
Abstract
This chapter summarizes and highlights advances from the last decade which have significantly contributed to our understanding of how endocannabinoid signaling is influenced during acute and chronic stress conditions, and in turn is able to importantly shape endocrine and behavioral stress responses through a variety of stress-responsive nuclei. The reviewed literature underscores a pivotal interaction of glucocorticoid-mediated changes during stress scenarios, and region-specific changes that display specialized responses depending on whether encountered stressors are experienced acutely or chronically. While the majority of reviewed content discusses our current understanding of in vitro and in vivo animal work, promising translational studies which have documented similar parallels in human literature are additionally spotlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Endocannabinoid
- Hypothalamic-pituitary-adrenal axis
- Glucocorticoid stress responses
- Stress adaptation
- Anandamide
- 2-arachidonoylglycerol
6.1 Introduction
More than a decade ago, cannabinoids were shown to act as novel retrograde messengers capable of synaptic modulation, which prompted interest in a possible application to stress-neurocircuitry (Auclair et al. 2000; Wilson and Nicoll 2001; Ohno-Shosaku et al. 2001) . Anecdotally, the stress-reducing effects of cannabinoids and cannabis usage are traced back to antiquity (Skaper and Di Marzo 2012) . And yet the examination of cannabinoids in the regulation of stress only seriously emerged following the identification of cannabinoid receptors in the brain (Devane et al. 1988; Herkenham et al. 1991) , and the ability to selectively stimulate or antagonize them through advances in genetics and pharmacology. These developments have since led to pivotal discoveries in the area of stress research and established that: (1) cannabinoids inhibit excitation of the hypothalamic-pituitary-adrenal (HPA) axis, which ultimately regulates endocrine stress responses and (2) this neurotransmitter system is activated by glucocorticoid elevations during stress, enabling cannabinoids to significantly shape the magnitude and duration of neural excitation imposed on the HPA axis. Thus, the cannabinoid system has quickly become a target of interest for stakeholders engaged in stress research including scientists, clinicians, and pharmaceutical corporations.
6.2 Endocannabinoid Basics
The endogenous cannabinoid system, denoted as the “endocannabinoid system,” is a neurotransmitter family composed of two lipid-based ligands and two G protein-coupled receptors. These receptors are activated by endogenous and exogenous cannabinoid molecules (i.e., THC or delta9-tetrahydrocannabinol) and are commonly referred to as cannabinoid receptors 1 and 2, or CB1 and CB2. CB1 receptors (CB1Rs) are widely distributed in the brain with notable distribution in stress-responsive regions like the hippocampus, amygdala , cortex, hypothalamus , septum, and brainstem (Herkenham et al. 1991; Marsicano and Lutz 1999; Egertova et al. 2003) . CB1Rs are coupled to Gi/Go proteins and as their expression is almost exclusively confined to axon terminals, activation of this receptor results in a suppression of voltage-gated calcium channels, activation of outward rectifying potassium channels, and a net inhibition of synaptic release of neurotransmitters (Katona and Freund 2012) . Initial perspectives thought that CB1Rs were exclusively found in the brain and its counterpart CB2R was isolated to peripheral immune-regulating cells or cells that had peripheral origins (e.g., leukocytes, macrophages, microglia), and peripheral organs (e.g., the spleen) (Munro et al. 1993; Parolaro 1999; Cabral and Marciano-Cabral 2005; Atwood and Mackie 2010) . However, although CB1R and CB2R distribution is still regarded as distinct and largely non-overlapping, views on the distribution of these receptors continues to change. CB1R has also been found in the spine, vascular tissue, adipocytes, and on peripheral organs including all endocrine glands (Herkenham et al. 1991; Parolaro 1999; Cota et al. 2003; Bellocchio et al. 2008) . Emerging evidence also indicates CB2R is limitedly expressed within neural tissue (Nunez et al. 2004; Van Sickle et al. 2005; Gong et al. 2006; Palazuelos et al. 2006; Onaivi 2011; Xi et al. 2011) . Based on the initial discoveries which suggested that CB1Rs were exclusively found in the brain, the effects of endocannabinoid signaling on HPA axis activity has been entirely focused on CB1R synaptic contributions. Therefore, the remainder of this chapter will discuss the effects of endocannabinoid signaling with attention specifically on the existing CB1R-related evidence.
6.3 Endocannabinoid Synthesis and Metabolism
Just as the lipid structure of glucocorticoid steroids allows easy passage through cell membranes and penetration throughout the brain and body, the two endocannabinoid ligands N-arachidonoyl-ethanolamine (anandamide (AEA)) (Devane et al. 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al. 1995; Sugiura et al. 1995) , are similarly composed of lipids, thus providing them ubiquitous systemic access. Contrary to typical neurotransmitters which usually move across synapses from a pre- to postsynaptic membrane surface, these modulators are instead made postsynaptically during neuronal activation through intracellular elevations in calcium and the activation of specific phospholipases in an “on demand” fashion, then released retrogradely, allowing them to act on presynaptic CB1Rs (Wilson and Nicoll 2001; Alger 2002) . Endocannabinoids are not packaged into synaptic vesicles like classic neurotransmitters, but are instantaneously released into the synaptic cleft following their membrane-based production. CB1Rs are also found on the axon terminals of many different neural phenotypes including glutaminergic, GABAergic, and monoaminergic neurons (Schlicker and Kathmann 2001; Freund et al. 2003) , thus it is not surprising that CB1R activation has region-specific effects, which is dictated by the excitatory or inhibitory nature of the cell populations involved.
Synthesis of AEA and 2-AG during neuronal depolarization, or as a result of postsynaptic signaling cascades, is thought to occur through enzyme-mediated cleavage of membrane-associated phospholipids. Although production of these coordinating enzymes is believed to be triggered by changes in intracellular calcium, activation of metabotropic receptors is also a major factor for endocannabinoid mobilization (Freund et al. 2003) . In the case of 2-AG, phospholipase C and D can both stimulate production of diacylglyerol (DAG), which is readily converted to 2-AG via enzymatic actions of DAG lipase (Hillard 2000; Sugiura et al. 2002; Di Marzo 2008) . The pathway coordinating AEA production however is less clear as three independent mechanisms have been reported (Liu et al. 2006; Simon and Cravatt 2006; Okamoto et al. 2007) . It also remains to be confirmed which possible pathways drive AEA synthesis in the brain (Ahn et al. 2008; Bisogno 2008) .
Following postsynaptic release, endocannabinoids exhibit a very transient lifespan and are metabolized quickly, which allows for tight regulation of their temporal influence on synaptic transmission. However, AEA and 2-AG are not uniformly metabolized by the same enzyme. Fatty acid amide hydrolase (FAAH), which is a postsynaptically expressed enzyme found on the membrane of the endoplasmic reticulum, is the only known catabolic enzyme capable of hydrolyzing AEA into ethanolamine and arachidonic acid (Deutsch et al. 2002; Ueda 2002) 2-AG can be metabolized by FAAH, however this appears to be an artifact of in vitro preparations, as in vivo testing has shown it is primarily degraded (85 %) by presynaptic monoacylglyceride (MAG) lipase into glycerol and arachidonic acid, while the rest (15%) is degraded by the recently identified postsynaptic enzymes ABHD6 and ABHD12 (Ueda 2002; Dinh et al. 2002; Blankman et al. 2007; Marrs et al. 2010) . The capacity that cells have to selectively metabolize 2-AG without altering AEA tone intriguingly suggests functional differences in these ligands—but the implications and the nature of these differences remain unresolved.
