et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet. 2011;20:202–10.
Lopez-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004;24:5439–44.
Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81:70–3.
Macias M, Fehr S, Dwornik A, Sulejczak D, Wiater M, Czarkowska-Bauch J, et al. Exercise increases mRNA levels for adhesion molecules N-CAM and L1 correlating with BDNF response. Neuroreport. 2002;13:2527–30.
Majocha RE, Jungalwala FB, Rodenrys A, Marotta CA. Monoclonal antibody to embryonic CNS antigen A2B5 provides evidence for the involvement of membrane components at sites of Alzheimer degeneration and detects sulfatides as well as gangliosides. J Neurochem. 1989;53:953–61.
Manev H, Favaron M, Vicini S, Guidotti A, Costa E. Glutamate-induced neuronal death in primary cultures of cerebeller granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther. 1990;252:419–27.
Mansson J-E, Vanier M-T, Svennerholm L. Changes in the fatty acid and sphingosine composition of the major gangliosides of human brain with age. J Neurochem. 1978;30:273–5.
Masliah E, Terry RD. Role of synaptic pathology in the mechanisms of dementia in Alzheimer’s disease. Clin Neurosci. 1993;1:192–8.
Matsumura S, Shinoda K, Yamada M, Yokojima S, Inoue M, Ohnishi T, et al. Two distinct amyloid β-protein (A β) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analysis. J Biol Chem. 2011;286:11555–62.
Mazulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher’s disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52.
McFarlane I, Georgopoulou N, Coughlan CM, Gillian AM, Breen KC. The role of the protein glycosylation state in the control of cellular transport of the amyloid β-precursor protein. Neuroscience. 1999;90:15–25.
Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol. 2009;66:571–6.
Mizutani T, Kasahara M. Hippocampal atrophy secondary to entorhinal cortical degeneration in Alzheimer-type dementia. Neurosci Lett. 1997;222:119–22.
Mutoh T, Tokuda A, Miyadai T, Hamaguchi M, Fujiki N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc Natl Acad Sci U S A. 1995;92:5087–91.
Nakamura H, Kobayashi S, Ohashi Y, Ando S. Age-changes of brain synapses and synaptic plasticity in response to an enriched environment. J Neurosci Res. 1999;56:307–15.
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Caims NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362–81.
Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Robin H, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132:1783–94.
Norton WT, Poduslo SE. Myelination in rat brain: changes in myelin composition during brain maturation. J Neurochem. 1973;21:759–73.
O’Nuallian B, Freir DB, Nicoll AJ, Risse E, Ferguson N, Herron CE, et al. Aβ dimers rapidly form stable synaptotoxic protofibrils. J Neurosci. 2010;30:14411–9.
Oderfeld-Nowak B, Casamenti F, Pepeu G. Gangliosides in the repair of brain cholinergic neurons. Acta Biochim Pol. 1993;40:395–404.
Ohmi Y, Ohkawa Y, Yamauchi Y, Tajima O, Furukawa K, Furukawa K. Essential roles of gangliosides in the formation and maintenance of membrane microdomains in brain tissues. Neurochem Res. 2012;37:1185–91.
Oikawa N, Yamaguchi K, Ogino K, Taki T, Yuyama K, Yamamoto N, et al. Gangliosides determine the amyloid pathology of Alzheimer’s disease. Neuroreport. 2009;20:1043–6.
Pahlsson P, Shakin-Eshleman SH, Spitalnik SL. N-Linked glycosylation of β-amyloid precursor protein. Biochem Biophys Res Commun. 1992;189:1667–73.
Palestini P, Masserini M, Sonnino S, Giuliani A, Tettamanti G. Changes in the ceramide composition of rat forebrain gangliosides with age. J Neurochem. 1990;54:230–5.
Palestini P, Masserini M, Fiorilli A, Calappi E, Tettamanti G. Age-related changes in the ceramide composition of the major gangliosides present in rat brain subcellular fractions enriched in plasma membranes of neuronal and myelin origin. J Neurochem. 1993;61:955–60.
Pender MP, Csurhes PA, Wolfe NP, Hooper KD, Good MF, McCombe PA, et al. Increased circulating T cell reactivity to GM3 and GQ1b gangliosides in primary progressive multiple sclerosis. J Clin Neurosci. 2003;10:63–6.
Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417:254–9.
Pestronk A, Adams RN, Clawson L, Cornblath D, Kuncl RW, Griffin D, et al. Serum antibodies to GM1 ganglioside in amyotrophic lateral sclerosis. Neurology. 1988;38:1457–61.
Pestronk A, Chaudhry V, Feldman EL, Griffin JW, Cornblath DR, Denys EH, et al. Lower motor neuron syndromes defined by patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid antibodies. Ann Neurol. 1990;27:316–26.
Pestronk A, Lopate G, Kornberg AJ, Elliott JL, Blume G, Yee WC, et al. Distal lower motor neuron syndrome with high-titer serum IgM anti-GM1 antibodies: improvement following immunotherapy with monthly plasma exchange and intravenous cyclophosphamide. Neurology. 1994;44:2027–31.
Peters A, Sethares C. Is there remyelination during aging of the primate central nervous system? J Comp Neurol. 2003;460:238–54.
Peters A, Sethares C, Killiany R. Effects of age on the thickness of myelin sheath in monkey primary visual cortex. J Comp Neurol. 2001;435:241–8.
Polo A, Kirschner G, Guidotti A, Costa E. Brain content of GSLs after oral administration of monosialoganglioside GM1 and LIGA20 to rats. Mol Chem Neuropathol. 1994;21:41–53.
Presse N, Shatenstein B, Kergoat MJ, Ferland G. Low vitamin K intakes in community-dwelling elders at an early stage of Alzheimer’s disease. J Am Diet Assoc. 2008;108:2095–9.
Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res. 1983;268:249–54.
Rabin SJ, Mocchetti I. GM1 ganglioside activates the high-affinity nerve growth factor receptor TrkA. J Neurochem. 1995;65:347–54.
Rabin SJ, Bachis A, Mocchetti I. Gangliosides activate Trk receptors by inducing the release of neurotrophins. J Biol Chem. 2002;277:49466–72.
Rhouma FB, Kallel F, Kefi R, Cherif W, Nagara M, Azaiez H, et al. Adult Gaucher disease in southern Tunisia: report of three cases. Diagn Pathol. 2012;7:4.
Richardson PJ, Walker JH, Jones RT, Whittaker VP. Identification of a cholinergic-specific antigen Chol-1 as a ganglioside. J Neurochem. 1982;38:1605–14.
Ryu BR, Choi DW, Hartley DM, Costa E, Jou I, Gway BJ. Attenuation of cortical neuronal apoptosis by gangliosides. J Pharmacol Exp Ther. 1999;290:811–6.
Saito M, Yu RK. Role of myelin-associated neuraminidase in the ganglioside metabolism of rat brain myelin. J Neurochem. 1992;58:83–7.
Saito M, Yu RK. Possible role of myelin-neuraminidase in membrane adhesion. J Neurosci Res. 1993;36:127–32.
Saito S, Kobayashi S, Ohashi Y, Igarashi M, Komiya Y, Ando S. Decreased synaptic density in aged brains and its prevention by rearing under enriched environment as revealed by synaptophysin content. J Neurosci Res. 1994;39:57–62.
Saito M, Tanaka Y, Tang C-P, Yu RK, Ando S. Characterization of sialidase activity in mouse synaptic plasma membranes and its age-related changes. J Neurosci Res. 1995;40:401–6.
Saito M, Hagita H, Ito M, Ando S, Yu RK. Age-dependent reduction in sialidase activity of nuclear membranes from mouse brain. Exp Gerontol. 2002;37:937–41.