6.4 Current Trends in Endocannabinoid-Stress Research
Initially, AEA and 2-AG were thought to have similar physiological and behavioral effects; however there exists differences in binding affinity, pharmacokinetics, and ligand signaling efficacy (Sugiura et al. 2006) , which has led researchers to suspect that AEA and 2-AG act during different temporal phases of neuronal activation and regulate different neuronal states. In applying this concept to activation of the HPA axis , an on-going hypothesis we and others are pursuing is the idea that constituent levels of AEA provide “tonic inhibition” on synaptic signaling allowing tight regulation of neurotransmitter release under normal basal conditions (Hill and Tasker 2012) . Conversely, it appears 2-AG is produced “on demand” and is robustly increased during scenarios of sustained neuronal activation, contributing to the onset of adaptive forms of synaptic plasticity (Ahn et al. 2008; Gorzalka et al. 2008) . This framework is importantly shaping how previous and emerging endocannabinoid research is being viewed. This categorization of roles for AEA and 2-AG also foreshadows the current trends in this field; which as discussed below, emphasizes a prominent role for increased 2-AG signaling during acute and mild repetitive stress conditions, whereby enhanced HPA axis inhibition could be adaptive and appropriate in the face of predictable, non-threatening scenarios to prevent HPA axis hyperactivation. Conversely, at the other end of the stress-scenario spectrum, when conditions involve chronic unpredictable physical and emotional stressors, the endocannabinoid system appears to respond with both ligand and receptor changes to promote HPA axis responsiveness downstream of the prefrontal cortex (PFC) , while enhancing the inhibitory strength of the PFC via CB1R upregulation. Although HPA axis sensitization provides certain survival advantages in the context of physical or predatory threats, it may be the case however, that chronic stress-induced adaptations to the central endocannabinoid system create a physiological state vulnerable to excitotoxicity, neuroinflammation, and stress-related disorders (Zoppi et al. 2011) .
6.5 Origins of Endocannabinoid-Stress Research
The first characterizations of CB1R expression revealed a wide distribution throughout the brain with notable expression in stress-sensitive regions communicating with the HPA axis, and low but detectable levels in the hypothalamus , median eminence, and anterior pituitary (Herkenham et al. 1991; Gonzalez et al. 1999; Marsicano and Lutz 1999; Egertova et al. 2003) . With the advent of receptor-specific pharmacological drugs, and the ability to measure stress-induced changes in endocannabinoid content, this neurotransmitter system has been an exciting new target in the field of stress research. As previously mentioned, cannabinoids have long been perceived as having anxiolytic effects, however it has only been in the last decade that the underlying mechanisms explaining these effects have been explored. Initial studies administering THC intracerebroventricularly to rodents in tandem with a CB1R antagonist, showed that CB1R blockade at high concentrations increased basal levels of adrenocorticotrophin (ACTH) and corticosterone (CORT), suggesting an inhibitory role of the endocannabinoid system over the HPA axis (Manzanares et al. 1999) .
In trying to further clarify the role of CB1R in the stress response, it was work from Jeff Tasker and colleagues who used a more isolated and direct approach involving hypothalamic rat slices to show that endocannabinoids can modulate neurosecretory cells within the command center of the HPA axis, the paraventricular nucleus (PVN) . This groundbreaking study was the first in vitro experiment to establish that endocannabinoids can inhibit HPA axis signaling, as they found that CB1R activation decreases presynaptic glutamate release onto PVN parvocellular populations, which included corticotropin releasing hormone (CRH) positive cells, and other stress-regulating oxytocin-, vasopressin-, and thyrotrophin-releasing hormone-positive cells (Di et al. 2003) . Continued work from Tasker’s group has shown that endocannabinoid signaling in the PVN does not merely rely on postsynaptic activation, but is contingent on rapid non-genomic glucocorticoid signaling (Tasker 2006) . This exciting work has contributed significantly to our understanding of glucocorticoid negative feedback by providing insight into how activation of the lower affinity glucocorticoid receptor (GR) actually coordinates an inhibitory influence on synaptic communication. These findings have also revealed that a downstream component of this long-established GR-mediated negative feedback cascade relies on endocannabinoids; opening up new and exciting avenues for investigating the etiology and treatment of diseases marked by glucocorticoid hypersecretion.
6.6 Early Studies in Acute Stress Literature
The seminal work of Di et al. (2003) have since set the stage for follow-up studies to confirm and further explore with in vitro and in vivo approaches how acute stress and glucocorticoids effect endocannabinoid synaptic transmission . These findings have also inspired the use of knockout approaches to examine the consequences of endocannabinoid dysregulation on stress-related endocrine and behavioral measures. Genetic deletion of CB1R in knockout models has been found to enhance stress-induced peak responses of ACTH and CORT under a variety of stress conditions including restraint (Uriguen et al. 2004) , tail suspension (Aso et al. 2008) , forced swim (Steiner et al. 2008), and novel cage stress (Barna et al. 2004; Haller et al. 2004) . CB1R knockout mice (CB1R-/-) also have enhanced HPA axis circadian peaks and impaired glucocorticoid feedback (Cota et al. 2007) . Although knockout models are susceptible to possible compensatory changes, the knowledge generated using this approach has been consistent with experiments using pharmacological manipulations, which also have underscored that CB1R antagonism potentiates peak ACTH, CORT, and cFos mRNA responses during noise stress (Newsom et al. 2012) ; potentiates CORT elevations during restraint recovery when administered locally into the PFC (Hill et al. 2011a) ; potentiates CORT responses during forced swim (Steiner et al. 2008) and social defeat (Steiner and Wotjak 2008); and increases basal circadian CORT levels (Atkinson et al. 2010) . This work has led to the suggestion that CB1Rs negatively influence activation of the HPA axis in two regards: (1) by dampening the initial activation of the HPA axis to attenuate peak increases and (2) by facilitating termination of HPA axis activity to reduce the overall duration that glucocorticoid elevations are experienced systemically (Barna et al. 2004; Haller et al. 2004; Uriguen et al. 2004; Steiner and Wotjak 2008; Hill et al. 2010a, 2011a) .
6.7 Endocannabinoid Changes During Acute Stress
In vitro studies modeling acute stress conditions have shown that bath application of CORT and dexamethasone increases CB1R-mediated inhibition of glutamate release in the PVN, supraoptic nucleus, basolateral amygdala , dorsal raphe, but not the cerebellum, suggesting a CORT-dependent relationship selective to stress-regulating circuits (Di et al. 2003, 2005; Malcher-Lopes et al. 2006; Karst et al. 2010; Wang et al. 2012a) . These studies have confirmed that CB1R-mediated inhibition of glutamate release occurs throughout the brain; and in examining the PVN specifically, that this effect is found in a variety of cell populations including parvo-, magno-, and pre-autonomic cells (Tasker 2006; Boychuk et al. 2013) . In modeling hemorrhage-stress, CB1R-mediated inhibition of PVN glutamate release has been shown to be activated by alpha-2-adrenergic receptors (Kuzmiski et al. 2009) . Tasker and colleagues have also revealed that glucocorticoid-induced biosynthesis of endocannabinoids in the PVN is blocked by the satiety hormone leptin (Malcher-Lopes et al. 2006). It additionally appears that endocannabinoids do not only modulate glutamate release in the PVN, but display CORT-dependent CB1R regulation of GABA synapses as well (Wamsteeker et al. 2010) . A similar relationship is also found outside the hypothalamus, as CORT-dependent inhibition of GABA release has been documented in the hippocampus (Wang et al. 2012b) and PFC (Hill et al. 2011a) . Taken together these studies have led to the consensus that the inhibitory effects of endocannabinoid signaling on stress responsivity show a prominent, although not exclusive, glucocorticoid dependence (Kuzmiski et al. 2009; Crosby et al. 2011) , and underscore that CB1R plays a prominent regulatory role on both glutamatergic and GABAergic neurons throughout the brain. Our knowledge of stress-induced CB1R signaling also continues to expand as microdialysis studies have shown that stress-induced CB1R activation in the hippocampus is able to limit acetylcholine transmission, in addition to GABA release (Degroot et al. 2006) .
Having established that glucocorticoids can significantly alter the endocannabinoid system, many studies in the last decade have focused on determining if stress scenarios alter endocannabinoid tone by testing for possible stress-induced changes to the receptor, ligands, and the metabolic enzymes composing this neuromodulatory family. During acute physical stressors like foot shock, AEA and 2-AG increases have been demonstrated in the periaqueductal gray (Hohmann et al. 2005) . However, when stressors are primarily psychological, such as, acute restraint, increases appear to be dominated by 2-AG rises in the PFC, hippocampus, and hypothalamus (Evanson et al. 2010; Hill et al. 2011a; Wang et al. 2012b) , with no change in the amygdala (Hill et al. 2009a; Patel et al. 2009) . 2-AG increases in the PFC, hippocampus, and hypothalamus are considered CORT-dependent (Hill et al. 2010b; Wang et al. 2012b) —unlike the rapid nongenomic effects observed in the hypothalamus (Di et al. 2003; Hill et al. 2010b) —as CORT application to the PFC elicits 2-AG rises with a slower onset (1 h) suggesting genomic actions (Hill et al. 2011a). Similarly, CORT application to the hippocampus also produces slower (30 min) 2-AG increases (Wang et al. 2012b) . When further tested in vivo, CB1R antagonist administered into the PFC does not alter restraint-induced CORT peak responses, but does potentiate post-stress recovery levels of CORT via a mechanism that is glucocorticoid-dependent (Hill et al. 2011a) . These data suggest that CORT-initiated 2-AG increases in the PFC have a greater contribution to the termination of the stress response, as opposed to its initiation and maintenance. These findings also beg the question as to whether antagonism of hippocampal CB1Rs would also have a greater influence during stress recovery, on the basis that lesion studies have revealed that its inhibitory HPA axis contribution is most apparent during the recovery phase (Herman et al. 2005) . As yet, the mechanisms causing acute 2-AG increases is unknown, but preliminary indications point to a CORT-mediated decrease in MAG lipase, which may have a facilitatory role by reducing 2-AG metabolism, herein enhancing its synaptic availability (Sumislawski et al. 2011) .
In many cases, a corresponding rapid AEA decrease is found in the PFC, hippocampus, and amygdala following forced swim stress (McLaughlin et al. 2012) or restraint stress (Hill et al. 2009a; Wang et al. 2012b) ; which in the case of the amygdala appears to coincide with increases in FAAH-mediated AEA metabolism (Hill et al. 2009a) . Given that CORT-dependent endocannabinoid mobilization and CB1R activation has mostly been studied in vitro, our laboratory has made attempts to study the in vivo effects of CORT elevations on AEA and 2-AG regional levels. Acute intraperitoneal CORT injections have a stimulatory effect on AEA content in the amygdala, hippocampus, and hypothalamus , and elicit increases in 2-AG content within the hypothalamus (Hill et al. 2010b) . These data would suggest that glucocorticoids on their own possess the ability to increase both AEA and 2-AG (consistent with in vitro studies) (Malcher-Lopes et al. 2006), but under conditions of stress, an additional stress-induced neural signal (possibly CRH or norepinephrine) seems to engage FAAH activity to instead reduce AEA content. Our working hypothesis is that CORT-mediated increases in AEA account for the recovery in AEA levels following cessation from stress, but that the reductions in AEA content following stress are through a CORT-independent mechanism.
With respect to CB1R function, acute restraint exposure does not appear to alter CB1R binding density (Rademacher et al. 2008; Hill et al. 2009a; Evanson et al. 2010) , while acute social defeat stress has been found to blunt CB1R-mediated inhibition of GABAergic transmission in the striatum (Rossi et al. 2008) . Additionally, 24 h food deprivation stress extinguishes CB1R-mediated inhibition of GABA synapses in the dorsomedial hypothalamus (DMH) in a manner that is CORT- and nitric oxide-dependent (Crosby et al. 2011) . Given that the DMH, striatum, and limited brainstem regions have been found to be vulnerable to stress-induced endocannabinoids changes, future research examining ligand and receptor changes in these regions, in addition to, and in comparison to the more typical target structures for stress research (i.e. PFC, hippocampus, hypothalamus, amygdala), should aid in rounding out our understanding of the neuroanatomical impact of emotional and physical stressors. Recent work from our laboratory also suggests measurement of inducible serum endocannabinoid changes may be an area for bridging and comparing rodent and human studies. Using the Trier social stress test entailing a mock job interview, female participants were found to exhibit rapid increases in plasma 2-AG levels with no change in circulating AEA (Hill et al. 2009b) . Together this literature has established that endocannabinoid levels do change in the brain and blood during acute stressors and indicate 2-AG rises during psychological stressors show a fair degree of consistency across rodents and humans thus far.
6.7.1 Circuit Implications
Based on our findings in the amygdala that AEA concentrations negatively correlate with stress-induced CORT (Hill et al. 2009a), the evolving model that our laboratory has proposed is that AEA in the amygdala serves as a gatekeeper—tonically inhibiting amygdalar glutamatergic projections to the PVN via both limited direct (Prewitt and Herman 1998; Csaki et al. 2000) , and more prominent indirect routes (Dong et al. 2001) . So far stress-induced FAAH increases have been localized to the amygdala, suggesting that FAAH-mediated hydrolysis of AEA may create a state of stress-hypersensitivity in the amygdala allowing it to play an enhanced role during the initial stages of stress detection and appraisal. In other regions like the hippocampus , PFC, and hypothalamus , where both AEA and 2-AG changes occur but in opposite directions (Hill et al. 2007; Rademacher et al. 2008; Evanson et al. 2010; Hill et al. 2011; McLaughlin et al. 2012; Wang et al. 2012b) , there may be differences in the temporal onset of these changes allowing for CB1R activation to be selectively decreased through rapid AEA reductions, but then later increased once HPA activation has been achieved, through CORT-dependent 2-AG rises (see Hill and McEwen 2010, for review) . From stress onset, glucocorticoid increases typically take 2–3 min to become significantly elevated within plasma, and 10–15 min to become significantly increased centrally (Vahl et al. 2005; Droste et al. 2008) . This suggests that the initial moments of HPA axis activation may favor early events coordinating FAAH-mediated AEA hydrolysis to facilitate HPA axis stimulation through disinhibition of the amygdala. Then following successful glucocorticoid mobilization, the effects of CORT-negative feedback likely initiate “on demand” 2-AG increases to inhibit glutamate release in the PVN and amygdala, while inhibiting GABA transmission in the PFC and hippocampus (Katona et al. 1999; Irving et al. 2000; Hill and Tasker 2012; Wang et al. 2012b) , to enhance activation of glutamatergic projections to downstream inhibitory PVN relays such as the bed nucleus of the stria terminalis (Cullinan et al. 1993; Radley et al. 2006b; Choi et al. 2008; Radley et al. 2009) (Table 6.1, Fig. 6.1). Notably, certain aspects of this proposed cascade still need to be elucidated—the mechanisms driving stress-induced FAAH increases remain unknown, as well the developmental onset of these mechanisms. Additionally, limited studies have examined these processes in female rodents (Cota et al. 2007; Reich et al. 2009; Atkinson et al. 2010) ; or fully explored the contributions of the lower affinity, membrane-bound mineralocorticoid receptor that was recently uncovered (Karst et al. 2005; de Kloet et al. 2008; Olijslagers et al. 2008; Karst et al. 2010) .