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A. 2011;108:12101–6.
Sardi SP, Clarke J, Kinnecom C, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A. 2013;110:3537–42.
Sato Y, Endo T. Alterations with age of the neurons expressing P0 in the rat spinal cord. Neurosci Lett. 2000;281:41–4.
Sato Y, Endo T. Alteration of brain glycoproteins during aging. Geriatr Gerontol Int. 2010;10:S32–40.
Sato S, Fujita S, Furukata K, Ogura H, Yoshimura S, Itoh M, et al. Synthesis of 2-(5-cholesten-3β-yloxy)glycosides of N-acetyl-D-neuraminic acid derivatives. Chem Pharm Bull. 1987;35:4043–8.
Sato Y, Kimura M, Yasuda C, Nakao Y, Tomita M, Kobata A, Endo T. Evidence for the presence of major peripheral myelin glycoprotein P0 in mammalian spinal cord and a change of its glycosylation state during aging. Glycobiology. 1999;9:655–60.
Sato Y, Akimoto Y, Kawakami H, Hirano H, Endo T. Location of sialylglycoconjugates containing the Siaα2-3Gal and Siaα2-6Gal groups in the rat hippocampus and the effect of aging on their expression. J Histochem Cytochem. 2001;49:1311–9.
Sato Y, Suzuki Y, Ito E, Shimazaki S, Ishida M, Yamammoto T, et al. Identification and characterization of an increased glycoprotein in aging: age-associated translocation of cathepsin D. Mech Ageing Dev. 2006;127:771–8.
Schachner M, Martini R. Glycans and the modulation of neural-recognition molecule function. Trends Neurosci. 1995;18:183–91.
Scheff SW, Sparks DL, Price DA. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease. Ann Neurol. 1993;34:356–61.
Scheibel AB, Tomiyasu U. Dendritic sprouting in Alzheimer’s presenile dementia. Exp Neurol. 1978;60:1–8.
Schneider JS, Pope A, Simpson K, Taggart J, Smith MG, DiStetano L. Recovery from experimental parkinsonism in primates with GM1 ganglioside treatment. Science. 1992;256:843–6.
Schulze ET, Geary EK, Susmaras TM, Paliga JT, Maki PM, Little DM. Anatomical correlates of age-related working memory declines. J Aging Res. 2011;2011:606871.
Segler-Stahl K, Webster JC, Brunngraber EG. Changes in the concentration and composition of human brain gangliosides with aging. Gerontology. 1983;29:161–8.
Seki T, Arai Y. The persistent expression of a highly polysialylated NCAM in the dentate gyrus of the adult rat. Neurosci Res. 1991;12:503–13.
Seren MS, Rubini R, Lazzaro A, Zanoni R, Fiori MG, Leon A. Protective effects of a monosialoganglioside derivative following transitory forebrain ischemia in rats. Stroke. 1990;21:1607–12.
Shen S, Sandoval J, Swiss VA, Li J, Dupree J, Franklin RJM. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat Neurosci. 2008;11:1024–34.
Shields SA, Gilson JM, Blakemore WF, Franklin RJ. Remyelination occurs as extensively but slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia. 1999;28:77–83.
Sidransky E, Nalls MA, Aasly JO, Aharon-Perez J, Annesi G, Barbose ER, et al. Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N Engl J Med. 2009;361:1651–61.
Silverberg GD, Messier AA, Miller MC, Machan JT, Majmudar SS, Stopa EG, et al. Amyloid efflux transporter expression at the blood–brain barrier declines in normal aging. J Neuropathol Exp Neurol. 2010;69:1034–43.
Sim FJ, Zhao C, Penderis J, Franklin RJ. The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci. 2002;22:2451–9.
Sugiura Y, Shimma S, Konishi Y, Yamada MK, Setou M. Imaging mass spectrometry technology and application of ganglioside study: visualization of age-dependent accumulation of C20- ganglioside molecular species in the mouse hippocampus. PLoS One. 2008;3(9):e3232.
Sun GY, Sun KL. Metabolism of arachidonyl phosphoglycerides in mouse brain subcellular fractions. J Neurochem. 1979;32:1053–9.
Sundaram KS, Lev M. Warfarin administration reduces synthesis of sulfatides and other sphingolipids in mouse brain. J Lipid Res. 1988;29:1475–9.
Sunwoo M-K, Kim S-M, Lee S, Lee RH. Parkinsonism associated with glucocerebrosidase mutation. J Clin Neurol. 2011;7:99–101.
Suzuki K. The pattern of mammalian brain gangliosides—III. Regional and developmental differences. J Neurochem. 1965;12:969–79.
Svennerholm L, Ställberg-Stenhagen S. Changes in the fatty acid composition of cerebrosides and sulfatides of human nervous tissue with age. J Lipid Res. 1968;9:215–25.
Svennerholm L, Boström K, Fredman P, Månsson J-E, Rosengren B, Rynmaek B-M. Human brain gangliosides: developmental changes from early fetal stage to advanced age. Biochim Biophys Acta. 1989;1005:109–17.
Svennerholm L, Boström K, Helander CG, Jungbjer B. Membrane lipids in the aging human brain. J Neurochem. 1991;56:2051–9.
Svennerholm L, Bostöm K, Jungbjer B, Olsson L. Membrane lipids of adult human brain: lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J Neurochem. 1994;63:1802–11.
Takahashi H, Hirokawa K, Ando S, Obata K. Immunohistological study on brains of Alzheimer’s disease using antibodies to fetal antigens, C-series gangliosides and microtubule-associated protein 5. Acta Neuropathol. 1991;81:626–31.
Taki T, Hirabayashi Y, Ichikawa H, Ando S, Kon K, Tanaka Y, et al. A ganglioside of rat ascites hepatoma AH7974F cells: occurrence of a novel disialoganglioside (GD1aα) with a unique N-acetylneuraminyl-α2,6-N-acetylgalactosamine structure. J Biol Chem. 1986;261:3075–8.
Tanaka Y, Ando S. Modulation of cholinergic synaptic functions by sialylcholesterol. Glycoconj J. 1996;13:321–6.
Tanaka Y, Hasegawa A, Ando S. Impaired synaptic functions with aging as characterized by decreased calcium influx and acetylcholine release. J Neurosci Res. 1996;43:63–70.
Tanaka Y, Waki H, Kon K, Ando S. Gangliosides enhance KCl-induced Ca2+ influx and acetylcholine release in brain synaptosomes. Neuro Report. 1997;8:2203–7.
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synaptic loss in the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80.
Uchida Y. Molecular mechanism of regeneration in Alzheimer’s disease brain. Geriatr Gerontol Int. 2010;10:S158–68.
Ulbanyi Z, Laszlo L, Yomasi TB, Toth E, Mekes E, Sass M, et al. Serum amyloid P component induces neuronal apoptosis and beta-amyloid immunoreactivity. Brain Res. 2003;988:67–77.
Vaccarino F, Giodotti A, Costa E. Ganglioside inhibition of glutamate-mediated protein kinase C translocation in primary cultures of cerebellar neurons. Proc Natl Acad Sci U S A. 1987;84:8707–11.
Vanier MT, Holm M, Öhman R, Svennerholm L. Developmental profiles of gangliosides in human and rat brain. J Neurochem. 1971;18:581–92.
Vogelgesang S, Warzok RW, Cascorbi I, Kunert-Keil C, Schroeder E, Kroemer HK, et al. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2004;1:121–5.
Waki H, Kon K, Tanaka Y, Ando S. Facile methods for isolation of gangliosides in a small scale: age-related changes of gangliosides in mouse brain synaptic plasma membranes. Anal Biochem. 1994;222:156–62.