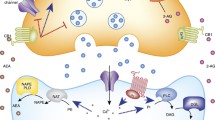
Acute effects of stress- and glucocorticoid-mediated changes in endocannabinoids. 1. Stress causes a decrease in anandamide (AEA) content in the BLA, through an increase in fatty acid amide hydrolase (FAAH) content within this region. This increase in FAAH and subsequent decrease in AEA content lessens the basal gate-keeping tone in the BLA—and through this excitatory facilitation of amygdalar projections, eventually their downstream projections lead to a removal of the GABAergic inhibition of the paraventricular nucleus (PVN) in the hypothalamus, thus driving the HPA response. 2. Corticotropin releasing hormone (CRH) is released from the PVN into the anterior pituitary, causing the release of adrenocorticotropin (ACTH), which is then released into circulation. 3. ACTH drives the release of corticosterone (CORT) from the adrenal cortex. CORT is released into circulation and exerts negative feedback on HPA axis signaling. There is direct negative feedback at the level of the pituitary and PVN and indirect feedback, both mediated by endocannabinoids at upstream limbic regions. 4. Circulating CORT causes an increase in 2-arachidonoylglycerol (2-AG) in multiple regions, including the PVN, prefrontal cortex (PFC), and hippocampus. 5. At the level of the PVN and amygdala, the rise in 2-AG content inhibits glutamate transmission, thus rapidly inhibiting the drive on the HPA axis. Additionally, the increase in 2-AG in the PFC and hippocampus, leads to a decrease in GABA transmission, which, in the case of the PFC and possibly in the case of the hippocampus, leads to an activation of glutamatergic projections to downstream inhibitory circuits on the PVN, thus providing a slower mechanism of shutting down the drive on the HPA axis. Finally, AEA content within the BLA is increased, thus restoring the basal inhibitory gate-keeping tone on the HPA axis
6.8 Endocannabinoid Changes During Repeated Homotypic Stress and Chronic Unpredictable Stress
The emerging pattern of endocannabinoid changes during repeated homotypic stress consistently shows 2-AG increases isolated to stress-sensitive relays like the hypothalamus, amygdala, and the PFC (Patel et al. 2004, 2005b; Rademacher et al. 2008; Patel et al. 2009) . Although 2-AG increases are known to be CORT-dependent in many stress structures, the mechanisms involved remain unknown (Malcher-Lopes et al. 2006; Hill et al. 2010b; Bowles et al. 2012) . While CORT-induced decreases in MAG lipase may contribute to acute stress 2-AG increases (Sumislawski et al. 2011) , upregulation of the 2-AG precursor DAG during repeated restraint appears to be an underlying contributing factor when looking in the BLA (Patel et al. 2009). Unlike 2-AG, repeated stress studies typically report stress-induced AEA reductions occurring in regions like the amygdala, PFC, hypothalamus, and hippocampus (Patel et al. 2004, 2005b; Hill et al. 2007, 2008a; Rademacher et al. 2008; Patel et al. 2009; Hill et al. 2010b) . Based on the discriminative expression of CB1R within the amygdala, such that it is predominately found in the basolateral aspect and less so in the medial and central divisions, it now appears that AEA and 2-AG induced changes, and their ensuing immediate effects on synaptic communication, have prominent effects in the BLA (Hill et al. 2009a; Patel et al. 2009). This , is supported by antagonist work confirming that CB1R blockade increases stress-induced CORT elevations when introduced locally into the BLA and not neighboring nuclei (Hill et al. 2009a). However, it should not be overlooked that CB1R activation also has downstream consequences for neuronal signaling in the central amygdala (Patel et al. 2005a). The induction of endocannabinoid changes during repeated restraint also show variations in temporal onset, which might be aligned with species differences and regional differences in the sensitivity of synapses to initiate 2-AG increases. Following 5 days of repeated restraint, mice show 2-AG increases in the amygdala, hypothalamus, and forebrain (Patel et al. 2004, 2005b) , although there are reports that the amygdala and PFC take 10 days, and not 7 to show increases in 2-AG (Rademacher et al. 2008) . In contrast, rats show increases in amygdalar 2-AG following 9 days of repeated stress (Hill et al. 2010a), with no detectable increases elsewhere. Patel et al. (2009) have found 2-AG increases in the amygdala following repeated restraint at 20 min following stress onset, but are non-detectable at 60 min, suggesting possible discrepancies among studies may be due to the transient nature of 2-AG increases. Similarly in the rat, 2-AG levels return to normal, 24 h following the final stressor (Hill et al. 2009c), suggesting that the ability of repeated stress to increase 2-AG content is a transient response.
Few repeated stress studies have quantified changes in CB1R binding or mRNA levels (Rademacher et al. 2008; Hill et al. 2012; Lee and Hill 2012) ; but in vitro tests indicate CB1R function is downregulated in the hypothalamus (Wamsteeker et al. 2010) , nucleus accumbens (Wang et al. 2010) , BLA (Patel et al. 2009), and hippocampus (Hu et al. 2011) . As stress paradigms shift from repeated homotypic stress to more intense chronic physical and emotional stressors, the resulting effects on the endocannabinoid system show a prominent shift, and a greater impact on CB1R levels. When looking at the effects of chronic unpredictable stress (CUS), the net effect of CB1R changes appears adaptive, in that it increases the efficiency by which the HPA axis is both activated and terminated, therein creating a faster “on” and “off” switch. Consistent across rodent studies CUS induces significant increases in PFC CB1R binding density, but prevalent CB1R decreases within downstream HPA axis relays including the hippocampus, amygdala, and hypothalamus (Hillard et al. 2006; Bortolato et al. 2007; Hill et al. 2008a; McLaughlin et al. 2013) . Given that CORT-dependent downregulation of CB1R has been reported in the hippocampus, amygdala , hypothalamus, and striatum (Hill et al. 2008b; Rossi et al. 2008; Wamsteeker et al. 2010; Bowles et al. 2012) , it is likely CUS-induced CB1R decreases are CORT-mediated, and quite possible that PFC CB1Rs are exceptionally sensitive to CORT-upregulation as well. Consistent with this, postmortem tissue of individuals with major depression also present with PFC CB1R elevations (Hungund et al. 2004) , which has highlighted CB1R forebrain increases as a potentially very important synaptic compensatory change during states of chronic stress. These findings are also complemented by evidence from selective knockout models generated by Beat Lutz and Giovanni Marsicano. The effects of CB1R knockout on cortical glutamatergic (Glu-CB1R-/-), just GABAergic (GAB-CB1R-/-), and all principal forebrain neurons (CaMK-CB1R-/-), have shown that removing CB1R from cortical glutamate and GABA synapses has no effect on CORT release during the forced swim test (FST), whereas CB1R deletion from principal forebrain neurons elevates FST endocrine stress response (Steiner et al. 2008) . These findings suggest that abolishing CB1R from cortical glutamatergic and CB1R-GABAergic expression throughout the brain results in a net change that does not significantly alter CORT output, whereas CB1Rs on principal neurons in the forebrain have the capacity to significantly inhibit stress-induced CORT responses (Steiner et al. 2008) . The PFC has long been regarded as an important inhibitory influence on the PVN (Diorio et al. 1993; Radley et al. 2006a) , however until now little has been known about the synaptic mechanisms coordinating this effect. Together, these data suggest CB1Rs are differently regulated in a site-specific manner with glucocorticoids negatively regulating CB1Rs in the hippocampus, amygdala , striatum, and hypothalamus, and possibly having an opposite effect on CB1Rs in the PFC (McLaughlin et al. 2013) . CUS may be associated with widespread AEA reductions across the hippocampus, hypothalamus, ventral striatum, amygdala, and midbrain (Hill et al. 2008a), although this possibility has yet to be consistently reported (Hill et al. 2005; Wang et al. 2010) . Similar to repeated restraint, CUS also induces 2-AG increases; however these increases have only been reported in the hypothalamus, midbrain, and thalamus (Bortolato et al. 2007; Hill et al. 2008a) . More studies are needed to confirm the effects of CUS on induced 2-AG levels, and particularly the temporal nature of these changes given that the effects of repeated stress seem to be temporally constrained to stress exposure.
In addition to stress-induced changes in endocannabinoid signaling, stress-induced structural changes also represent an important influence on synaptic transmission during chronic stress. FAAH-dependent amygdalar changes in excitability are associated with stress-induced increases in dendritic arborization, complexity, and spine density, which parallel increases in anxiety behavior (Hill et al. 2011b). These effects are abolished in FAAH-knockout mice—verifying that FAAH activity within the BLA increases amygdalar excitability and promotes a hyper-anxious state during chronic stress. Similarly CB1R-/- mice are also vulnerable to stress-induced dendritic changes in the amygdala, and under nonstressed conditions show prelimbic structural changes which mirror the dendritic retraction and reductions in branch points typically induced by chronic stress (Hill et al. 2011b) . Together these data suggest PFC CB1Rs are critical for maintaining normal synaptic function and structure, and are an important point of comparison when investigating the hallmark changes of depression and chronic stress. It additionally appears that amygdalar synaptic changes induced by stress are multifaceted, entailing structural, ligand, and receptor changes, paired with altered endocannabinoid anabolic and catabolic capacities.
6.8.1 Circuit Implications
As neurons sense their external environment changing and consistently experience glucocorticoid elevations, repeated restraint appears to cause AEA reductions paired with 2-AG elevations throughout the limbic-HPA axis. Widespread AEA declines likely prime the HPA axis and its afferents for future anticipated stress by lowering the activation threshold of HPA axis relays to enhance synaptic communication. While at the same time “on demand” CORT-dependent increases in 2-AG become heightened to provide a more robust “brake” on activated stress-circuitry, leading to faster and efficient termination of behavioral and endocrine stress responses. In contrast to repeated restraint which favors an upregulation of ligands to enhance CB1R-activated HPA inhibition, the utility of significantly reducing CB1R expression during CUS in subcortical regions is likely necessary for maintaining HPA axis responsiveness. CORT-dependent CB1R declines in the amygdala are poised to enhance glutamatergic amygdalar activation, thus promoting and maintaining HPA axis responsivity. Similarly, hippocampal CB1R declines may promote HPA axis activation by enhancing hippocampal GABA release, thus silencing the hippocampus and reducing its capacity to provide indirect inhibition on the PVN (Sapolsky et al. 1984; Herman et al. 1992, 2005) . Thus it appears that CB1R is necessary for promoting adaptation during repeated homotypic stress conditions, but under chronic stress conditions, subcortical downregulation of CB1R is more favorable. CB1R decreases could be beneficial in the face of life-threatening physical stressors and especially adaptive when repeated stressors are unpredictable, but still highly anticipated. Based on the conditional knockout models which have shown that forebrain CB1Rs are essential for dampening endocrine stress responses (Steiner and Wotjak 2008) , the data seem to suggest that CUS-induced CB1R increases in the PFC should protect individuals from HPA axis hyperactivation. In the PFC, CB1Rs are almost entirely expressed on GABAergic terminals in the prelimbic division (Hill and Tasker 2012) , indicating stress-induced CB1R increases are positioned to promote activation of PFC projections to downstream inhibitory PVN afferents like the bed nucleus (Radley et al. 2006a, 2009) . Based on the evidence that depressed, suicidal individuals show higher CB1R levels in the PFC (Hungund et al. 2004) , and that this is a similar hallmark of rodent CUS models, CB1R PFC increases could be a compensatory change aimed at preventing hyper-glucocorticoid secretion and promoting termination of the stress response once the threatening stimulus is removed. This is consistent with a recent report which suggests that upregulation of prefrontal cortical CB1R is an adaptive response aimed at limiting the adverse effects of stress (McLaughlin et al. 2013) (Table 6.2, Fig. 6.2).

Chronic effects of stress- and glucocorticoid-mediated changes in endocannabinoids. 1. Repeated restraint leads to a decrease of the anandamide (AEA) tone in the BLA, through an increase in fatty acid amide hydrolase (FAAH) activity, which possibly lowers the activation threshold for HPA axis activation. 2. Upon loss of the gate-keeping tone in the primed BLA, the paraventricular nucleus (PVN) is activated to release corticotropin releasing hormone (CRH), which is released into the anterior pituitary causing the release of adrenocorticotropin (ACTH). 3. ACTH is released into circulation and causes the adrenal cortex to release corticosterone (CORT). In the case of repeated stress, there is a habituation in the amount of CORT released. 4. CORT-induced 2-arachidonoylglycerol (2-AG) increases in the prefrontal cortex (PFC), hypothalamus, and hippocampus are elevated, which may be causing a more effective and quicker termination of the HPA axis response to repeated homotypic stressors. 5. This is in contrast to chronic unpredictable stressors. Animals exposed to CUS do not show CORT habituation. Furthermore, after CUS, there is a decrease in cannabinoid receptor 1 (CB1R) in the amygdala and hippocampus. These declines could promote HPA axis signaling through different mechanisms. In the amygdala, a decrease in CB1R would lead to an enhancement of glutamatergic amygdalar activation, which would promote HPA axis signaling. In the hippocampus, it is through enhancing GABA signaling on hippocampal interneurons, which silences the hippocampus and its inhibitory relays to the PVN. 6. In the PFC, CB1R is upregulated under chronic stress conditions. This is in contrast to the subcortical decreases in CB1R, which facilitate HPA axis activation. CB1R upregulation in the PFC could serve to protect against hyperactivation of the HPA axis and by terminating the stress response through downstream inhibitory projections to the PVN.
6.9 Future Considerations
6.9.1 Psychological Versus Physical Stress Circuits
Restraint is primarily a psychological stress, thus studies are currently needed to confirm that restraint induced 2-AG increases are indeed isolated to prominent limbic-HPA axis regions such as the hippocampus. It also has yet to be shown if physical and psychological stimuli induce similar or anatomically distinct endocannabinoid responses. Since limbic-PVN circuits are primarily recruited during psychological stress, and brainstem-PVN circuits are differently responsive to physical stress (Herman and Cullinan 1997; Dayas et al. 2001) it may be the case that physical stressors elicit distinct regional changes within the brainstem and spine that warrant more detailed investigation.