Walhovd KB, Fjell AM, Reinvang I, Lundervold A, Dale AM, Eilertsen DE, et al. Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiol Aging. 2005;26:1261–70.
Westbroek W, Gustafson AM, Sidransky E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol Med. 2011;17:485–93.
Wieraszko A, Seifert W. Evidence for the functional role of monosialoganglioside GM1 in synaptic transmission in rat hippocampus. Brain Res. 1986;371:305–13.
Wu G, Lu Z-H, Wang J, Wang Y, Xie X, Meyenhofer MF, et al. Enhanced susceptibility to kainite-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA20, a membrane-permeant analogue GM1. J Neurochem. 2005;25:11014–22.
Wu G, Lu Z-H, Kulkarni N, Amin R, Ledeen RW. Mice lacking major gangliosides develop parkinsonism. Neurochem Res. 2011;36:1706–14.
Wu G, Lu Z-H, Kulkarni N, Ledeen RW. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J Neurosci Res. 2012;90:1997–2008.
Yamada S, Mizutani T, Asano T, Enomoto M, Sakata M, Esaki Y, et al. Age-related brain atrophy with a constant cortical thickness in the normal elderly. Neuropathology. 1998;18:276–83.
Yamamoto N, Matsubara E, Maeda S, Minagawa H, Takashima A, Maruyama W, et al. A ganglioside-induced toxic soluble Α β assembly, its enhanced formation from Α β bearing the Arctic mutation. J Biol Chem. 2007;282:2646–55.
Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid β-protein (A β): a possible form of preamyloid in Alzheimer’s disease. Nat Med. 1995;1:1062–6.
Yap TL, Grushus JM, Velayati A, Westbroek W, Goldin E, Moaven N, et al. α-Synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem. 2011;286:28080–8.
Yasojima K, Schwab C, McGeer EG, McGeer PL. Human neurons generate C-reactive protein and amyloid P: upreguration in Alzheimer’s disease. Brain Res. 2000;887:80–9.
Yazaki T, Miura M, Asou H, Kitamura K, Toya S, Uyemura K. Glycopeptide of P0 protein inhibits hemophilic cell adhesion: competition assay with transformants and peptides. FEBS Lett. 1992;307:361–6.
Yohe HC, Roark DE, Rosenberg A. C20 sphingosine as a relevant factor in determining aggregative properties of gangliosides. J Biol Chem. 1976;251:7083–7.
Yu RK, Iqbal K. Sialosylgalactosyl ceramide as a specific marker for human myelin and oligodendroglial perikarya : gangliosides of human myelin oligodendroglia and neurons. J Neurochem. 1979;32:293–300.
Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J Lipid Res. 2004;45:783–93.
Yu RK, Tsai YT, Ariga T. Functional roles of gangliosides in neurodevelopment: an overview of recent advances. Neurochem Res. 2012;37:1230–44.
Yusuf HKM, Dickerson JWT. Disialoganglioside GD1a of rat brain subcellular particles during development. Biochem J. 1978;174:655–7.
Zappia M, Crescibene L, Bosco D, Arabia G, Nicoletti G, Bagalia A, et al. Anti-GM1 ganglioside antibodies in Parkinson’s disease. Acta Neurol Scand. 2002;106:54–7.
Zhang J, Kramer EG, Asp L, Dutta DJ, Navrazhina K, Pham T, et al. Promoting myelin repair and return of function in multiple sclerosis. FEBS Lett. 2011;585:3813–20.
Conflict of Interest
The author declares no conflicts of interest in preparing this article.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Ando, S. (2014). Glycoconjugate Changes in Aging and Age-Related Diseases. In: Yu, R., Schengrund, CL. (eds) Glycobiology of the Nervous System. Advances in Neurobiology, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1154-7_19
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1154-7_19
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1153-0
Online ISBN: 978-1-4939-1154-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)