6.9.2 CB1R Quantification Tools
There is some indication during CUS paradigms that larger hippocampal decreases exist in the dorsal versus ventral zone, and that females may in fact show CUS-induced CB1R hippocampal increases (Reich et al. 2009). However, these data have been limited to western blot analysis and there is a current lack of specific CB1R antibodies which have been validated in knockout tissue (Grimsey et al. 2008) . These findings do raise tremendous interest though as to possible underlying sex differences in the endocannabinoid system which should be explored with additional binding and mRNA approaches. Already the circadian CORT rhythm of male rats has been found to be more sensitive to CB1R antagonism, suggesting additional sex differences are probable (Atkinson et al. 2010) .
6.9.3 Methodology and Controls
Discrepancies do arise when comparing the effects of CUS across studies, but these differences may be linked to methodology. In particular, CB1R changes reported by Bortolato et al. (2007) may be different compared to other reports since the control rats in this experiment were exposed to isolation as well as food and water deprivation stress which may have generated unintended stress-mediated CB1R changes, making it difficult to separate out, and detect CUS-induced treatment effects. Studies which have been subsequent to Hungund et al. (2004) in examining CB1R changes in depressed, suicidal individuals are also difficult to apply to existing rodent findings as these studies are usually restricted to alcoholic populations without the inclusion of nonalcoholic controls (Vinod et al. 2005, 2010) .
6.9.4 Permanence and Plasticity
Proving that stress-induced changes display a great deal of plasticity , the permanence of stress-induced changes have been tested to a limited extent. Looking at repeated social defeat stress Rossi et al. (2008) have found that glucocorticoid-dependent CB1R-mediated inhibition of GABAergic transmission in the striatum arises after 3 and 7 days of stress exposure, and that they were able to reverse these effects by providing rodents access to running wheels, sucrose, and cocaine. These data have importantly shown that changes to the efficacy of synaptic signaling can be recovered through physical and metabolic experiences which are known to activate central reward systems (Rossi et al. 2008). As well, simple cessation of repeated restraint for 1 week is also sufficient to reverse signs of long-term depression at inhibitory BLA synapses and behavioral changes in feeding latency (Sumislawski et al. 2011) . Recently our laboratory has shown that repeated restraint results in a reduction in CB1 receptor binding in the hippocampus and increased CB1 receptor binding in the PFC, and that following a 4-week recovery period the PFC returns to normal, while in the hippocampus there is actually a surprising rebound effect where CB1R densities increase significantly above what is seen in control animals (Lee and Hill 2012) . These findings highlight the plasticity of synaptic changes, enabling neural systems to dynamically respond with reversible changes as situational changes arise. Although the structural consequences of CUS stress have yet to be examined, this synaptic flexibility may be compromised in chronic conditions creating a vulnerable state of hyper-excitable stress centers, exacerbating an individual’s susceptibility to glucocorticoid hypersecretion .
6.10 Conclusion
In summary, the role of endocannabinoids within stress neural-circuitry aligns with the inhibitory and excitatory influences of each structure. Under acute conditions, HPA axis stimulatory regions such as the PVN and amygdala show CORT-mediated recruitment of endocannabinoids to inhibit presynaptic glutamate release, leading to reduced neural activation. Whereas in HPA axis inhibitory structures, like the PFC and hippocampus, CORT-mediated recruitment of endocannabinoids inhibits GABA release to increase neural activation of glutamatergic projections which communicate with intermediate inhibitory PVN afferents (i.e. the bed nucleus of the stria terminalis and PVN surround). The effects of chronic stress on this neurotransmitter system lead to widespread receptor and ligand alterations whereby CB1R activity is reduced throughout the brain, but selectively increased in the PFC to provide an increased descending inhibitory input, while enhancing the stress-sensitivity of subcortical relays. Evidently, endocannabinoid and glucocorticoid signaling robustly interact at the synaptic level to regulate endocrine stress responses; however the full breadth of this relationship and its application to stress-linked disorders remains to be elucidated.
Abbreviations
- 2-AG:
-
2-arachidonoylglycerol
- ACTH:
-
Adrenocorticotropin
- AEA:
-
Anandamide
- CB1R:
-
Cannabinoid receptor 1
- CB2R:
-
Cannabinoid receptor 2
- CUS:
-
Chronic unpredictable stress
- CORT:
-
Corticosterone
- CRH:
-
Corticotropin releasing hormone
- THC:
-
Delta9-tetrahydrocannabinol
- DAG:
-
Diacylglyerol
- FAAH:
-
Fatty acid amide hydrolase
- FST:
-
Forced swim test
- GR:
-
Glucocorticoid receptor
- HPA axis:
-
Hypothalamic-pituitary-adrenal axis
- MAG lipase:
-
Monoacylglyceride lipase
- PVN:
-
Paraventricular nucleus
- PFC:
-
Prefrontal cortex
- DMH:
-
Dorsomedial hypothalamus
- BLA:
-
Basolateral amygdala
- GABA:
-
Gamma-aminobutyric acid
References
Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108(5):1687–707.
Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68(4):247–86.
Aso E, Ozaita A, Valdizan EM, Ledent C, Pazos A, Maldonado R, et al. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J Neurochem. 2008;105(2):565–72.
Atkinson HC, Leggett JD, Wood SA, Castrique ES, Kershaw YM, Lightman SL. Regulation of the hypothalamic-pituitary-adrenal axis circadian rhythm by endocannabinoids is sexually diergic. Endocrinology. 2010;151(8):3720–7.
Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160(3):467–79.
Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. J Neurophysiol. 2000;83(6):3287–93.
Barna I, Zelena D, Arszovszki AC, Ledent C. The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci. 2004;75(24):2959–70.
Bellocchio L, Cervino C, Vicennati V, Pasquali R, Pagotto U. Cannabinoid type 1 receptor: another arrow in the adipocytes’ bow. J Neuroendocrinol. 2008;20(Suppl 1):130–38.
Bisogno T. Endogenous cannabinoids: structure and metabolism. J Neuroendocrinol. 2008;20(Suppl 1):1–9.
Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14(12):1347–56.
Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, et al. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62(10):1103–10.
Bowles NP, Hill MN, Bhagat SM, Karatsoreos IN, Hillard CJ, McEwen BS. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience. 2012;204:83–9.
Boychuk CR, Zsombok A, Tasker JG, Smith BN. Rapid glucocorticoid-induced activation of TRP and CB1 receptors causes biphasic modulation of Glutamate release in gastric-related hypothalamic preautonomic neurons. Front Neurosci. 2013;7:3.
Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78(6):1192–7.
Choi DC, Furay AR, Evanson NK, Ulrich-Lai YM, Nguyen MM, Ostrander MM, et al. The role of the posterior medial bed nucleus of the stria terminalis in modulating hypothalamic-pituitary-adrenocortical axis responsiveness to acute and chronic stress. Psychoneuroendocrinology. 2008;33(5):659–69.
Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112(3):423–31.
Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, et al. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148(4):1574–81.
Crosby KM, Inoue W, Pittman QJ, Bains JS. Endocannabinoids gate state-dependent plasticity of synaptic inhibition in feeding circuits. Neuron. 2011;71(3):529–41.
Csaki A, Kocsis K, Halasz B, Kiss J. Localization of glutamatergic/aspartatergic neurons projecting to the hypothalamic paraventricular nucleus studied by retrograde transport of [3H]D-aspartate autoradiography. Neuroscience. 2000;101(3):637–55.
Cullinan WE, Herman JP, Watson SJ. Ventral subicular interaction with the hypothalamic paraventricular nucleus: evidence for a relay in the bed nucleus of the stria terminalis. J Comp Neurol. 1993;332(1):1–20.
Dayas CV, Buller KM, Crane JW, Xu Y, Day TA. Stressor categorization: acute physical and psychological stressors elicit distinctive recruitment patterns in the amygdala and in medullary noradrenergic cell groups. Eur J Neurosci. 2001;14(7):1143–52.
de Kloet ER, Karst H, Joels M. Corticosteroid hormones in the central stress response: quick-and-slow. Front Neuroendocrinol. 2008;29(2):268–72.
Degroot A, Kofalvi A, Wade MR, Davis RJ, Rodrigues RJ, Rebola N, et al. CB1 receptor antagonism increases hippocampal acetylcholine release: site and mechanism of action. Mol Pharmacol. 2006;70(4):1236–45.
Deutsch DG, Ueda N, Yamamoto S. The fatty acid amide hydrolase (FAAH). Prostaglandins Leukot Essent Fatty Acids. 2002;66(2–3):201–10.
Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–9.
Devane WA, Dysarz FA 3rd, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34(5):605–13.
Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24.
Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci. 2003;23(12):4850–7.
Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, et al. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005;569(Pt 3):751–60.
Dinh TP, Freund TF, Piomelli D. A role for monoglyceride lipase in 2-arachidonoylglycerol inactivation. Chem Phys Lipids. 2002;121(1–2):149–58.
Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13(9):3839–47.
Dong HW, Petrovich GD, Swanson LW. Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Brain Res Rev. 2001;38(1–2):192–246.
Droste SK, de Groote L, Atkinson HC, Lightman SL, Reul JM, Linthorst AC. Corticosterone levels in the brain show a distinct ultradian rhythm but a delayed response to forced swim stress. Endocrinology. 2008;149(7):3244–53.
Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb(1) cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119(2):481–96.
Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151(10):4811–9.
Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83(3):1017–66.
Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071(1):10–23.
Gonzalez S, Manzanares J, Berrendero F, Wenger T, Corchero J, Bisogno T, et al. Identification of endocannabinoids and cannabinoid CB(1) receptor mRNA in the pituitary gland. Neuroendocrinology. 1999;70(2):137–45.
Gorzalka BB, Hill MN, Hillard CJ. Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci Biobehav Rev. 2008;32(6):1152–60.
Grimsey NL, Goodfellow CE, Scotter EL, Dowie MJ, Glass M, Graham ES. Specific detection of CB1 receptors; cannabinoid CB1 receptor antibodies are not all created equal! J Neurosci Methods. 2008;171(1):78–86.
Haller J, Varga B, Ledent C, Barna I, Freund TF. Context-dependent effects of CB1 cannabinoid gene disruption on anxiety-like and social behaviour in mice. Eur J Neurosci. 2004;19(7):1906–12.
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11(2):563–83.
Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20(2):78–84.
Herman JP, Cullinan WE, Young EA, Akil H, Watson SJ. Selective forebrain fiber tract lesions implicate ventral hippocampal structures in tonic regulation of paraventricular nucleus corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) mRNA expression. Brain Res. 1992;592(1–2):228–38.
Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(8):1201–13.
Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(5):791–7.
Hill MN, Tasker JG. Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience. 2012;204:5–16.
Hill MN, Patel S, Carrier EJ, Rademacher DJ, Ormerod BK, Hillard CJ, et al. Downregulation of endocannabinoid signaling in the hippocampus following chronic unpredictable stress. Neuropsychopharmacology. 2005;30(3):508–15.
Hill MN, Barr AM, Ho WS, Carrier EJ, Gorzalka BB, Hillard CJ. Electroconvulsive shock treatment differentially modulates cortical and subcortical endocannabinoid activity. J Neurochem. 2007;103(1):47–56.
Hill MN, Carrier EJ, Ho WS, Shi L, Patel S, Gorzalka BB, et al. Prolonged glucocorticoid treatment decreases cannabinoid CB1 receptor density in the hippocampus. Hippocampus. 2008b;18(2):221–6.
Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, et al. Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem. 2008a;106(6):2322–36.
Hill MN, Miller GE, Ho WSV, Gorzalka BB, Hillard CJ. Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry. 2008c; 41(2):48-53.
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, et al. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009a;34(13):2733–45.
Hill MN, Miller GE, Carrier EJ, Gorzalka BB, Hillard CJ. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology. 2009b;34(8):1257–62.
Hill MN, Hunter RG, McEwen BS. Chronic stress differentially regulates cannabinoid CB1 receptor binding in distinct hippocampal subfields. Eur J Pharmacol. 2009c;614(1–3):66–9.
Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010a;107(20):9406–11.
Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010b;35(9):1333–8.
Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011a;31(29):10506–15.
Hill MN, Hillard CJ, McEwen BS. Alterations in corticolimbic dendritic morphology and emotional behavior in cannabinoid CB1 receptor-deficient mice parallel the effects of chronic stress. Cereb Cortex. 2011b;21(9):2056–64.
Hill MN, Kumar SA, Filipski SB, Iverson M, Stuhr KL, Keith JM, et al. Disruption of fatty acid amide hydrolase activity prevents the effects of chronic stress on anxiety and amygdalar microstructure. Mol Psychiatry. 2012 July 10.
Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61(1–2):3–18.
Hillard CJ, Hill MN, Carrier EJ, Shi L, Cullinan WE, Gorzalka BB. Regulation of cannabinoid receptor expression by chronic, unpredictable stress in rats and mice. Soc Neurosci Abstr. 2006;746:19.
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, et al. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435(7045):1108–12.
Hu W, Zhang M, Czeh B, Zhang W, Flugge G. Chronic restraint stress impairs endocannabinoid mediated suppression of GABAergic signaling in the hippocampus of adult male rats. Brain Res Bull. 2011;85(6):374–9.
Hungund BL, Vinod KY, Kassir SA, Basavarajappa BS, Yalamanchili R, Cooper TB, et al. Upregulation of CB1 receptors and agonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Mol Psychiatry. 2004;9(2):184–90.
Irving AJ, Coutts AA, Harvey J, Rae MG, Mackie K, Bewick GS, et al. Functional expression of cell surface cannabinoid CB(1) receptors on presynaptic inhibitory terminals in cultured rat hippocampal neurons. Neuroscience. 2000;98(2):253–62.
Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102(52):19204–7.
Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107(32):14449–54.
Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, et al. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19(11):4544–58.
Katona I and Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529-58.
Kuzmiski JB, Pittman QJ, Bains JS. Metaplasticity of hypothalamic synapses following in vivo challenge. Neuron. 2009;62(6):839–49.
Lee TT, Hill MN. Age of stress exposure modulates the immediate and sustained effects of repeated stress on corticolimbic cannabinoid CB(1) receptor binding in male rats. Neuroscience. 2012 Nov 27.
Liu J, Wang L, Harvey-White J, Osei-Hyiaman D, Razdan R, Gong Q, et al. A biosynthetic pathway for anandamide. Proc Natl Acad Sci U S A. 2006;103(36):13345–50.
Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, et al. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26(24):6643–50.
Manzanares J, Corchero J, Fuentes JA. Opioid and cannabinoid receptor-mediated regulation of the increase in adrenocorticotropin hormone and corticosterone plasma concentrations induced by central administration of delta(9)-tetrahydrocannabinol in rats. Brain Res. 1999;839(1):173–9.
Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13(8):951–7.
Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11(12):4213–25.
McLaughlin RJ, Hill MN, Dang SS, Wainwright SR, Galea LA, Hillard CJ, et al. Upregulation of CB(1) receptor binding in the ventromedial prefrontal cortex promotes proactive stress-coping strategies following chronic stress exposure. Behav Brain Res. 2013;237:333–7.
McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, et al. Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol. 2012;22(9):664–71.
Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50(1):83–90.
Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–5.
Newsom RJ, Osterlund C, Masini CV, Day HE, Spencer RL, Campeau S. Cannabinoid receptor type 1 antagonism significantly modulates basal and loud noise induced neural and hypothalamic-pituitary-adrenal axis responses in male Sprague-Dawley rats. Neuroscience. 2012;204:64–73.
Nunez E, Benito C, Pazos MR, Barbachano A, Fajardo O, Gonzalez S, et al. Cannabinoid CB2 receptors are expressed by perivascular microglial cells in the human brain: an immunohistochemical study. Synapse. 2004;53(4):208–13.
Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29(3):729–38.
Okamoto Y, Wang J, Morishita J, Ueda N. Biosynthetic pathways of the endocannabinoid anandamide. Chem Biodivers. 2007;4(8):1842–57.
Olijslagers JE, de Kloet ER, Elgersma Y, van Woerden GM, Joels M, Karst H. Rapid changes in hippocampal CA1 pyramidal cell function via pre- as well as postsynaptic membrane mineralocorticoid receptors. Eur J Neurosci. 2008;27(10):2542–50.
Onaivi ES. Commentary: functional neuronal CB2 cannabinoid receptors in the CNS. Curr Neuropharmacol. 2011;9(1):205–8.
Palazuelos J, Aguado T, Egia A, Mechoulam R, Guzman M, Galve-Roperh I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006;20(13):2405–7.
Parolaro D. Presence and functional regulation of cannabinoid receptors in immune cells. Life Sci. 1999;65(6–7):637–44.
Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145(12):5431–8.
Patel S, Cravatt BF, Hillard CJ. Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology. 2005a;30(3):497–507.
Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005b;21(4):1057–69.
Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34(13):2699–709.
Prewitt CM, Herman JP. Anatomical interactions between the central amygdaloid nucleus and the hypothalamic paraventricular nucleus of the rat: a dual tract-tracing analysis. J Chem Neuroanat. 1998;15(3):173–85.
Rademacher DJ, Meier SE, Shi L, Ho WS, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum, and medial prefrontal cortex in mice. Neuropharmacology. 2008;54(1):108–16.
Radley JJ, Arias CM, Sawchenko PE. Regional differentiation of the medial prefrontal cortex in regulating adaptive responses to acute emotional stress. J Neurosci. 2006a;26(50):12967–76.
Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, et al. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb Cortex. 2006b;16(3):313–20.
Radley JJ, Gosselink KL, Sawchenko PE. A discrete GABAergic relay mediates medial prefrontal cortical inhibition of the neuroendocrine stress response. J Neurosci. 2009;29(22):7330–40.
Reich CG, Taylor ME, McCarthy MM. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav Brain Res. 2009;203(2):264–9.
Rossi S, De Chiara V, Musella A, Kusayanagi H, Mataluni G, Bernardi G, et al. Chronic psychoemotional stress impairs cannabinoid-receptor-mediated control of GABA transmission in the striatum. J Neurosci. 2008;28(29):7284–92.
Sapolsky RM, Krey LC, McEwen BS. Glucocorticoid-sensitive hippocampal neurons are involved in terminating the adrenocortical stress response. Proc Natl Acad Sci U S A. 1984;81(19):6174–7.
Schlicker E, Kathmann M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci. 2001;22(11):565–72.
Simon GM, Cravatt BF. Endocannabinoid biosynthesis proceeding through glycerophospho-N-acyl ethanolamine and a role for alpha/beta-hydrolase 4 in this pathway. J Biol Chem. 2006;281(36):26465–72.
Skaper SD, Di Marzo V. Endocannabinoids in nervous system health and disease: the big picture in a nutshell. Philos Trans R Soc Lond B Biol Sci. 2012;367(1607):3193–200.
Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432.
Steiner MA, Marsicano G, Wotjak CT, Lutz B. Conditional cannabinoid receptor type 1 mutants reveal neuron subpopulation-specific effects on behavioral and neuroendocrine stress responses. Psychoneuroendocrinology. 2008;33(8):1165–70.
Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, et al. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215(1):89–97.
Sugiura T, Kobayashi Y, Oka S, Waku K. Biosynthesis and degradation of anandamide and 2-arachidonoylglycerol and their possible physiological significance. Prostaglandins Leukot Essent Fatty Acids. 2002;66(2–3):173–92.
Sugiura T, Kishimoto S, Oka S, Gokoh M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res. 2006;45(5):405–46.
Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: a potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36(13):2750–61.
Tasker JG. Rapid glucocorticoid actions in the hypothalamus as a mechanism of homeostatic integration. Obesity (Silver Spring). 2006;14(Suppl 5):259S–65S.
Ueda N. Endocannabinoid hydrolases. Prostaglandins Other Lipid Mediat. 2002;68–69:521–34.
Uriguen L, Perez-Rial S, Ledent C, Palomo T, Manzanares J. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology. 2004;46(7):966–73.
Vahl TP, Ulrich-Lai YM, Ostrander MM, Dolgas CM, Elfers EE, Seeley RJ, et al. Comparative analysis of ACTH and corticosterone sampling methods in rats. Am J Physiol Endocrinol Metab. 2005;289(5):E823–8.
Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310(5746):329–32.
Vinod KY, Arango V, Xie S, Kassir SA, Mann JJ, Cooper TB, et al. Elevated levels of endocannabinoids and CB1 receptor-mediated G-protein signaling in the prefrontal cortex of alcoholic suicide victims. Biol Psychiatry. 2005;57(5):480–6.
Vinod KY, Kassir SA, Hungund BL, Cooper TB, Mann JJ, Arango V. Selective alterations of the CB1 receptors and the fatty acid amide hydrolase in the ventral striatum of alcoholics and suicides. J Psychiatr Res. 2010;44(9):591–7.
Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30(33):11188–96.
Wang J, Shen RY, Haj-Dahmane S. Endocannabinoids mediate the glucocorticoid-induced inhibition of excitatory synaptic transmission to dorsal raphe serotonin neurons. J Physiol. 2012a;590(Pt 22):5795–808.
Wang M, Hill MN, Zhang L, Gorzalka BB, Hillard CJ, Alger BE. Acute restraint stress enhances hippocampal endocannabinoid function via glucocorticoid receptor activation. J Psychopharmacol. 2012b;26(1):56–70.
Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, et al. Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology. 2010;35(11):2249–61.
Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410(6828):588–92.
Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, et al. Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci. 2011;14(9):1160–6.
Zoppi S, Perez Nievas BG, Madrigal JL, Manzanares J, Leza JC, Garcia-Bueno B. Regulatory role of cannabinoid receptor 1 in stress-induced excitotoxicity and neuroinflammation. Neuropsychopharmacology. 2011;36(4):805–18.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Gray, J., Vecchiarelli, H., Hill, M. (2014). Endocannabinoid Signaling and Synaptic Plasticity During Stress. In: Popoli, M., Diamond, D., Sanacora, G. (eds) Synaptic Stress and Pathogenesis of Neuropsychiatric Disorders. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1056-4_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1056-4_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1055-7
Online ISBN: 978-1-4939-1056-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)